

Abstract
Astrocytes in the hippocampus express high-affinity glutamate transporters that are important for lowering the concentration of extracellular glutamate after release at excitatory synapses. These transporters exhibit a permeability to chaotropic anions that is associated with transport, allowing their activity to be monitored in cell-fee patches when highly permeant anions are present. Astrocyte glutamate transporters are highly temperature sensitive, becausel-glutamate-activated, anion-potentiated transporter currents in outside-out patches from these cells exhibited larger amplitudes and faster kinetics at 36°C than at 24°C. The cycling rate of these transporters was estimated by using paired applications of either l-glutamate or d-aspartate to measure the time necessary for the peak of the transporter current to recover from the steady-state level. Transporter currents in patches recovered with a time constant of 11.6 msec at 36°C, suggesting that either the turnover rate of native transporters is much faster than previously reported for expressed EAAT2 transporters or the efficiency of these transporters is very low. Synaptically activated transporter currents persisted in astrocytes at physiological temperatures, although no evidence of these currents was found in CA1 pyramidal neurons in response to afferent stimulation. l-glutamate-gated transporter currents were also not detected in outside-out patches from pyramidal neurons. These results are consistent with the hypothesis that astrocyte transporters are responsible for taking up the majority of glutamate released at Schaffer collateral–commissural synapses in the hippocampus.
Keywords: glutamate transporter, astrocyte, GLT-1, GLAST, EAAC1, hippocampus, CA1
Removal of glutamate from the extracellular space is achieved via the activity of high-affinity Na+-dependent transporters. This clearance of extracellular glutamate is necessary to maintain low ambient levels, reducing the tonic activation of receptors so that excitotoxic neuronal degeneration does not occur (Rothstein et al., 1996; Tanaka et al., 1997). Members of the glutamate transporter family exhibit cell type-specific expression, with GLT-1 and GLAST expressed in glial cells (primarily astrocytes and Bergmann glial cells) and EAAC1 and EAAT4 expressed in neurons (Kanai et al., 1995; Yamada et al., 1996; but see Conti et al., 1998). However, the relative contribution of neuronal versus glial transport in the uptake of synaptically released glutamate is not known. A dramatic demonstration of the importance of the two glial transporters in glutamate homeostasis has emerged from studies in which their expression has either been reduced or blocked entirely. These animals have elevated glutamate in the CSF, exhibit extensive neuronal degeneration, suffer from spontaneous seizures, and ultimately die prematurely (Rothstein et al., 1996; Tanaka et al., 1997; Watase et al., 1997). The role of the neuronal EAAC1 transporter is less certain. Animals lacking EAAC1 transporters develop essentially normally, do not suffer seizures, and exhibit no neurodegeneration (Peghini et al., 1997). Although this phenotype contrasts with animals treated with antisense (Rothstein et al., 1996), the spontaneous seizures observed in antisense-treated animals may result from dysfunction of GABA metabolism (Sepukty et al., 1997).
By recording the net charge movement that accompanies glutamate transport, it has been possible to monitor the activity of these transporters in glial cells after release of glutamate at excitatory synapses, both in culture (Mennerick and Zorumski, 1994; Linden, 1997) and in acute slices of hippocampus (Bergles and Jahr, 1997) and cerebellum (Bergles et al., 1997; Clark and Barbour, 1997). These studies indicate that some of the glutamate released into the synaptic cleft reaches glial membranes, despite evidence that glutamate transporters are expressed by the postsynaptic neurons (Rothstein et al., 1994). At climbing fiber synapses in the cerebellum, less than one-quarter of the glutamate released from a vesicle is recovered postsynaptically (Otis et al., 1997), providing further evidence that glial cells are primarily responsible for the uptake of synaptically released glutamate. However, these studies were performed at room temperature. Given the reportedly high-temperature dependence of glutamate flux (Wadiche et al., 1995a) and the apparent rapid turnover rate of endogenous transporters (Bergles and Jahr, 1997), it is possible that at physiological temperatures neuronal transporters may be more effective in restricting the diffusion of glutamate from the cleft (Tong and Jahr, 1994; Asztely et al., 1997).
To investigate the temperature dependence of glial glutamate transport in the hippocampus, we recorded synaptically evoked transporter currents from astrocytes located in stratum radiatum of area CA1. The persistence of these currents at physiological temperatures and the lack of detectable transporter currents in pyramidal neurons suggest that astrocyte transporters are primarily responsible for the uptake of glutamate released from the terminals of Schaffer collateral–commissural fibers in the hippocampus.
MATERIALS AND METHODS
Hippocampal slices were prepared from 12- to 15-d-old male Sprague Dawley rats in accordance with a protocol approved by the Department of Animal Care at Oregon Health Sciences University. Rats were anesthetized with halothane and decapitated, and the hippocampi were removed and placed in ice-cold artificial CSF (ACSF) consisting of (in mm): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose, saturated with 95% O2–5% CO2. Hippocampi were mounted in an agar block, and 400-μm-thick slices were made by cutting the tissue transversely using a Vibratome (TPI Instruments). Slices were allowed to recover on a gauze net submerged in ACSF for 30 min at 34°C and at room temperature thereafter.
