

Abstract
It is well established that the microtubules of the mitotic spindle are organized by a variety of motor proteins, and it appears that the same motors or closely related variants organize microtubules in the postmitotic neuron. Specifically, cytoplasmic dynein and the kinesin-related motor known as CHO1/MKLP1 are used within the mitotic spindle, and recent studies suggest that they are also essential for the establishment of the axonal and dendritic microtubule arrays of the neuron. Other motors are required to tightly regulate microtubule behaviors in the mitotic spindle, and it is attractive to speculate that these motors might also help to regulate microtubule behaviors in the neuron. Here we show that a homolog of the mitotic kinesin-related motor known as Eg5 continues to be expressed in rodent neurons well after their terminal mitotic division. In neurons, Eg5 is directly associated with the microtubule array and is enriched within the distal regions of developing processes. This distal enrichment is transient, and typically lost after a process has been clearly defined as an axon or a dendrite. Strong expression can resume later in development, and if so, the protein concentrates within newly forming sprouts at the distal tips of dendrites. We suggest that Eg5 generates forces that help to regulate microtubule behaviors within the distal tips of developing axons and dendrites.
Keywords: microtubule, neuron, Eg5, axon, dendrite, motor protein
Microtubules are essential for the differentiation of axons and dendrites. Throughout the axon and in the distal region of the dendrite, microtubules are uniformly oriented with their plus-ends distal to the cell body (Heidemann et al., 1981; Baas et al., 1989). In contrast, microtubules in the proximal and middle regions of the dendrite are nonuniformly oriented (Baas et al., 1988,1989). Given that the polarity of a microtubule is relevant to both its dynamic and transport properties, these distinct patterns could provide a basis for the morphological and compositional differences that distinguish axons and dendrites from one another (Black and Baas, 1989). Most efforts to understand how cells regulate their microtubule arrays have focused on dynamic events such as microtubule assembly, disassembly, and stabilization. However, it is now clear that cells have another powerful strategy for organizing their microtubules. Specifically, motor proteins can generate forces on microtubules, and thereby move them into specific locations within the cell and into specific orientations. This strategy is particularly important in cells such as neurons that must establish and regulate arrays of microtubules in locations far from their nucleation sites within the cell body. Recent studies suggest that microtubules are transported into axons and dendrites with the appropriate polarity orientations by the motor proteins known as cytoplasmic dynein and CHO1/MKLP1 (Sharp et al., 1997; Ferhat et al., 1998b; Ahmad et al., 1998).
Might other motor proteins generate forces on microtubules in the neuron, and if so, might such forces be relevant to axonal and dendritic differentiation? It is compelling to contemplate that the growth of a neuronal process might be modulated by antagonistic and complementary forces generated by a variety of motor proteins. Precedent for this scenario derives from the mitotic spindle, the formation and functioning of which involve a host of motor proteins that impose such forces on specific regions of the microtubule array (for review, see Walczak and Mitchison, 1996). In fact, the two motor proteins thus far implicated in the transport of neuronal microtubules, cytoplasmic dynein and CHO1/MKLP1, are known to play key roles in organizing microtubules during mitosis (Nislow et al., 1992; Heald et al., 1996).
Here we sought to determine whether postmitotic neurons express a homolog of the kinesin-related protein known as Eg5. This motor and related members of the bimC family are critical for generating forces on microtubules that separate the duplicated centrosomes or spindle poles early in prophase (Enos and Morris, 1990;LeGuellec et al., 1991; Hoyt et al., 1992; Roof et al., 1992; Hagan and Yanagida, 1992; Sawin et al., 1992, Sawin and Mitchison, 1995; Blangy et al., 1995; Barton et al., 1995). In addition, these motors may help organize the bipolar spindle later in mitosis by providing counterforces to those generated by cytoplasmic dynein (Gaglio et al., 1996). Our studies demonstrate that rodent neurons express a homolog of Eg5 well past their terminal mitotic division, and that this protein is localized in discrete and functionally important regions of developing neuronal processes.
MATERIALS AND METHODS
cDNA library screening. A cDNA library constructed by H. Okayama (unpublished data) using mRNA from MCA16 cells (C3H10T1/2 mouse cells transformed by 3-methylcholanthrene; Shih et al., 1979) was screened with a 32P-labeled 782 bp PCR fragment (106 cpm/ml) coding for the motor domain of HsEg5 (human Eg5; nt 327–1109, accession number X85137; Blangy et al., 1995). A total of 1.5 × 105 colonies were transferred to nitrocellulose filters (Schleicher and Schuell, Keene, NH) and hybridized at 60°C for 24 hr in a hybridization buffer (6× SSC, 0.5× Denhardt’s solution, 0.1% SDS). Filters were washed four times at 60°C for 15 min in a solution containing 6× SSC and 0.5% SDS. Positive colonies were purified, and inserts were subcloned into pBluescript KS+ plasmid (Stratagene, La Jolla, CA). The remaining 120 bp at the 5′ end of the cDNA were obtained with a RT-PCR-based method using mRNA from mouse L cells primed with 5′ primer (5′-ATCTCGAGAACCATGGCGTCCCAGCCGAGTTC-3′) derived from genomic sequences and the 3′ primer (5′-CTCAACAATTTGTTCCTCCTG-3′) derived from cDNA sequence corresponding to amino acids (aa) 414–420. Nucleotide sequence determination was performed by the dideoxy chain termination method, using specific oligonucleotides as primers. Sequence data treatment was performed using computer facilities at the Pôle de Bioinformatique de Villejuif (Dessen et al., 1990). The cDNA nucleotide sequence encoding mouse Eg5 (termed MmEg5, Mus musculus) reported in this paper has been submitted to the European Molecular Biology Laboratory/GenBank data bank under accession number AJ223293.
Recombinant vector constructions. The XhoI fragment of the longest cDNA clone was inserted into pBluescript KS+ plasmid (Stratagene) at the SalI site (pBSEg5). Plasmid pBSEg5S containing the stalk domain and part of the tail (aa 349–881) of MmEg5 was obtained by digestion of pBSEg5 by EcoRI andBglII restriction enzymes and religated. For removal of the 3′ UTR containing repetitive sequences, the plasmid pBSEg5S was digested by EcoRV and XhoI and religated pBSEg5S(-R). For Northern blot analyses, the double-stranded, 1.6 kb-purified cDNA insert encoding the stalk domain and part of the tail, obtained by digestion of pBSEg5S9(-R) with XbaI andApaI restriction enzymes, was labeled with [α-32P]dCTP as described below.
mRNA isolation and Northern blot analyses. Total RNA from mouse whole embryos at embryonic day 10.5 (E10.5), E11.5, E13.5, and E15.5 and from mouse brains at postnatal day 0 (P0), P7, P14, P21, and adult, respectively, was purified by the Trizol (Life Technologies, Grand Island, NY) extraction method as described in the manufacturer’s protocol. After isopropanol RNA precipitation, pellets were washed with 75% ethanol and resuspended in diethylpyrocarbonate-treated water. Aliquots of the RNA were used for quantification by optical density scanning (210–320 nm), and the integrity of the extracted RNA was confirmed by running 2 μg total RNA on a denaturing (formaldehyde 2.2 m) agarose gel (1%) in 1× MAE buffer (in mm: 20 4-morpholinepropanesulfonic acid, pH 7.0, and 8 sodium acetate, 1 EDTA, pH 8.0). For Northern blot analysis, RNA (30 μg/lane) was separated on a 1% agarose formaldehyde gel and capillary-transferred with 10× SSC onto noncharged nylon membrane (Micron Separations, Inc., Westborough, MA). The 1.6 kb Eg5 cDNA probe described above was labeled with [α-32P]dCTP to >109 cpm per μg of DNA using klenow enzyme and a random hexanucleotide kit (Promega, Madison, WI). The blots were hybridized using 2.5 × 106 cpm/ml labeled probe in QuickHyb (Stratagene) according to the manufacturer’s protocol. The blots were washed with 2× SSC, 0.1% SDS at room temperature for 10 min (twice), and then at high stringency at 68°C with 0.1× SSC, 0.1% SDS for 15 min (twice), as recommended in the manufacturer’s protocol. Finally, the washed membranes were directly exposed at −70°C to X-Omat AR film (Eastman Kodak, Rochester, NY) with two intensifying screens for 7 d.
