

Abstract
Structural plasticity of nerve cells is a requirement for activity-dependent changes in the brain. The growth-associated protein GAP-43 is thought to be one determinant of such plasticity, although the molecular mechanism by which it mediates dynamic structural alterations at the synapse is not known. GAP-43 is bound by calmodulin when Ca2+ levels are low, and releases the calmodulin when Ca2+ levels rise, suggesting that calmodulin may act as a negative regulator of GAP-43 during periods of low activity in the neurons. To identify the function of GAP-43 during activity-dependent increases in Ca2+ levels, when it is not bound to calmodulin, we sought proteins with which GAP-43 interacts in the presence of Ca2+. We show here that rabaptin-5, an effector of the small GTPase Rab5 that mediates membrane fusion in endocytosis, is one such protein. We demonstrate that GAP-43 regulates endocytosis and synaptic vesicle recycling. Modulation of endocytosis by GAP-43, in association with rabaptin-5, may constitute a common molecular mechanism by which GAP-43 regulates membrane dynamics during its known roles in activity-dependent neurotransmitter release and neurite outgrowth.
Keywords: GAP-43, growth-associated protein, calmodulin, rabaptin-5, endocytosis, synaptic vesicle
Dynamic restructuring of the synaptic membrane is thought to underlie activity-dependent changes in the mature brain. GAP-43 (B-50, F1, neuromodulin) is a phosphoprotein of the presynaptic membrane that has been proposed to play a special role in such synaptic reorganization, as well as during neural development. In the mature nervous system, the correlation of GAP-43 phosphorylation by protein kinase C (PKC) with neurotransmitter release (Dekker et al., 1989) and with the establishment and persistence of long-term potentiation (Nelson et al., 1989; Gianotti et al., 1992) indicates that this molecule plays a role in synaptic plasticity. In developing or regenerating neurons, GAP-43 is an integral constituent of the growth cone (Katz et al., 1985; Meiri et al., 1986; Skene et al., 1986; De Graan et al., 1989). However, the molecular mechanism by which GAP-43 regulates activity-dependent changes in the mature brain and neurite outgrowth during development remains obscure.
GAP-43 binds calmodulin in the absence of Ca2+ and releases calmodulin at micromolar Ca2+concentrations (Andreasen et al., 1983; Alexander et al., 1987; De Graan et al., 1990; Liu and Storm, 1990), suggesting that calmodulin is a negative regulator of GAP-43 during periods of reduced activity in the neurons. To identify proteins that may interact with GAP-43 during activity-dependent increases in intracellular Ca2+, we screened a human fetal brain expression cDNA library in the presence of 1.2 mm Ca2+ with radiolabeled GAP-43. One of the cDNA clones retrieved encoded rabaptin-5, a signaling molecule that is an effector of the small G-protein Rab5 (Stenmark et al., 1995) and is involved in expansion of early endosomes, helping to dock and fuse incoming vesicles. We demonstrate below that GAP-43 interacts with rabaptin-5 in a Ca2+-dependent manner, and that GAP-43, like rabaptin-5, participates in endocytosis.
MATERIALS AND METHODS
Library screening. We expressed human GAP-43 in frame in a variant of pGEX, GSTag, which has a built-in phosphorylation site for the catalytic subunit of cAMP-dependent protein kinase (PKA) adjacent to the multiple cloning sites (Ron and Dressler, 1992). The purified fusion protein was used to screen a human fetal brain library exactly as described (Chow et al., 1996). Full-length clones were obtained by PCR screening of the library as described (Chow et al., 1996).
Antibodies. The following antibodies were used: the monoclonal antibody 91E12 against GAP-43 (Boehringer Mannheim, Indianapolis, IN), a polyclonal antibody against Rab5A (Santa Cruz Biotechnology, Santa Cruz, CA), a monoclonal antibody against SV2 [gift of Dr. Kathleen Buckley (Buckley and Kelly, 1985)], the monoclonal antibody 53FC3 against mannosidase II (Babco, Richmond, CA), and a polyclonal antisera made against rabaptin-5, for which the bacterially expressed 11–2 protein fragment was removed from the glutathione S-transferase (GST)-11–2 fusion protein by thrombin cleavage and used as an antigen for the production of polyclonal antibodies (Babco). For the immunocytochemical experiments, the monoclonal antibody 7B10 against GAP-43 (Sigma, St. Louis, MO) and a polyclonal antibody against GAP-43 (Gamby et al., 1996a,b) were used to confirm independently the specificity of the staining obtained with 91E12.
In vitro binding assays and immunoprecipitation. The PCR was used to fuse the sequence GCCGCCACCATG in frame to the 5′ end of the original 11–2 cDNA, encoding the C-terminal 311 amino acids of rabaptin-5, after which it was cloned into pBS SKII+ (Stratagene, La Jolla, CA) and used for in vitro transcription (Ambion) and translation (IVT) in a wheat germ lysate (Ambion). [35S]methionine-labeled 11–2 IVT was incubated with control GST or GST–GAP-43 fusion proteins on beads in binding buffer (20 mm Tris, pH 7.5, 1 mm DTT, 40 μg/ml BSA, 1 mm EDTA, 5 mmMgCl2, 1.2 mm CaCl2) at 4°C overnight. Complexes were washed with 3 × 1 ml of PBS containing 1.2 mm CaCl2 and 0.5% Nonidet P-40, immediately boiled in SDS sample buffer, and analyzed by PAGE. The gels were stained with Coomassie blue to verify that equal amounts of GST fusion proteins had been loaded in the lanes, dried, and exposed to Hyperfilm MP (Amersham, Arlington Heights, IL) for 5–10 d. In some experiments CaCl2 was omitted from the binding and wash buffers.
Immunoprecipitations were performed according to Nishimoto et al. (1993) with the following modifications. Six days after plating, primary cortical neurons from embryonic day 18 (E18) rat embryos were washed twice with PBS, harvested in buffer A (10 mm HEPES, pH 7.4, 1 mm EDTA, 10 mm ascorbic acid, 250 mm sucrose) plus 1.2 mm CaCl2 (in some experiments the CaCl2 was omitted from all the buffers) and protease inhibitors (1 mm PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mmNaVO4, 1 mm benzamidine, 10 mm β-glycerophosphate) and homogenized with 10 strokes in a dounce homogenizer. The homogenate was centrifuged at 100,000 ×g for 1 hr, and the supernatant was called the “cytosolic fraction.” Pellets were resuspended in 500 μl of buffer B (10 mm HEPES, pH 7.4, 1 mm EDTA, 120 mmNaCl, 0.5% CHAPS) plus 1.2 mm CaCl2 and protease inhibitors as above and stirred at 4°C for 1 hr. This material was centrifuged at 16,000 × g for 10 min, and the supernatant was called the “membrane fraction.” Both fractions were precleared with protein A Sepharose CL-4B (Pharmacia, Piscataway, NJ) for 30 min. Primary antibody was mixed with protein A Sepharose in buffer D (20 mm HEPES, pH 7.4, 1 mm EDTA, 120 mm NaCl) plus 1.2 mm CaCl2 and protease inhibitors as above, for 1 hr at 4°C. Approximately 250 μg of protein was used for each sample, which was incubated overnight with the beads coated with the appropriate primary antibody (or protein A alone) plus 2% BSA. The following day, the beads were washed four times with buffer D plus 1.2 mm CaCl2 and protease inhibitors. Samples were separated electrophoretically on SDS-PAGE (4–15%; Bio-Rad, Hercules, CA) and transferred to polyvinylidene difluoride membrane (Millipore, Bedford, MA). Immunoblots were processed using the Western light-enhanced chemiluminescence (ECL) protocol (Tropix), with the following modifications. Five percent Tween-20 was included in the I-block, and membranes were blocked overnight at 4°C. Membranes were incubated with antibody 91E12 (Boehringer Mannheim) at a 1:1000 dilution, and the secondary antibody was used at a 1:15,000 dilution.
