

Abstract
Evidence has accumulated to suggest that the NMDA glutamate receptor subtype plays an important role in neuronal degeneration evoked by hypoxia, ischemia, or trauma. Cerebellar granule cells in culture are vulnerable to NMDA-induced neuronal excitotoxicity. In these cells, brain-derived neurotrophic factor (BDNF) and basic fibroblast growth factor (FGF2) prevent the excitotoxic effect of NMDA. However, little is known about the molecular mechanisms underlying the protective properties of these trophic factors. Using cultured rat cerebellar granule cells, we investigated whether BDNF and FGF2 prevent NMDA toxicity by downregulating NMDA receptor function. Western blot and RNase protection analyses were used to determine the expression of the various NMDA receptor subunits (NR1, NR2A, NR2B, and NR2C) after BDNF or FGF2 treatment. FGF2 and BDNF elicited a time-dependent decrease in the expression of NR2A and NR2C subunits. Because NMDA receptor activation leads to increased intracellular Ca2+ concentration ([Ca2+]i), we studied the effect of the BDNF- and FGF2-induced reduction in NR2A and NR2C synthesis on the NMDA-evoked Ca2+ responses by single-cell fura-2 fluorescence ratio imaging. BDNF and FGF2 reduced the NMDA-mediated [Ca2+]i increase with a time dependency that correlates with their ability to decrease NR2A and NR2C subunit expression, suggesting that these trophic factors also induce a functional downregulation of the NMDA receptor. Because sustained [Ca2+]i is believed to be causally related to neuronal injury, we suggest that BDNF and FGF2 may protect cerebellar granule cells against excitotoxicity by altering the NMDA receptor–Ca2+ signaling via a downregulation of NMDA receptor subunit expression.
Keywords: BDNF, FGF2, glutamate, NMDA receptor subunits, NR1, NR2A, Ca2+
Neurotrophic factors are polypeptides that induce differentiation and maturation of neuronal cells (Mocchetti and Wrathall, 1995). The spectrum of action of neurotrophic factors has expanded in recent years to include protection against glutamate-mediated excitotoxicity (Mattson and Scheff, 1994), because glutamate is thought to play an important role in neuronal cell death in a variety of acute and chronic neurodegenerative diseases (Wielock, 1985; Rothman and Olney, 1986; Choi, 1988; Coyle and Puttfarcken, 1993). Among various neurotrophic factors, brain-derived neurotrophic factor (BDNF) and basic fibroblast growth factor (FGF2) have been shown to prevent glutamate-mediated neuronal cell death in several neuronal populations both in vitro (Mattson et al., 1989; Fernandez-Sanchez and Novelli, 1993; Lindholm et al., 1993;Zirrgiebel et al., 1995; Kume et al., 1997; Marini et al., 1997a) andin vivo (Freese et al., 1992; Frim et al., 1993; Peterson et al., 1996). However, the molecular mechanisms by which BDNF and FGF2 prevent glutamate excitotoxicity are still poorly understood.
Cerebellar granule neurons in culture express a number of glutamate receptor subtypes (Burgoyne and Cambray-Deakin, 1988; Cox et al., 1990;Van der Valk et al., 1991). Consequently, these cells have been used to study possible mechanisms of excitotoxicity evoked by excessive activation of glutamate receptors and, in particular, the NMDA receptor subtype (Favaron et al., 1988; Novelli et al., 1988; Marini and Paul, 1992; Resink et al., 1994). Exposure of cerebellar granule cells to FGF2 and BDNF for at least 6 hr increases neuronal survival after glutamate- or NMDA-mediated neurotoxicity (Fernandez-Sanchez and Novelli, 1993; Lindholm et al., 1993; Marini et al., 1997a), indicating that these cells are also a good in vitro model for studying the mechanisms of neuroprotection against excessive stimulation of NMDA receptors.
The NMDA receptor (NR) is an ion channel formed by distinct subunits. The obligatory subunit termed NR1 contains the binding site for ligands (Moriyoshi et al., 1991). The NR2 regulatory subunit consists of four homologous isoforms (NR2A–D) (Kutsuwada et al., 1992; Monyer et al., 1992). When coexpressed with NR1, each of the NR2 subunits can form an ion channel (Meguro et al., 1992; Ishii et al., 1993) characterized by high Ca2+ conductance (MacDermott et al., 1986). After overactivation of this channel, a sustained rise in cytosolic Ca2+ concentration ([Ca2+]i) occurs that is believed to trigger neurotoxicity (Garthwaite et al., 1986; Choi, 1987;Rothman et al., 1987; Hahn et al., 1988; Anegawa et al., 1995). Thus, preventing elevated [Ca2+]i after NMDA receptor activation may limit the neuropathological processes associated with the excitotoxic effects of glutamate.
FGF2 has been shown to decrease the glutamate-mediated surge in [Ca2+]i in hippocampal neurons (Mattson et al., 1989), suggesting that the neuroprotective properties of this and potentially other neurotrophic factors may rely on their ability to reduce [Ca2+]i. However, the mechanisms underlying the effect of FGF2 on [Ca2+]i remain primarily unknown. Using cerebellar granule cells, we tested the hypothesis that FGF2 and BDNF exert their neuroprotective effect by decreasing NMDA receptor subunit expression and Ca2+ signaling. We report that BDNF and FGF2 selectively decrease the expression of NR2A and NR2C subunits and reduce receptor sensitivity to NMDA activation.
MATERIALS AND METHODS
Cerebellar granule cell preparation. Granule cells were prepared from postnatal day 8 Sprague Dawley rat pups (Taconic, Germantown, NY) as described previously (Longone et al., 1993; Marini et al., 1997b). Neurons were plated onto poly-l-lysine– (1%) precoated 100 mm plastic dishes at a density of 2.5 × 106 cells/ml and were grown in Basal Medium Eagle (Life Technologies, Gaithersburg, MD) containing glutamine (2 mm), fetal calf serum (10%), KCl (25 mm), gentamycin (100 μg/ml), and penicillin/streptomycin (10,000 U/ml). Cells were maintained at 37°C in 5% CO2/95% air. Cytosine arabinoside (10 μm) was added 24 hr after cell plating to inhibit glial proliferation. Neurons were exposed to BDNF (50 ng/ml), FGF2 (50 ng/ml), or vehicle (0.19% BSA in PBS) in serum-free growth medium starting on day 8 in vitro (8 DIV) for all experiments.
Probe preparation. Plasmids pFPR2AR, pFPR2BR, and pFPR2R contain cDNA fragments for the rat NR2A, NR2B, and NR2C subunits, respectively, inserted into the polylinker region of pAMP1 cloning vector (Follesa and Ticku, 1995). These plasmids were linearized withEcoRI, and SP6 RNA polymerase was used to generate [32P]CTP-labeled cRNAs. cRNAs used in the protection assay for detection of NR2A, NR2B, and NR2C mRNAs were 662, 530, and 404 bases long, respectively, including 92 bases of the polylinker region of the cloning vector. The cRNA for cyclophilin wasin vitro transcribed with SP6 polymerase fromEcoRI-linearized plasmid pIG15 (Follesa et al., 1994). This probe was used as a standard control to correct for potential artifacts caused by RNA extraction and gel loading (Follesa et al., 1994;Mocchetti et al., 1996).
RNase protection assay. The RNase protection assay was performed as described previously (Follesa et al., 1994; Mocchetti et al., 1996). In brief, total RNA extracted from cerebellar granule cells was dissolved in hybridization solution containing32P-labeled NR2A, NR2B, and NR2C cRNAs (150,000 cpm of each; specific activity, >1 × 108 cpm per μg of RNA) and pIGl5 cRNA, used at a lower specific activity (∼1 × 106 cpm per μg) to balance the relatively high abundance of the corresponding mRNA. Hybridization was performed at 50°C overnight. RNA was digested with RNase A (1 U/ml) and T1 (200 U/ml) for 30 min at 37°C. The reaction was stopped, RNA was precipitated with isopropanol [1:1 (v/v)], and the pellet, containing the RNA:RNA hybrids, was dissolved in loading buffer (80% formamide, 0.1% xylene cyanol, 0.1% bromophenol blue, and 2 mm EDTA), boiled at 95°C, and separated on a 5% polyacrylamide/urea sequencing gel. The gel was dried, and the protected fragments were visualized by autoradiography on x-ray film with the use of an intensifying screen (Hyperscreen; Amersham, Arlington Heights, IL). Autoradiographic bands were analyzed with a densitometer (Hoefer GS 300; Hoefer Scientific, San Francisco, CA). The content of mRNA was calculated in arbitrary units expressed as a ratio between the densitometric area of the NMDA receptor subunit band and that of the cyclophilin band as described previously (Follesa et al., 1994; Mocchetti et al., 1996).