Individual slices that had recovered for at least 1 hr were transferred to a Lucite chamber with a coverslip bottom, where they were held stationary with a “harp” made from 0.5 mm silver wire and single nylon threads. Whole-cell recordings were made from astrocytes located in the stratum radiatum region of area CA1, which were visualized using an upright microscope (Axioskop; Zeiss) equipped with infrared–Nomarski optics. Astrocytes were identified as described previously (Bergles and Jahr, 1997), by their small size, high-resting potentials, and low-input resistances. The internal solution for whole-cell recordings of transporter currents contained (in mm): 130 K-methanesulfonate, 20 HEPES, 10 EGTA, and 1 MgCl2, pH 7.2. To increase the likelihood of detecting transporter currents in pyramidal neurons, K-thiocyanate (KSCN) (130 mm) was substituted for K-methanesulfonate in some recordings. SCN− was not used as the permeant anion in whole-cell recordings from astrocytes, because it induced a holding current and caused them to visibly shrink over time, affecting the stability of the recordings. Whole-cell recordings of EPSCs were made from CA1 pyramidal neurons using an internal solution composed of (in mm): 100 Cs-methanesulfonate, 20 TEA-Cl, 20 HEPES, 10 EGTA, 1 MgCl2, 4 ATP-Mg, and 0.3 GTP-Na, pH 7.2. Holding potentials have been corrected for the different junction potentials.
Evoked responses were elicited with a constant-current stimulator (Winston Electronics) using bipolar stainless steel electrodes (tip separation, 200 μm) placed in stratum radiatum >100 μm from the cell. Stimulus parameters were 40–100 μA, 100 μsec for all experiments, except those testing for transporter currents in pyramidal neurons in which the parameters were 100–200 μA, 200 μsec. Whole-cell currents were amplified using an Axopatch 200A (Axon Instruments), filtered at 1–5 kHz and sampled at 10–20 kHz. Series resistance was <10 MΩ, and 80–90% compensation was used. Field electrodes had resistances of 1–3 MΩ when filled with 3m NaCl and were placed in stratum radiatum of area CA1. Field EPSPs (fEPSPs) were recorded with an Axoclamp-2A (Axon Instruments). Slope measurements were made from the initial rise of the fEPSP using linear regression. To avoid contamination of the rising phase of the fEPSP with the fiber volley at 36°C, 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxalie-7-sulfonamide (NBQX) (10 μm) and 3-(2-carboxypiperazin-4-yl)-propyl-l-phosphonic acid (CPP) (5 μm) were applied at the end of the experiment to allow the stimulus artifact and fiber volley to be subtracted from the synaptic response. A cut was made at the CA3–CA2 border when picrotoxin and SR-95531 were included in the ACSF to reduce the propagation of seizure discharges to area CA1. Stimulus artifacts have been truncated for clarity.
Outside-out patches were removed from astrocytes and pyramidal neurons located in area CA1. Rapid applications of solutions to these patches were performed as described previously, using a four-barrel flow pipe attached to a piezoelectric bimorph (Tong and Jahr, 1994). The junction potential between the control and test solutions was used to monitor the speed and duration of the solution application. The rise and decay of these “open tip” responses occurred in <200 μsec (20–80%). Nucleated patches were obtained by maintaining slight negative pressure when pulling the electrode away from the cell. Patch recordings were made with an internal solution containing (in mm): 130 KSCN, 20 HEPES, 10 EGTA, and 1 MgCl2, pH 7.2. Patch currents were filtered at 5 kHz and sampled at 50 kHz.
The bath temperature was raised by allowing heated water to circulate through a jacket surrounding a 5 inch length of the inflow tubing. The temperature of the solutions applied to patches was maintained at the bath temperature by raising the meniscus formed by the objective so that the tips of the flow pipes were submerged by >5 mm. Temperature changes were measured with a miniature thermistor probe (TeleThermometer; YSI) placed in the center of the chamber or in front of the flow pipes.
Half-decay measurements were made relative to steady state. For measurements of peak amplitudes during paired-pulse experiments, a 30 msec control response was subtracted from the first three (l-glutamate) or all (d-aspartate) of the paired responses before measuring the amplitude of the second response. This adjusted for currents arising on the decay of the control response.
Data are expressed as mean ± SD, and all comparisons between 24°C and 36°C were significant at p < 0.01 (Student’s paired t test), unless otherwise noted. When percentages are listed, the mean values for the two temperatures are also listed in parenthesis.
RESULTS
Temperature-dependent changes of transporter currents in patches
In addition to the stoichiometrically defined movement of Na+, K+, and H+ that are coupled to glutamate translocation (Zerangue and Kavanaugh, 1996), glutamate transporters exhibit a permeability to anions that is uncoupled from glutamate flux (Fairman et al., 1995). By including chaotropic anions, such as SCN−, in the patch pipette that are highly permeable through these transporters (Kavanaugh et al., 1997), the activity of the transporters can be monitored by recording the glutamate-gated movement of SCN. These anion-potentiated transporter currents are ∼10-fold larger than currents mediated solely by the net movement cations that accompanies transport (Bergles and Jahr, 1997), allowing kinetic measurements of transporter activity to be made. To measure the change in the intrinsic kinetics of astrocyte transporters, outside-out patches were removed from the somata of CA1 astrocytes, and transporter currents activated in response to a saturating dose (10 mm) of l-glutamate were recorded first at room temperature (22–24°C) and then at 36°C. Raising the temperature of the solutions to 36°C caused marked changes in the transporter current (Fig. 1A), increasing the peak amplitude by 28.7 ± 19.7% (24°C, −39.5 pA; 36°C, −50.2 pA), decreasing the rise time (20–80%) by 21.5 ± 20.9% (24°C, 121.9 μsec; 36°C, 95.0 μsec), and decreasing the decay from the peak (half-decay) by 50.6 ± 11.0% (24°C, 1.04 msec; 36°C, 0.51 msec) (n = 30). The change in rise time is likely to be an underestimate, limited by the speed of solution exchange. A small increase in the steady-state current was sometimes visible in these patches (8 of 30 patches); however, the steady-state transporter current was small under these conditions (−6.5 ± 5.2 pA) (n = 30), which decreased the precision of this measurement.