RNA blots. RNA blots were performed as described (Neve et al., 1986). Total RNA (20 μg) was electrophoresed in each lane, and the 11–2 cDNA was used as probe. The blot was exposed to film for 72 hr.
Generation of recombinant HSV vectors and infection of primary rat cortical cultures. PCR mutagenesis (Neve and Neve, 1995) was used to generate three different mutations in the human GAP-43 cDNA. In the G19 mutant, Ser 41 was changed to a Gly, eliminating the normal site for phosphorylation by PKC. In the CAM mutant, the calmodulin-binding domain of GAP-43 (amino acids 43–58) was deleted. In the Asp mutant, Ser 41 was changed to an Asp, to mimic the presence of a phosphate group at this residue.
We prepared replication-defective HSV vectors expressing the wild-type (HSV/GAP-43) and mutant (HSV/G19, HSV/CAM, and HSV/Asp) human GAP-43 cDNAs and Escherichia coli β-galactosidase (HSV/Lac; negative control) in the expression vector pHSVPrpUC as described (Carlezon et al., 1997). The titer of the helper virus component of each stock was 1–1.2 × 106 plaque-forming units (pfu)/ml on 2–2 cells. The titer of the recombinant virus component of each stock, as assayed by expression of the exogenous gene in PC12 cells, was consistently 3 × 107infectious units (iu)/ml.
Primary cortical cultures dissociated from E18 rat embryos were plated at a density of 10 × 106 viable cells per 15 cm poly-d-lysine-coated dish or 2–5 × 105 viable cells per 35 mm dish containing poly-d-lysine-coated ACLAR coverslips (Ted Pella, Inc.) in Neurobasal medium supplemented with B27 (Life Technologies, Gaithersburg, MD), 1% fetal bovine serum, and 1% horse serum. Between 6 and 8 d after plating, some of the cultures that were used for immunochemistry or for endocytosis assays were infected with the indicated virus stocks at a multiplicity of infection (moi) of ∼1. Under these conditions 50–75% of the neurons expressed the transgene, as assayed by immunocytochemistry. Some experiments used PC12 cells, which were maintained as described (Yankner et al., 1989). Neuronal and PC12 cultures infected with HSV vectors were harvested for fractionation or fixed for immunocytochemistry 9–14 hr after infection.
Biochemical fractionation. Primary rat cortical cultures were infected at an moi of 0.1 with HSV/GAP-43 or HSV/Lac vectors, or were mock-infected, and 16 hr later the cultures were harvested for biochemical fractionation (Gray and Whittaker, 1962). After a 1000 × g spin to remove nuclei and cell debris (pellet is P1), the supernatant, S1, was centrifuged at 10,000 × g for 20 min to generate S2 and P2. The S2 was subjected to centrifugation at 100,000 × g to produce P3 (microsomes) and S3 (ribosomes: post-microsomal fraction) fractions. The P2 pellet was resuspended in 0.32 m sucrose solution and subjected to discontinuous sucrose gradient centrifugation, yielding fractions enriched for myelin (P2–0.32 m), synaptosomes (P2–0.8m), and mitochondria and lysosomes (P2–1.2 m). Five or 10 μl of each of the fractions [S1 and S2: 10 μl (5 μg) from a total of 7.5 ml each; S3: 10 μl (2 μg) from 11.5 ml; P2: 5 μl (10 μg) from 4.3 ml; P2–0.32 m, P2–0.8m, P2–1.2 m, and P3: 5 μl (1, 1.6, 7, and 5 μg, respectively) from 50 μl] was subjected to SDS-PAGE followed by immunoblot analysis with the GAP-43 or rabaptin-5 (11–2) antibodies.
To determine whether GAP-43 and rabaptin-5 localized to synaptic vesicle membrane or plasma membrane, within the synaptosome fraction, adult rat brain was subfractionated exactly as described (Whittaker et al., 1964). The fractions comprised the following morphological structures (Whittaker et al., 1964): D, synaptic vesicles, occasional microsomes; E, microsomes, some synaptic vesicles, occasional myelin fragments; F, synaptosome ghosts, myelin fragments, nonvesicular membrane fragments; G, synaptosome ghosts, membrane fragments; H, damaged synaptosomes; and I, small mitochondria, some shrunken synaptosomes. Immunoblot analysis of the fractions was performed as described above. Two micrograms of protein were loaded from fraction D, whereas 4 μg of protein was loaded from each of the other fractions.
Immunoelectron microscopy, immunofluorescence, and confocal microscopy. For immunoelectron microscopy (immuno-EM), adult rats were perfused with a modified Karnovsky fixative containing 0.025% CaCl2 and 5% sucrose. Post-embedding immunocytochemistry with anti-GAP-43 monoclonal 91E12 and anti-rabaptin-5 (11–2 polyclonal) was performed as described in the Biocell protocol for the use of gold conjugates. Secondary antibodies (anti-mouse IgG conjugated to 10 nm colloidal gold and anti-rabbit IgG conjugated to 20 nm colloidal gold) were purchased from Zymed. The sections were post-fixed with 1% glutaraldehyde and counterstained with uranyl acetate. Grains were virtually absent on sections in which the primary antibodies were omitted.
For immunofluorescence, cell cultures were permeabilized for 5 min with 0.05% saponin in PBS and fixed for 45 min in freshly made 4% paraformaldehyde. Fixed cells were rinsed with PBS, blocked for 30 min at room temperature in PBS with 0.1% Triton X-100 and 5% normal goat serum, and incubated for 2 hr at room temperature with primary antibody in PBS with 5% normal goat serum. Cells were rinsed with PBS and incubated for 90 min at room temperature with secondary antibody (Cappel, West Chester, PA) in PBS with 5% normal goat serum. After a final rinse in PBS, cells on coverslips were mounted into glass microscope slides using Gel/Mount (Biomeda). Omission of the primary antibodies resulted in only nonpunctate background fluorescence.
Quantification of endosome size. Neurons and PC12 cells were immunofluorescently stained for the endosomal marker Rab5 and for GAP-43, after treatment with 0.5% saponin to visualize endosomes. Confocal images were generated using a Leica TCS-NT laser confocal microscope. Stored digital images were quantified using MCID image analysis software by measuring endosome diameter at the widest point for each endosome. t tests and ANOVAs were performed with the cell as the unit of measure.
To create the histograms shown in Table 1, endosome diameters within each experimental group were collapsed across cells and sorted into histogram bins. Frequency counts per bin were then normalized across groups by dividing each bin by endosome count and regraphing the normalized values as relative frequency histograms.
Table 1.