Western blot. Cells were washed twice in PBS, harvested in 50 mm Tris HCl, pH 7.0, and pelleted. Pellets were homogenized in the same buffer with a polytron, centrifuged at 100,000 × g for 20 min, resuspended in 1 ml of Tris HCl, and stored at −20°C. After removal of cellular debris by centrifugation, protein levels in the lysates were measured by the Bradford Coomassie blue colorimetric assay (Bio-Rad, Hercules, CA). Equal amounts of proteins were loaded onto an 8.5% SDS-polyacrylamide gel. Proteins were transferred on a nitrocellulose membrane and blocked with Tris-buffered saline (TBS-T) (25 mm Tris and 1% Tween) containing 5% milk powder. Blots were incubated with rabbit affinity-purified polyclonal antibodies anti-NR2A (AB1555; Chemicon, Temecula, CA) or anti-NR1 (AB1516; Chemicon), both at a dilution of 1:2500 in TBS-T plus 5% milk. After several washes with TBS-T, blots were then incubated with the secondary antibody, peroxidase-conjugated anti-rabbit IgG (dilution 1:10,000; Boehringer Mannheim, Indianapolis, IN). Immunoreactivity was detected by the use of an enhanced chemiluminescence system (Amersham).
Fluorescence Ca2+ imaging. The [Ca2+]i was measured by single-cell fura-2 fluorescence ratio imaging as described previously (De Bernardi et al., 1996). For this purpose, cells were plated onto 25-mm-round, 1-mm-thick glass coverslips (Fisher Scientific, Houston, TX) precoated with poly-l-lysine (1%). At the end of the trophic factor treatment, cells were loaded with the cell-permeable fura-2 AM (2–5 μm; Molecular Probes, Eugene, OR) at 37°C and in an atmosphere of 5% CO2. Loading was performed in either serum-free growth medium or Locke’s solution (154 mm NaCl, 5.6 mm KCl, 3.6 mmNaHCO3, 2.3 mm CaCl2, 1.2 mm MgCl2, 5.6 mmglucose, and 15 mm HEPES, pH 7.4) for 30 and 45 min, respectively. Cells were then washed with Mg2+-free Locke’s solution and allowed to sit for 10 min to complete fura-2 de-esterification. Ca2+ imaging was performed at room temperature while neurons were bathed in Mg2+-free Locke’s solution. Neurons were imaged using an Attofluor RatioVision digital fluorescence microscopy system (Atto Instruments, Rockville, MD) equipped with a Zeiss Axiovert 135 microscope and a F-Fluar 40×, 1.3 numerical aperture oil-immersion objective, as described previously (De Bernardi et al., 1996). Briefly, fura-2 was excited at 334 and 380 nm with its emission monitored at 510–530 nm; the 334/380 nm excitation ratio increases as a function of the [Ca2+]i. Each day before the experiments, the instrument was calibrated (calibration was done in vitro with fura-2 pentapotassium salt in the presence of a high concentration of Ca2+ or EGTA), and the 334/380 nm excitation ratio was converted to [Ca2+]i nm values (Grynkiewicz et al., 1985). For each coverslip, 50–99 neurons were simultaneously imaged in a given microscopic field, and single-cell Ca2+ responses were collected. Flat [Ca2+]i traces from cells nonresponding to NMDA (1–5%) both in control and neurotrophic factor-treated cells were identified and not included in the calculation of the mean Ca2+ response.
RESULTS
To test our working hypothesis that BDNF and FGF2 protect cerebellar granule cells from glutamate toxicity by downregulating NMDA receptor expression and function, we exposed these neurons to either neurotrophic factor for different times. The levels of mRNA and/or protein for NR1, NR2A, NR2B, and NR2C subunits, as well as the NMDA-evoked Ca2+ responses, were analyzed. NR2D, the other known isoform of the NR2 subunit component, was not included in our study because this subunit appears not to be expressed in rat cerebellar granule cells (Ishii et al., 1993; Monyer et al., 1994).
Lack of effect of BDNF and FGF2 on NR1 subunit protein levels
Cerebellar granule cells were exposed to BDNF and FGF2 at the concentration of 50 ng/ml that has been shown previously to protect these cells against glutamate-induced cell death (Fernandez-Sanchez and Novelli, 1993; Lindholm et al., 1993; Marini et al., 1997a). NR1 subunit protein levels were determined by Western blot analysis using a specific antibody that recognizes only one NR1-immunoreactive band (Fig. 1A). Time course studies performed at 3, 6, 24, and 48 hr after the addition of BDNF, FGF2, or vehicle (0.19% BSA in PBS) to the serum-free culture medium revealed that neither neurotrophic factor changed NR1 subunit levels (Fig. 1B). To eliminate the possibility that the expression of NR1 subunit cannot be regulated under our culture conditions, cerebellar granule cells were also exposed to a subtoxic concentration of NMDA (100 μm) that has been shown to downregulate NR1 expression in these cells (Resink et al., 1995, 1996). NMDA elicited a time-dependent decrease in NR1 immunoreactivity starting at 6 hr and lasting up to 48 hr (Fig.1A,B). This effect was blocked by the NMDA-receptor antagonist MK-801 (1 μm) (data not shown), confirming findings by other investigators (Resink et al., 1995, 1996). Thus, it appears that the mechanism by which BDNF and FGF2 exert a neuroprotective effect in cerebellar granule cells does not involve an alteration in NR1 subunit protein levels.
Fig. 1.
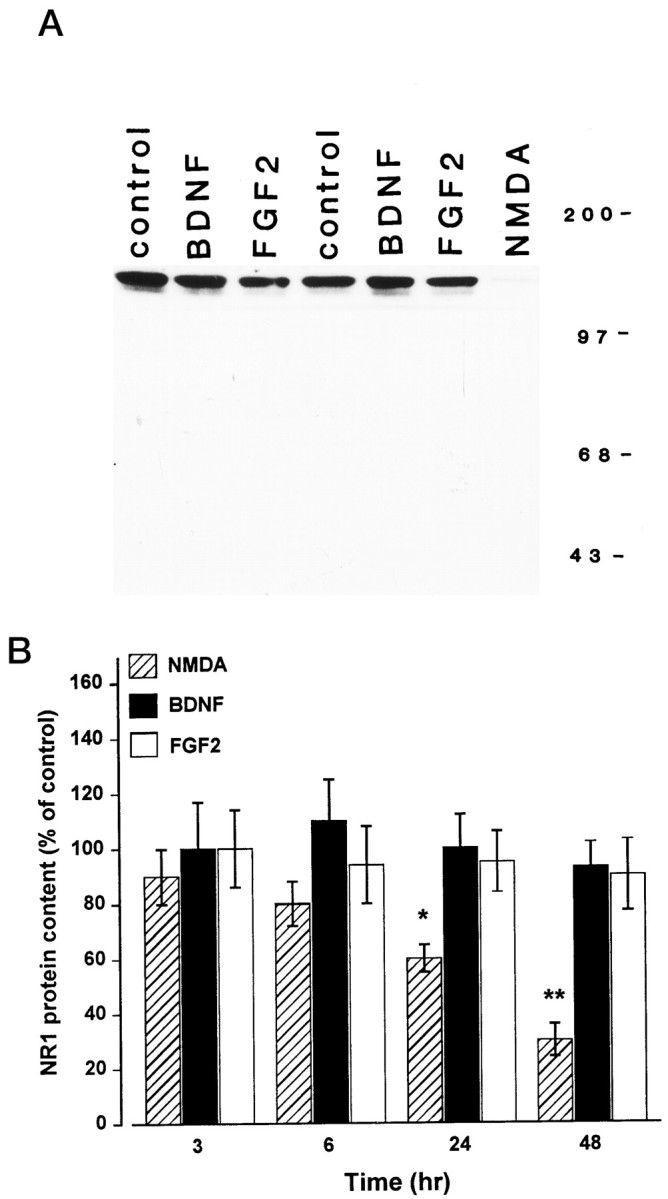
NMDA but not BDNF or FGF2 elicits a time-dependent decrease in NR1 subunit protein levels. Cerebellar granule cells (8 DIV) were exposed to NMDA (100 μm), BDNF (50 ng/ml), or FGF2 (50 ng/ml) for 3, 6, 24, and 48 hr. Cells were harvested, and the content of the NR1 subunit protein was determined by Western blot analysis using NR1 antibody. A, Representative blot showing NR1 immunoreactivity in cerebellar granule cells 24 hr after various treatments. Molecular weight markers are in kilodaltons.B, Time course of the effect of BDNF, FGF2, and NMDA on the NR1 subunit protein levels. Levels of NR1 were calculated by densitometric analysis of the NR1-immunoreactive band. Data, expressed as percent of control, are the mean ± SEM of three separate preparations of granule cells (n = 6). *p < 0.05; **p < 0.01 versus control (ANOVA and Dunnett’s test). Similar results were obtained using cells at 12 DIV.