Fig. 1.

Temperature-dependent changes in astrocyte transporter currents. A, Outside-out patch removed from an astrocyte located in stratum radiatum of area CA1. Solid lines in A and B are the control responses recorded at room temperature, and dotted linesare the responses of the same patch recorded at 36°C.Inset, Response at 36°C has been scaled to that in control and shown at a faster time base to illustrate the time course of the two responses. B, Response of a different patch to d-aspartate (10 mm). Tracesare illustrated as in A. Traces are averages of 8–12 consecutive responses recorded at −90 mV. KSCN-based internal solution. The open tip response above eachtrace indicates the duration of the agonist application.
To gain a better estimate of this increase in steady-state current, temperature-dependent changes in the response of patches to saturatingd-aspartate were also recorded. d-aspartate elicits currents with a larger steady-state component (Fig.1B) (Bergles and Jahr, 1997), consistent with the greater steady-state anion flux per transport cycle observed ford-aspartate than for l-glutamate (Wadiche et al., 1995b) and the higher affinity of d-aspartate for EAAT1 and EAAT2 transporters (Arriza et al., 1994). The amplitude of the steady-state current measured at the end of a 30 msec pulse of 10 mmd-aspartate increased by 11.1 ± 7.9% (n = 9); however, this was not statistically significant. Given the previously reported temperature dependence of glutamate flux (Wadiche et al., 1995a), an increase in temperature should increase the steady-state current produced by the flux of cations that are stoichiometrically coupled to transport because of the increased cycling of the transporters. In these recordings, however, ∼90% of the current is mediated by the uncoupled movement of anions through the transporters (Bergles and Jahr, 1997). The lack of change in the steady-state anion current suggests that the proportion of time that transporters spend in the conducting state per transport cycle is unchanged at 36°C.
The steady state/peak ratio for d-aspartate decreased from 0.42 ± 0.11 to 0.36 ± 0.10 (n = 9) at 36°C, as a result of the larger increase in peak amplitude (30.0 ± 11.9%; n = 9; 24°C, −23.9 pA; 36°C, −31.1 pA). A decrease in steady state/peak ratio was also observed forl-glutamate (24°C, 0.16 ± 0.06; 36°C, 0.085 ± 0.03) (n = 30). The rise time, half-decay time, and deactivation time (decay of the current from the end of the glutamate pulse) also decreased with the jump in temperature for responses elicited by d-aspartate (rise time, 24.5 ± 23.4%; 24°C, 133.3 μsec; 36°C, 91.1 μsec; half-decay time, 45.2 ± 5.7%; 24°C, 0.90 msec; 36°C, 0.50 msec; deactivation time, 54.5 ± 10.7%; 24°C, 24.4 msec; 36°C, 11.03 msec) (n = 9) (Fig. 1B).
The cycling rate of the glutamate transporter EAAT2 (GLT-1) has been reported to exhibit a Q10 of 2.5–3 (Wadiche et al., 1995a). The turnover rate of astrocyte transporters in patches was estimated by measuring the time necessary for the peak amplitude of the transporter current to recover from the steady-state level, by applying paired applications of l-glutamate separated by different intervals. At room temperature, transporter currents recovered with a time constant of 23.7 ± 1.0 msec (n = 4; single exponential fit) (Fig.2A,C). This time constant of recovery decreased at 36°C to 11.6 ± 0.3 msec (n = 11) (Fig.2B,C), a Q10of >2, suggesting that the turnover rate of these native glial transporters approaches ∼100 sec−1, which is two to three times faster than turnover rates estimated for EAAT2 transporters expressed in oocytes (Wadiche et al., 1995a). The recovery of transporter currents in patches in response tod-aspartate was slower than for l-glutamate (Fig.3A,C), as expected from the lower equilibrium Imaxmeasured for d-aspartate in EAAT1- and EAAT2-expressing oocytes (Arriza et al., 1994). Increasing the temperature decreased the time constant of recovery from 35.0 ± 1.8 (n = 4) to 16.5 ± 0.6 msec (n = 10) (Fig.3B,C), similar to the approximately twofold decrease observed for l-glutamate.
Fig. 2.

Recovery time course ofl-glutamate-evoked transporter currents. A,B, l-glutamate (10 mm) was applied for 30 msec to an outside-out patch from an astrocyte and then reapplied for 20 msec after a variable delay, at both 24°C (top traces) and 36°C (bottom traces).Traces are averages of six consecutive responses recorded at −90 mV. KSCN-based internal solution. C, Summary plot of the ratio of the peak amplitude of the second pulse (P2) over the peak amplitude of the control response (P1) for recordings made at both 24°C (n = 4) and 36°C (n = 11). The four patches used to measure the recovery at 24°C were also used to measure the recovery at 36°C.
Fig. 3.

Recovery time course ofd-aspartate-evoked transporter currents. A,B, d-aspartate (10 mm) was applied to an outside-out patch from astrocytes for 30 msec and then reapplied for 20 msec after a variable delay, at both 24°C (top traces) and 36°C (bottom traces).Traces are averages of five consecutive responses recorded at −90 mV. KSCN-based internal solution. C, Summary plot of the ratio of the peak amplitude of the second pulse (P2) over the peak amplitude of the control response (P1) for recordings made at both 24°C (n = 4) and 36°C (n = 10). The four patches used to measure the recovery at 24°C were also used to measure the recovery at 36°C.