Effects of wild-type and mutant GAP-43 on endosome size

Assay for endocytosis. To label early endosomes, primary neurons infected 12–14 hr earlier with HSV/Lac or HSV/GAP-43, or mock-infected, were incubated with 15 nm BSA-gold (OD520∼2.5) for 8 min at 37°C (Griffiths et al., 1989). The cells were quickly washed, fixed, scraped up, and pelleted before being processed for EM. Note that the scraping caused mechanical shearing of the neuronal processes, so that endocytosis in the cell soma was primarily detected by EM. EM photomicrographs were taken randomly through areas of labeled endosomes, yielding 34 endosomes from 24 HSV/GAP-43-infected cells and 17 endosomes from 14 HSV/Lac-infected cells. Gold particles within endosomes were then counted, and histograms were calculated based on the count of particles/endosome.
FM1–43 loading and destaining. Cells were rinsed in low-K+ saline buffer (128 mm NaCl, 25 mm HEPES, 4.8 mm KCl, 1.3 mmCaCl2, 1.2 mm MgSO4, 1.2 mm potassium phosphate buffer, pH 7.4, 0.56% glucose). They were then incubated at 37°C in high-K+ saline buffer (110 mm KCl, 22.8 mm NaCl; other components the same as low-K+ buffer) in the presence of 10 μm FM1–43 for 2 min, washed eight times with low-K+ buffer, incubated at 37°C in high-K+ saline buffer for 1 min twice, with a room temperature 30 sec incubation between the two high-K+ incubations, and washed with low-K+ buffer eight times. Control cultures were treated identically except that they were incubated twice with low-K+ buffer rather than high-K+buffer, or they were incubated with high-K+ buffer once, followed by a second incubation with low-K+buffer.
RESULTS
Retrieval of a cDNA encoding a binding protein for GAP-43
We screened a human fetal brain expression cDNA library in the presence of 1.2 mm Ca2+ with bacterially produced 32P-labeled GAP-43. A cDNA clone (termed 11–2) identified as positively binding to GAP-43 comprised a 1.9 kilobase (kb) fragment that contained the coding sequence for amino acids 539–849 of rabaptin-5 (Stenmark et al., 1995), together with approximately 1000 base pairs (bp) representing the 3′ untranslated region of the rabaptin-5 mRNA. The full-length coding sequence was obtained by additional screening of the cDNA library using the PCR and was found to be identical to the published sequence (Stenmark et al., 1995) of the cDNA for human rabaptin-5, a signaling molecule that is an effector of the activated (GTP-bound) small G-protein Rab5 and is involved in expansion of early endosomes, helping dock and fuse incoming vesicles.
Co-precipitation of GAP-43 and rabaptin-5
To confirm the interaction of the product of the 11–2 cDNA with GAP-43, the cDNA was transcribed and translated in vitro in the presence of [35S]methionine, and the 30 kDa radiolabeled translation product was incubated with a GST fusion protein (Smith and Johnson, 1988) of GAP-43 or with GST alone immobilized on glutathione-agarose beads. The beads were pelleted, washed, and subjected to SDS-PAGE. An autoradiogram of the gel (Fig.1A) demonstrates the precipitation of radiolabeled 11–2 polypeptide by GST-GAP-43 but not by GST alone. The 11–2 polypeptide was not precipitated by other, unrelated GST fusion proteins, nor was it precipitated by GST-GAP-43 in the absence of Ca2+ (data not shown).
Fig. 1.

A, The protein product (arrow) of the 11–2 cDNA binds to GAP-43 in a solid phase assay. The 30 kDa radiolabeled 11–2 polypeptide (arrow) is precipitated by GST-GAP-43 (lane 2) but not by GST alone (lane 1).B, The 11–2 antibody immunodetects a 115 kDa protein in human fetal brain homogenates (lane 1). Lanes 2 and 3, 11–2 antibody preabsorbed with GST-11–2 or with an unrelated GST fusion protein, respectively;lane 4, preimmune serum. C, GAP-43 (arrow) co-precipitates with rabaptin-5 from rat primary cortical culture homogenates in the presence of calcium (lanes 1–6) but not in the absence of calcium (lanes 7–9). Lanes 1–3, Cytosolic fraction (+1.2 mm CaCl2); lanes 4–6, membrane fraction (+1.2 mm CaCl2);lanes 7–9, membrane fraction (no CaCl2). Lanes 1, 4, and 7, Immunoprecipitation with the 11–2 antibody;lanes 2, 5, and 8, primary antibody omitted from immunoprecipitation; lanes 3,6, and 9, immunoprecipitation with an irrelevant antibody (directed against glutathioneS-transferase). The blot was probed with an anti-GAP-43 antibody. The GAP-43 protein band is indicated with anarrow. The bands above it that are present in all lanes represent nonspecific immunoglobulins. D, Rabaptin-5 mRNA is expressed at highest levels in the brain. The 11–2 cDNA was used as a probe.
To test by co-immunoprecipitation whether the GAP-43 interaction with rabaptin-5 occurs in the brain, we generated an antibody to rabaptin-5. The 11–2 protein fragment was cleaved from the GST fusion protein and used as an antigen for the production of polyclonal antibodies. As shown in Figure 1B, an affinity-purified antibody reacts with an ∼115 kDa protein in human fetal brain homogenates at a 1:10,000 dilution (lane 1). Preabsorption of the antibody with GST-11–2 (lane 2) abolishes its immunoreactivity with the 115 kDa band; however, preabsorption with an unrelated GST fusion protein (lane 3) does not affect its immunoreactivity with this band. No immunoreactivity is seen with the preimmune serum (lane 4).
We then performed immunoprecipitations of cytosolic and membrane fractions of rat neuronal culture homogenates with the 11–2 antibody, separated the proteins in the precipitate by SDS-PAGE, and blotted with the antibody to GAP-43 (Fig. 1C). A 43 kDa band corresponding to the known size of GAP-43 was immunodetected in the anti-11–2 precipitation products from both cytosolic (lane 1) and membrane (lane 4) fractions, but was not present in immunoprecipitations performed in the absence of anti-11–2 antibody (lanes 2 and 5) or in the presence of an irrelevant antibody to GST (lanes 3 and 6). GAP-43 was co-precipitated with rabaptin-5 in the presence of 1.2 mm Ca2+, or in the presence of 1.2 mm Ca2+ and 8 μm calmodulin (data not shown), but not in the absence of Ca2+ (lane 7), whether calmodulin was present or not, confirming the Ca2+ dependence of the interaction of GAP-43 with rabaptin-5. GAP-43 was not co-precipitated with rabaptin-5 from extracts of neuronal cultures solubilized with SDS.
Pattern of expression of the rabaptin-5 mRNA
Rabaptin-5 has been reported to be expressed ubiquitously in cell lines obtained from numerous different tissues (Stenmark et al., 1995). Northern blot analysis of RNAs from a range of human 20-week-old fetal tissues (Fig. 1D) indicated that the rabaptin-5 cDNA represented a 6.5 kb mRNA expressed at highest levels in the brain, with traces of expression in muscle and kidney. Thus, the expression of rabaptin-5 is more restricted in primary tissues than in cell lines, suggesting that its expression is upregulated during the establishment of cell lines.
