

Abstract
The nucleus accumbens is a key component of the reward pathway that plays a role in addiction to many drugs of abuse, including psychostimulants and opioids. The effects of withdrawal from chronic morphine were examined in the nucleus accumbens using brain slices from morphine-treated animals. Recordings were made from interneurons in the shell of nucleus accumbens, and the presynaptic inhibition of GABA-A IPSCs by opioids was examined. In slices from control animals, opioids caused a maximal inhibition of 50%, forskolin increased the IPSC amplitude by less than twofold, and the maximal inhibition by opioids in the presence of forskolin was not changed. During withdrawal, however, forskolin caused approximately a fourfold increase in the amplitude of the IPSC, and the maximal inhibition by opioids was increased to 80%. The results indicate that transmitter release is increased during opioid withdrawal, particularly after the activation of adenylyl cyclase. The cAMP-dependent increase in transmitter release is potently inhibited by opioids, such that the overall effect of opioids is augmented during withdrawal. The induction of an opioid-sensitive cAMP-dependent mechanism that regulates transmitter release may be a critical component of acute opioid withdrawal.
Keywords: μ-opioid, adenosine, cAMP, adenylyl cyclase, electrophysiology, A-kinase
The nucleus accumbens plays an important role in the process of opioid addiction and withdrawal, as indicated by behavioral, biochemical, and molecular studies (Wise, 1987; Koob and Bloom, 1988; Maldonado et al., 1992; Harris and Aston-Jones, 1994; Self and Nestler, 1995). Opioids inhibit the activity of medium spiny output neurons in the nucleus accumbens by presynaptic inhibition of EPSPs (Martin et al., 1997). A similar presynaptic inhibition of EPSPs was observed in striatum, as well as a direct hyperpolarization of a subpopulation of neurons (Jiang and North, 1992). Thus, opioids have presynaptic and postsynaptic actions in the nucleus accumbens mediated by both μ- and δ-subtype receptors. Little is known, however, about the effects of chronic morphine treatment on the excitability or synaptic regulation of neurons within the nucleus accumbens.
Chronic morphine treatment results in an upregulation of the cAMP-dependent cascade in opioid-sensitive cells that is most evident during acute withdrawal (Sharma et al., 1975; Terwilliger et al., 1991;Avidor-Reiss et al., 1996, 1997). One physiological consequence of this upregulation is an increased release of transmitter (Bonci and Williams, 1997). The nucleus accumbens is an area enriched in type V adenylyl cyclase (Glatt and Snyder, 1993; Mons and Cooper, 1995). This isoform belongs to a group of adenylyl cyclases (isoforms I, V, VI, and VIII) that are acutely inhibited by opioid receptor activation and upregulated with chronic morphine treatment (Avidor-Reiss et al., 1996,1997). The inhibition of adenylyl cyclase by opioids has been known since the early 1970s (Sharma et al., 1975); however, there are few reports of the physiological consequences of this action (Ingram and Williams, 1994). For example, inhibition of synaptic transmission by opioids appears to be cAMP-independent (Schoffelmeer et al., 1986), although activation of adenylyl cyclase increases transmitter release at many synapses (Cameron and Williams, 1993; Chavez-Noriega and Stevens, 1994; Salin et al., 1996; Bonci and Williams, 1997; Chen and Regehr, 1997; Kondo and Marty, 1997).
This study examines the action of opioids on GABA-A IPSCs on interneurons in the nucleus accumbens during withdrawal. Interneurons of the nucleus accumbens play a key role in the integration of afferent input and regulation of the output of the nucleus (Heimer et al., 1995). Anatomical data indicate that interneurons not only innervate but receive collateral innervation from the GABAergic medium spiny projection neurons (Wilson, 1990; Meredith et al., 1993). Thus, this GABA input onto interneurons regulates intrinsic excitability within the nucleus accumbens and may indicate how GABA release is affected in projection areas. The purpose of this study was as follows: (1) to determine whether the cAMP-dependent increase in GABA IPSCs during withdrawal (Bonci and Williams, 1997) is common to opioid-sensitive synapses, and (2) to examine the mechanism of presynaptic inhibition by opioids during withdrawal in the absence and presence of agents that stimulate adenylyl cyclase.
MATERIALS AND METHODS
Unless otherwise stated, the following protocol was used to treat animals chronically with morphine. Male Wistar rats (150–250 gm) were anesthetized with halothane, and morphine pellets (75 mg each) were implanted subcutaneously, one on day 1 and two on days 3 and 5. Experiments were done 7–10 d after the start of treatment (2–5 d after the last set of pellets). This treatment protocol has been shown to produce strong opioid dependence (Chieng and Christie, 1995). On the day of the experiment, rats were anesthetized with halothane and killed. Horizontal brain slices containing the nucleus accumbens (250 μm) were cut with a vibratome at 4°C and maintained in physiological saline. Slices were then transferred to a bath and superfused with physiological saline (morphine-free) at 34°C (1.5 ml/min); the tissue content of morphine after 1–2 hr in morphine-free solutions is negligible (Chieng and Christie, 1995). The physiological saline contained (in mm): 126 NaCl, 2.5 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 11 glucose, and 24 NaHCO3; it was gassed with 95% O2-5% CO2. Drugs were applied to the slice by superfusion.