Animal dissection and tissue preparation. For all of the studies presented here, we used samples obtained from rodents. For the studies on MmEg5 expression in vivo, we used mice because the clone was isolated from mouse cells (Shih et al., 1979). We reasoned that using mice would optimize the signal-to-noise ratio in the in situ hybridization analyses. For the studies on prenatal animals, pregnant mice were euthanized, and embryos were removed by caesarean section on E10.5, E11.5, E13.5, or E15.5. Postnatal studies were performed on animals at ages P0, P7, P14, P21, and adult (ad). The whole embryos were rapidly removed and dissected from the amniotic membrane in ice-cold 1× PBS, pH 7.4, and fixed overnight at 4°C in freshly prepared cold 4% paraformaldehyde (PFA). The embryos were then rinsed in 1× PBS, dehydrated through an ascending ethanol series, embedded in paraffin (Paraplast; Oxford Labware, St. Louis, MO), and stored at room temperature. The postnatal animals (from P0 pups to adult) were decapitated, and their brains were rapidly removed and treated as described above and then stored at room temperature until needed. Sagittal sections (6 μm) of the whole embryos and brains of P0 pups to adults were cut, mounted onto gelatin-coated slides, and then kept desiccated at room temperature until used.
Cell cultures. For most of our studies on cultured neurons, we obtained the neuronal tissue from rats, because cultures of rat hippocampal and sympathetic neurons are well characterized, and also our studies showed sufficient cross-reactivity of cultured rat neurons with the mouse probe and the affinity-purified polyclonal antibody against the motor domain of HsEg5 described below to provide good signal-to-noise ratio. Cultures of embryonic rat hippocampal neurons were prepared as previously described (Goslin and Banker, 1991; Sharp et al., 1995). Briefly, hippocampi were dissected from 18 d rat embryos, treated with trypsin for 15 min at 37°C, and triturated with fire-polished Pasteur pipettes. The cells were plated at a density of 1000 cells/cm2 onto glass coverslips coated with 1 mg/ml poly-d-lysine in Minimum Essential Medium (MEM, Life Technologies) containing 10% horse serum. After 2–4 hr, the coverslips plated with neurons were cocultured into plastic tissue-culture dishes containing a monolayer of astroglial cells. The astroglial cells had been grown in medium containing MEM and 10% fetal bovine serum. One day before coculture, the medium was changed to a fresh medium containing MEM, the N2 supplements described by Bottenstein (Goslin and Banker, 1991), 0.1% ovalbumin, and 0.01 mg/ml sodium pyruvate.
Cultures of sympathetic neurons from the superior cervical ganglia were prepared from newborn rat pups. After dissection, the ganglia were treated with 0.25% collagenase for 1 hr followed by 0.25% trypsin for 45 min, and then triturated with fire-polished Pasteur pipettes into a single cell dispersion as previously described (Baas and Ahmad, 1993). Before plating the cells, the glass coverslips were coated for 3 hr with 1 mg/ml poly-d-lysine, rinsed extensively, and then treated with 10 μg/ml laminin for 4 hr as described by Higgins et al. (1991). Cells were then plated in Leibovitz’s L15 medium (Sigma, St. Louis, MO) supplemented with 0.6% glucose, 2 mml-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10% fetal bovine serum, and 100 μg/ml nerve growth factor for 24 hr. For long-term culture, the medium was replaced the next morning by N2 medium (Baas and Ahmad, 1993) supplemented with 5% fetal bovine serum and 100 ng/ml nerve growth factor. Cytosine arabinoside was added at 10 μm to reduce the proliferation of non-neuronal cells.
For one set of studies, primary neuron cultures were generated from hamster cerebral cortex. The methods for generating these cultures have been described in detail (Szebenyi et al., 1998).
Cultures of mouse neuroblastoma cells (N2a) and human HeLa cells were maintained as previously described (Blangy et al., 1995; Yu et al., 1997).
In situ hybridization probes. In vitrotranscription of 35S-UTP- or digoxigenin-UTP-labeled MmEg5 riboprobes was performed from linearized pBSEg5S(-R) plasmids using an Ambion (Austin, TX) or Boehringer Mannheim (Indianapolis, IN) in vitro transcription kit, respectively, according to each manufacturer’s protocol. The sense and antisense riboprobes were prepared from the 1.6 kb mouse cDNA fragment cloned into pBSEg5S(-R) vector described above flanked by T3 and T7 promoters. The sense riboprobes (radioactive and nonradioactive) were transcribed in vitro from an ApaI linearized plasmid using T7 RNA polymerase purchased from Ambion and Boehringer Mannheim, respectively. The antisense riboprobes (radioactive and nonradioactive) were transcribed from a XbaI linearized plasmid using T3 RNA polymerase (Ambion).
In situ hybridization on brain sections. In situ hybridization was performed by a modification of the protocol of Lyons et al. (1996). Briefly, sections were deparaffinized in xylene, rehydrated through a descending ethanol series, fixed in 4% PFA in 1× PBS for 15 min, rinsed in 1× PBS, and treated with proteinase K (20 mg/ml, Boehringer Mannheim) for 7.5 min at room temperature. After post-fixation with 4% PFA for 5 min, acetylation in triethanolamine for 10 min, dehydration in 30, 50, 70, 85, 95, and 100% ethanol, and delipidation in chloroform for 5 min, the sections were prehybridized for 2 hr in 4× SSC buffer containing 50% formamide, 1× Denhardt’s solution, 300 μg/ml yeast RNA, 300 μg/ml salmon sperm DNA, and 100 mm dithiothreitol (DTT). The sections were hybridized with 5 × 105 cpm/100 μl of the antisense or sense riboprobe overnight at 50°C. The tissue was then rinsed three times in 2× SSC for 15 min at room temperature, treated with 20 μg/ml RNase A (Boehringer Mannheim), and finally washed in increasingly stringent conditions up to 0.1× SSC at 60°C for 30 min. All rinse and wash buffers contained 0.25 gm/ml sodium thiosulfate. The sections were processed for both film (Hyperfilm-βmax; Amersham, Arlington Heights, IL) and emulsion autoradiography (NTB2, Eastman Kodak), with exposure times of 30 d and 8 weeks, respectively. After development of emulsion autoradiograms, the sections were counterstained with cresyl violet and mounted with Permount. In the case of the film autoradiography, photographs were digitized by scanning the films. In the case of the emulsion autoradiography, photomicrographs were taken with a Zeiss Axiophot (Carl Zeiss Incorporated, Thornwood, NY) microscope equipped with dark-field illumination. Hybridization of adjacent sections with the sense riboprobe was used as a control.
In situ hybridization on primary neuron cultures. In situ hybridization was performed on cultured hippocampal and sympathetic neurons that had been grown on glass coverslips. The cells were fixed for 15 min at room temperature in 4% PFA in 1× PBS and dehydrated in graded alcohols (30, 50, 70, 85, 95, and 100%), after which they were hybridized with antisense or sense riboprobes that had been either radioactively or digoxygenin-labeled. In the case of the radioactively labeled probes, hybridization was performed overnight at 50°C with the same hybridization mixture described above using 5 × 105 cpm/100 μl of the sense or antisense riboprobe. Subseqent steps and visualization of the radioactive signal were performed as previously described (Ferhat et al., 1997, 1998a,b). In the case of the digoxygenin-labeled probes, hybridization was performed overnight at 50°C with the same hybridization using 7.5 ng/100 μl of the sense or antisense riboprobe. After hybridization, cells were rinsed, treated with RNase and then subjected to high stringency washes as described above. The cells were then washed twice for 10 min each in Tris-HCl buffer (100 mm Tris-HCl, pH 7.4, and 150 mm NaCl). After exposure for 30 min to a blocking solution containing 0.1% Triton X-100 and 2% normal sheep serum (Sigma) in Tris-HCl buffer, the cells were incubated overnight at 4°C with sheep antidigoxigenin alkaline phosphatase antibody (Boehringer Mannheim) diluted 1:1000 in blocking buffer. The coverslips were rinsed twice for 10 min in Tris-HCl buffer and then exposed for 10 min to color development buffer (in mm: 100 Tris-HCl, pH 9.5, 100 NaCl, and 50 MgCl2), after which they were incubated with Tris-HCl buffer substrate solution (100 mm Tris-HCl, pH 9.5, and 50 mm MgCl2) containing nitro-blue tetrazolium (NBT, 340 μg/ml) and bromochloroindolylyl phosphate (BCIP, 170 μg/ml). For reduction of the endogenous phosphatase activity, 5 mm levamisole was added to the color development buffer. The color signal was monitored by microscopy, and the reaction was stopped when a strong cellular signal was developed against a low background. After transferring them to buffer containing 10 mm Tris-HCl, pH 8.0, and 1 mmEDTA, the coverslips were washed twice for 10 min in distilled water, air-dried, and mounted in mounting aqueous solution. Cells were visualized, and photographs were taken using bright-field microscopy to reveal the reddish alkaline-phosphate reaction product.