Co-localization of GAP-43 and rabaptin-5 within neuronal cells
To determine whether GAP-43 and rabaptin-5 coexist in specific cellular compartments, we infected primary rat cortical cultures with an HSV vector expressing human GAP-43 and harvested the cultures for biochemical fractionation (Gray and Whittaker, 1962). Rabaptin-5 and GAP-43 were enriched in certain particulate fractions, most notably the synaptosome-enriched fraction of the P2 pellet (Fig.2A, P2-0.8m) and the high-speed microsomal pellet (Fig.2A, P3). To determine whether these proteins were part of synaptic vesicles or of the plasma membrane within the synaptosome-enriched fractions, subfractionation of synaptosomes (Whittaker et al., 1964) was performed (Fig.2B). Neither GAP-43 nor rabaptin-5 was immunodetected in fraction D, which is greatly enriched for synaptic vesicles and lacks plasma membrane. These results suggest co-localization of rabaptin-5 and GAP-43 to a nonsynaptic vesicle membrane such as plasma membrane, Golgi apparatus, or endoplasmic reticulum. Detectable amounts of rabaptin-5 are also seen in the post-microsomal fraction (Fig.2A, S3), consistent with the previous observation of rabaptin-5 in cytosolic fractions (Stenmark et al., 1995).
Fig. 2.

A, Biochemical fractionation studies suggest that GAP-43 and rabaptin-5 coexist in certain membranous cellular compartments. See Materials and Methods for description of each fraction. Note that an antibody to the integral synaptic vesicle membrane protein SV2 detects high levels of antigen in the synaptosome fraction (P2-0.8 m), indicating the presence of synaptic vesicles in this fraction. The two bands immunodetected with the antibody to GAP-43 represent human GAP-43 (arrowhead) expressed from the HSV vector and endogenous rat GAP-43 (arrowhead). Because the cultures were infected at a low moi, the human GAP-43 increases the overall expression of GAP-43 by <50%. Primary cultures infected with HSV/Lac vectors alone showed a distribution of endogenous GAP-43 and rabaptin-5 identical to that seen in the cultures infected with HSV/GAP-43 vectors (data not shown). B, Subfraction of rat brain synaptosomes reveals that GAP-43 and rabaptin-5 are present in a nonsynaptic vesicle membrane fraction of the synaptosome. See Materials and Methods for description of each fraction. Note that antibodies to these proteins fail to immunodetect bands in subfraction D, which is enriched for synaptic vesicle proteins as shown by SV2 immunostaining.
To map the cellular locations of rabaptin-5 and GAP-43 in finer detail, immuno-EM was performed on dual-labeled sections from adult rat prefrontal cortex and the CA1 region of the hippocampus, using the 91E12 monoclonal antibody to GAP-43 and the polyclonal 11–2 antibody directed against rabaptin-5. The proteins were most readily detected in the neuropil (Fig. 3), where they were associated with endosome-like structures (Fig. 3A) and with the plasma membrane (Fig. 3B). GAP-43 and rabaptin-5 were less frequently detected among clusters of synaptic vesicles in axons (Fig. 3C).
Fig. 3.

GAP-43 and rabaptin-5 immunoreactivity are detected most readily in the neuropil. A, GAP-43 (arrowheads, 10 nm gold particles) and rabaptin-5 (arrows, 20 nm gold particles) are both seen most frequently on endosome-like structures, most often at what appears to be the junctions of these structures with the plasma membrane.B, GAP-43 and rabaptin-5 are also present at the plasma membrane. C, GAP-43 and rabaptin-5 are observed occasionally in association with clusters of synaptic vesicles in axons. Scale bars, 200 nm.
Confocal microscopy was performed on primary cortical cultures immunostained with antibodies to GAP-43, rabaptin-5, and Rab5. The first set of experiments (Fig.4A–D) confirmed the co-localization of GAP-43 and rabaptin-5 in primary cortical neurons. As has been described previously, neurons displayed intense immunoreactivity for GAP-43 in axons, with lighter staining in the cell soma (Fig. 4D). Rabaptin-5, on the other hand, was present at higher levels in the cell body than in the process (Fig.4D). Nevertheless, areas of co-localization were detected in both the cell body (Fig. 4A–C,D, arrowhead) and the growth cone (Fig.5D–F). This co-localization was particularly distinct (Fig. 4A,B) when cells were pretreated with saponin (Bucci et al., 1992) to enhance visualization of membranous organelles in the cell. Immunoreactivity for GAP-43 co-localized with that for Rab5, an endosomal marker (Fig.4E), suggesting that the major organelle in which GAP-43 and rabaptin-5 co-localize is the endosome.
Fig. 4.
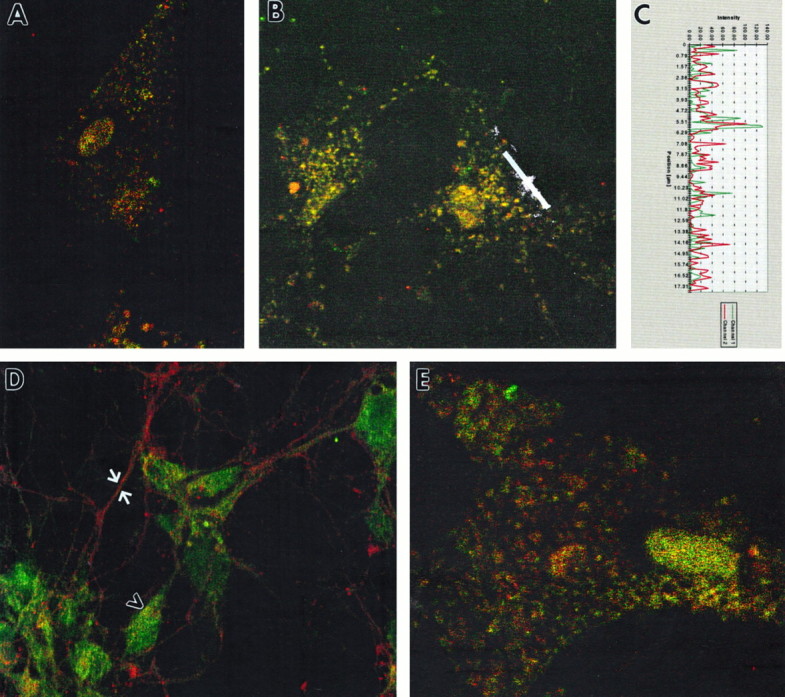
Immunofluorescent co-localization of GAP-43 and rabaptin-5 in noninfected primary neuronal cultures. GAP-43 is represented by red fluorescence in A–E. Rabaptin-5 is represented by green fluorescence inA–D, and Rab5 is represented by greenfluorescence in E. Cells in A,B, and E were pretreated with saponin to enhance visualization of endosomes. A, B, Dual labeling of endosomes with antibodies to GAP-43 and rabaptin-5 was revealed with confocal microscopy. C, Fluorescence intensity profile measured along white line in B. Areas of overlapping as well as nonoverlapping fluorescence are evident.D, Cells not treated with saponin, to preserve neuronal morphology, displayed intense staining of axons with GAP-43 (arrows) and scattered areas of co-localization within cell bodies (arrowheads). E, Saponin pretreatment reveals that GAP-43 co-localizes with Rab5, an endosomal marker. Scale bar: A, B, D, 10 mm; E, 5 mm.
Fig. 5.