Temperature-dependent changes in synaptic transporter currents
To test whether glutamate transporters were activated in astrocyte membranes at physiological temperatures, transporter currents elicited via single (100 μsec) stimuli to the Schaffer collateral–commissural projections in stratum radiatum were recorded from astrocytes at room temperature and at 36°C. Ionotropic glutamate receptors were blocked in these experiments with 10 μmNBQX and 5 μmd,l-CPP. At room temperature, transporter currents recorded under these conditions had a rise time of 4.4 ± 0.8 msec and a half-decay time of 17.7 ± 1.8 msec (n = 14) (Fig.4A). Raising the bath temperature to 36°C decreased the latency of the response and decreased the rise time (2.2 ± 0.4 msec) and half-decay time (7.7 ± 0.8 msec) (n = 14) of the synaptic transporter current (Fig. 4A). These results indicate that at near physiological temperatures glutamate escapes from these synaptic clefts and reaches transporters on glial membranes, suggesting that the increased activity of neuronal transporters at this temperature is not sufficient to sequester the released glutamate. The rise and half-decay times of these currents decreased at the higher temperature by 49.2 ± 8.3 and 56.5 ± 5.1% (n = 14), respectively. The effect of raising the temperature on the peak amplitudes of these evoked currents was more variable, but on average, they were increased by 35.9 ± 34.3% (24°C, −39.5 pA; 36°C, −53.8 pA) (n = 14). The approximately twofold decrease in the rise and decay times of these evoked transporter currents at 36°C is consistent with the faster kinetics of the transporter currents observed in patches at this temperature and suggests that the time course of the synaptic transporter current cannot be accounted for by free diffusion of glutamate to these sites, because free diffusion has aQ10 of 1.2–1.3 (Hille, 1992). The total charge transfer was less at 36°C, suggesting that less glutamate is transported by the astrocyte. However, the slow tail of the response also decreased (Fig. 4A), presumably reflecting an increase in the rate of redistribution of extracellular potassium, making a quantitative comparison of the amount of glutamate transported under the two conditions problematic.
Fig. 4.

Temperature-dependent changes in evoked responses.A, Synaptically activated transporter current (STC) recorded from a stratum radiatum astrocyte.Vm = −96 mV. K-methanesulfonate-based internal solution. B, AMPA receptor-mediated EPSCs recorded from a CA1 pyramidal neuron. Vm = −80 mV. Cs-methanesulfonate-based internal solution. C, Field EPSPs (fEPSP) recorded in stratum radiatum of area CA1. A–C, Solid lines are the responses recorded at room temperature, and the dotted lines are the responses recorded at 36°C.
Increasing the bath temperature to 36°C produced similar changes in AMPA receptor-mediated EPSCs in pyramidal neurons, decreasing the rise time by 43.8 ± 12.2% (24°C, 1.54 msec; 36°C, 0.84 msec), decreasing the half-decay time by 45.9 ± 10.0% (24°C, 7.58 msec; 36°C, 4.03 msec), and increasing the peak by 55.27 ± 67.6% (24°C, −133.7 pA; 36°C, −194.4 pA) (n = 19). These temperature-dependent changes in EPSCs were reflected in the 73.3 ± 39.6% (24°C, 1.01 mV/msec; 36°C, 1.81 mV/msec) (n = 16) increase in the slope of fEPSPs recorded in stratum radiatum (Fig. 4C).
Transporter currents in CA1 pyramidal neurons
In situ hybridization and immunocytochemical studies suggest that EAAC1 transporters are expressed in rat CA1 pyramidal neurons during the second postnatal week (Shibata et al., 1996; Furuta et al., 1997). The human homolog of EAAC1, EAAT3 also exhibits a permeability to anions (Wadiche et al., 1995b), suggesting that glutamate-gated anion currents might be elicited in SCN−-loaded pyramidal neurons with synaptic stimulation. However, when EPSCs elicited via Schaffer collateral stimulation were blocked with antagonists of AMPA and NMDA receptors (NBQX, GYKI-52466, and d,l-CPP), no remaining evoked inward current was detected (n = 8) (Fig.5). Because the turnover rates of transporters are strongly dependent on temperature, single and paired responses (50 msec interval) were also recorded at 36°C but similarly failed to elicit transporter currents. It is possible that there were too few transporters activated with these synaptic stimuli to elicit a detectable anion current, because the mean amplitude of the AMPA receptor EPSCs recorded under these conditions was −274 pA (Vm = −90 mV), much smaller than the climbing fiber EPSCs associated with synaptic transporter currents in Purkinje cells (Otis et al., 1997). However, large amplitude (>2 nA) autaptic EPSCs elicited in microisland cultures of CA1 pyramidal neurons also were not accompanied by detectable transporter currents (J. S. Diamond and C. E. Jahr, unpublished observations).
Fig. 5.

Transporter currents are not associated with EPSCs in CA1 pyramidal neurons. The ionotropic glutamate receptor antagonists NBQX (10 μm) and d,l-CPP (10 μm) completely blocked evoked responses, at both 24°C and 36°C. The brief inward current observed in NBQX and CPP is attributable to the increased amplitude and slower decay of the stimulus artifact at 36°C. Vm = −90 mV. ACSF contained picrotoxin (100 μm) and SR-95531 (5 μm). KSCN-based internal solution.