Whole-cell recordings were made using a patch-clamp amplifier (Axopatch 1D). Cells were visualized with a 40× water immersion lens using Normaski optics and infrared illumination. All neurons were sampled close to the medial boundary of the nucleus accumbens just lateral to the “islands of Calleja” (“major island”) and were considered to be primarily in the shell region of the nucleus accumbens (Paxinos and Watson, 1986). Patch pipettes of 1.5–2.5 MΩ resistance were filled with intracellular solution containing (in mm): 120 KCl, 0.3 CaCl2, 1 MgCl2, 1 EGTA, 2 MgATP, 0.25 GTP, and buffered with 10 HEPES, pH 7.3. Acceptable access resistance was <10 MΩ and was periodically monitored with repetitive 10 mV steps (20 msec duration), and membrane potential was voltage clamped at −70 or −80 mV. Series resistance compensation of 80% was used throughout experiments. Synaptic currents were evoked with bipolar tungsten stimulating electrodes placed near (100–200 μm) the cell body. Such stimulation most likely activated local fibers rather than afferent pathways. Electrical stimulation (1 msec duration) was applied at 20 sec intervals. The intensity of stimulation was adjusted to obtain initial IPSCs having an amplitude of 200–400 pA, such that both inhibition and augmentation could be observed with the superfusion of drugs. The stimulus intensity was not changed once the amplitude of the IPSC was adjusted to obtain a steady value. Iontophoreses of GABA was done with an Axoclamp 2 using glass microelectrodes (30 MΩ; 0.1 or 1 m GABA in water) placed near (15–30 μm) the cell. The iontophoretic current used was adjusted (30–100 nA, with pulse durations of 50–200 msec) to result in GABA currents that were reproducible with repeated applications over the course of a 30 min experiment. The amplitude (800–1200 pA) and duration (0.5–2 sec) of the resulting GABA currents were somewhat larger and longer than IPSCs but were a reasonable approximation. GABA currents were more consistent when no backing current was applied. Current balancing was not used. Data were acquired using pClamp6 and analyzed with Axograph 3.0 (Axon Instruments). Summarized results were presented as mean ± SEM, with statistical significance atp < 0.05.
Morphine base pellets and morphine sulfate were obtained from National Institute on Drug Abuse (Bethesda, MD). [D-Ala2,MePhe4,Gly5-ol]enkephalin (DAMGO), 1,9-dideoxyforskolin, forskolin, GABA, kynurenic acid, [Met5]enkephalin (ME), picrotoxin, isoproterenol, and staurosporin were obtained from Sigma (St Louis, MO). 8-Cyclopentyl-1,3-propylxanthine (DPCPX), RO201724, SKF82958, SCH23390, Sp-cAMPS, and naloxone were obtained from Research Biochemicals (Natick, MA).
RESULTS
Whole-cell recordings were made from visually identified interneurons, and GABA-A IPSCs were electrically evoked. All experiments were done in the presence of kynurenic acid (1 mM) to block ionotropic glutamate receptors. GABA-A-mediated IPSCs were completely blocked by picrotoxin (100 μm). In addition to the distinctive size and shape (Phelps and Vaughn, 1986; Meredith et al., 1989) (Fig. 1), interneurons were identified physiologically. All cells were spontaneously active (2.8 ± 0.5 Hz; n = 19), had a membrane potential of −54 ± 1 mV (n = 10), and had anIh current at negative potentials. These observations were consistent with reports describing the membrane properties of interneurons (Wilson, 1990; Wilson et al., 1990;Kawaguchi et al., 1995). Unless otherwise stated, chronic morphine treatment was performed using subcutaneous implantation of time-released morphine pellets. All experiments were done in the absence of morphine such that slices from morphine-treated animals were termed opioid-withdrawn.
Fig. 1.

Interneurons were identified visually. A typical brain slice showing images in two different focal planes. Many medium spiny cells are visible (asterisks), and one single interneuron (arrow) is shown. Scale bar, 10 μm.
Superfusion with a high concentration of ME (10 μm) did not affect the holding current but decreased the GABA-A IPSC by ∼50% (Fig. 2). When a paired pulse protocol was used (two stimuli separated by 100 msec), the second IPSC was smaller than the first, indicating a paired pulse depression. As shown in Figure 2, the paired pulse ratio was shifted toward facilitation in the presence of ME, suggesting a presynaptic mechanism for the depression of the IPSC (Mennerick and Zorumski, 1995). In addition, the GABA current induced by iontophoretically applied GABA was not affected by ME. Therefore, the release of GABA at the synapse between medium spiny cells and interneurons in the nucleus accumbens is depressed by opioids.
Fig. 2.