Affinity purification of anti-Eg5 antibodies. AnEcoRI–BglII cDNA restriction fragment of 494 bp encoding 161 amino acid residues (17.9 kDa) of HsEg5 amino-terminal region was cloned downstream from the trpE gene into theEcoRI-BamHI of the pATH10 expression vector (Koerner et al., 1991). The Eg5 fusion protein was resolved by SDS-PAGE, purified, and injected into New Zealand white rabbits. Affinity-purified Eg5 motor antibodies were obtained by elution of Igs bound to the MalE–HsEg5 fusion protein. In brief, the MalE–HsEg5 fusion protein was resolved by SDS-PAGE and transferred to an Immobilon P filter (Millipore, Bedford, MA). The strip of Immobilon P filter that carried the HsEg5 protein was incubated with the polyclonal antibody for 16 hr at 4°C. After an extensive washing step, Igs bound to the protein were recovered by brief treatment with 0.1 mglycine, pH 2.8, followed by rapid neutralization with 0.1 volume of 1m Tris-HCl, pH 8. The antibody was stored at 4°C after addition of 5 mg of bovine serum albumin per milliliter (Sambrook et al., 1989).
Preparation of protein samples for Western blotting.Cultures were washed three times with 1× PBS, scraped, and homogenized at 4°C in (in mm:) 50 Tris-HCl, pH 7.5, 250 NaCl, 0.1% NP40, and 5 EDTA with 1 PMSF and 10 μg/ml each of aprotinin and leupeptin. Samples were centrifuged at 15,000 × g for 20 min at 4°C. Extracts were clarified by centrifugation at 15,000 × g for 30 min. Finally, protein concentrations of cultures and tissues extracts were determined by the DC protein assay (Bio-Rad, Hercules, CA) according to the manufacturer’s protocol.
SDS-PAGE and Western blotting. The protein samples were boiled for 10 min, and the same amounts were loaded into each well and resolved on 8% SDS–polyacrylamide gels. After electrophoresis, the proteins were transferred to nitrocellulose membranes (Micron Separations, Inc.). Blots were blocked with 5% nonfat dried milk and 0.2% Tween 20 in 1× PBS (PBS–milk) for 3 hr at room temperature and incubated overnight at 4°C in the Eg5 antibody described above at 1:1000 in PBS–milk. The membranes were washed six times for 15 min each with a solution containing 1× PBS and 0.1% Tween 20, incubated with horseradish peroxidase goat anti-rabbit Ig at 1/2500 in PBS–milk for 2 hr at room temperature, washed, and immunodetected using the enhanced chemiluminescence system (ECL; Amersham).
Immunofluorescence microscopy. For immunofluorescence analyses, the cultures were fixed for 6 min in cold methanol (−20°C), rehydrated three times for 5 min each in 1× PBS, and incubated for 30 min in blocking solution containing 5% normal goat serum in 1× PBS. The cells were then exposed overnight at 4°C to a mouse monoclonal antibody that specifically recognizes β-tubulin (used at 1:500; Amersham), a mouse monoclonal antibody that specifically recognizes a poorly phosphorylated neurofilament protein enriched in the somatodendritic domain of the neuron (RMDO9.6, used at 1:500, provided as a kind gift from Dr. V. Lee, Philadelphia, PA), or to the human polyclonal Eg5 antibody described above (used at 1:500). The cells were washed extensively in 1× PBS and incubated either with an FITC anti-mouse second antibody or with a combination of a biotinylated anti-rabbit secondary antibody followed by streptavidin-conjugated with Cy3. Fluorescent second antibodies and probes were purchased from Jackson ImmunoResearch (West Grove, PA). Double-immunostaining for tubulin and Eg5 or neurofilament and Eg5 were performed using appropriate combinations of the antibodies listed above. After washes in 1× PBS, cells were mounted in a medium that reduces photobleaching, and were then viewed with a confocal microscope (LSM 410, Carl Zeiss).
RESULTS
Isolation and DNA sequence analysis of mouse Eg5
To isolate the mouse Eg5 gene, we screened a cDNA library from MCA16 cells using as a probe a cDNA fragment corresponding to the amino-terminal motor domain of human Eg5 (see Materials and Methods). Two positive clones were isolated, and the corresponding insert of the longest clone (4412 nt) was subcloned in plasmid vectors and subjected to DNA sequence analysis. The sequence of the longest cDNA contains a single open reading frame encoding a polypeptide of 1014 amino acids. This cDNA lacks the 5′ end sequence. To complete the sequence, we performed RT-PCR using specific oligonucleotides as primers (for details, see Materials and Methods). Figure1A shows the comparison of the MmEg5-predicted protein sequence with the HsEg5-predicted protein sequence (Blangy et al., 1995). The predicted sequences of the mouse and human proteins are 80% identical and 87% similar, and show greatest homology within their amino-terminal domains. However, appreciable sequence conservation is also found within other domains of the molecules, as shown in Figure 1B, suggesting that the two proteins are functional homologs. MmEg5 also shows considerable homology with Xenopus Eg5, but less so compared with human Eg5. The predicted sequences between mouse and Xenopus are 56% identical and 71% similar, with most of the additional divergence appearing within the C-terminal regions of the molecule. Using the method of Lupas et al. (1991), we determined that amino acid residues 325–440, 451–480, and 625–653 of MmEg5 should form an extensive coiled coil conformation (probability >50%, Fig. 1C). The amino-terminal domain contains the consensus motifs that are normally found in motor domains of kinesin-related proteins, including YGQTXXGK(T/S), NXXSSRSH, and DLAGXE (Fig. 1A), indicating that this domain is responsible for force generation against microtubules, as is the case with Eg5 homologs from other species. The central α-helical region contains leucine zipper motifs at amino acid residues 408–436, which are probably involved in the association of MmEg5 molecules into complexes. The C-terminal domain contains a cdc2 consensus site corresponding to Thr(887). Such a site has been shown to be essential for the interaction of the motor with microtubules in bothXenopus (Sawin and Mitchison, 1995) and human (Blangy et al., 1995).
Fig. 1.

Cloning and characterization of Eg5 in mouse cells (MmEg5). A, Alignment of MmEg5 and HsEg5 protein sequences. Amino acids are shown using the single-letter code. The human sequence (Blangy et al., 1995) is shown only when it differs from the mouse sequence. Identities are indicated by dashes, and conservative substitutions (T/S, E/N/D/Q, K/R, Y/F/W, L/V/I/M) are shown by dots. The amino-terminal domain contains the consensus motifs that are normally found in motor domains of kinesin-related proteins, including YGQTXXGK(T/S), NXXSSRSH, and DLAGXE (boxes). Vertical bars mark the boundaries of the motor, link, stalk, and tail domains. Another group has recently published partial sequence information from the N-terminal region of MmEg5 that is almost identical to ours but contains a small number of nucleotide differences that result in six amino acid substitutions and one additional amino acid (Nakagawa et al., 1997). The asterisk indicates threonine (T), a site that can be phosphorylated presumably by cdc2 kinase. The consensus motifs of kinesin-like proteins are boxed, and the leucine zipper motif is underlined. B, Diagram showing the homologies (similar/identical amino acids) in different domains of MmEg5 and HsEg5. BESTFIT was used to find the best segments of similarities between the two sequences (Devereux et al., 1984).C, Coiled coil structure predicted by the algorithm ofLupas et al. (1991).