Large outside-out patches from CA1 pyramidal cells were also used to test for functional transporters in neuronal membranes. With SCN− (130 mm) in the pipette,l-glutamate (10 mm) elicited large currents (Fig. 6A1), which were blocked by the glutamate receptor antagonists NBQX, GYKI-52466, and d,l-CPP (Fig.6A2), demonstrating that they were mediated by AMPA and NMDA receptors (n = 10). In the presence of these antagonists, no inward current remained at either negative (Vm = −90 mV) or positive (Vm = 90 mV) potentials at room temperature (Fig. 6A2) or at 36°C (n = 6), as would be expected if anion-permeable transporters were present. Transporter currents were also not detected in six nucleated patches from CA1 pyramidal cells that had l-glutamate-activated inward currents >1 nA (Vm = −60 mV) in the absence of antagonists. It is unlikely that the lack of transporter currents in these patches is caused by blockade by the glutamate receptor antagonists, because these antagonists (at the same concentrations) did not affect l-glutamate-gated transporter currents in patches from astrocytes (n = 6) (Fig. 6B1,B2), consistent with previously published results (Mennerick and Zorumski, 1994;Bergles and Jahr, 1997). These data also indicate that astrocytesin situ do not express ionotropic glutamate receptors in their somatic membranes.
Fig. 6.
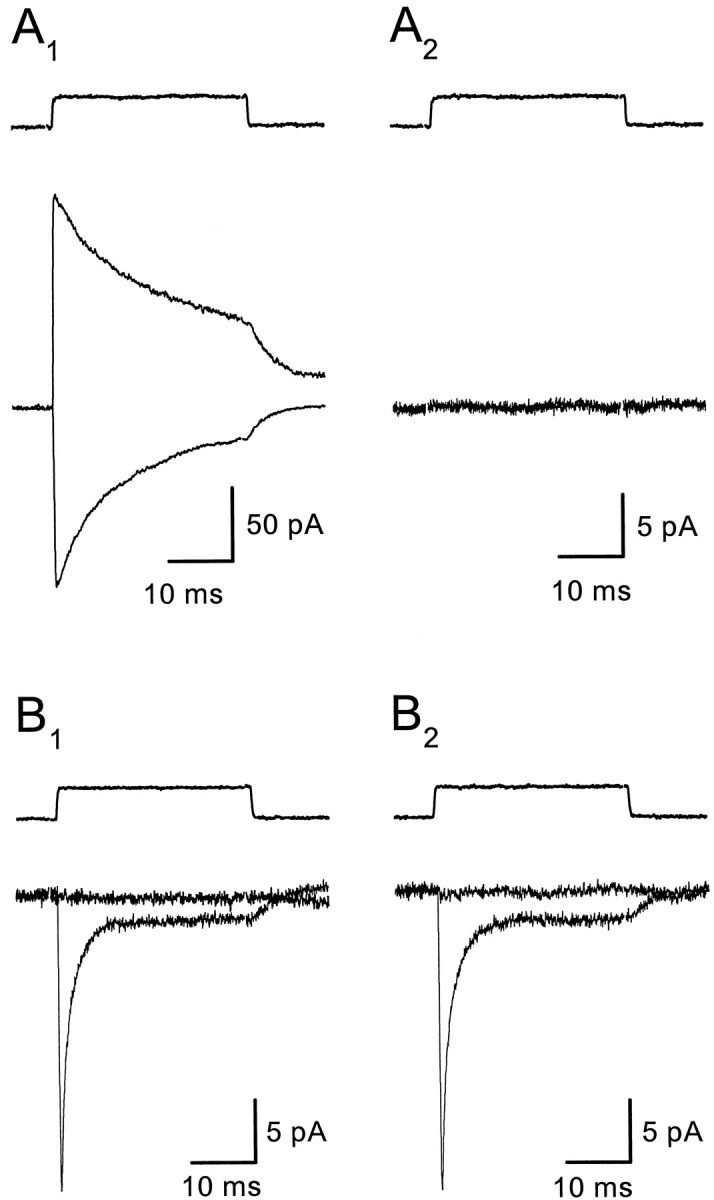
Transporter currents are not detected in pyramidal cell patches. A, Outside-out patch from a CA1 pyramidal neuron. l-glutamate (10 mm) activates inward (Vm = −90 mV) and outward (Vm = 90 mV) (A1) currents that are completely blocked by NBQX (10 μm), GYKI-52466 (25 μm), and d,l-CPP (10 μm) (A2). B,l-glutamate (10 mm) activates transporter currents in outside-out patches from CA1 astrocytes (B1), which were not affected by NBQX (10 μm), GYKI-52466 (25 μm), andd,l-CPP (10 μm) (B2). Responses were recorded at −90 mV and +90 mV, as in A. All solutions contained 20 μmglycine. Traces are averages of 8–12 consecutive responses. A KSCN-based internal solution was used for both recordings.
DISCUSSION
Astrocytes in the mammalian CNS have highly branched processes that come in close apposition to synaptic contacts (Spacek, 1985;Kosaka and Hama, 1986), they express glutamate transporters at a high density (Lehre et al., 1995; Furuta et al., 1997), and they have a high capacity for glutamate uptake (Attwell et al., 1993). The two transporters expressed in astrocytes, GLT-1 and GLAST, are critically important for maintaining a low extracellular concentration of glutamate (Rothstein et al., 1996; Tanaka et al., 1997). The current generated by the electrogenic cycling of these transporters can be detected in astrocytes after stimulation of Schaffer collateral–commissural fibers in acute hippocampal slices (Bergles and Jahr, 1997). We report here that at near physiological temperatures these transporter currents persist, becoming faster and larger, indicating that glutamate continues to diffuse out of these synaptic clefts and reach glial membranes. Stimulation of Schaffer collateral–commissural fibers did not elicit transporter currents in CA1 pyramidal neurons, nor were they present in patches removed from these cells, suggesting that glutamate transporters are expressed at a very low density at this age in pyramidal neurons. Because there is no direct evidence for the expression of glutamate transporters presynaptically at these synapses (Rothstein et al., 1994; Furuta et al., 1997; but see Taxt and Storm-Mathisen, 1984), these results suggest that astroglial transporters are primarily responsible for the uptake of glutamate released from Schaffer collateral–commissural fibers.