BDNF and FGF2 decrease NR2A subunit protein levels
Western blot analyses were then used to determine whether BDNF and FGF2 alter the levels of NR2A, one of the four known isoforms of the NR2 subunit involved in the functional assembly of the NMDA receptor channel. Time course studies revealed that BDNF evoked a time-dependent decrease in NR2A protein levels beginning at 6 hr (30% decrease), peaking at 24 hr (50% decrease), and returning to control levels by 48 hr (Fig.2A,B). FGF2 also induced a decrease in NR2A levels, but its effect was transient (only at 6 hr) and overall weaker than that elicited by BDNF (Fig. 2B). Moreover, each trophic factor failed to change the total protein content, determined either by Bio-Rad assay on cell lysates or Coomassie staining on Western blots (data not shown). Because NMDA has been shown to alter the expression of the NR2A subunit (Resink et al., 1995, 1996), cerebellar granule cells were also incubated with NMDA for 24 hr. NMDA (100 μm) decreased NR2A protein levels to an extent greater than that elicited by BDNF (Fig. 2A).
Fig. 2.

BDNF and FGF2 decrease NR2A subunit protein levels. Cerebellar granule cells were exposed to BDNF, FGF2, or NT-3 (all at 50 ng/ml) for 1, 3, 6, 24, and 48 hr or to NMDA (100 μm) for 24 hr. Lysates were prepared, and Western blot analysis was performed using NR2A antibody. A, Representative blot showing NR2A immunoreactivity of cells 24 hr after various treatments. Molecular weight markers are in kilodaltons.B, Time course of the effect of BDNF, FGF2, and NT-3 on the NR2A protein levels. Protein content was calculated by densitometric analysis of the NR2A-immunoreactive band. Data, expressed as percent of control, are the mean ± SEM of three separate experiments (n = 6), using different preparations of granule cells. *p < 0.05; **p < 0.01 versus control (ANOVA and Dunnett’s test).
BDNF binds primarily to the receptor tyrosine kinase TrkB, the only known high-affinity neurotrophin receptor functionally expressed in rat cerebellar granule cells at 8 DIV (Marini et al., 1997a). To eliminate the possibility that the BDNF-mediated reduction of the NR2A subunit occurs via a nonspecific mechanism, we then exposed neurons to a member of the neurotrophin family known to activate TrkC receptors (NT-3). NT-3 (50 ng/ml) failed to change NR2A levels at any time tested (Fig.2B), indicating that the BDNF effect is specific.
BDNF and FGF2 reduce NR2A and NR2C subunit mRNA levels
To determine whether the decrease in NR2A protein content might result from a decrease in mRNA levels, steady-state NR2A mRNA levels were analyzed after treatment with either BDNF or FGF2. To test for specificity of the effect, we performed the RNase protection assay using simultaneously three distinct cRNAs encoding for NR2A, NR2B, and NR2C subunits. Analysis of RNA from cerebellar granule cells revealed that all three subunits are expressed in these neurons, with the NR2A mRNA being the predominant species (Fig.3), in accordance with earlier findings (Ishii et al., 1993; Monyer et al., 1994). BDNF (50 ng/ml) evoked a time-dependent reduction in NR2A mRNA content, beginning at 6 hr (50% reduction) and lasting at least up to 24 hr (Fig.4A). FGF2 (50 ng/ml) elicited a faster (within 3 hr) decrease in NR2A mRNA levels (Fig.4A), although this decrease was transient because NR2A mRNA returned to control levels by 6 hr (Fig.4A). These results, by showing that the decrease in NR2A mRNA content precedes the decrease in protein levels, suggest that FGF2 and BDNF affect NR2A subunit synthesis.
Fig. 3.

Reduction of NR2A and NR2C subunit mRNA levels by BDNF. RNase protection assay of 25 μg of total RNA extracted from cerebellar granule cells exposed to serum-free medium in the absence (CT) or presence of BDNF (50 ng/ml) for 6 hr.DP, Digested probe; P, aliquot of the hybridization solution containing the cRNA probes for NR2A (R2A), NR2B (R2B), NR2C (R2C), and cyclophilin (cyc). Thecyc probe was labeled at a lower specific activity.M, Molecular weight markers (MspI-digested pBR322). Protected fragments (indicated by arrows) were visualized by overnight exposure on x-ray film.
Fig. 4.
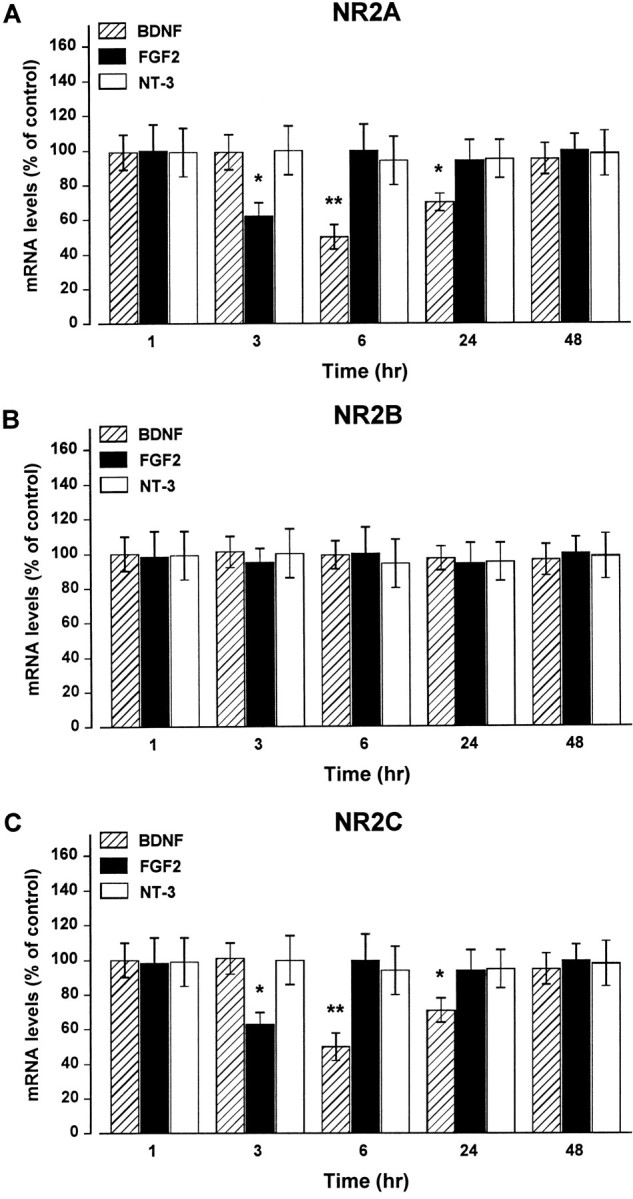
BDNF and FGF2 induce a time-dependent decrease in NR2A and NR2C subunit mRNA content. Cerebellar granule cells were exposed to BDNF, FGF2, or NT-3 for the indicated times. NR2A (A), NR2B (B), and NR2C (C) mRNA levels were determined by RNase protection assay and were calculated as described in Materials and Methods. Data, expressed as percent of control, are the mean ± SEM of three independent experiments (n = 6). *p < 0.05; **p < 0.01 versus control (ANOVA and Dunnett’s test).