Expression of Eg5 in tissues of the mouse determined by Northern blot and in situ hybridization
Having obtained the above sequence information, our next goal was to determine whether MmEg5 is expressed only in cells undergoing mitosis or alternatively, whether it is also expressed in differentiated cells such as neurons. To investigate this issue we first used Northern blot analyses to study Eg5 expression in mouse whole embryos at E10.5, E11.5, E13.5, E15.5, and mouse brain at P0, P7, P14, P21, and adult. Tissue from the small intestine was also analyzed at P0 and adult. As a positive control, we used mitotic mouse neuroblastoma cells during their exponential growth phase in culture. Cultured human HeLa cells were used as a negative control because under high stringency conditions we would not expect the MmEg5 probe to cross-hybridize with the human sequence. Equal amounts (30 μg) of total RNA were loaded per lane. When these RNAs were hybridized with the cDNA probe for MmEg5 (see Materials and Methods), we observed three transcripts (5.0 kb, 5.6 kb, and 6.5 kb) in the whole embryos at all stages of brain development, in the P0 intestine, and in the neuroblastoma cells (see Fig.2A,B). Quantitative analyses indicate that the three transcripts are roughly equally expressed in all samples studied, although slight variations were observed in some cases (Fig. 2A′,B′). During the development of the whole embryo and the brain these transcripts were downregulated (Figs.2A′,B′). A similar downregulation of expression was observed in the case of other structures such as the developing small intestine (Fig. 2A′). These transcripts may represent alternative splicing, different 5′,3′-untranslated regions, or different poly (A+) signals used in protein synthesis. However, the multiple transcripts could not be the result of multiple Eg5 genes, given that Southern blot analyses demonstrate the presence of only one Eg5 gene in the mouse and human (M. Kress, unpublished data). As expected based on sequence divergence in the stalk and tail regions of the molecule, no transcripts were visualized in HeLa cells using the MmEg5 probe. However, three transcripts with a similar pattern have been detected in HeLa cells using a probe specific to the human Eg5 sequence (M. Kress, unpublished data).
Fig. 2.

Expression of MmEg5 mRNAs in mouse tissues and cultured cells determined by Northern blot analyses. Total RNA (30 μg/lane) isolated from whole embryo at E10.5, E11.5, E13.5, E15.5 (A, lanes 1–4), from small intestine at P0 and adult (lanes 5,6), from whole brain at P0, P7, P14, P21, and adult (B, lanes 2–6), and from cultured mouse neuroblastoma cells (used as a positive control; A,lane 7) was electrophoresed in a formaldehyde 1% agarose gel, transferred to a nylon membrane, and then probed with radioactively labeled MmEg5 cDNA (see Materials and Methods). The transcripts (5.0, 5.6, and 6.5 kb) detected in whole embryo, small intestine, and whole brain were identical in size to those found in neuroblastoma cells. The histograms A′ andB′ show the changes in the levels of Eg5 mRNAs in different tissues during their development.
Having established the presence of Eg5 transcripts in mouse tissues, we next used in situ hybridization to study the regional and cellular distribution of Eg5 mRNA in developing mouse tissues. For these analyses, we used sense and antisense riboprobes that were synthesized from the same 1.6 kb MmEg5 cDNA fragment described above. The specificity of the MmEg5 antisense riboprobe was first assessed in analyses on neuroblastoma cells used as a positive control and HeLa cells used as a negative control. In neuroblastoma cells, hybridization signal was observed both during interphase and mitosis, but was clearly higher in dividing cells (Fig.3A). Consistent with the specificity of the antisense probe, hybridization signal was barely detectable within the HeLa cells, with levels no higher than the very low background detected in neuroblastoma (Fig. 3B) and HeLa cells using the sense riboprobe. Figure 3C shows an embryo at E15.5 hybridized with the antisense riboprobe. Prominent signal was detected in structures including the submandibular gland, epithelium surrounding the eye (better visualized in other sections), the liver, kidney, lung, thymus gland, cartilage primordium of the body of the hyoid bone, and the gut. Emulsion analyses indicate that the hybridization signal is present in postmitotic cells, such as the smooth muscle cells of the gut, as well as in mitotic cells, such as the mucosal cells of the gut (data not shown). Lower levels of signal were detected in structures such as the tongue, the heart, and the epithelium of the hindlimbs. Hybridization of sagittal sections of whole embryo E15.5 (and all other ages) with the sense riboprobe resulted in only background labeling with low density and equal grain distribution over the embryo (Fig. 3D). Consistent with the results of the Northern blot analyses on whole brain, MmEg5 mRNAs are also strongly expressed in CNS structures such as the E15.5 epithalamus (Fig. 3E) and cerebral cortex (Fig.3E,F) but their expression is downregulated during development. By P7, the signal is low within the hippocampus, but is high within the cerebellum and the olfactory bulb (Fig. 3G). At P21 and in the adult, the signal is low throughout most of the brain (Fig.3H,I). In the adult, detectable signal is again visible within the olfactory bulb (Fig.3I). No such signal was apparent with the sense control (Fig. 3J). These patterns of expression are consistent with the different temporal patterns of development of these various brain structures and the fact that neurons within the olfactory bulb remain plastic even in the adult.
Fig. 3.
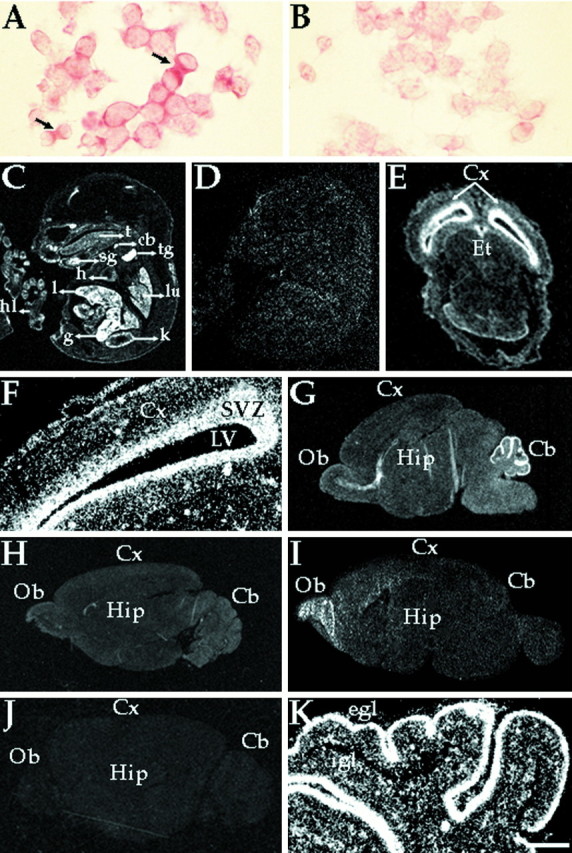
Expression of MmEg5 mRNAs in mouse tissues and cultured cells determined by in situ hybridization.A and B show cultured mouse neuroblastoma cells hybridized with the MmEg5 antisense and sense riboprobes, respectively. Autoradiographs C and D are representative of the hybridization pattern obtained with MmEg5 antisense and sense riboprobes, respectively, at E15.5. Hybridization signal was detected within the submandibular salivary gland (sg), cartilage primordium of the body of the hyoid bone (cb), thymus gland (tg), liver (l), gut (g), heart (h), tongue (t), kidney (k), lung (lu), and epithelial cells of the hindlimbs (hl). Shown inE and F, respectively, are a film autoradiograph and corresponding dark-field illumination of a transverse section of the cerebral cortex (Cx) at E15.5 obtained with the MmEg5 antisense riboprobe. Etindicates epithalamus, and SVZ indicates subventricular zone. LV indicates lateral ventricle. AutoradiographsG–I are representative of the hybridization patterns obtained with MmEg5 antisense riboprobe at P7, P21, and adult mouse brain (Ad), respectively.Cb, cerebellum; Hip, hippocampus;Ob, olfactory bulb. Autoradiograph J is representative of the adult mouse brain hybridization pattern obtained with MmEg5 sense riboprobe. K, Dark-field illumination of the cerebellum hybridized for MmEg5 at P7. At P7, the external granular cell layer (egl) and the internal granular cell layer (igl) are labeled. Scale bar:A, B, 6 μm; C,D, 0.3 cm; E, 0.1 cm;F–K, 0.2 cm.