A–C, GAP-43 immunofluorescence is present in soma of noninfected cells, where it is partially co-localized with the Golgi apparatus marker mannosidase II.A, GAP-43 immunofluorescence; B, mannosidase immunofluorescence; C, dual labeling showing superimposition of the images in A and B. The arrow in B points to perinuclear Golgi body staining, also seen as an area of strong overlap inC. D–F, GAP-43 and rabaptin-5 co-localize in neuronal growth cones. GAP-43 is represented bygreen fluorescence and rabaptin-5 by redfluorescence. Arrows indicate heaviest rabaptin-5 immunoreactivity in body of growth cones, proximal to leading edge and filopodia. Scale bar: A–E, 10 μm; F, 5 μm.
Interestingly, expression of GAP-43 was also evident in Golgi bodies (Fig. 5A–C, arrow), where it also co-localized with rabaptin-5 (data not shown). The two proteins were also found together in growth cones (Fig. 5D–F). The heaviest rabaptin-5 immunoreactivity was in the body of growth cones, proximal to the leading edge and filopodia.
Effect of overexpression of GAP-43 on endosome size
Overexpression of rabaptin-5 in cell lines causes the accumulation of enlarged endosomes (Stenmark et al., 1995), suggesting that its interaction with Rab5 modulates fusion and expansion of early endosomes. Therefore, we asked whether overexpression of GAP-43 in cells would affect endosome size. PC12 cells or primary neurons infected with HSV vectors expressing GAP-43 were dual-labeled with antibodies to GAP-43 and Rab5. The sizes of Rab5-positive structures were quantified in those cells that were brightly labeled with the antibody to GAP-43. As indicated in Table1, endosomes were 16% smaller in diameter (representing a 41% decrease in volume) in primary neurons, and 8% smaller in diameter in PC12 cells overexpressing GAP-43 via an HSV vector than in cells infected with the control vector HSVlac. These data suggest that if the GAP-43 interaction with rabaptin-5 plays a functional role in endocytosis, it may act at an earlier stage than the fusion and expansion of the endosomes.
GAP-43 is associated with calmodulin in neurons (Gamby et al., 1996b). This interaction is regulated by intracellular Ca2+concentration and phosphorylation, and GAP-43 regulation of neurotransmitter release is dependent on this interaction (Gamby et al., 1996b). Mutagenesis of Ser 41 abolishes phosphorylation of GAP-43 by protein kinase C (Nielander et al., 1990) and causes it to retain calmodulin in the presence of increasing Ca2+concentrations (Meiri et al., 1996). We expressed such a mutant, termed G19, in primary neurons and quantified the sizes of Rab5-positive endosomes (Table 1) in HSV/G19-infected neurons. Average endosome size in these cells was larger than that in HSV/GAP-infected neurons and was not significantly different from that in control HSV/Lac-infected cultures, suggesting that the role played by GAP-43 in endocytosis is dependent on its ability to release calmodulin. Expression of a GAP-43 mutant (CAM) deleted for the calmodulin binding domain resulted in a reduction of average endosome size that exceeded that caused by overexpression of wild-type GAP-43. The histograms shown in Table 1demonstrate that the smaller average endosome size in cells expressing GAP-43 or the CAM mutant reflects a shift in the overall size distribution of the endosomes. These results indicate that GAP-43 has a specific effect on endosome size that is altered by mutations modulating its ability to bind calmodulin.
Participation of GAP-43 in an early stage of endocytosis
To examine the question of whether GAP-43 participates actively in an earlier stage of endocytosis than the fusion of the endosomes, we performed electron microscopy on neurons that were infected with HSV/GAP-43 or HSV/Lac and 16 hr later were exposed to colloidal gold-BSA in the medium for 8 min at 37°C to label early endosomes (Griffiths et al., 1989). The number of gold particles in randomly selected endosomes was counted and analyzed (Fig.6), revealing that endosomes in the HSV/GAP-43-infected neurons contained, on the average, more grains than did endosomes in the control cultures. These data suggest that overexpression of GAP-43 in neurons enhances the uptake of particles into endosomes.
Fig. 6.

Histogram showing that the number of gold particles/ endosome in HSV/GAP-43-infected neurons is greater than it is in HSV/Lac-infected neurons. The difference is significant:p < 0.035 by the one-tailed ttest.
Regulation of synaptic vesicle recycling by GAP-43
We and others (Dekker et al., 1991; Hens et al., 1993, 1995; Ivins et al., 1993; Kumagai-Tohda et al., 1993; Gamby et al., 1996a,b) have shown that GAP-43 modulates activity-dependent neurosecretion. The results presented herein suggest that GAP-43 may be involved in the endocytic stage of neurotransmitter release, when vesicular membrane is retrieved for reuse (Ceccarelli et al., 1973; Heuser and Reese, 1973). To test this possibility, we examined the effect of overexpression of GAP-43 on synaptic vesicle recycling (Fig.7) using the fluorescent membrane dye FM1–43 (Betz and Bewick, 1992; Betz et al., 1992; Ryan and Smith, 1995). When vesicular membrane internalization occurs in the presence of FM1–43, the dye is carried into the cell inside the endocytosed vesicles. When subsequent exocytosis occurs, the dye is expelled from the cell.
Fig. 7.

GAP-43 and its mutants exert differential effects on synaptic vesicle recycling as measured with FM1–43. HSV recombinants for each infection are listed on the left. Each column represents a different stimulation paradigm: low K+ followed by low K+, high K+ followed by low K+, and high K+ followed by high K+. Note that each stimulation paradigm for a given recombinant uses a different coverslip of neurons. The CAM mutation deletes the calmodulin binding domain, the G19 mutation changes Ser 41 to a Gly, and the Asp mutation changes Ser 41 to an Asp.
Primary cortical neurons were infected with HSV/Lac or HSV/GAP-43. In the presence of FM1–43, the cultures were treated with low K+ twice, with high K+ and then low K+, or with high K+ twice (Fig. 7, top two rows). In control HSV/Lac-infected cultures (and in mock-infected cultures; data not shown), only faint background labeling of neurons with FM1–43 was detected in the basal state (low K/low K). Depolarization of the neurons with high K+(high K/low K) caused the appearance of bright fluorescent spots in cells throughout the field, whereas a second high K+treatment (high K/high K) caused the loss of the fluorescence from the cells, presumably caused by exocytosis of stained synaptic vesicles, because it does not occur when the cells are maintained in low K+ after the depolarization with high K+ (high K/low K). In HSV/GAP-43-infected cultures, FM1–43 fluorescence was high even in cultures exposed only to low K+ (low K+/low K+). Depolarization of the cells with high K+ caused a robust increase in fluorescence that exceeded that of control cultures, and a second treatment with high K+ caused a return of the fluorescence to basal levels. These data suggest that active internalization of membranes occurs in these cells even in the resting state and is enhanced when the cells are depolarized and internalization of recycling synaptic vesicle membranes occurs.