Kinetics of astrocyte transporters at physiological temperatures
Astrocyte transporter currents in patches had more rapid kinetics at 36°C than at room temperature (22–24°C), activating, desynchronizing, and deactivating more rapidly, as would be expected for underlying conformational changes that require high-activation energies (Hille, 1992). The turnover or cycling rate of the transporters was estimated by measuring the time necessary for the peak of the anion current to recover from the steady-state level. The time constant of this recovery was 23.7 ± 1.0 msec forl-glutamate at room temperature and decreased to 11.6 ± 0.3 msec at 36°C. This rapid recovery suggests a physiological turnover rate of 86 sec−1 for these endogenous transporters, which is two to three times faster than that previously measured for the cloned EAAT2 transporters expressed in oocytes (14.6 sec−1 at room temperature, with a reportedQ10 of 2.5–3) (Wadiche et al., 1995a). The estimate of turnover rate based on recovery of the anion conducting state assumes that once glutamate binds the transporter it undergoes translocation, with negligible unbinding occurring to the outside. An alternative explanation for this rapid recovery is that some glutamate unbinds before being transported, resulting in a lower transport efficiency but a faster recovery of the anion conductance. Although this efficiency may be temperature-dependent, it is unlikely to entirely account for the faster recovery of the transporter current at 36°C, because transporters also cycle faster at higher temperatures (Wadiche et al., 1995a). The previous estimate of the turnover rate of EAAT2 transporters was based on the steady-state accumulation of radiolabeled l-glutamate (Wadiche et al., 1995a), an assay that does not provide information about the efficiency of transport. Nevertheless, a model involving significant unbinding of glutamate to the outside is consistent with a lower equilibrium flux rate and may provide an explanation for the discrepancy between the two results.
Glutamate transporters in CA1 pyramidal neurons
Hippocampal pyramidal neurons in juvenile rats (postnatal days 14–16) express the EAAC1 glutamate transporter (Shibata et al., 1996;Furuta et al., 1997). The human homolog of EAAC1, EAAT3 (Arriza et al., 1994) exhibits a permeability to anions similar to other glutamate transporters (Wadiche et al., 1995b), and C6 glioma cells that express only EAAC1 transporters (Palos et al., 1996) have anion permeable glutamate transporters (M. P. Kavanaugh, personal communication). The conductance of the EAAT3 transporter, relative to others in the family and based on the reversal potentials of the combined transport and anion current, suggests that the permeability of EAAT3 to anions is less than GLAST but greater than GLT-1 (Wadiche et al., 1995b). Although transporter currents were not detected in pyramidal neurons after stimulation of Schaffer collateral–commissural fibers, the quantal content of these EPSCs was small relative to those used to resolve transporter currents in Purkinje neurons (Otis et al., 1997), raising the possibility that not enough transporters were activated to be detected. Nevertheless, transporter currents also were not detected in pyramidal neurons in microisland cultures after autaptic stimulation.
l-glutamate-gated transporter currents were also not resolved in large outside-out patches from CA1 pyramidal neurons. Anionic currents similar to those observed in astrocyte patches (Bergles and Jahr, 1997) would be expected in pyramidal cell patches if neuronal transporters were (1) expressed at similar densities, (2) had comparable anion permeabilities, and (3) were present in somatic membranes. Because recent results indicate that EAAC1 is present in the somatic membrane of pyramidal neurons at this developmental age (Furuta et al., 1997), these results suggest that the combination of the lower density and lower conductance of these transporters to anions accounts for the inability to detect glutamate-activated transport currents in CA1 pyramidal neurons. These results also suggest that GLT-1 (Mennerick et al., 1998) or other as yet unidentified transporters that have an associated anion permeability are not expressed at a high density in CA1 pyramidal neurons at this age. Our results are consistent with the phenotype of animals in which the expression of EAAC1 transporters has been selectively reduced by transgenic means. Mice lacking EAAC1 transporters appear essentially normal, exhibiting no neurodegeneration, no increased susceptibility to induced seizures, and no increased mortality (Peghini et al., 1997). This phenotype contrasts strongly with animals that lack GLT-1 expression. These transgenic animals have spontaneous seizures, an increased susceptibility to ischemic insults, and elevated glutamate levels in the extracellular space (Tanaka et al., 1997), consistent with results obtained through treatment with antisense for GLT-1 (Rothstein et al., 1996). Although animals treated with antisense EAAC1 have a higher incidence of spontaneous seizures (Rothstein et al., 1996), this may be attributable to a breakdown in GABA metabolism rather than a consequence of reduced uptake of glutamate by pyramidal neurons (Sepukty et al., 1997). These results suggest that EAAC1 transporters are not critical for maintaining a low ambient glutamate concentration.
Unlike the other glutamate transporters (GLT-1, GLAST, and EAAT4), the expression of EAAC1 transporters is not restricted to the CNS (Pines et al., 1992; Storck et al., 1992; Fairman et al., 1995), with substantial EAAC1 found in peripheral tissues, including the heart, muscle, kidney, and intestine (Kanai and Hediger, 1992; Mukainaka et al., 1995). This wide distribution of EAAC1 transporters suggests that they may perform more of a metabolic function, providing additional glutamate necessary for the detoxification of ammonia and synthesis of proteins. This hypothesis is supported by the lack of colocalization of EAAC1 and other synaptic markers (Coco et al., 1997).