Opioids inhibit GABA-A IPSCs by a presynaptic mechanism. Top left, IPSC, Representative current records of IPSCs evoked using a paired pulse protocol. The depression of IPSC in the presence of ME is accompanied by a shift in the paired pulse ratio to facilitation. Top right,GABA iontophoresis, Current traces resulting from iontophoretic application of GABA. The GABA currents were not affected by ME but were blocked by picrotoxin (data not shown).Middle, IPSC, Summary of results demonstrating the inhibition of the IPSC by ME (10 μm). The inhibition was the same in control and withdrawn slices. In this and other plots, the amplitude of the IPSC is normalized for each cell using the mean amplitude recorded during the first 10 min and is plotted as a function of time. Bottom, GABA iontophoreses, Summary of results indicating that ME had no effect on exogenously applied GABA.
Increased IPSCs with adenylyl cyclase stimulation
The amplitude of the IPSC measured in interneurons in the nucleus accumbens was increased by activation of adenylyl cyclase using forskolin (10 μm), the D1 agonist SKF82958 (1 μm), and the β-adrenoceptor agonist isoproterenol (1 μm) (Fig. 3). The augmentation caused by SKF82958 was completely blocked by the D1 receptor antagonist SCH23390 (1 μm) (Fig. 3). In addition, the inactive analog of forskolin, 1,9-dideoxyforskolin (10 μm), had no effect (1 ± 20%; n = 3) (Fig. 3b). The increase in the ISPC caused by forskolin, SKF82958, and isoproterenol were all significantly greater in withdrawn slices than in controls. The postsynaptic sensitivity of GABA in the absence and presence of forskolin was tested with iontophoretic application of GABA (Fig. 3c). The current induced by iontophoretic application of GABA was not affected by forskolin (control, 6 ± 4%; n = 3; withdrawn, 6 ± 10%; n = 7), indicating that the postsynaptic sensitivity to GABA was not affected by forskolin. Superfusion with picrotoxin (100 μm; n = 5) completely blocked the GABA-induced current. Two conclusions can be drawn from these results. First, presynaptic facilitation of transmitter release mediated by the activation of adenylyl cyclase is augmented during acute opioid withdrawal. Second, the cAMP-dependent increase in release is functionally coupled to receptor activation. This mechanism appears to be common to many opioid-sensitive synapses. Forskolin was used for further characterization of this augmentation of transmitter release, because it caused the most robust facilitation.
Fig. 3.
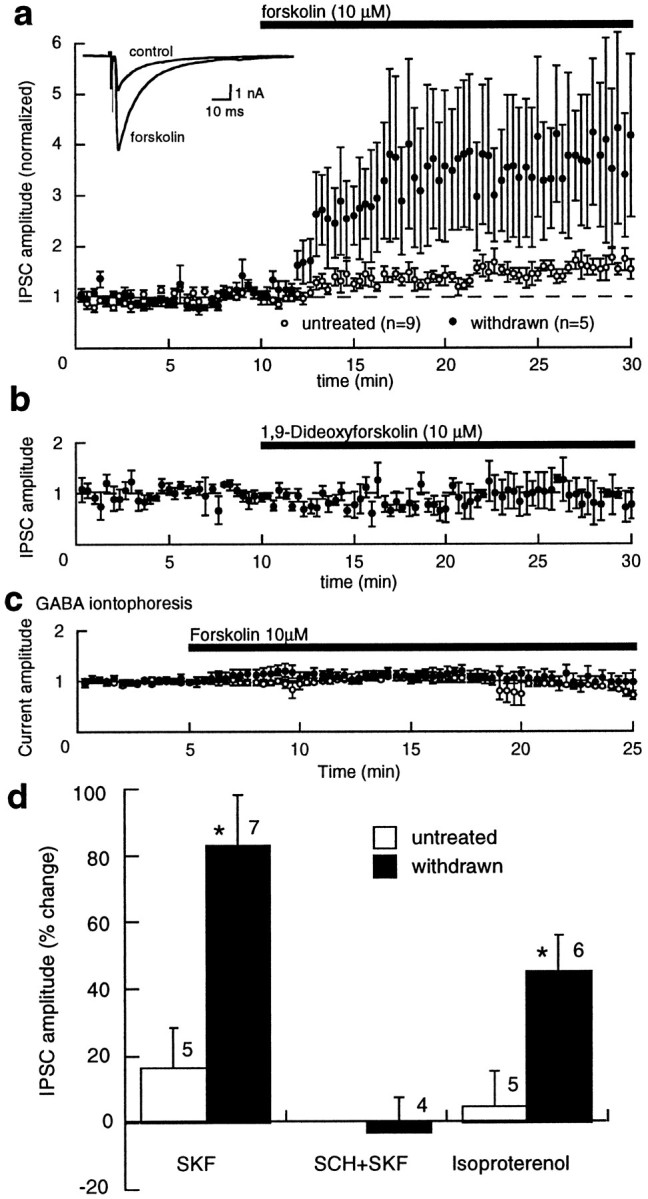
Upregulated adenylyl cyclase activity in morphine-withdrawn slices. a, Summary of results showing that forskolin (10 μm) caused a greater increase in the amplitude of the IPSC in morphine-withdrawn slices than in untreated controls. Inset, Two superimposed IPSCs from a withdrawn slice indicating that forskolin increased the amplitude without affecting the time course of the current. b, Dideoxyforskolin had no effect on the IPSC amplitude in withdrawn slices. c, Iontophoretic application of GABA induced currents that were not changed during superfusion with forskolin (10 μm) from control or withdrawn slices. d, IPSCs were increased by the D1 agonist SKF82958 (SKF) (1 μm) and the β-adrenoceptor agonist isoproterenol (1 μm). The increase caused by each was greater in withdrawn slices than in control. The D1 antagonist SCH23390 (SCH) (1 μm) completely blocked the augmentation caused by SKF82958.