Analysis of NR2B and NR2C mRNA revealed that exposure of cerebellar granule cells to either BDNF or FGF2 did not alter NR2B mRNA levels (Fig. 4B), whereas both trophic factors elicited a time-dependent decrease in NR2C mRNA levels similar to that observed for NR2A. As shown in Figure 4C, BDNF caused a 50% reduction in NR2C mRNA levels by 6 hr, and the effect lasted up to 24 hr. FGF2 elicited a 30% decrease in NR2C mRNA levels by 3 hr that was over by 6 hr. Again, NT-3 failed to change NR2A, NR2B, or NR2C mRNA levels at any time point tested (Fig. 4). The levels of cyclophilin mRNA did not change under our experimental conditions, suggesting that the BDNF- and FGF2-mediated decrease in NR2A and NR2C mRNA levels is not attributable to a generalized inhibition of mRNA synthesis.
BDNF and FGF2 reduce NR2A expression via a receptor-mediated mechanism
The biological activity of BDNF and FGF2 depends on the activation of tyrosine kinase receptors. To examine whether the reduction of NR2A synthesis after either BDNF or FGF2 is tyrosine kinase receptor-mediated, we tested whether the blockade of TrkB or FGF2 receptor signaling by the tyrosine kinase inhibitor K252a (Berg et al., 1992; Kaplan and Stevens, 1994) would inhibit BDNF and FGF2 effects. We have used this tyrosine kinase inhibitor because it has been shown not to affect cell viability (Marini et al., 1997a). Cerebellar granule cells were exposed to K252a (100 nm) 10 min before the addition of BDNF or FGF2 (each at 50 ng/ml). K252a prevented the BDNF-mediated decrease in NR2A mRNA subunit at 6 hr (Fig.5A) and the FGF2- and BDNF-mediated downregulation of NR2A protein levels observed at 6 and 24 hr, respectively (Fig. 5B). K252a alone did not alter NR2A mRNA or protein content (Fig.5A,B) or the NMDA-mediated decrease in NR2A mRNA (Fig. 5A). These results suggest that the activation of tyrosine kinase receptor signaling is involved in the reduced expression of NR2A and, most likely, NR2C subunits evoked by BDNF and FGF2.
Fig. 5.

K252a prevents the BDNF- and FGF2-mediated downregulation of NR2A expression. Cerebellar granule cells were preincubated with K252a (100 nm) 10 min before the addition of BDNF (50 ng/ml), FGF2 (50 ng/ml), or NMDA (100 μm). NR2A mRNA (A) and protein (B) levels were determined by RNase protection assay and Western blot analysis at 6 and 24 hr, respectively (NR2A protein levels in FGF2-treated cells were measured at 6 hr). Data, expressed as percent of control, are the mean ± SEM of three determinations from three separate cell preparations (n = 6). *p < 0.05 versus control; p < 0.05 versus BDNF or FGF2.
BDNF and FGF2 decrease the NMDA-induced Ca2+response in cerebellar granule neurons
The decrease in the expression of NR2A and NR2C subunits induced by BDNF and FGF2 may have a physiological relevance if it is followed by a reduction in receptor function. Because activation of NMDA receptor promotes influx of extracellular Ca2+through its own channel, the functional state of the NMDA receptor can be assessed by measuring its ability to evoke an [Ca2+]i increase after stimulation with a proper ligand. Therefore, we tested whether the decrease in the expression of NR2A and NR2C subunits induced by BDNF and FGF2 results in an alteration in NMDA receptor-mediated Ca2+responses. To this end, time course experiments, similar to those performed for the analysis of NR subunit proteins, were performed with either growth factor, and the NMDA-induced [Ca2+]i increase was measured.
Because nerve growth factor, a member of the neurotrophin family closely related to BDNF, has been shown to increase [Ca2+]i via either voltage-dependent Ca2+ channels (Levine et al., 1995) or mobilization of intracellular Ca2+ (De Bernardi et al., 1996), we first tested BDNF and FGF2 for any direct effect on the [Ca2+]i in cerebellar granule neurons. Neither growth factor (50–100 ng/ml and up to 30 min) was found to affect [Ca2+]i either in the presence or absence of Mg2+ (data not shown). Neurons were then exposed to BDNF (50 ng/ml for 3, 24, and 48 hr) or FGF2 (50 ng/ml for 6 and 24 hr), and the [Ca2+]iincrease evoked by NMDA (100 μm) was monitored in Mg2+-free conditions.
Resting [Ca2+]i showed no statistically significant differences between control and BDNF- or FGF2-treated cultures (in nm: control, 38.8 ± 6.6; BDNF, 44.0 ± 9.1; and FGF2, 34.2 ± 5.4). In neurons exposed to BDNF for 24 hr (treatment that maximally decreased NR2A protein levels, see Fig. 2), the NMDA-evoked [Ca2+]i increase was consistently reduced by 50% compared with that in the control (Fig.6A,B). However, in neurons exposed to BDNF for 3 or 48 hr (treatments that failed to change NR2A expression), the magnitude as well as the kinetics of the [Ca2+]i increase elicited by NMDA was found to be comparable with that in control cells (Fig. 6B).
Fig. 6.
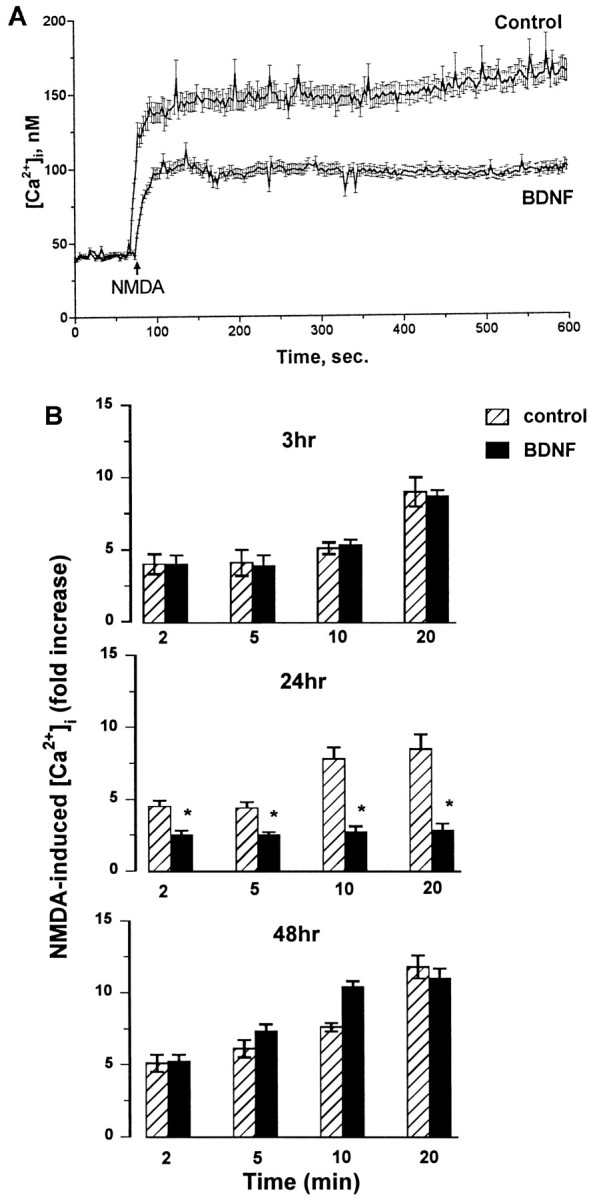
BDNF elicits a time-dependent inhibition of the NMDA-evoked [Ca2+]i increase in cerebellar granule cells. Neurons (8 DIV) were exposed to serum-free medium for 3, 24, or 48 hr in the absence (control, vehicle-treated) or presence of BDNF (50 ng/ml). Neurons were then loaded with fura-2, and Ca2+ imaging was performed in Mg2+-free Locke’s solution. Resting [Ca2+]i was recorded, and NMDA (100 μm) was applied. Single-cell [Ca2+]i was measured and analyzed as described in Materials and Methods. A, Representative of NMDA-induced Ca2+ response in neurons exposed to BDNF or vehicle (control) for 24 hr. B, Time course of the effect of BDNF on the NMDA-induced [Ca2+]i increase. The single-cell [Ca2+]i rise after NMDA was measured in neurons exposed to BDNF for 3, 24, or 48 hr. Data are expressed as an NMDA-induced fold [Ca2+]i increase plotted against time after NMDA addition. Results represent the mean ± SEM of four separate preparations of cerebellar granule cells (each preparation included at least three coverslips; 50–99 neurons being imaged per coverslip in a single microscopic field). *p < 0.01 (ANOVA and Dunnett’s test).