We showed above that mutations in GAP-43 that modulate its ability to bind calmodulin have differential effects on endosome size. We tested the effect of these same mutants on synaptic vesicle recycling as measured by FM1–43 fluorescence (Fig. 7). Expression of the G19 mutant, in which Ser 41 was replaced by Gly and which retains calmodulin in the presence of increasing Ca2+concentrations, resulted in a high basal level of FM1–43 fluorescence. However, when the HSV/G19-infected neurons were depolarized with high K+, their fluorescence did not increase and was comparable to that of controls, whereas a second treatment with high K+ caused a decrease of the fluorescence to basal levels that was similar in intensity to that of control HSV/Lac-infected cultures. Expression of the CAM mutant, in which the calmodulin binding domain is deleted, enhanced FM1–43 fluorescence in a manner comparable to wild-type GAP. We also tested a point mutant of GAP-43 (Asp) in which Ser 41 was replaced by Asp, mimicking the phosphorylated state of that Ser. Binding of calmodulin to GAP-43 has been shown to be abolished by this mutation (Chapman et al., 1991). Expression of the Asp mutant in neurons enhanced FM1–43 fluorescence just as expression of the CAM mutant did, but in contrast, the second high-K+ treatment eliminated detectable fluorescence, suggesting that exocytosis is enhanced relative to controls in these cultures.
DISCUSSION
GAP-43 has been linked to multiple signal transduction pathways, and it is known that GAP-43 regulates neurotransmitter release and neurite outgrowth. However, we know very little about how the involvement of GAP-43 in each signal transduction pathway is related to these two functions. Taken together, the data presented here suggest that endocytic events involving GAP-43 and rabaptin-5 underlie the dynamic membrane alterations required for release of neurotransmitter and growth cone extension. It is of particular interest that overexpression of GAP-43 in neurons causes a decrease in the size of Rab5-containing endosomes, accompanied by an apparent acceleration of uptake of material into endosomes and of synaptic vesicle recycling. These data implicate GAP-43 in an earlier step of endocytosis than the fusion and expansion of endosomes that is mediated by the recruitment of rabaptin-5 by activated Rab5 (Stenmark et al., 1995). GAP-43 binds to rabaptin-5 within its C-terminal 311 amino acids, the region that also contains the site of interaction of rabaptin-5 with Rab5 (Stenmark et al., 1995). The interactions of GAP-43 and activated Rab5 with rabaptin-5 may be mutually exclusive and may constitute a molecular switch regulated by calmodulin binding to GAP-43. We have demonstrated that endosomes are smaller in neurons expressing a mutant of GAP-43 (CAM) that cannot bind calmodulin than in neurons expressing wild-type GAP-43, whereas endosomes are larger in neurons expressing a mutant of GAP-43 (G19) that constitutively binds to calmodulin. Our data support a model in which calmodulin binding to GAP-43 imposes a negative regulation, preventing GAP-43 interaction with rabaptin-5, which occurs when calmodulin is released by GAP-43. We propose that the internalization of vesicles by GAP-43-facilitated endocytosis progresses to fusion of the vesicles with each other under the control of activated Rab5. Rabaptin-5, in such a scenario, would participate both in an early step of endocytosis and also in endocytic membrane fusion, dependent on its interaction with GAP-43 or Rab5, respectively.
These molecules may play similar roles in the cell body, where they localize together in the Golgi. Others have observed immunoreactivity for GAP-43 in the Golgi (Van Hooff et al., 1989; Goslin et al., 1990; Van Lookeren Campagne et al., 1992; Liu et al., 1994) and have indicated that passage through the Golgi apparatus may be an obligatory step for the transport of GAP-43 into axons. However, GAP-43 and rabaptin-5 may do more than simply pass through this structure. An emerging view of the transport of proteins from the endoplasmic reticulum to the Golgi is that it resembles the dynamics of endocytosis (Presley et al., 1997), suggesting that rabaptin-5 and GAP-43 may play a functional role in these structures as well as at the presynaptic terminal.
Mutants of GAP-43 that we analyzed (Table2) have been assessed previously with respect to their phenotypic behavior in neuronal process outgrowth (Widmer and Caroni, 1993; Aigner et al., 1995). A Ser 41-to-Ala mutant (which binds calmodulin in a calcium-independent manner and is equivalent to the G19 mutant described herein) and a Ser 41-to-Asp mutant (which does not bind calmodulin and is identical to our Asp mutant) have opposite effects on cell morphology in vitro (Widmer and Caroni, 1993) and on neurite outgrowthin vivo. Expression of the Ser-to-Ala mutant in cell lines produced small, poorly spreading cells with numerous short filopedia, and in transgenic mice it caused less nerve sprouting than did wild-type GAP-43. Our equivalent mutant caused an increase in endosome size. In contrast, cells expressing the Ser-to-Asp mutant displayed large membraneous extensions with few filopedia and had a sprouting activity comparable to wild type in vivo. Our CAM mutant, which has the same effect in inhibiting binding of GAP-43 to calmodulin, caused a decrease in endosome size. Although it is not clear how these phenotypes correlate mechanistically, it is notable that these mutants have opposite effects on cell morphology, just as they have opposite effects on endosome size.
Table 2.
Summary of phenotypic effects of GAP-43 mutations
| Mutation | Functional consequences | |
|---|---|---|
| G19 | Ser 41 → Gly | Cannot be phosphorylated by PKC |
| (equivalent to Ser 41 → Ala) | Binds calmodulin constitutively | |
| Enlarged endosomes | ||
| Deficient in depolarized-induced FM1-43 fluorescence | ||
| CAM | Delete calmodulin | Cannot bind calmodulin |
| binding domain | Smaller endosomes | |
| Enhancement of depolarization-induced FM1-43 fluorescence | ||
| Asp | Ser 41 → Asp | Mimics phosphorylation of Ser 41 by PKC |
| Cannot bind calmodulin | ||
| Enhancement of depolarization-induced FM1-43 fluorescence | ||
| Greater depletion of FM1-43 fluorescence after second high-K+treatment |
Neurons overexpressing wild-type GAP-43 show enhanced FM1–43 fluorescence, both in the basal state and after depolarization with high K+, relative to control neurons infected with HSV/Lac or mock-infected. Neurons expressing the G19 mutant, which cannot be phosphorylated by PKC and which binds calmodulin in a Ca2+-independent manner, do not show enhanced FM1–43 fluorescence relative to HSV/Lac-infected controls when they are depolarized, and a second treatment with high K+is not as effective in depleting FM1–43 fluorescence in these cells as in controls. Thus, this mutant seems to be defective in regulating both activity-dependent vesicular membrane internalization and exocytosis. Neurons expressing the CAM mutant, which is deleted for the calmodulin binding domain and does not bind calmodulin, show enhanced FM1–43 fluorescence relative to HSV/Lac-infected controls on depolarization with high K+, and partial depletion of FM1–43 fluorescence when treated with high K+ a second time. The Asp mutant mimics PKC-phosphorylated GAP-43 and does not bind calmodulin. Cultures infected with HSV/Asp, like those infected with HSV/CAM, show enhanced FM1–43 fluorescence relative to HSV/Lac-infected controls on depolarization with high K+, but exhibit much greater depletion of FM1–43 fluorescence than HSV/CAM-infected cultures when treated with high K+ a second time, suggesting enhanced activity-dependent exocytosis.