Increased opioid inhibition
To determine how the upregulation of cAMP-dependent processes during withdrawal affected the sensitivity to opioids, the inhibition by opioids was examined in the absence and presence of forskolin. For these experiments, the selective adenosine A1 receptor antagonist DPCPX was included in the superfusion solution to eliminate the effects of increased extracellular adenosine after the activation of adenylyl cyclase (Bonci and Williams, 1996; Brundege et al., 1997). In the absence of forskolin, the maximum inhibition induced by ME (10 μm) was 48 ± 7% (n = 6) in controls and 52 ± 5% (n = 8) in withdrawn slices (Fig. 2). In the presence of forskolin, the inhibition caused by ME was unchanged in control slices (45 ± 9%; n = 4) (Fig. 4). In withdrawn slices, however, the ME-induced inhibition was increased to 82 ± 7% (n = 4; unpaired t = 3.17;p < 0.02) (Fig. 4). ME had no effect on the holding current, nor did it affect the current induced by iontophoretic application of GABA (control, 8 ± 8%; n = 3; withdrawn, 8 ± 5%; n = 5) (Fig. 2). Thus, rather than finding tolerance to opioids, treatment of withdrawn slices with forskolin revealed an unexpected increase in the presynaptic inhibition caused by ME.
Fig. 4.

The opioid-induced inhibition of IPSCs is larger in morphine-withdrawn slices. a, i, Superimposed traces of the IPSC in control and in the presence of forskolin and forskolin plus DPCPX. a,ii, Three superimposed traces of the same cell in forskolin and DPCPX before and after the addition of ME (10 μm). b, Summarized results indicating the augmented effect of forskolin and DPCPX and the increased inhibition caused by ME in morphine-withdrawn slices.
The augmented opioid inhibition was further characterized using the metabolically stable μ-selective agonist DAMGO. In the absence of forskolin, a maximal concentration of DAMGO (10 μm) caused an inhibition of 46 ± 4% (n = 4) in control, the same as was found in withdrawn slices (46 ± 5%;n = 6). In the absence of forskolin, the concentration response curves to DAMGO were also the same in control and withdrawn slices (Fig. 5a). In the presence of forskolin, however, the maximum inhibition induced by DAMGO was significantly greater in morphine-withdrawn slices (control, 51 ± 7%; n = 5; morphine-withdrawn, 73 ± 6%; n = 7; unpaired t = 2.24;p < 0.05) (Fig. 5b). The EC50to DAMGO was unchanged (control, 60 ± 17 nm; withdrawn, 65 ± 20 nm), and the inhibition was completely antagonized by naloxone (1 μm;n = 9). Thus, during withdrawal, the efficacy, but not the potency, of DAMGO was increased.
Fig. 5.

The maximum opioid inhibition is greater in withdrawn slices after stimulation of adenylyl cyclase.a, Concentration response curve to DAMGO measuring the amplitude of the IPSC in the absence of forskolin. DAMGO caused an inhibition that was the same in control untreated and morphine-withdrawn slices. b, In the presence of forskolin and DPCPX, DAMGO caused a larger inhibition of the IPSC in morphine-withdrawn slices than in controls. The sensitivity to DAMGO was not changed, whereas the maximum inhibition was increased. The stimulus intensity was adjusted such that initial amplitude of the IPSC was between 400 and 600 pA in each group.
Kinase dependence
The increased sensitivity to opioids was dependent on kinase activity, because when slices were pretreated with the nonselective kinase inhibitor staurosporin (3 μm), forskolin neither increased the IPSCs (control, −24 ± 4%; n = 3; withdrawn, 9 ± 25%; n = 5) nor changed the maximal inhibition of the IPSC by ME (10 μm). In fact, the inhibition caused by ME (10 μm) in staurosporin and forskolin (control, 45 ± 6%; n = 4; morphine-withdrawn, 44 ± 9%; n = 6) was the same as that caused by ME alone (control, 48 ± 7%; n= 4; withdrawn, 52 ± 5%; n = 8). Thus, ME inhibited the IPSC by a kinase-independent mechanism in both control and withdrawn slices. With the activation of adenylyl cyclase, however, opioids caused an additional inhibition that was dependent on protein kinase activity. The necessity to stimulate adenylyl cyclase suggests that basal activity is relatively low in the slice preparation under the conditions of this experiment.