A 6 hr exposure of cerebellar granule cells to FGF2 (which also maximally reduced NR2A protein levels, see Fig. 2) resulted in a decreased NMDA-induced [Ca2+]i rise (Fig.7A,B). Overall, the extent of the effect exerted by FGF2 on the NMDA-elicited [Ca2+]i rise was weaker than that produced by BDNF, in agreement with the results showing that BDNF inhibits NR2A subunit expression to a greater extent than does FGF2. In contrast, a 24 hr exposure of the neurons to FGF2 failed to reduce the magnitude of the NMDA-evoked [Ca2+]iincrease (Fig. 7B), consistent with the lack of effect by FGF2 at this time point on the NR subunit expression (see Fig. 2). These data, by providing a temporal correlation between the reduction of NR2A expression by BDNF and FGF2 and the NMDA-elicited [Ca2+]i increase, suggest that the decreased responsiveness to NMDA in cells exposed to BDNF and FGF2 may be the result of NMDA receptor downregulation.
Fig. 7.
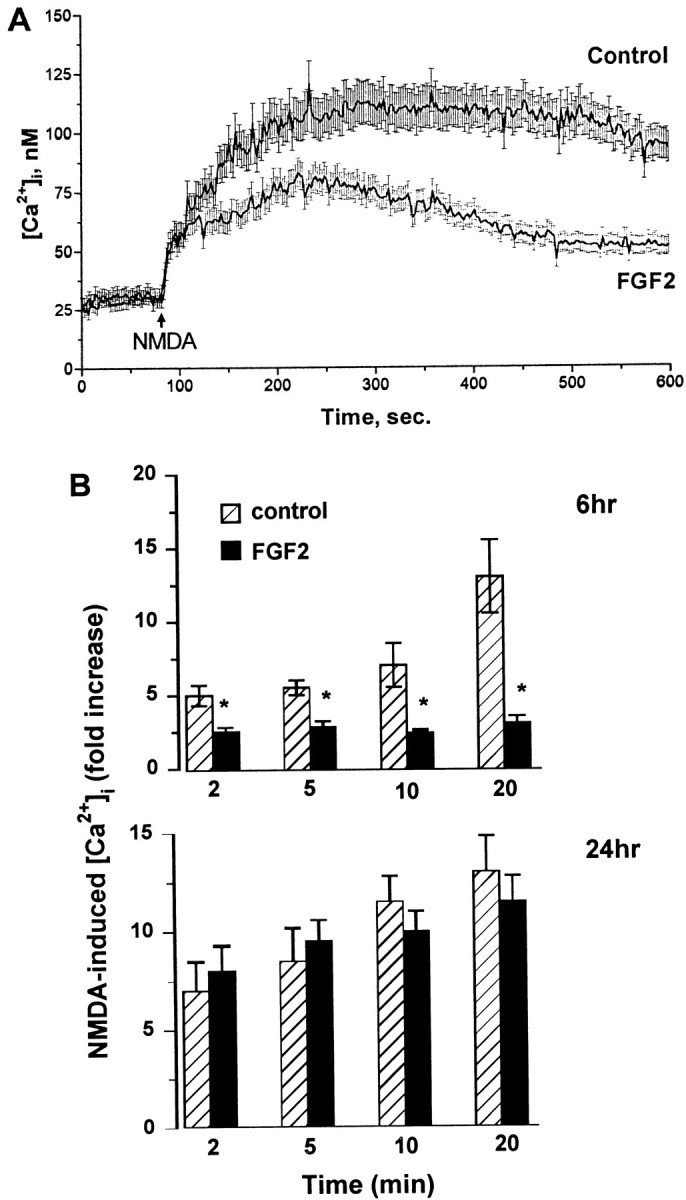
Inhibitory effect of FGF2 on the NMDA-evoked [Ca2+]i increase. Cerebellar granule cells were exposed to serum-free medium for 6 or 24 hr in the absence (control, vehicle-treated) or presence of FGF2 (50 ng/ml). [Ca2+]i imaging was performed as described in the legend of Figure 6. A, Representative of NMDA-induced Ca2+ response in neurons exposed to FGF2 or vehicle for 6 hr. B, Time course of the effect of FGF2 on the NMDA-induced [Ca2+]iincrease. The single-cell [Ca2+]iincrease after NMDA was measured in neurons exposed to FGF2 for 6 or 24 hr. Data are expressed as an NMDA-induced fold [Ca2+]i increase plotted against time after NMDA addition and represent the mean ± SEM from a population of 65 (control) and 70 (FGF2-treated) neurons per coverslip from four separate preparations of cerebellar granule cells (each preparation included at least three coverslips). *p< 0.05 (ANOVA and Dunnett’s test).
DISCUSSION
BDNF and FGF2 are two neurotrophic factors known to prevent and limit glutamate-induced neuropathological damages in selected neuronal populations including rat cerebellar granule cells (Fernandez-Sanchez and Novelli, 1993; Lindholm et al., 1993; Marini et al., 1997a). Although the neuroprotective properties of these trophic factors are rather well established, little is known about the molecular mechanism(s) of neuroprotection. To gain insight into the neuroprotective effects of BDNF and FGF2, we tested the hypothesis that these trophic factors prevent excitotoxicity by downregulating NMDA receptor subunit expression and function in cultures of cerebellar granule cells. Neither BDNF nor FGF2 affected the content of the obligatory NR1 subunit protein that contains the binding site for NMDA. This finding suggests that the total number of NMDA receptors capable of binding the proper ligand is not a likely target for the downregulation of the NMDA receptor that, according to our working hypothesis, could account for the neuroprotective activity of BDNF and FGF2. On the other hand, both trophic factors elicited a time-dependent decrease in mRNA and protein levels of the NR2A subunit. Importantly, the decrease in NR2A protein content is preceded by a quantitatively comparable decrease in NR2A mRNA levels, suggesting that BDNF and FGF2 very likely affect the synthesis of the NR2A subunit rather than the stability of the protein. In addition, both trophic factors decreased NR2C (but not NR2B) subunit mRNA with a time course similar to that observed for NR2A mRNA, suggesting that BDNF and FGF2 also reduce NR2C subunit synthesis. Because homomeric NR1 receptors are less responsive to NMDA than are heteromeric receptors composed of NR1 and NR2 subunits (for review, see Hollmann and Heinemann, 1994), it appears that, by reducing NR2A/NR2C expression, BDNF and FGF2 might change the NR1 to NR2A/NR2C subunit ratio and, thus, alter the functional state of the NMDA receptor. Taken together, these findings suggest that BDNF and FGF2 might modulate the expression of distinct subunits of the NMDA receptor and support the specificity of the effect of BDNF and FGF2 on the expression of the various subunits.