We next explored the expression of MmEg5 mRNAs in developing cells of the CNS. One possibility is that the expression and downregulation of these mRNAs relate to the mitotic divisions of undifferentiated neuroblasts rather than terminally postmitotic neurons. Another possibility is that developing neurons continue to express MmEg5 mRNAs after their terminal mitotic division. As a first measure toward exploring this issue, we focused our attention on the laminar structure of the developing cerebellum. The external granular layer contains mitotic neuroblasts that gradually become postmitotic. Then, these postmitotic neurons migrate into the internal granular layer in which they continue to differentiate (Hatten et al., 1997). Analyses of sections exposed to emulsion indicate that in the P7 cerebellum, the external granule cells are highly labeled by the MmEg5 probe and that cells of the internal granular layer are labeled as well (Fig.3K). Although the labeling in the external granular layer might reflect the residual mitotic activity of some of these cells, it is unlikely that the labeling in the internal granular layer can be attributed to such activity. Thus, these observations suggest that postmitotic neurons continue to express MmEg5 as they differentiate.
Expression of Eg5 in neuronal cultures determined by in situ hybridization
To confirm that Eg5 is expressed in postmitotic neurons as well as in dividing neuroblasts, we performed in situ hybridization analyses on two well characterized culture systems of terminally postmitotic neurons, one from the central and one from the peripheral nervous system. Hippocampal and sympathetic neurons were obtained from rat fetuses and newborn rat pups at times when most of them had completed their terminal mitotic division (Goslin and Banker, 1991;Higgins et al., 1991). In situ hybridization was performed using both radioactively labeled probes and probes labeled with digoxygenin. Sympathetic neurons form axons within the first few hours in culture and dendrites within the first few days. The mRNAs encoding MmEg5 were expressed in sympathetic neurons at 1 d (Fig.4A), 3 d (Fig.4B), and 7 d (Fig. 4C) but were not detected at 14 d (Fig. 4D). At 1 d, most cells displayed high levels of expression. At 3 d, all of the cells exhibited their highest levels of expression. At 7 d, expression levels were lower than at 1 or 3 d. At 14 d, the signal was significantly decreased and was no higher than the low background signal obtained with the sense riboprobe at all time points (Fig. 4E).
Fig. 4.

Expression of Eg5 mRNAs in cultured rat sympathetic neurons determined by in situ hybridization. Cultured sympathetic neurons were hybridized with either the radioactive (large panels) or with the digoxygenin-labeled (small panels) antisense (A–D) or sense (E) riboprobe for MmEg5. Sympathetic neurons were obtained from superior cervical ganglia of newborn rat pups and were grown for 1, 3, 7, and 14 d. In situ hybridization analyses show that mRNAs encoding Eg5 were expressed at 1, 3, and 7 d. At 14 d, the hybridization signal was similar to that detected at 3 d with the sense riboprobe control. Note also that Eg5 mRNAs are downregulated during in vitro development. Scale bar, 10 μm.
The hippocampal cultures are useful for developmental studies because they differentiate axons and dendrites in a well characterized sequence of stages that presumably reflects their in vivo development (Dotti et al., 1988). The cells initially extend lamellipodia (stage 1) which coalesce into immature processes within a few hours after plating (stage 2). One of these immature processes becomes the axon by 1.5 d in culture (stage 3), after which those remaining differentiate into dendrites by 3–4 d in culture (stage 4). By 1 week, the neurons have developed many mature characteristics, such as the presence of dendritic sprouts (stage 5). Hybridization signal for MmEg5 mRNAs was present at all of these stages (data not shown). Levels varied from cell to cell at stage 1, but were high in all cells at stage 2. At stage 3, stage 4, and in some cells at stage 5, expression levels were substantially decreased compared with those at stages 1 or 2. At stage 5, some cells displayed levels of expression that were as high as those at stages 1 or 2. Hybridization of neurons with the sense riboprobe at all stages resulted only in low background labeling. These results on cultured hippocampal and sympathetic cultures indicate that neurons continue to express MmEg5 mRNAs well past their terminal mitotic division. The fact that older hippocampal but not sympathetic neurons express detectable levels of MmEg5 mRNAs may relate to the fact that hippocampal neurons are more plastic later in development.
Identification of MmEg5 protein in neurons
Western blot analyses were performed on samples extracted from cultured sympathetic neurons at 3 d because the levels of mRNAs for Eg5 were highest at this stage of development (Fig.5). These analyses were performed using an affinity-purified polyclonal antibody raised against a region of the motor domain of HsEg5 that is highly conserved in MmEg5 (see Materials and Methods). HeLa cells, used as a positive control, showed a single major band at 135 kDa when 10 μg of total protein were loaded (data not shown), and an additional minor band of 130 kDa when at least 50 μg were loaded. Similar results have been obtained with a polyclonal antibody against the tail region of HsEg5 (Blangy et al., 1995; M. Kress, unpublished data). We also obtained similar results with the polyclonal antibody against the motor domain in studies on chinese hamster ovary (CHO) cells. Cultured neuroblastoma cells showed the same major 135 kDa band when 50 μg of total protein were loaded. At 3 d in culture, the sympathetic neurons showed a comparable 135 kDa band. Overexposure of the blots revealed an additional band at 93 kDa within the 3 d cultures (data not shown). No bands were observed in control studies in which the primary antibody was deleted. These results indicate that neurons express protein recognized by a polyclonal antibody specific for Eg5.
Fig. 5.

Western blot analyses using a polyclonal antibody against Eg5 on extracts prepared from cultured cells. Western blot analyses were performed on samples extracted from rat cultured sympathetic neurons (SN) at 3 d using an affinity-purified polyclonal antibody raised against a region of the motor domain of HsEg5 that is highly conserved in MmEg5. The mitotic form of the Eg5 protein focuses as a 135 kDa band in CHO, HeLa, and neuroblastoma (N2a) cells, used as positive controls. HeLa cells also show a minor 130 KDa band. The 135 kDa protein is also expressed in postmitotic sympathetic neurons. Arrowsindicate protein ladder (Life Technologies).
Distribution of Eg5 protein in mitotic cells and developing neurons
To further confirm the specificity of the polyclonal Eg5 antibody in mouse cells, we performed immunofluorescence analyses on the cultured neuroblastoma cells. The results of these analyses were entirely similar to those obtained on HeLa cells using either the tail polyclonal (Blangy et al., 1995) or the motor polyclonal HsEg5 antibody (data not shown). Specifically, staining is low and diffuse in the cytoplasm during interphase (Fig. 6,arrows), after which it becomes concentrated in the region of the centrosomes during their separation in prophase (Fig.6A) and in the half-spindles near each centrosome during metaphase (Fig. 6B). Then during anaphase, the staining becomes weaker and more diffuse (Fig. 6C), after which it localizes to the postmitotic bridges during telophase (Fig.6D). Thus immunofluorescence staining with the polyclonal antibody results in a pattern consistent with its specific recognition of the Eg5 protein.
Fig. 6.

The polyclonal HsEg5 antibody recognizes MmEg5 protein in neuroblastoma cells. The polyclonal antibody generated from the N-terminal motor region of the human Eg5 molecule reveals the same distribution of Eg5 protein during different phases of mitosis in mouse neuroblastoma cells as observed in HeLa cells with an antibody directed against the tail region of HsEg5 (Blangy et al., 1995).Arrows indicate interphase cells in various panels. Each panel also shows one or more cell in a particular stage of mitosis.A, Prophase; B, Metaphase;C, Anaphase; D, Telophase. Scale bar, 10 μm.