Staurosporin alone had no effect on IPSCs in control slices (1 ± 11%; n = 3), whereas it caused an inhibition of 36 ± 8% (n = 5; unpaired t = 2.62; p < 0.05) in withdrawn slices. The inhibition in withdrawn slices is comparable to that found in dopamine cells of the ventral tegmental area (VTA) (45%) in withdrawn slices (Bonci and Williams, 1997). This observation suggests that there was a small tonic kinase-dependent effect on transmitter release during withdrawal.
cAMP, phosphodiesterase, and adenosine
If the increase in IPSC amplitude caused by forskolin was dependent on an upregulation of protein kinase A (PKA), then application of cAMP analogs should be more effective in withdrawn slices than in controls. The cAMP analog Sp-cAMP-S, which activates PKA directly produced inconsistent increases in the amplitude of the IPSC in both control (13 ± 11%; n = 4) and withdrawn slices (25 ± 10%; n = 4;p > 0.05) that were not significantly different. This result suggests that an increase in cAMP production rather than augmented kinase activity may be the primary mechanism for the increased transmitter release in withdrawn slices. This suggestion was further examined with the use of a phosphodiesterase inhibitor to reduce the metabolism of cAMP.
The phosphodiesterase inhibitor RO201724 (200 μm) had little effect on the IPSC amplitude in control slices (3 ± 6%;n = 5) but significantly increased the IPSC in withdrawn slices (31 ± 6%; n = 9), suggesting that adenylyl cyclase activity was elevated in withdrawn slices. The action of RO201724 could result from two mechanisms: an increased activation of PKA through increasing cAMP levels or a decrease in adenosine tone by inhibition of cAMP metabolism to adenosine. The results suggest the latter, because the increase in IPSC amplitude caused by RO201724 was the same as that observed by blocking A1 adenosine receptors with DPCPX (Fig. 6). In addition, the augmentation of the IPSC by DPCPX was occluded after treatment of withdrawn slices with RO201724 (DPCPX alone, −39 ± 12%; n = 9; DPCPX after RO201724, −4 ± 5%;n = 3) (Fig. 6). All effects appear to be presynaptic, because the current induced by iontophoretically applied GABA was not affected by DPCPX (control, 5 ± 13%; n = 4; withdrawn, 2 ± 11%; n = 6).
Fig. 6.

Adenosine tone is increased as a result of an increase in cAMP during opioid withdrawal. a, Blockade of adenosine A1 receptors with DPCPX (200 nm) caused an increase in IPSC amplitude in withdrawn slices relative to the untreated controls. b, IPSCs from a single cell.Insets, IPSCs taken during the period indicated bynumbers. The amplitude of the IPSC was increased by RO201724. Addition of DPCPX caused no further effect on the IPSC, whereas forskolin caused a potent increase in the IPSC.
Time course of withdrawal
The time course of withdrawal from chronic morphine treatment has phases that are both acute (hours) and long-term (days to weeks) (Nestler and Aghajanian, 1997). The duration of the upregulated adenylyl cyclase was examined in two additional groups of morphine-treated animals. Both groups were given single daily injections of morphine (20 mg/kg, i.p., for 7 d). In one group of animals, experiments were done 1–2 d after the last morphine injection. In this group, similar to animals treated with morphine pellets, forskolin caused a larger increase in IPSCs (182 ± 7%;n = 4) compared with controls (45 ± 9%;n = 4; unpaired t = 3.17;p < 0.02) (Fig. 7). In the second group of morphine-injected animals, experiments were performed 1 week after the last injection. The effect of forskolin in this long-term (1 week) withdrawal group was the same as that in untreated control animals (63 ± 35%; n = 4;p > 0.05). Thus, the adaptation resulting from morphine treatment is closely associated with the acute phase (hours to days) of opioid withdrawal.
Fig. 7.

A summary of experiments indicating that the increased effect of forskolin was greater in withdrawn slices from animals treated with morphine pellets (withdrawn for 1–4 hr) and single daily injections (withdrawn for 1–2 d), whereas 7 d after the last morphine injection, the effect of forskolin returned to control. The IPSC amplitude was plotted as percentage of change from baseline, such that 0 indicates no change.
DISCUSSION
Together, the results suggest that the upregulation of adenylyl cyclase during withdrawal is a key component in the modulation of GABA release from these opioid-sensitive synapses. The primary observation made in this study is that withdrawal from morphine reveals an new opioid effector, inhibition of cAMP-dependent transmitter release, that is not present in slices from control animals. This new effector links opioid receptors, adenylyl cyclase, and PKA with an increased inhibition of GABA release in the shell region of the nucleus accumbens. The time course of the increased adenylyl cyclase activity suggests that this mechanism plays a role in aspects of morphine withdrawal that last for hours to days but that dissipate within 1 week of the termination of drug treatment.