The increased basal FM1–43 fluorescence relative to HSV/Lac-infected cultures that is caused by all of the mutants in addition to wild-type GAP-43 suggests that GAP-43 participates in a type of membrane internalization that is independent of its interactions with calmodulin. However, the mutants do differ from each other in depolarization-induced FM1–43 fluorescence, indicating that that step of neurosecretion is dependent on GAP-43 interaction with calmodulin. In addition, the CAM and Asp mutants, neither of which can bind calmodulin but only one of which in addition mimics phosphorylation by PKC, have differential effects on depletion of FM1–43 fluorescence after the second high-K+ treatment of the neurons. These data suggest that PKC phosphorylation of GAP-43 (mimicked by the Asp mutant) plays more of a role in GAP-43 function than simply causing GAP-43 to dissociate from calmodulin. For example, it may enhance interaction of GAP-43 with rabaptin-5. It will be important to measure the ability of each of these mutants to bind to rabaptin-5, and to determine the effect of overexpression of rabaptin-5 on synaptic vesicle recycling, to decipher the distinct roles of rabaptin-5 and calmodulin relative to GAP-43 in neurosecretion and neurite outgrowth.
Footnotes
Received May 15,1998; revised July 20, 1998; accepted July 23, 1998.
This work was supported by a grant from National Institutes of Health (National Institute of Child Health and Human Development) to R.L.N. We thank Dr. Marianne Wessling-Resnick for helpful discussions, Drs. Frederick Boyce and Kathleen Buckley for critical reading of this manuscript, Dr. Lawrence Baizer for his gift of the GAP-43 polyclonal antibody, Kathleen Buckley for her gift of the SV2 antibody, Dr. Anne Cataldo for technical advice, and Bartek Konieczny for photographic work.
R.L.N. and R.C. contributed equally to this paper.
Correspondence should be addressed to Rachael L. Neve, 202 Mailman Research Center, McLean Hospital, 115 Mill Street, Belmont, MA 02178.
REFERENCES
- 1.Aigner L, Caroni P. Depletion of 43-kD growth-associated protein in primary sensory neurons leads to diminished formation and spreading of growth cones. J Cell Biol. 1993;123:417–429. doi: 10.1083/jcb.123.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander KA, Cimler BM, Meier KE, Storm DR. Regulation of calmodulin binding to P-57. J Biol Chem. 1987;262:6108–6113. [PubMed] [Google Scholar]
- 3.Andreasen TJ, Luetje CW, Heiderman W, Storm DR. Purification of a novel calmodulin binding protein from bovine cerebral cortex membranes. Biochemistry. 1983;22:4615–4618. doi: 10.1021/bi00289a001. [DOI] [PubMed] [Google Scholar]
- 4.Betz WJ, Bewick GS. Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- 5.Betz WJ, Mao F, Bewick WJ. Activity dependent staining and destaining of living motor nerve terminals. J Neurosci. 1992;12:363–375. doi: 10.1523/JNEUROSCI.12-02-00363.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bucci C, Parton RG, Mather IH, Stunnenberg H, Simons K, Hoflack B, Zerial M. The small GTPase rab5 functions as a regulatory factor in the early endocytic pathway. Cell. 1992;70:715–728. doi: 10.1016/0092-8674(92)90306-w. [DOI] [PubMed] [Google Scholar]
- 7.Buckley KM, Kelly RB. Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J Cell Biol. 1985;100:1284–1294. doi: 10.1083/jcb.100.4.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlezon WA, Jr, Boundy VA, Haile CN, Kalb RG, Neve RL, Nestler EJ. Sensitization to morphine induced by viral-mediated gene transfer. Science. 1997;277:812–814. doi: 10.1126/science.277.5327.812. [DOI] [PubMed] [Google Scholar]
- 9.Ceccarelli B, Hurlbut WP, Mauro A. Turnover of transmitter and synaptic vesicles at the frog neuromuscular junction. J Cell Biol. 1973;57:499–524. doi: 10.1083/jcb.57.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chapman ER, Au D, Alexander KA, Nicolson TA, Storm DR. Characterization of the calmodulin binding domain of neuromodulin. Functional significance of serine 41 and phenylalanine 42. J Biol Chem. 1991;266:207–213. [PubMed] [Google Scholar]
- 11.Chow N, Korenberg JR, Chen X-N, Neve RL. APP-BP1, a novel protein that binds to the carboxyl-terminal region of the amyloid precursor protein. J Biol Chem. 1996;271:11339–11346. doi: 10.1074/jbc.271.19.11339. [DOI] [PubMed] [Google Scholar]
- 12.De Graan PNE, Van Hooff COM, Tilly BC, Oestreicher AB, Schotman P, Gispen WH. Phosphoprotein B-50 in nerve growth cones from fetal rat brain. Neurosci Lett. 1989;61:235–241. doi: 10.1016/0304-3940(85)90470-7. [DOI] [PubMed] [Google Scholar]
- 13.De Graan PNE, Oestreicher AB, De Wit M, Kroef M, Schrama LH, Gispen WH. Evidence for the binding of calmodulin to endogenous B-50 (GAP-43) in native synaptosomal plasma membranes. J Neurochem. 1990;55:2139–2141. doi: 10.1111/j.1471-4159.1990.tb05808.x. [DOI] [PubMed] [Google Scholar]
- 14.Dekker LV, De Graan PNE, Versteeg DHG, Oestreicher AB, Gispen WH. Phosphorylation of B-50 (GAP43) is correlated with neurotransmitter release in rat hippocampal slices. J Neurochem. 1989;52:24–30. doi: 10.1111/j.1471-4159.1989.tb10893.x. [DOI] [PubMed] [Google Scholar]
- 15.Dekker LV, De Graan PNE, Pijnappel P, Oestreicher AB, Gispen WH. Noradrenaline release from streptolysin O-permeated rat cortical synaptosomes: effects of calcium, phorbol esters, protein kinase inhibitors, and antibodies to the neuron-specific protein kinase C substrate B-50 (GAP-43). J Neurochem. 1991;56:1146–1153. doi: 10.1111/j.1471-4159.1991.tb11404.x. [DOI] [PubMed] [Google Scholar]
- 16.Gamby C, Waage MC, Allen RG, Baizer L. Growth-associated protein-43 (GAP-43) facilitates peptide hormone secretion in mouse anterior pituitary AtT-20 cells. J Biol Chem. 1996a;17:10023–10028. doi: 10.1074/jbc.271.17.10023. [DOI] [PubMed] [Google Scholar]
- 17.Gamby C, Waage MC, Allen RG, Baizer L. Analysis of the role of calmodulin binding and sequestration in neuromodulin (GAP-43) function. J Biol Chem. 1996b;271:26698–26705. doi: 10.1074/jbc.271.43.26698. [DOI] [PubMed] [Google Scholar]
- 18.Gianotti C, Nunzi MG, Gispen WH, Corradetti R. Phosphorylation of the presynaptic protein B-50 (GAP-43) is increased during electrically induced long-term potentiation. Neuron. 1992;8:843–848. doi: 10.1016/0896-6273(92)90198-m. [DOI] [PubMed] [Google Scholar]
- 19.Goslin K, Schreyer DJ, Skene JHP, Banker GA. Changes in the distribution of GAP-43 during the development of neuronal polarity. J Neurosci. 1990;10:588–602. doi: 10.1523/JNEUROSCI.10-02-00588.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gray E, Whittaker V. The isolation of nerve endings from brain: an electron-microscopic study of cell fragments derived by homogenization and centrifugation. J Anat. 1962;96:79–87. [PMC free article] [PubMed] [Google Scholar]