At stage 1 of development, cultured hippocampal neurons show Eg5 immunoreactivity within the cell body and lamellipodia (data not shown). At stage 2, the protein is localized within the cell body and within most of the immature processes (Fig.7A). Most typically the protein was concentrated at the distal tips of the processes, but sometimes along their lengths. At stage 3, the protein is still present within the cell body and minor processes. In some axons, the protein was no longer observed at the distal tip of the early axon (Fig.7B, arrow). In most cases, the protein was observed at the distal tips of the early axon (Fig.7C,D) and within branches of the axons (Fig. 7D). At stage 4, protein levels were significantly diminished throughout the neuron (Fig. 7E). Very low levels of protein were sometimes detected at dendrite tips (Fig.7E, arrow). At stage 5, the protein levels in most cells were notably increased. Figure8 shows three such cells double-labeled with a β-tubulin antibody to reveal cellular morphology (Fig.8A–C) and the polyclonal Eg5 antibody (Fig. 8A′–C′). Figure 8, A andA′, shows a neuron early in stage 5 before the development of dendritic sprouts. Eg5 staining is apparent in the distal tips of the dendrites. The remaining panels of the figure show two neurons later in stage 5 after the development of dendritic sprouts. Eg5 staining is concentrated within the sprouts.
Fig. 7.

Immunofluorescence analyses on the distribution of Eg5 in cultured rat hippocampal neurons at early stages of development. At stage 2 (A), the protein is localized within cell bodies and within most minor processes. Most typically, it is present at the tips of the minor processes, but sometimes along their lengths. At stage 3, the protein is still present within the cell body and minor processes. In some axons, the protein was no longer observed at the distal tip of the process (B,arrow). In most cases, the protein was observed at the distal tips of the early axon (C, D) and within branches of the axons (D). At stage 4 (E), protein levels were significantly diminished throughout the neuron. Very low levels of protein were sometimes detected at dendrite tips (arrow). Scale bar, 10 μm.
Fig. 8.

Immunofluorescence analyses on the distribution of Eg5 in cultured rat hippocampal neurons at stage 5 of development. Shown are stage 5 hippocampal cultures double-immunostained for β-tubulin in A–C to reveal cellular morphology and in A′–C′ to show Eg5 distribution. Eg5 protein levels are significantly higher than at stage 4. The protein is localized within the tips of dendrites (A′), as well as within newly forming sprouts of dendrites (B′, C′). Scale bar, 5.5 μm.
Figure 9A shows cultured sympathetic neurons stained for Eg5 6 hr after plating. Immunoreactivity is localized within the cell body, lamellipodia, and distal regions of developing processes. Figure 9, B andB′, shows a neuron with longer axons from a 6 hr culture double-labeled for β-tubulin to reveal cellular morphology and Eg5, respectively. Eg5 is localized within the cell body and distal tips of the axons. The remaining panels of the figure show cells double-labeled with a neurofilament antibody to reveal cellular morphology (Fig.9C–E) and the polyclonal Eg5 antibody (Fig.9C′–E′). The neurofilament antibody recognizes a poorly phosphorylated epitope that is enriched in cell bodies and dendrites and, hence, is particularly useful for discerning dendrites from axons. At 3 d, Eg5 is localized in the distal tips of the dendrites (Fig. 9C,C′). Unlike the case with cultured hippocampal neurons, the dendrites of cultured sympathetic neurons do not branch as extensively and tend not to form sprouts later in development. In the rare instances in which we were able to observe a single short branch extending from a dendrite, Eg5 staining appeared within the branch (Fig. 9D,D′). At 7 d (data not shown) and 14 d (Fig.9E,E′), Eg5 was observed within the cell body. Staining was also found along the length of the dendrite, but this staining was weak and diffuse, and never enriched in their distal tips.
Fig. 9.

Immunofluorescence analyses on the distribution of Eg5 in developing cultured rat sympathetic neurons. A,B, and B′ show neurons cultured for 6 hr.A shows that Eg5 is present within cell bodies, lamellipodia, short processes resulting from coalescence of lamellipodia, and within the distal tips of early axons.B is a β-tubulin double-stain to reveal morphology.B′ shows that Eg5 is present within the cell bodies and distal tips of somewhat longer axons. The remaining panels show older cultures (3 and 14 d) double-immunostained for a dendrite-enriched neurofilament protein in C–E to reveal morphology and for Eg5 in C′–E′. At 3 d, Eg5 is concentrated at the dendrite tip (C′,D′) as well as in dendritic branches (D′). At 14 d, Eg5 is still detected in the cell body, but is no longer concentrated at the dendrite tip (E′). Scale bar: A, 15 μm;B–E′, 10 μm.
Association of Eg5 with microtubules
The immunofluorescence images of the cultured rat hippocampal and sympathetic neurons do not provide sufficient resolution to determine whether Eg5 is directly associated with microtubules in the distal regions of neuronal processes. To obtain better resolution, we performed double-label immunostain analyses for tubulin and Eg5 on cultured hamster cortical neurons, which we have found to generate unusually broad growth cones with splayed microtubules. Figure10 shows two examples of the distal regions of developing axons. Eg5 immunostaining is concentrated in the most distal region of the growth cone (Fig.10A′,B′) and shows colocalization with a subpopulation of the microtubule polymer (Fig.10A,B).
Fig. 10.

Immunofluorescence analyses on the distribution of Eg5 in cultured hamster cortical neurons. Shown are hamster cortical neurons double-immunostained for β-tubulin in A andB and for Eg5 in A′ andB′. Eg5 immunostain colocalizes with a subpopulation of microtubule polymer within the growth cone. Scale bar, 13 μm.
DISCUSSION
The organization of microtubule arrays within living cells cannot be explained entirely by the association of individual microtubules with their sites of nucleation. Recent studies have identified some of the molecular mechanisms by which microtubules are organized into a bipolar spindle in mitotic cells. These studies demonstrate that microtubules are organized by forces generated by a variety of molecular motor proteins that are expressed during mitosis. We have proposed that the microtubule arrays of the postmitotic neuron are established by forces generated by the same or closely motor proteins. Studies from our laboratory have shown that cytoplasmic dynein, a multifunctional motor required for spindle formation, is also important for organizing microtubules in developing neuronal processes (Ahmad et al., 1998). Other studies from our laboratory have shown that CHO1/MKLP1, which is thought to generate forces against oppositely oriented microtubules in the spindle midzone, is essential for establishing the nonuniform microtubule polarity pattern of developing dendrites (Sharp et al., 1997; Yu et al., 1997; Ferhat et al., 1998b). Microtubule organization in the mitotic spindle requires additional forces to those generated by cytoplasmic dynein and CHO1/MKLP1, and it seems reasonable that this may also be the case in the postmitotic neuron.
In the present study, we sought to determine whether rodent neurons express a homolog of Eg5, a member of the bimC family of kinesin-related motors known to be essential for mitotic spindle formation. We cloned from mitotic cells a cDNA encoding the mouse homolog, which we have called MmEg5. The sequence shares homology with other members of the BimC family, which have been isolated from widely divergent organisms from yeast to humans. These homologs share 50–60% identity within the motor domain and relatively little homology elsewhere in the molecule. Indeed, the deduced amino acid MmEg5 sequence is 80% identical to that of the HsEg5 sequence derived from HeLa cells (Blangy et al., 1995). In addition, MmEg5 localizes to the same regions of the mitotic spindle as its homologs, suggesting identical functions. We have documented that Eg5 is also expressed within developing neurons well past their terminal mitotic division. Northern blot analyses revealed similar transcripts in both mitotic cells and nervous tissue, and in situ hybridization analyses confirmed the presence of Eg5 mRNAs in postmitotic neurons. Western blot analyses also showed a similar polypeptide in mitotic cells and postmitotic neurons. Samples obtained from mouse, rat, and hamster all showed good cross-reactivity and cross-hybridization with the available probes.
It has been suggested that all of the motor proteins expressed in postmitotic neurons are involved in the transport of membranous organelles rather than of microtubules (Hirokawa, 1997). However, the Eg5 homologs do not appear to interact with membranous organelles, but instead appear to associate primarily with microtubules (Chang et al., 1996). In the neuron, we have found Eg5 to be tightly concentrated within discrete regions of the processes. This pattern is more reminiscent of the localization of Eg5 and other motor proteins along microtubules within the mitotic spindle and of fibrous MAPs that bind to microtubules along their lengths. High-resolution images of flattened growth cones with splayed microtubules reveal a tight colocalization of Eg5 with a subpopulation of the microtubule polymer. Together, these observations suggest that Eg5 is unlikely to be involved in the transport of membranous organelles and is more likely to be involved in organizing microtubules themselves.