Implications for synapse specificity in the hippocampus
Glutamate released at excitatory synapses in the CA1 region of the hippocampus is not restricted to the synaptic space after release but diffuses rapidly out the cleft and reaches nearby glial membranes within a millisecond (Bergles and Jahr, 1997). The slow time course of the transporter current recorded from astrocytes suggests that glutamate may remain elevated for many milliseconds after release in the extrasynaptic space, diffusing to transporters distant from the cleft (Chaudhry et al., 1995). Neighboring synapses in this region of the hippocampus are on average <0.5 μm apart (Rusakov and Kullmann, 1998), suggesting that high-affinity ionotropic NMDA and metabotropic receptors may be activated and AMPA receptors desensitized by this spillover of glutamate. Although a direct demonstration of glutamate spillover in the hippocampus has been elusive, this phenomenon has been proposed to explain why NMDA receptors sense a larger number of quanta than AMPA receptors, based on the lower amplitude variability of NMDA receptor-mediated responses at these synapses (Asztely et al., 1997). Diffusion of glutamate to adjacent sites could also explain why NMDA receptor-only EPSCs are sometimes visible at low stimulus strengths (for review, see Kullman and Asztely, 1998). If glutamate transporters expressed by astrocytes are primarily responsible for the uptake of glutamate released at these synapses, they may be an important determinant in maintaining synapse specificity by restricting the diffusion of glutamate to neighboring synapses, although barriers to diffusion and additional binding sites may also play an important role (Barbour and Hausser, 1997). The pivotal role of glial transporters is suggested by a recent study showing that the GLT-1-selective transporter antagonist dihydrokainate can increase the incidence of spillover at Schaffer collateral–commissural synapses at physiological temperatures; this conclusion was based on the ability of dihydrokainate to increase the difference in the variance between AMPA and NMDA receptor-mediated components of EPSCs in CA1 pyramidal neurons (Asztely et al., 1997). The results presented here showing that glutamate reaches astrocyte transporters at physiological temperatures, but does not elicit detectable transporter currents in pyramidal cells, suggests that astrocytes are primarily responsible for the uptake of glutamate released from Schaffer collateral–commissural synapses.
Footnotes
This work was supported by a grant from National Institutes of Health.
Correspondence should be addressed to Dwight E. Bergles, Vollum Institute, L474, Oregon Health Sciences University, Portland, Oregon 97201.
REFERENCES
- 1.Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- 3.Attwell D, Barbour B, Szatkowski M. Nonvesicular release of neurotransmitter. Neuron. 1993;11:401–407. doi: 10.1016/0896-6273(93)90145-h. [DOI] [PubMed] [Google Scholar]
- 4.Barbour B, Hausser M. Intersynaptic diffusion of neurotransmitter. Trends Neurosci. 1997;20:377–384. doi: 10.1016/s0166-2236(96)20050-5. [DOI] [PubMed] [Google Scholar]
- 5.Bergles DE, Jahr CE. Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron. 1997;19:1297–1308. doi: 10.1016/s0896-6273(00)80420-1. [DOI] [PubMed] [Google Scholar]
- 6.Bergles DE, Dzubay J, Jahr CE. Glutamate transporter currents in Bergmann glial cells follow the time course of extrasynaptic glutamate. Proc Natl Acad Sci USA. 1997;94:14821–14825. doi: 10.1073/pnas.94.26.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15:711–720. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 8.Clark BA, Barbour B. Currents evoked in Bergmann glial cells by parallel fibre stimulation in rat cerebellar slices. J Physiol (Lond) 1997;502:335–350. doi: 10.1111/j.1469-7793.1997.335bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coco S, Verderio C, Trotti D, Rothstein JD, Volterra A, Matteoli M. Non-synaptic localization of the glutamate transporter EAAC1 in cultured hippocampal neurons. Eur J Neurosci. 1997;9:1902–1910. doi: 10.1111/j.1460-9568.1997.tb00757.x. [DOI] [PubMed] [Google Scholar]
- 10.Conti F, DeBiasi S, Minelli A, Rothstein JD, Melone M. EAAC1, a high-affinity glutamate transporter, is localized to astrocytes and gabaergic neurons besides pyramidal cells in the rat cerebral cortex. Cereb Cortex. 1998;8:108–116. doi: 10.1093/cercor/8.2.108. [DOI] [PubMed] [Google Scholar]
- 11.Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG. An excitatory amino acid transporter with properties of a ligand-gated chloride channel. Nature. 1995;375:599–603. doi: 10.1038/375599a0. [DOI] [PubMed] [Google Scholar]
- 12.Furuta A, Rothstein JD, Martin LJ. Glutamate transporter subtypes are expressed differentially during rat CNS development. J Neurosci. 1997;17:8363–8375. doi: 10.1523/JNEUROSCI.17-21-08363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hille B. Ion channels of excitable membranes. Sinauer; Sunderland, MA: 1992. [Google Scholar]
- 14.Kanai Y, Hediger MA. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature. 1992;360:467–471. doi: 10.1038/360467a0. [DOI] [PubMed] [Google Scholar]
- 15.Kanai Y, Nussberger S, Romero MF, Boron WF, Hebert SC, Hediger MA. Electrogenic properties of the epithelial and neuronal high affinity glutamate transporter. J Biol Chem. 1995;270:16561–16568. doi: 10.1074/jbc.270.28.16561. [DOI] [PubMed] [Google Scholar]