Opioids and adenylyl cyclase
Experiments investigating opioid inhibition of cAMP accumulation often activate adenylyl cyclase with forskolin or PGE2 rather than measuring basal activity, presumably to increase the sensitivity of the assay (Sharma et al., 1975; Law et al., 1982; Puttfarken et al., 1988;Terwilliger et al., 1991; Ammer and Schulz, 1996; Avidor-Reiss et al., 1996, 1997). Similarly, preactivation of adenylyl cyclase with forskolin or PGE2 was required to observe an opioid modulation of the cAMP-dependent current Ih in primary afferent neurons (Ingram and Williams, 1994). These results suggest that opioid actions mediated by inhibition of adenylyl cyclase depend on the initial activity of adenylyl cyclase. The upregulation of adenylyl cyclase in response to chronic morphine treatment increases both the forskolin-mediated augmentation of GABA release and opioid inhibition of that increased release. During acute opioid withdrawal, there is an increase in noradrenaline release in terminal fields of the locus ceruleus that results, at least in part, from an increase in firing (Akaoka and Aston-Jones, 1991; Done et al., 1992). The activation of β-adrenoceptors resulting from this increased release of noradrenaline could be one stimulus for cAMP-dependent effectors at other opioid-sensitive sites in widespread areas of the CNS, although the role of the locus ceruleus in the overt signs and symptoms of opioid withdrawal is controversial (Christie et al., 1997).
cAMP and adenosine
The forskolin-induced rise of cAMP had two effects on the GABA-A IPSC: a kinase-dependent increase in amplitude and a simultaneous decrease in amplitude resulting from an increase in adenosine tone. Inhibition of kinase activity with staurosporin decreased the IPSC in withdrawn slices, indicating a kinase-dependent component of GABA release. On the other hand, blocking A1 adenosine receptors with DPCPX or decreasing adenosine tone by inhibition of phosphodiesterase activity with RO201724 increased the IPSC, suggesting a tonic adenosine-dependent inhibition of GABA release. The relative role that each mechanism plays can vary considerably from site to site. For example, GABA-A and GABA-B IPSPs are differentially affected in the ventral tegmental area during withdrawal (Bonci and Williams, 1996,1997). GABA-A IPSCs were augmented by a cAMP-dependent mechanism and were not sensitive to adenosine antagonists during acute withdrawal (Bonci and Williams, 1997). In contrast, the amplitude of GABA-B IPSPs was increased by DPCPX in slices taken from drug-treated animals (Bonci and Williams, 1996). In fact, forskolin and D1 receptor agonists depressed the GABA-B IPSP in slices from drug-treated animals before treatment with DPCPX and increased the IPSP after blockade of A1 receptors. The balance between augmented release by a kinase-dependent mechanism and depression by adenosine therefore appears to be dependent on the receptors present at individual terminals.
Presynaptic or postsynaptic modulation
The results were interpreted to indicate that withdrawal from chronic morphine changed the presynaptic regulation of GABA release within the nucleus accumbens. The primary evidence supporting this interpretation was that the currents induced by iontophoretic application of GABA were not affected by forskolin in cells from either control or morphine-withdrawn slices. A further suggestion of a fundamental change in presynaptic regulation was a significant shift in the paired pulse ratio from depression in control slices and toward facilitation in withdrawn slices (data not shown). An increase in GABA release during acute withdrawal from morphine has also been found in the VTA (Bonci and Williams, 1997) and periaqeductal gray area (S. L. Ingram and M. J. Christie, personal communication). There is evidence, however, that the postsynaptic sensitivity to GABA can be affected through a cAMP-dependent pathway in acutely isolated striatal cholinergic interneurons (Yan and Surmeier, 1997). Activation of D5 dopamine receptors increased GABA currents that were selectively sensitive to Zn2+. Although this mechanism was not detected in the present study, a postsynaptic increase in GABA inhibition during opioid withdrawal would have the same qualitative effect as an augmented presynaptic release of GABA.
cAMP and adenosine in acute opioid withdrawal
A role of both cAMP and adenosine in acute opioid withdrawal has been suggested at the behavioral level. A series of symptoms were induced in animals treated with theophylline, an inhibitor of phosphodiesterase (Collier et al., 1974) and an adenosine receptor antagonist. These behaviors were termed the quasimorphine abstinence syndrome, because they appeared to mimic opioid withdrawal. More recently, selective adenosine antagonists were found to exacerbate some signs and symptoms of morphine withdrawal, whereas agonists alleviated others (Dionyssopoulos et al., 1992; Kaplan and Sears, 1996; Salem and Hope, 1997). The present results provide a cellular mechanism that may explain these results. The quasimorphine abstinence syndrome induced by theophylline probably resulted from the blockade of adenosine receptors rather than inhibition of phosphodiesterase activity. The increased adenylyl cyclase activity observed during acute withdrawal not only increases the sensitivity to opioids but indirectly increases endogenous adenosine that acts to counteract some signs of withdrawal.
Footnotes
This work was supported by National Institutes of Health Grant DA08163 and a C. J. Martin Fellowship. We thank Drs. M. J. Christie, C. D. Fiorillo, S. L. Ingram, and O. Manzoni for their helpful comments.
Correspondence should be addressed to Dr. John T. Williams, The Vollum Institute, Oregon Health Services University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97201.
Dr. Chieng’s present address: Division of Neuroscience, John Curtin School of Medical Research, Mills Road, Acton ACT 2601, Australia.