We suspect that the precise functions of Eg5 in the neuron are in some way analogous to its functions in mitotic cells. Several lines of evidence indicate that Eg5 and other members of the bimCfamily are essential for separating the duplicated centrosomes or spindle pole bodies during prophase (for review, see Kashina et al., 1997), but the precise mechanisms for this are not fully understood. At least in the case of Drosophila, the Eg5 homolog forms a homotetramer with all four motor domains directed outward (Kashina et al., 1996). Because Eg5 moves toward plus-ends of microtubules, it has been suggested that the homotetramer could drive apart the two poles by generating forces against oppositely oriented microtubules emanating from each pole. Another possibility is that the tail end of the molecule might be tethered to the centrosome or spindle pole body while the motor end moves toward the plus-ends of microtubules from the opposite pole. This would also drive the two poles apart. During metaphase, Eg5 localizes within each half-spindle near the pole, suggesting that an additional function of the motor might be to hold the minus-ends of microtubules near the pole after their release from it (Sawin et al., 1992). Such forces would antagonize those generated by cytoplasmic dynein, which would otherwise transport the microtubules with plus-ends leading away from each spindle pole (Gaglio et al., 1996).
In light of the manner by which Eg5 functions during mitosis, there would appear to be multiple possibilities for the means by which Eg5 could modulate microtubule organization in the distal regions of neuronal processes. First, the motor might form a homotetramer that is not tethered to any other structure. In this case, the motor complex translocates toward the plus-ends of neighboring microtubules, thus zippering them together but not inducing the transport of either. Second, the motor might form a homotetramer that is tethered to some other structure in the cytoplasm that has a greater resistance to movement than the microtubules. This structure may be a component of the cell cortex, for example, and would be functionally analogous to the structure that has been proposed to tether the motor to the centrosome. In this case, movement of the motor complex toward the plus-ends of the microtubules would cause the microtubules to move in a retrograde direction within the process. Third, the motor might exist as a dimer or monomer that generates forces between neighboring microtubules, with the longer microtubule associated with the motor domain and the shorter microtubule associated with the tail. In this case, the shorter microtubule would move in an anterograde direction. The fourth possibility is similar to the third, except that the shorter microtubule is associated with the motor domain. In this case, the shorter microtubule would move in a retrograde direction. The final possibility is that the motor exists as a dimer or monomer whose tail is associated with a nonmicrotubule structure with greater resistance to movement. In this case, the microtubules would move in a retrograde direction.
The concentration of Eg5 at the tips of developing processes suggests an important role for the protein in regulating their growth. At least in the case of hippocampal neurons, the cells initially generate several immature processes that remain roughly the same length until one differentiates into the axon (Dotti et al., 1988). The others maintain their short length for a few days, after which they begin to grow longer and become dendrites. Notably, the axon ceases its rapid growth until after the dendrites have completed their elongation. Then, as indicated by studies on a variety of different types of neurons, the growth of the axon is marked by intermittent forward movements, backward movements, and pauses (Halloran and Kalil, 1994). We strongly suspect that these various behaviors relate to the transport of microtubules within the distal regions of these processes (Tanaka and Kirschner, 1991). As discussed above, Eg5 has the appropriate properties to produce forces that could modulate the anterograde transport of microtubules by cytoplasmic dynein. But, does Eg5 complement or antagonize anterograde microtubule transport? If it is the former, then we would conclude that in the neuron, Eg5 is activated in processes undergoing rapid phases of process growth and inactivated in processes undergoing retraction or pauses in their growth. If it is the latter, we would conclude that Eg5 is activated in processes undergoing retraction or pauses and inactivated in processes undergoing bouts of rapid growth. The latter explanation seems more satisfactory because it can explain all of the observed behaviors, whereas the former does not explain why processes retract or pause. In addition, the latter explanation is more consistent with the enrichment later in development of Eg5 within dendritic sprouts, which tend to remain short. In either case, however, the modulation of microtubule transport by Eg5 would be a major factor in regulating the growth properties of a developing neuronal process and thereby defining it as an axon or a dendrite.
How might Eg5 be activated or inactivated in select regions of developing neurons? Although there are numerous ways in which the function of a motor might be regulated, a particularly compelling possibility is suggested by studies on Eg5 homologs in other species. These studies indicate that the association of the motor with microtubules is mediated by phosphorylation of a single amino acid (Blangy et al., 1995; Sawin and Mitchison, 1995). MmEg5 has a similar potential phosphorylation site, which probably regulates its association with microtubules. It is possible that the capacity of Eg5 to influence microtubule organization depends on the binding to the microtubules of a certain number of motor molecules and that this association is regulated by phosphorylation. If this is correct, regulation of Eg5 phosphorylation may be an important means by which the development of axons and dendrites is integrated with external and intrinsic cues, both of which are known to affect protein phosphorylation as well as neuronal differentiation (Ferhat et al., 1993).
Footnotes
This work was supported by grants from the National Institutes of Health and the National Science Foundation to P.W.B., and from the Association de la Recherche sur le Cancer to M.K. We thank Hassan Bousbaa and Pierre d’Hérin for their assistance in the isolation of the murine Eg5 cDNAs. We thank John Callaway, Erik Dent, and Katherine Kalil for advice and assistance in the preparation of cultures of hamster cortical neurons.
Correspondence should be addressed to Dr. Peter W. Baas, Department of Anatomy, The University of Wisconsin Medical School, 1300 University Avenue, Madison, WI 53706.
REFERENCES
- 1.Ahmad FJ, Echeverri CJ, Vallee RB, Baas PW. Cytoplasmic dynein and dynactin are required for the transport of microtubules into the axon. J Cell Biol. 1998;140:246–256. doi: 10.1083/jcb.140.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baas PW, Ahmad FJ. The transport properties of axonal microtubules establish their polarity orientation. J Cell Biol. 1993;120:1427–1437. doi: 10.1083/jcb.120.6.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baas PW, Deitch JS, Black MM, Banker GA. Polarity orientation of microtubules in hippocampal neurons: uniformity in the axon and nonuniformity in the dendrite. Proc Natl Acad Sci USA. 1988;85:8335–8339. doi: 10.1073/pnas.85.21.8335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baas PW, Black MM, Banker GA. Changes in microtubule polarity orientation during the development of hippocampal neurons in culture. J Cell Biol. 1989;109:3085–3094. doi: 10.1083/jcb.109.6.3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barton NR, Pereira AJ, Goldstein LSB. Motor activity and mitotic spindle localization of the Drosophila kinesin-like protein KLP61F. Mol Biol Cell. 1995;6:1563–1574. doi: 10.1091/mbc.6.11.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Black MM, Baas PW. The basis of polarity in the neuron. Trends Neurosci. 1989;12:211–214. doi: 10.1016/0166-2236(89)90124-0. [DOI] [PubMed] [Google Scholar]
- 7.Blangy A, Lane HA, d’Hérin P, Harper M, Kress M, Nigg EA. Phosphorylation by p34(cdc2) regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83:1159–1169. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 8.Chang P, LeGuellec K, Houliston E. Immunodetection of cytoskeletal structures and the Eg5 motor protein on deep-etch replicas of Xenopus egg cortices isolated during the cortical rotation. Biol Cell. 1996;88:89–98. [PubMed] [Google Scholar]