- 16.Kavanaugh MP, Bendahan A, Zerangue N, Zhang Y, Kanner BI. Mutation of an amino acid residue influencing potassium coupling in the glutamate transporter GLT-1 induces obligate exchange. J Biol Chem. 1997;272:1703–1708. doi: 10.1074/jbc.272.3.1703. [DOI] [PubMed] [Google Scholar]
- 17.Kosaka T, Hama K. Three-dimensional structure of astrocytes in the rat dentate gyrus. J Comp Neurol. 1986;249:242–260. doi: 10.1002/cne.902490209. [DOI] [PubMed] [Google Scholar]
- 18.Kullmann DM, Asztely F. Extrasynaptic glutamate spillover in the hippocampus: evidence and implications. Trends Neurosci. 1998;21:8–14. doi: 10.1016/s0166-2236(97)01150-8. [DOI] [PubMed] [Google Scholar]
- 19.Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Linden DJ. Long-term potentiation of glial synaptic currents in cerebellar culture. Neuron. 1997;18:983–994. doi: 10.1016/s0896-6273(00)80337-2. [DOI] [PubMed] [Google Scholar]
- 21.Mennerick S, Zorumski CF. Glial contribution to excitatory transmission in cultured hippocampal cells. Nature. 1994;368:59–62. doi: 10.1038/368059a0. [DOI] [PubMed] [Google Scholar]
- 22.Mennerick S, Dhond RP, Benz A, Xu W, Rothstein JD, Danbolt NC, Isenberg KE, Zorumski CF. Neuronal expression of GLT-1 glutamate transporter in hippocampal cultures. J Neurosci. 1998;15:4490–4499. doi: 10.1523/JNEUROSCI.18-12-04490.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukainaka Y, Tanaka K, Hagiwara T, Wada K. Molecular cloning of two glutamate transporter subtypes from mouse brain. Biochem Biophys Acta. 1995;1224:233–237. doi: 10.1016/0304-4165(95)00062-g. [DOI] [PubMed] [Google Scholar]
- 24.Otis TS, Kavanaugh MP, Jahr CE. Postsynaptic glutamate transport at the climbing fiber–Purkinje cell synapse. Science. 1997;277:1515–1518. doi: 10.1126/science.277.5331.1515. [DOI] [PubMed] [Google Scholar]
- 25.Palos TP, Ramachandran B, Boado R, Howard BD. Rat C6 and human astrocytic tumor cells express a neuronal type of glutamate transporter. Brain Res Mol Brain Res. 1996;37:297–303. doi: 10.1016/0169-328x(95)00331-l. [DOI] [PubMed] [Google Scholar]
- 26.Peghini P, Janzen J, Stoffel W. Glutamate transporter EAAC-1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J. 1997;16:3822–3832. doi: 10.1093/emboj/16.13.3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pines G, Danbolt N, Bjoras M, Zhang Y, Bendahan A, Eide L, Koepsell H, Storm-Mathisen J, Seeberg E, Kanner B. Cloning and expression of a rat brain l-glutamate transporter. Nature. 1992;360:464–467. doi: 10.1038/360464a0. [DOI] [PubMed] [Google Scholar]
- 28.Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 29.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kunci RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 30.Rusakov DA, Kullmann DM. Extrasynaptic glutamate diffusion in the hippocampus: ultrastructural constraints, uptake, and receptor activation. J Neurosci. 1998;18:3158–3170. doi: 10.1523/JNEUROSCI.18-09-03158.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sepukty J, Eccles CU, Lesser RP, Dykes-Hoberg M, Rothstein JD. Molecular knockdown of neuronal glutamate transporter EAAT3 produces epilepsy and dysregulation of GABA metabolism. Soc Neurosci Abstr. 1997;23:1484. [Google Scholar]
- 32.Shibata T, Watanabe M, Tanaka K, Wada K, Inoue Y. Dynamic changes in expression of glutamate transporter mRNAs in developing brain. NeuroReport. 1996;7:705–709. doi: 10.1097/00001756-199602290-00006. [DOI] [PubMed] [Google Scholar]
- 33.Spacek J. Three-dimensional analysis of dendritic spines. III. Glial sheath. Anat Embryol. 1985;171:245–252. doi: 10.1007/BF00341419. [DOI] [PubMed] [Google Scholar]
- 34.Storck T, Schulte S, Hofmann D, Stoffel W. Structure, expression, and functional analysis of a Na(+)-dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci USA. 1992;89:10955–10959. doi: 10.1073/pnas.89.22.10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- 36.Taxt T, Storm-Mathisen J. Uptake of d-aspartate and l-glutamate in excitatory axon terminals in hippocampus: autoradiographic and biochemical comparison with γ-aminobutyrate and other amino acids in normal rats and in rats with lesions. Neuroscience. 1984;11:79–100. doi: 10.1016/0306-4522(84)90215-x. [DOI] [PubMed] [Google Scholar]
- 37.Tong G, Jahr CE. Block of glutamate transporters potentiates postsynaptic excitation. Neuron. 1994;13:1195–1203. doi: 10.1016/0896-6273(94)90057-4. [DOI] [PubMed] [Google Scholar]
- 38.Wadiche JI, Arriza JL, Amara SG, Kavanaugh MP. Kinetics of a human glutamate transporter. Neuron. 1995a;14:1019–1027. doi: 10.1016/0896-6273(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 39.Wadiche JI, Amara SG, Kavanaugh MP. Ion fluxes associated with excitatory amino acid transport. Neuron. 1995b;15:721–728. doi: 10.1016/0896-6273(95)90159-0. [DOI] [PubMed] [Google Scholar]
- 40.Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Hori S, Takimoto M, Wada K, Tanaka K. Targeted disruption of the murine GLAST gene. Soc Neurosci Abstr. 1997;23:981. [Google Scholar]
- 41.Yamada K, Watanabe M, Shibata T, Tanaka K, Wada K, Inoue Y. EAAT4 is a post-synaptic glutamate transporter at Purkinje cell synapses. NeuroReport. 1996;7:2013–2017. doi: 10.1097/00001756-199608120-00032. [DOI] [PubMed] [Google Scholar]
- 42.Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]