REFERENCES
- 1.Akaoka H, Aston-Jones G. Opiate withdrawal-induced hyperactivity of locus ceruleus neurons is substantially mediated by augmented excitatory amino acid input. J Neurosci. 1991;11:3830–3839. doi: 10.1523/JNEUROSCI.11-12-03830.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ammer H, Schulz R. Morphine dependence in human neuroblastoma SH-SY5Y cells is associated with adaptive changes in both the quantity and functional interaction of PGE1 receptors and stimulatory G proteins. Brain Res. 1996;707:235–244. doi: 10.1016/0006-8993(95)01265-6. [DOI] [PubMed] [Google Scholar]
- 3.Avidor-Reiss T, Nevo I, Pfeuffer T, Vogel Z. Chronic opioid treatment induces adenylyl cyclase V superactivation: involvement of Gβγ. J Biol Chem. 1996;271:21309–21315. doi: 10.1074/jbc.271.35.21309. [DOI] [PubMed] [Google Scholar]
- 4.Avidor-Reiss T, Nevo I, Saya D, Bayewitch M, Vogel Z. Opiate-induced adenylyl cyclase superactivation is isozyme-specific. J Biol Chem. 1997;272:5040–5047. doi: 10.1074/jbc.272.8.5040. [DOI] [PubMed] [Google Scholar]
- 5.Bonci A, Williams JT. A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron. 1996;16:631–639. doi: 10.1016/s0896-6273(00)80082-3. [DOI] [PubMed] [Google Scholar]
- 6.Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brundege JM, Diao L, Proctor WR, Dunwiddie TV. The role of cyclic AMP as a precursor of extracellular adenosine in the rat hippocampus. Neuropharmacology. 1997;36:1201–1210. doi: 10.1016/s0028-3908(97)00102-0. [DOI] [PubMed] [Google Scholar]
- 8.Cameron DL, Williams JT. Dopamine D1 receptors facilitate transmitter release. Nature. 1993;366:344–347. doi: 10.1038/366344a0. [DOI] [PubMed] [Google Scholar]
- 9.Chavez-Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci. 1994;14:310–317. doi: 10.1523/JNEUROSCI.14-01-00310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen C, Regehr WG. The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci. 1997;17:8687–8694. doi: 10.1523/JNEUROSCI.17-22-08687.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chieng B, Christie MJ. Lesions to terminals of noradrenergic locus coeruleus neurones do not inhibit opiate withdrawal behaviour in rats. Neurosci Lett. 1995;186:37–40. doi: 10.1016/0304-3940(95)11276-3. [DOI] [PubMed] [Google Scholar]
- 12.Christie MJ, Williams JT, Osborne PB, Bellchambers CE. Where is the locus in opioid withdrawal? Trends Pharmacol Sci. 1997;18:134–140. doi: 10.1016/s0165-6147(97)01045-6. [DOI] [PubMed] [Google Scholar]
- 13.Collier HOJ, Francis DL, Henderson G, Schneider C. Quasi morphine-abstinence syndrome. Nature. 1974;249:471–473. doi: 10.1038/249471a0. [DOI] [PubMed] [Google Scholar]
- 14.Dionyssopoulos T, Hope W, Coupar IM. Effect of adenosine analogues on the expression of opiate withdrawal in rats. Pharmacol Biochem Behav. 1992;42:201–206. doi: 10.1016/0091-3057(92)90516-i. [DOI] [PubMed] [Google Scholar]
- 15.Done C, Silverstone P, Sharp T. Effect of naloxone-precipitated morphine withdrawal on noradrenaline release in rat hippocampus in vivo. Eur J Pharmacol. 1992;215:333–336. doi: 10.1016/0014-2999(92)90052-6. [DOI] [PubMed] [Google Scholar]
- 16.Glatt CE, Snyder SH. Cloning and expression of an adenylyl cyclase localized to the corpus striatum. Nature. 1993;361:536–538. doi: 10.1038/361536a0. [DOI] [PubMed] [Google Scholar]
- 17.Harris GC, Aston-Jones G. Involvement of D2 dopamine receptors in the nucleus accumbens in the opiate withdrawal syndrome. Nature. 1994;371:155–157. doi: 10.1038/371155a0. [DOI] [PubMed] [Google Scholar]
- 18.Heimer L, Zahm DS, Alheid GF. Basal ganglia. In: Paxinos G, editor. The rat nervous system. Academic; San Diego: 1995. pp. 579–628. [Google Scholar]
- 19.Ingram SL, Williams JT. Opioid inhibition of Ih via adenylyl cyclase. Neuron. 1994;13:179–186. doi: 10.1016/0896-6273(94)90468-5. [DOI] [PubMed] [Google Scholar]
- 20.Jiang ZG, North RA. Pre- and postsynaptic inhibition by opioids in rat striatum. J Neurosci. 1992;12:356–361. doi: 10.1523/JNEUROSCI.12-01-00356.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaplan GB, Sears MT. Adenosine receptor agonists attenuate and adenosine receptor antagonists exacerbate opiate withdrawal signs. Psychopharmacology. 1996;123:64–70. doi: 10.1007/BF02246282. [DOI] [PubMed] [Google Scholar]
- 22.Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- 23.Kondo S, Marty A. Protein kinase A-mediated enhancement of miniature IPSC frequency by noradrenaline in rat cerebellar stellate cells. J Physiol (Lond) 1997;498:165–176. doi: 10.1113/jphysiol.1997.sp021849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science. 1988;242:715–723. doi: 10.1126/science.2903550. [DOI] [PubMed] [Google Scholar]