- 9.Dessen P, Fondrat C, Valencien C, Mugnier C. BISANCE: a French service for access to biomolecular sequence databases. Comput Appl Biosci. 1990;6:355–356. doi: 10.1093/bioinformatics/6.4.355. [DOI] [PubMed] [Google Scholar]
- 10.Devereux J, Haeberli P, Smithies O. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 1984;12:387–395. doi: 10.1093/nar/12.1part1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:121–130. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Enos AP, Morris NR. Mutation of a gene that encodes a kinesin-like protein blocks nuclear division in A. nidulans. Cell. 1990;60:1019–1027. doi: 10.1016/0092-8674(90)90350-n. [DOI] [PubMed] [Google Scholar]
- 13.Ferhat L, Khrestchatisky M, Roisin M, Barbin G. Basic fibroblast growth factor-induced increase in zif/268 and c-fos mRNA levels is calcium dependent in primary cultures of hippocampal neurons. J Neurochem. 1993;61:1105–1112. doi: 10.1111/j.1471-4159.1993.tb03626.x. [DOI] [PubMed] [Google Scholar]
- 14.Ferhat L, Represa A, Zouaoui-Aggoun D, Ferhat W, Ben-Ari Y, Khrestchatisky M. FGF-2 induces nerve growth factor expression in cultured rat hippocampal neurons. Eur J Neurosci. 1997;9:1282–1289. doi: 10.1111/j.1460-9568.1997.tb01483.x. [DOI] [PubMed] [Google Scholar]
- 15.Ferhat L, Represa A, Ferhat W, Ben-Ari Y, Khrestchatisky M. MAP2d mRNA is expressed in identified neuronal populations in the developing and adult rat brain and its subcellular distribution differs from that of MAP2b in hippocampal neurones. Eur J Neurosci. 1998a;10:161–171. doi: 10.1046/j.1460-9568.1998.00044.x. [DOI] [PubMed] [Google Scholar]
- 16.Ferhat L, Kuriyama R, Lyons GE, Micales B, Baas PW. Expression of the mitotic motor protein CHO1/MKLP1 in postmitotic neurons. Eur J Neurosci. 1998b;10:1383–1393. doi: 10.1046/j.1460-9568.1998.00159.x. [DOI] [PubMed] [Google Scholar]
- 17.Gaglio T, Saredi A, Bingham JB, Hasbani MJ, Gill SR, Shroer TA, Compton DA. Opposing motor activities are required for the organization of the mammalian mitotic spindle pole. J Cell Biol. 1996;135:399–414. doi: 10.1083/jcb.135.2.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goslin K, Banker G. Rat hippocampal neurons in low-density culture culture. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT; Cambridge, MA: 1991. pp. 251–281. [Google Scholar]
- 19.Hagan I, Yanagida M. Kinesin-related cut7 protein associates with mitotic and meiotic spindles in fission yeast. Nature (Lond) 1992;356:74–76. doi: 10.1038/356074a0. [DOI] [PubMed] [Google Scholar]
- 20.Halloran MC, Kalil K. Dynamic behaviors of growth cones extending in the corpus callosum of living cortical brain slices observed with video microscopy. J Neurosci. 1994;14:2161–2177. doi: 10.1523/JNEUROSCI.14-04-02161.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hatten ME, Alder J, Zimmerman K, Heintz N. Genes involved in cerebellar cell specification and differentiation. Curr Opin Neurobiol. 1997;7:40–47. doi: 10.1016/s0959-4388(97)80118-3. [DOI] [PubMed] [Google Scholar]
- 22.Heald R, Tournebize R, Blank T, Sandaltzopoulos R, Becker P, Hyman A, Karsenti E. Self-organization of microtubules into bipolar spindles around artificial chromosomes in Xenopus egg extracts. Nature (Lond) 1996;382:420–425. doi: 10.1038/382420a0. [DOI] [PubMed] [Google Scholar]
- 23.Heidemann SR, Landers JM, Hamborg MA. Polarity orientation of axonal microtubules. J Cell Biol. 1981;91:661–665. doi: 10.1083/jcb.91.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higgins D, Lein PJ, Osterhout DJ, Johnson MI. Tissue culture of mammalian autonomic neurons. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT; Cambridge, MA: 1991. pp. 177–205. [Google Scholar]
- 25.Hirokawa N. The mechanisms of fast and slow transport in neurons: identification and characterization of the new kinesin superfamily of motors. Curr Opin Neurobiol. 1997;7:605–614. doi: 10.1016/s0959-4388(97)80079-7. [DOI] [PubMed] [Google Scholar]
- 26.Hoyt A, He L, Loo KK, Saunders WS. Saccharomyces cerevisiae kinesin related gene products required for mitotic spindle assembly. J Cell Biol. 1992;118:109–120. doi: 10.1083/jcb.118.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kashina AS, Baskin RJ, Cole DG, Wedaman KP, Saxton WP, Scoley JM. A bipolar kinesin. Nature (Lond) 1996;379:270–272. doi: 10.1038/379270a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kashina AS, Rogers GC, Scholey JM. The bimC family of kinesins: essential bipolar mitotic motors driving centrosome separation. Biochim Biophys Acta. 1997;1357:257–271. doi: 10.1016/s0167-4889(97)00037-2. [DOI] [PubMed] [Google Scholar]
- 29.Koerner TJ, Hill JE, Myers AM, Tzagoloff A. High-expression vectors with multiple cloning sites for construction of trpE fusion genes: pATH vectors. Methods Enzymol. 1991;194:477–490. doi: 10.1016/0076-6879(91)94036-c. [DOI] [PubMed] [Google Scholar]
- 30.LeGuellec R, Paris J, Couturier A, Roghi C, Philippe M. Cloning by differential sequencing of a Xenopus cDNA that encodes a kinesin-related protein. Mol Cell Biol. 1991;11:3395–3398. doi: 10.1128/mcb.11.6.3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lupas A, Vandyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 32.Lyons GE, Micales BK, Kim S, Herr M, Swanson BJ. In situ analysis of muscle gene expression in mouse embryos. J Animal Sci [Suppl 2] 1996;74:1–8. [Google Scholar]
- 33.Nakagawa T, Tanaka Y, Matsuoka E, Kondo S, Okada Y, Noda Y, Kanai Y, Hirokawa N. Identification and classification of 16 new kinesin superfamily (KIF) proteins in mouse genome. Proc Natl Acad Sci USA. 1997;94:9654–9659. doi: 10.1073/pnas.94.18.9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nislow C, Lombillo VA, Kuriyama R, McIntosh JR. A plus-end-directed motor that moves anti-parallel microtubules in vitro localizes to the interzone of mitotic spindles. Nature (Lond) 1992;359:543–547. doi: 10.1038/359543a0. [DOI] [PubMed] [Google Scholar]
- 35.Roof DM, Meluh PB, Rose MD. Kinesin-related proteins required for assembly of the mitotic spindle. J Cell Biol. 1992;118:95–108. doi: 10.1083/jcb.118.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sambrook J, Fritsch E, Maniatis T. Molecular cloning. A laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 37.Sawin KE, LeGuellec K, Philippe M, Mitchison TJ. Mitotic spindle organization by a plus-end-directed microtubule motor. Nature (Lond) 1992;359:540–543. doi: 10.1038/359540a0. [DOI] [PubMed] [Google Scholar]
- 38.Sawin KE, Mitchison TJ. Mutations in the kinesin-like protein Eg5 disrupt localization to the mitotic spindle. Proc Natl Acad Sci USA. 1995;92:4289–4293. doi: 10.1073/pnas.92.10.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharp DJ, Yu W, Baas JW. Transport of dendritic microtubules establishes their nonuniform polarity orientation. J Cell Biol. 1995;130:93–104. doi: 10.1083/jcb.130.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharp DJ, Yu W, Ferhat L, Kuriyama R, Rueger DC, Baas PW. Identification of a motor protein essential for dendritic differentiation. J Cell Biol. 1997;138:833–843. doi: 10.1083/jcb.138.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shih C, Shilo BZ, Goldfarb MP, Dannenberg A, Weinberg RA. Passage of phenotypes of chemically transformed cells via transfection of DNA and chromatin. Proc Natl Acad Sci USA. 1979;76:5714–5718. doi: 10.1073/pnas.76.11.5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szebenyi G, Callaway JL, Dent EW, Kalil K. Interstitial branches develop from active regions of the axon demarcated by the primary growth cone during pausing behavior. J Neurosci. 1998;18:7912–7922. doi: 10.1523/JNEUROSCI.18-19-07930.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka E, Kirschner MW. Microtubule behavior in the growth cones of living neurons during axon elongation. J Cell Biol. 1991;115:345–363. doi: 10.1083/jcb.115.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walczak CE, Mitchison TJ. Kinesin-related proteins at mitotic spindle poles: function and regulation. Cell. 1996;85:943–946. doi: 10.1016/s0092-8674(00)81295-7. [DOI] [PubMed] [Google Scholar]
- 45.Yu W, Sharp DJ, Kuriyama R, Mallik P, Baas PW. Inhibition of a mitotic motor protein compromises the formation of dendrite-like processes from neuroblastoma cells. J Cell Biol. 1997;136:659–668. doi: 10.1083/jcb.136.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]