Additional findings support the specificity of the reduction in NR2A subunit synthesis by BDNF and FGF2. The effect of either trophic factor on the expression of the various NR subunits was compared with that of NMDA, which has been shown to modulate NR subunit levels in cerebellar granule cells (Resink et al., 1995, 1996), and NT-3, a member of the neurotrophin family that is devoid of neuroprotective activity in these neurons at 8 DIV (Marini et al., 1997a). Although NT-3 failed to change all the NR2 subunit subtypes tested, NMDA decreased NR1 and NR2A protein levels, and its effect was specifically blocked by MK-801. Thus, consistent with a recent report (Resink et al., 1996), NMDA was able to modulate the expression of NR1 and NR2A subunits, whereas BDNF and FGF2 were not. The biological effect of modifying NR subunit composition might be the decreased ability of glutamate to induce cell death via NMDA receptors. We therefore propose that the neuroprotective activity of BDNF or FGF2 against glutamate may rely on their ability to reduce the sensitivity of a selected neuronal population to glutamate by decreasing NR subunit synthesis and function.
A number of reports has shown that NMDA receptors play a key role in mediating glutamate excitotoxicity in cerebellar granule cells (Favaron et al., 1988; Novelli et al., 1988; Marini and Paul, 1992; Resink et al., 1994). It is known that the NMDA receptor is a Ca2+ channel that, after activation, increases [Ca2+]i. Abnormally sustained [Ca2+]i after excessive stimulation of NMDA receptors has been implicated in neuronal cell death (Garthwaite et al., 1986; Choi, 1987; Rothman et al., 1987; Hahn et al., 1988;Anegawa et al., 1995). Both NR2A and NR2C subunits are crucial in the formation of the NMDA channel and appear to be essential for the NMDA-mediated Ca2+ influx (Hollmann and Heinemann, 1994). Thus, a decreased synthesis of these subunits by BDNF and FGF2 might result in a decreased NMDA-evoked Ca2+ entry and, consequently, reduced [Ca2+]i. Our data indicate that FGF2 and BDNF elicit a time-dependent inhibition of the NMDA-mediated increase in [Ca2+]i, suggesting a decreased “sensitivity” of the NMDA receptor to NMDA activation. This effect shows a time dependency that correlates with the decreased synthesis of NR2A and, very likely, NR2C subunits, suggesting that NMDA subunit downregulation evoked by either trophic factor may account for the reduced Ca2+ response to NMDA. Although we cannot completely eliminate the involvement of other cellular mechanisms in the BDNF- and FGF2-mediated inhibition of [Ca2+]i by NMDA, such as an effect on a 71 kDa protein (Mattson et al., 1993) that is believed to be a necessary component of NMDA receptor signaling (Kumar et al., 1991), or other routes of Ca2+ entry, the decreased sensitivity of these neurons to NMDA may explain why BDNF and FGF2 prevent glutamate excitotoxicity.
A decreased Ca2+ response to NMDA after FGF2 treatment is not unique to cerebellar granule cells as has been reported in hippocampal neurons, which FGF2 protects against glutamate excitotoxicity (Mattson et al., 1989, 1995; Cheng et al., 1995). Thus, the cascade of events culminating in a decreased Ca2+ response to glutamate could represent a general mechanism by which certain neurotrophic factors can prevent glutamate-mediated neuronal cell death caused by excessive NMDA receptor stimulation. Although only speculative at the present time, the possibility exists that, by reducing the intracellular responses to glutamate, neurotrophic factors with neuroprotective properties, such as BDNF and FGF2, might prevent the formation of cytotoxic metabolites such as nitric oxide, peroxidases, and free radicals (Maiese et al., 1993; Mattson et al., 1995; Kume et al., 1997) that are thought to be responsible for the final toxic effect of glutamate.
The specific cellular and molecular mechanisms by which BDNF and FGF2 reduce NR2A and NR2C subunit expression are still under investigation. BDNF and FGF2 exert their biological activity by binding to specific receptors, FGFR1 and TrkB, respectively, that possess an intrinsic tyrosine kinase activity (for review, see Mocchetti and Wrathall, 1995). In addition, BDNF may also activate the nonselective neurotrophin receptor p75 (Chao and Hempstead, 1995), whose expression has been detected in cerebellar granule cells (Segal et al., 1995;Courtney et al., 1997). However, p75 does not activate a tyrosine kinase pathway (Chao and Hempstead, 1995). Thus, to distinguish which signaling transduction mechanism is involved in the action of BDNF and FGF2, we first examined the role of the tyrosine kinase pathway. K252a is a nonselective kinase inhibitor (Kase et al., 1987), which blocks TrkB tyrosine kinase signaling (Tapley et al., 1992). K252a per se failed to affect expression of NR subunit proteins or the NMDA-mediated downregulation of NR2A subunit mRNA. In contrast, K252a blocked the decrease in NR2A mRNA and protein levels induced by BDNF and FGF2, suggesting that the activation of the tyrosine kinase signal transduction pathway is required for the BDNF- and FGF2-mediated regulation of NR subunit expression to occur. This is in agreement with recent findings (Knusen et al., 1997; S. Rabin and I. Mocchetti, unpublished observations) indicating that a prolonged treatment of cerebellar granule cells with BDNF results in downregulation of TrkB receptors. Because activation of TrkB is essential for BDNF activity, the latter finding could explain the short-term effect of BDNF (within 24 hr) on the NR subunit levels and, perhaps, why BDNF neuroprotective properties are over within 24 hr (Courtney et al., 1997). Importantly and consistent with our results, a recent report has shown that the protection against glutamate-induced cell death exerted by BDNF in cerebellar granule cells is also blocked by K252a (Marini et al., 1997a). Because tyrosine kinase receptors activate phosphatidylinositol 3-kinase, shown previously to mediate survival of cerebellar granule cells exposed to insulin-like growth factor I (Miller et al., 1997), our data support the hypothesis that the neuroprotective effect of BDNF and FGF2 may involve the activation of a tyrosine kinase signaling pathway. It remains to be established which tyrosine kinase substrate(s), either common or specific for FGF2 and BDNF, is involved in and might account for the neuroprotective effect of these trophic factors. Future studies will also examine whether these trophic factors activate a tyrosine kinase-independent pathway known to prevent cell death in cerebellar granule cell neurons (Courtney et al., 1997).
Glutamate has been suggested to play a major role in acute neurodegenerative processes, such as those after hypoxic and ischemic neuronal injury. Thus, it is appealing to propose that BDNF and FGF2 may reduce glutamate excitotoxicity after ischemia or traumatic lesion by downregulating NMDA receptor function in vivo as well. It has been suggested that neurotrophic factors can be effective neuroprotective agents only when added to neurons in culture several hours before glutamate exposure (Prehn, 1996). By demonstrating that BDNF and FGF2 downregulate NMDA receptor with a time lag of at least 6 hr, our data may explain why neurotrophic factors prevent NMDA or glutamate toxicity when delivered several hours before the excitotoxic insult. If this is the case, the use of neurotrophic factors to prevent acute neuropathological conditions in humans may indeed be difficult. On the other hand, FGF2 has been shown to limit secondary injury processes that occur at a later time after spinal cord trauma (Teng et al., 1998) and may continue to exacerbate neuronal loss triggered by the initial traumatic (or ischemic) insult. Therefore, neurotrophic factors might be potential therapeutic tools in chronic neurodegenerative diseases.
Footnotes
This work was supported by a Research Career Development Award NS 01675 to I.M., by National Institutes of Health Grants NS 32671 and HL 28940, and by the American Heart Association Nation’s Affiliate.
Correspondence should be addressed to Dr. Italo Mocchetti, Department of Cell Biology, Medical/Dental Building, Georgetown University, 3900 Reservoir Road Northwest, Washington, DC 20007.