- 25.Law PY, Hom DS, Loh HH. Opiate regulation of adenosine 3′:5′-cyclic monophosphate level in neuroblastoma x glioma NG108–15 hybrid cells. Relationship between receptor occupance and effect. Mol Pharmacol. 1982;23:26–35. [PubMed] [Google Scholar]
- 26.Maldonado R, Stinus L, Gold LH, Koob GF. Role of different brain structures in the expression of the physical morphine withdrawal syndrome. J Pharmacol Exp Ther. 1992;261:669–677. [PubMed] [Google Scholar]
- 27.Martin G, Nie S, Siggins GR. μ-opioid receptors modulate NMDA receptor-mediated responses in nucleus accumbens neurons. J Neurosci. 1997;17:11–22. doi: 10.1523/JNEUROSCI.17-01-00011.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mennerick S, Zorumski CF. Paired-pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. J Physiol (Lond) 1995;488:85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meredith GE, Blank B, Groenewegen HJ. The distribution and compartmental organization of the cholinergic neurons in nucleus accumbens of the rat. Neuroscience. 1989;31:327–345. doi: 10.1016/0306-4522(89)90377-1. [DOI] [PubMed] [Google Scholar]
- 30.Meredith GE, Pennartz CMA, Groenewegen HJ. The cellular framework for chemical signalling in the nucleus accumbens. Prog Brain Res. 1993;99:3–24. doi: 10.1016/s0079-6123(08)61335-7. [DOI] [PubMed] [Google Scholar]
- 31.Mons N, Cooper DMF. Adenylate cyclases: critical foci in neuronal signaling. Trends Neurosci. 1995;18:536–542. doi: 10.1016/0166-2236(95)98375-9. [DOI] [PubMed] [Google Scholar]
- 32.Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 33.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; Sydney: 1986. [DOI] [PubMed] [Google Scholar]
- 34.Phelps PE, Vaughn JE. Immunocytochemical localization of choline acetyltransferase in rat ventral striatum: a light and electron microscopic study. J Neurocytol. 1986;15:595–617. doi: 10.1007/BF01611860. [DOI] [PubMed] [Google Scholar]
- 35.Puttfarcken PS, Werling LL, Cox BM. Effects of chronic morphine exposure on opioid inhibition of adenylyl cyclase in 7315c cell membranes: a useful model for the study of tolerance at μ opioid receptors. Mol Pharmacol. 1988;33:520–527. [PubMed] [Google Scholar]
- 36.Salem A, Hope W. Effect of adenosine receptor agonists and antagonist on the expression of opiate withdrawal in rat. Pharmacol Biochem Behav. 1997;57:671–679. doi: 10.1016/s0091-3057(96)00393-0. [DOI] [PubMed] [Google Scholar]
- 37.Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- 38.Self DW, Nestler EJ. Molecular mechanisms of drug reinforcement and addiction. Annu Rev Neurosci. 1995;18:463–495. doi: 10.1146/annurev.ne.18.030195.002335. [DOI] [PubMed] [Google Scholar]
- 39.Sharma SK, Klee WA, Nirenberg M. Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc Natl Acad Sci USA. 1975;72:3092–3096. doi: 10.1073/pnas.72.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schoffelmeer AN, Wierenga EA, Mulder AH. Role of adenylate cyclase in presynaptic a2-adrenoreceptor- and μ-opioid receptor-mediated inhibition of [3H]-noradrenaline release from rat brain cortex slices. J Neurochem. 1986;46:1711–1717. doi: 10.1111/j.1471-4159.1986.tb08488.x. [DOI] [PubMed] [Google Scholar]
- 41.Terwilliger RZ, Beitner-Johnson D, Sevarino KA, Crain SM, Nestler EJ. A general role for adaptations in G-proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function. Brain Res. 1991;548:100–110. doi: 10.1016/0006-8993(91)91111-d. [DOI] [PubMed] [Google Scholar]
- 42.Wilson CJ. Basal ganglia. In: Shepherd GM, editor. The synaptic organization of the brain. Oxford UP; Oxford: 1990. pp. 279–316. [Google Scholar]
- 43.Wilson CJ, Chang HT, Kitai ST. Firing patterns and synaptic potentials of identified giant aspiny interneurons in the rat neostriatum. J Neurosci. 1990;10:508–519. doi: 10.1523/JNEUROSCI.10-02-00508.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wise RA. The role of reward pathways in the development of drug dependence. Pharmacol Ther. 1987;35:227–263. doi: 10.1016/0163-7258(87)90108-2. [DOI] [PubMed] [Google Scholar]
- 45.Yan Z, Surmeier DJ. D5 dopamine receptors enhance Zn2+-sensitive GABAA currents in striatal cholinergic interneurons through a PKA/PP1 cascade. Neuron. 1997;19:1115–1126. doi: 10.1016/s0896-6273(00)80402-x. [DOI] [PubMed] [Google Scholar]