- 21.Griffiths G, Back R, Marsh M. A quantitative analysis of the endocytic pathway in baby hamster kidney cells. J Cell Biol. 1989;109:2703–2720. doi: 10.1083/jcb.109.6.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hens JJH, Ghijsen WEJM, Dimjati W, Wiegant VM, Oestreicher AB, Gispen WH, De Graan PNE. Evidence for a role of protein kinase C substrate B-50 (GAP-43) in Ca2+-induced neuropeptide cholecystokinin-8 release from permeated synaptosomes. J Neurochem. 1993;61:602–609. doi: 10.1111/j.1471-4159.1993.tb02164.x. [DOI] [PubMed] [Google Scholar]
- 23.Hens JJH, De Wit M, Boomsma F, Mercken M, Oestreicher AB, Gispen WH, De Graan P. N-terminal-specific anti-B-50 (GAP-43) antibodies inhibit Ca2+-induced noradrenaline release, B-50 phosphorylation and dephosphorylation, and calmodulin binding. J Neurochem. 1995;64:1127–1136. doi: 10.1046/j.1471-4159.1995.64031127.x. [DOI] [PubMed] [Google Scholar]
- 24.Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivins KJI, Neve KA, Feller DJ, Fidel SA, Neve RL. Antisense GAP-43 inhibits the evoked release of dopamine from PC12 cells. J Neurochem. 1993;60:626–633. doi: 10.1111/j.1471-4159.1993.tb03194.x. [DOI] [PubMed] [Google Scholar]
- 26.Katz F, Ellis L, Pfenninger KH. Nerve growth cones isolated from fetal rat brain. III. Calcium-dependent protein phosphorylation. J Neurosci. 1985;5:1402–1411. doi: 10.1523/JNEUROSCI.05-06-01402.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumagai-Tohda C, Tohda M, Nomura Y. Increase in neurite formation and acetylcholine release by transfection of growth-associated protein-43 cDNA into NG108–15 cells. J Neurochem. 1993;61:526–532. doi: 10.1111/j.1471-4159.1993.tb02155.x. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Storm DR. Regulation of free calmodulin levels by neuromodulin: neuron growth and regeneration. Trends Pharmacol Sci. 1990;11:107–111. doi: 10.1016/0165-6147(90)90195-e. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Fisher DA, Storm DR. Intracellular sorting of neuromodulin (GAP-43) mutants modified in the membrane targeting domain. J Neurosci. 1994;14:5807–5817. doi: 10.1523/JNEUROSCI.14-10-05807.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meiri KF, Pfenninger KH, Willard MB. Growth-associated protein, GAP-43, a polypeptide that is induced when neurons extend axons, is a component of growth cones and corresponds to pp46, a major polypeptide of a subcellular fraction enriched in growth cones. Proc Natl Acad Sci USA. 1986;83:3537–3541. doi: 10.1073/pnas.83.10.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meiri KJ, Hammang JP, Dent EW, Baetge EE. Mutagenesis of ser41 to ala inhibits the association of GAP-43 with the membrane skeleton of GAP-43-deficient PC12B cells: effects on cell adhesion and the composition of neurite cytoskeleton and membrane. J Neurobiol. 1996;29:213–232. doi: 10.1002/(SICI)1097-4695(199602)29:2<213::AID-NEU7>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 32.Nelson RB, Linden DJ, Hyman C, Pfenninger KH, Routtenberg A. The two major phosphoproteins in growth cones are probably identical to two protein kinase C substrates correlated with persistence of long-term potentiation. J Neurosci. 1989;9:381–389. doi: 10.1523/JNEUROSCI.09-02-00381.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neve RL, Neve KA. Receptor expression in mammalian cells. In: Sealfon SC, editor. Receptor molecular biology, methods in neuroscience, Vol 25. Academic; San Diego, CA: 1995. pp. 163–174. [Google Scholar]
- 34.Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TD. Identification of cDNA clones for the human microtubule associated protein tau and chromosomal localization of the genes for tau and microtubule associated protein 2. Mol Brain Res. 1986;1:271–280. doi: 10.1016/0169-328x(86)90033-1. [DOI] [PubMed] [Google Scholar]
- 35.Nielander HB, Schrama LH, van Rozen AJ, Kasperaitis M, Oestreicher AB, Gispen WH, Schotman P. Mutation of serine 41 in the neuron-specific protein B-50 (GAP-43) prohibits phosphorylation by protein kinase C. J Neurochem. 1990;55:1442–1445. doi: 10.1111/j.1471-4159.1990.tb03159.x. [DOI] [PubMed] [Google Scholar]
- 36.Nishimoto I, Okamoto T, Matsuura Y, Okamoto T, Murayama Y, Ogata E. Alzheimer amyloid protein precursor forms a complex with brain GTP binding protein Go. Nature. 1993;362:75–79. doi: 10.1038/362075a0. [DOI] [PubMed] [Google Scholar]
- 37.Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. ER-to-Golgi transport visualized in living cells. Nature. 1997;389:81–85. doi: 10.1038/38001. [DOI] [PubMed] [Google Scholar]
- 38.Ron D, Dressler H. pGSTag: a versatile bacterial expression plasmid for enzymatic labeling of recombinant proteins. BioTechniques. 1992;13:866–869. [PubMed] [Google Scholar]
- 39.Ryan TA, Smith SJ. Vesicle pool mobilization during action potential firing at hippocampal synapses. Neuron. 1995;14:983–989. doi: 10.1016/0896-6273(95)90336-4. [DOI] [PubMed] [Google Scholar]
- 40.Skene JHP, Jacobson RD, Snipes GJ, McGuire CB, Norden J, Freeman JA. A protein induced during nerve growth (GAP-43) is a major component of growth-cone membranes. Science. 1986;233:783–785. doi: 10.1126/science.3738509. [DOI] [PubMed] [Google Scholar]
- 41.Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- 42.Stenmark H, Vitale G, Ullrich O, Zerial M. Rabaptin-5 is a direct effector of the small GTPase Rab5 in endocytic membrane fusion. Cell. 1995;15:423–432. doi: 10.1016/0092-8674(95)90120-5. [DOI] [PubMed] [Google Scholar]
- 43.Van Hooff COM, Holthuis J, Oestreicher AB, Boonstra J, De Graan PNE, Gispen WH. Nerve growth factor-induced changes in the intracellular localization of the protein kinase C substrate B-50 in pheochromocytoma PC12 cells. J Cell Biol. 1989;108:1115–1125. doi: 10.1083/jcb.108.3.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Lookeren Campagne M, Dotti CG, Verkleij AJ, Gispen WH, Oestreicher AB. B-50/GAP43 localization on membranes of putative transport vesicles in the cell body, neurites and growth cones of cultured hippocampal neurons. Neurosci Lett. 1992;137:129–132. doi: 10.1016/0304-3940(92)90314-w. [DOI] [PubMed] [Google Scholar]
- 45.Whittaker VP, Michaelson IA, Kirkland RJA. The separation of synaptic vesicles from nerve-ending particles (“synaptosomes”). Biochem J. 1964;90:293–303. doi: 10.1042/bj0900293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Widmer F, Caroni P. Phosphorylation-site mutagenesis of the growth-associated protein GAP-43 modulates its effects on cell spreading and morphology. J Cell Biol. 1993;120:503–512. doi: 10.1083/jcb.120.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yankner BA, Dawes LR, Fisher S, Villa-Komaroff L, Oster-Granite ML, Neve RL. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer’s disease. Science. 1989;245:417–420. doi: 10.1126/science.2474201. [DOI] [PubMed] [Google Scholar]