REFERENCES
- 1.Anegawa NJ, Lynch DR, Verdoorn TA, Pritchett DB. Transfection of N-methyl-d-aspartate receptors in a nonneuronal cell line leads to cell death. J Neurochem. 1995;64:2004–2012. doi: 10.1046/j.1471-4159.1995.64052004.x. [DOI] [PubMed] [Google Scholar]
- 2.Berg MM, Sterneberg DW, Parada LF, Chao MV. K-252a inhibits nerve growth factor-induced trk proto-oncogene tyrosine phosphorylation and kinase activity. J Biol Chem. 1992;267:13–16. [PubMed] [Google Scholar]
- 3.Burgoyne RD, Cambray-Deakin MA. The cellular neurobiology of neuronal development: the cerebellar granule cell. Brain Res Rev. 1988;13:77–101. doi: 10.1016/0165-0173(88)90006-9. [DOI] [PubMed] [Google Scholar]
- 4.Chao M, Hempstead B. p75 and Trk: a two-receptor system. Trends Neurosci. 1995;18:321–326. [PubMed] [Google Scholar]
- 5.Cheng B, Furukawa K, O’Keefe JA, Goodman Y, Kihiko M, Fabian T, Mattson MP. Basic fibroblast growth factor selectively increases AMPA-receptor subunit GluR1 protein level and differentially modulates Ca2+ responses to AMPA and NMDA in hippocampal neurons. J Neurochem. 1995;65:2525–2536. doi: 10.1046/j.1471-4159.1995.65062525.x. [DOI] [PubMed] [Google Scholar]
- 6.Choi DW. Ionic dependence of glutamate neurotoxicity in cortical cell cultures. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 8.Courtney MJ, Akeman KEO, Coffey ET. Neurotrophins protect cultured cerebellar granule neurons against the early phase of cell death by a two-component mechanism. J Neurosci. 1997;17:4201–4211. doi: 10.1523/JNEUROSCI.17-11-04201.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cox JA, Felder CC, Hennebury RC. Differential expression of excitatory amino acid receptor subtypes in cultured cerebellar neurons. Neuron. 1990;4:941–947. doi: 10.1016/0896-6273(90)90147-8. [DOI] [PubMed] [Google Scholar]
- 10.Coyle JT, Puttfarcken P. Oxidative stress, glutamate and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 11.De Bernardi MA, Rabin SJ, Colangelo AM, Brooker G, Mocchetti I. TrkA mediates the nerve growth factor-induced intracellular calcium accumulation. J Biol Chem. 1996;271:6092–6098. doi: 10.1074/jbc.271.11.6092. [DOI] [PubMed] [Google Scholar]
- 12.Favaron M, Manev H, Alho H, Bertolino M, Ferret B, Guidotti A, Costa E. Gangliosides prevent glutamate and kainate neurotoxicity in primary cultures of neonatal rat cerebellum and cortex. Proc Natl Acad Sci USA. 1988;85:7351–7355. doi: 10.1073/pnas.85.19.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandez-Sanchez MT, Novelli A. Basic fibroblast growth factor protects cerebellar neurons in primary cultures from NMDA and non-NMDA receptor-mediated neurotoxicity. FEBS Lett. 1993;335:124–131. doi: 10.1016/0014-5793(93)80453-2. [DOI] [PubMed] [Google Scholar]
- 14.Follesa P, Ticku MK. Chronic ethanol treatment differentially regulates NMDA receptor subunit mRNA expression in rat brain. Mol Brain Res. 1995;29:99–106. doi: 10.1016/0169-328x(94)00235-7. [DOI] [PubMed] [Google Scholar]
- 15.Follesa P, Gale K, Mocchetti I. Regional and temporal pattern of expression of nerve growth factor and basic fibroblast growth factor mRNA in rat brain following electroconvulsive shock. Exp Neurol. 1994;127:37–44. doi: 10.1006/exnr.1994.1077. [DOI] [PubMed] [Google Scholar]
- 16.Freese A, Finklestein SP, Difiglia M. Basic fibroblast growth factor protects striatal neurons in vitro from NMDA receptor-mediated excitotoxicity. Brain Res. 1992;575:351–355. doi: 10.1016/0006-8993(92)90104-h. [DOI] [PubMed] [Google Scholar]
- 17.Frim DM, Uhler TA, Short PM, Ezzedine DZ, Klagsbrun M, Breakefield XO, Isacson O. Effects of biologically delivered NGF, BDNF, and bFGF on striatal excitotoxic lesions. NeuroReport. 1993;4:367–370. doi: 10.1097/00001756-199304000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Garthwaite G, Hajos F, Garthwaite J. Ionic requirements for neurotoxic effects of excitatory amino acid analogues in rat cerebellar slices. Neuroscience. 1986;18:437–447. doi: 10.1016/0306-4522(86)90164-8. [DOI] [PubMed] [Google Scholar]
- 19.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 20.Hahn JS, Aizenman E, Lipton S. Central mammalian neurons normally resistant to glutamate toxicity are made sensitive by elevated Ca2+: toxicity is blocked by N-methyl-d-aspartate antagonist, MK801. Proc Natl Acad Sci USA. 1988;85:6556–6560. doi: 10.1073/pnas.85.17.6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hollmann M, Heinemann H. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 22.Ishii T, Moryoshi K, Sugihara H, Sakurada K, Kadotani H, Yokoi M, Akazawa C, Shigemoto R, Mizuno N, Masu M, Nakanishi N. Molecular characterization of a family of N-methyl-d-aspartate receptor subunits. J Biol Chem. 1993;268:2834–2843. [PubMed] [Google Scholar]
- 23.Kaplan DR, Stevens RM. Neurotrophin signal transduction by the trk receptor. J Neurobiol. 1994;25:1404–1417. doi: 10.1002/neu.480251108. [DOI] [PubMed] [Google Scholar]
- 24.Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent proterin kinases. Biochem Biophys Res Commun. 1987;142:436–440. doi: 10.1016/0006-291x(87)90293-2. [DOI] [PubMed] [Google Scholar]
- 25.Knusen B, Gao H, Okazaki T, Yoshida T, Mori N, Hefti F, Kaplan D. Ligand-induced down-regulation of Trk messenger RNA, protein and tyrosine phosphorylation in rat cortical neurons. Neuroscience. 1997;78:851–862. doi: 10.1016/s0306-4522(96)00616-1. [DOI] [PubMed] [Google Scholar]
- 26.Kumar KN, Tilakaratne N, Johnson PS, Allen AE, Michaelis EK. Cloning of cDNA for the glutamate-binding subunit of an NMDA receptor complex. Nature. 1991;354:70–73. doi: 10.1038/354070a0. [DOI] [PubMed] [Google Scholar]
- 27.Kume T, Kouchiyama H, Kaneko S, Maeda T, Kaneko S, Akaike A, Shimohama S, Kihara T, Kimura J, Wadfa K, Koizumi S. BDNF prevents NO mediated glutamate cytotoxicity in cultured cortical neurons. Brain Res. 1997;756:200–204. doi: 10.1016/s0006-8993(97)00195-9. [DOI] [PubMed] [Google Scholar]
- 28.Kutsuwada T, Kashiwabuchi N, Mori H, Sakimura K, Kushiya E, Araki K, Meguro H, Masaki H, Kumanishi T, Arakawa M, Mishima M. Molecular diversity of the NMDA receptor channel. Nature. 1992;358:36–42. doi: 10.1038/358036a0. [DOI] [PubMed] [Google Scholar]
- 29.Levine ES, Dreyfus CF, Black IB, Plummer MR. Differential effects of NGF and BDNF on voltage-gated calcium currents in embryonic forebrain neurons. J Neurosci. 1995;15:3084–3091. doi: 10.1523/JNEUROSCI.15-04-03084.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindholm D, Dechant G, Heisenberg CP, Thoenen H. Brain derived neurotrophic factor is a survival factor for cultured rat cerebellar granule neurons and protects against glutamate-induced neurotoxicity. Eur J Neurosci. 1993;5:1455–1464. doi: 10.1111/j.1460-9568.1993.tb00213.x. [DOI] [PubMed] [Google Scholar]
- 31.Longone P, Mocchetti I, Riva MA, Wojcik WJ. Characterization of a decrease in muscarinic m2 mRNA in cerebellar granule cells by carbachol. J Pharmacol Exp Ther. 1993;265:441–446. [PubMed] [Google Scholar]
- 32.MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-receptor activation increases cytoplasmic calcium concentrations in cultured spinal cord neurons. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 33.Maiese K, Boniece I, DeMeo D, Wagner JA. Peptide growth factors protect against ischemia in culture by preventing nitric oxide toxicity. J Neurosci. 1993;13:3034–3040. doi: 10.1523/JNEUROSCI.13-07-03034.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marini AM, Paul SM. N-Methyl-d-aspartate receptor-mediated neuroprotection in cerebellar granule cells requires new RNA and protein synthesis. Proc Natl Acad Sci USA. 1992;89:6555–6559. doi: 10.1073/pnas.89.14.6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marini AM, Rabin SJ, Lipsky RH, Mocchetti I. Activity-dependent stimulation of TrkB receptors underlies the neuroprotective effect of N-methyl-d-aspartate. Soc Neurosci Abstr. 1997a;23:650. [Google Scholar]
- 36.Marini AM, Spiga G, Mocchetti I. Toward the development of strategies to prevent ischemic neuronal injury: in vitro studies. Ann NY Acad Sci. 1997b;825:209–219. doi: 10.1111/j.1749-6632.1997.tb48431.x. [DOI] [PubMed] [Google Scholar]
- 37.Mattson MP, Scheff SW. Endogenous neuroprotection factors and traumatic brain injury mechanisms of action and implication for therapy. J Neurotrauma. 1994;11:3–33. doi: 10.1089/neu.1994.11.3. [DOI] [PubMed] [Google Scholar]
- 38.Mattson MP, Murrain M, Guthrie PB, Kater SB. Fibroblast growth factor and glutamate: opposing roles in the generation and degeneration of hippocampal neuroarchitecture. J Neurosci. 1989;9:3728–3732. doi: 10.1523/JNEUROSCI.09-11-03728.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mattson MP, Kumar K, Cheng B, Wang H, Michaelis EK. Basic fibroblast growth factor regulates the expression of a functional 71 kDa NMDA receptor protein that mediates calcium influx and neurotoxicity in cultured hippocampal neurons. J Neurosci. 1993;13:4575–4588. doi: 10.1523/JNEUROSCI.13-11-04575.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mattson MP, Lovell MA, Furukawa K, Markesbery WR. Neurotrophic factors attenuate glutamate-induced accumulation of peroxidase, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J Neurochem. 1995;65:1740–1751. doi: 10.1046/j.1471-4159.1995.65041740.x. [DOI] [PubMed] [Google Scholar]
- 41.Meguro H, Mori H, Araki K, Kushiya E, Kutsuwada T, Yamazaki M, Kumanishi T, Arakawa M, Sakimura K, Mishina M. Functional characterization of heteromeric NMDA receptor channel expressed from cloned cDNAs. Nature. 1992;357:70–74. doi: 10.1038/357070a0. [DOI] [PubMed] [Google Scholar]
- 42.Miller TM, Tansey MG, Johnson EM, Creedon DJ. Inhibition of phosphatidylinositol 3-kinase activity blocks depolarization and insulin-like growth factor I-mediated survival of cerebellar granule cells. J Biol Chem. 1997;272:9847–9853. doi: 10.1074/jbc.272.15.9847. [DOI] [PubMed] [Google Scholar]
- 43.Mocchetti I, Wrathall JR. Neurotrophic factors in central nervous system trauma. J Neurotrauma. 1995;12:853–870. doi: 10.1089/neu.1995.12.853. [DOI] [PubMed] [Google Scholar]
- 44.Mocchetti I, Spiga G, Hayes VY, Isackson PJ, Colangelo AM. Glucocorticoids differentially increase nerve growth factor and basic fibroblast growth factor expression in the rat brain. J Neurosci. 1996;16:2141–2148. doi: 10.1523/JNEUROSCI.16-06-02141.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuci M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 46.Monyer H, Burnashev H, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of the four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 47.Moriyoshi K, Masu M, Ishii T, Shigemono R, Mizuno N, Nakanishi S. Molecular cloning and characterization of the rat NMDA receptor. Nature. 1991;354:31–37. doi: 10.1038/354031a0. [DOI] [PubMed] [Google Scholar]
- 48.Novelli A, Reilly JA, Lysko PG, Henneberry RC. Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- 49.Peterson DA, Lucidi-Philippi CA, Murphy DP, Ray J, Gage FH. Fibroblast growth factor-2 protects entorhinal cortex layer II glutamatergic neurons from axotomy-induced death. J Neurosci. 1996;16:886–898. doi: 10.1523/JNEUROSCI.16-03-00886.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prehn J. Marked diversity in the action of growth factors on N-methyl-d-aspartate induced neuronal degeneration. Eur J Pharmacol. 1996;306:81–88. doi: 10.1016/0014-2999(96)00225-7. [DOI] [PubMed] [Google Scholar]
- 51.Resink A, Hack N, Boer GJ, Balázs R. Growth conditions differentially modulate the vulnerability of developing cerebellar granule cells to excitatory amino acids. Brain Res. 1994;655:222–232. doi: 10.1016/0006-8993(94)91617-9. [DOI] [PubMed] [Google Scholar]
- 52.Resink A, Villa M, Boer GJ, Möhler H, Balázs R. Agonist-induced down regulation of NMDA receptors in cerebellar granule cell cultures. Eur J Neurosci. 1995;7:1700–1706. doi: 10.1111/j.1460-9568.1995.tb00691.x. [DOI] [PubMed] [Google Scholar]
- 53.Resink A, Villa M, Benke D, Hidaka H, Möhler H, Balázs R. Characterization of agonist-induced down regulation of NMDA receptors in cerebellar granule cell cultures. J Neurochem. 1996;66:369–377. doi: 10.1046/j.1471-4159.1996.66010369.x. [DOI] [PubMed] [Google Scholar]
- 54.Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Ann Neurol. 1986;19:105–111. doi: 10.1002/ana.410190202. [DOI] [PubMed] [Google Scholar]
- 55.Rothman SM, Thurston JH, Hauhart RE. Delayed neurotoxicity of excitotoxic amino acids in vitro. Neuroscience. 1987;22:472–480. doi: 10.1016/0306-4522(87)90347-2. [DOI] [PubMed] [Google Scholar]
- 56.Segal RA, Pomeroy SL, Stiles CD. Axonal growth and fasciculation linked to differential expression of BDNF and NT-3 in developing cerebellar granule cells. J Neurosci. 1995;15:4970–4981. doi: 10.1523/JNEUROSCI.15-07-04970.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suen PC, Wu K, Levine ES, Mount HT, Xu JL, Lin SY, Black IB. Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic N-methyl-d-aspartate receptor subunit 1. Proc Natl Acad Sci USA. 1997;94:8191–8195. doi: 10.1073/pnas.94.15.8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tapley P, Lamballe F, Barbacid M. K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene. 1992;7:371–381. [PubMed] [Google Scholar]
- 59.Teng YD, Mocchetti I, Wrathall JR. Basic and acidic fibroblast growth factor protect spinal motor neurons in vivo after experimental spinal cord injury. Eur J Neurosci. 1998;10:798–802. doi: 10.1046/j.1460-9568.1998.00100.x. [DOI] [PubMed] [Google Scholar]
- 60.Vallano ML, Lambolez B, Audinat E, Rossier J. Neuronal activity differentially regulates NMDA receptor subunit expression in cerebellar granule cells. J Neurosci. 1996;16:631–639. doi: 10.1523/JNEUROSCI.16-02-00631.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van der Valk JBF, Resink A, Balazs R. Membrane depolarization and the expression of glutamate receptors in cerebellar granule cells. Eur J Pharmacol. 1991;201:247–250. doi: 10.1016/0014-2999(91)90354-s. [DOI] [PubMed] [Google Scholar]
- 62.Wielock T. Hypoglycemia-induced neuronal damage prevented by an N-methyl-d-aspartate antagonist. Science. 1985;230:681–683. doi: 10.1126/science.2996146. [DOI] [PubMed] [Google Scholar]
- 63.Zirrgiebel U, Ohga Y, Carter B, Berninger B, Inagaki N, Thoenen H, Lindholm D. Characterization of TrkB receptor-mediated signaling pathways in rat cerebellar granule neurons: involvement of protein kinase C in neuronal survival. J Neurochem. 1995;65:2241–2250. doi: 10.1046/j.1471-4159.1995.65052241.x. [DOI] [PubMed] [Google Scholar]