

Abstract
The temporal pattern of apoptosis in the adult rat brain after lateral fluid-percussion (FP) brain injury was characterized using terminal deoxynucleotidyl-transferase-mediated biotin-dUTP nick end labeling (TUNEL) histochemistry and agarose gel electrophoresis. Male Sprague Dawley rats were subjected to brain injury and killed for histological analysis at intervals from 12 hr to 2 months after injury (n = 3/time point). Sham (uninjured) controls were subjected to anesthesia with (n = 3) or without (n = 3) surgery. Apoptotic TUNEL-positive cells were defined using stringent morphological criteria including nuclear shrinkage and fragmentation and condensation of chromatin and cytoplasm. Double-labeled immunocytochemistry was performed to identify TUNEL-positive neurons (anti-neurofilament monoclonal antibody RM044), astrocytes (anti-glial fibrillary acidic protein polyclonal antibody), and oligodendrocytes (anti-cyclic nucleotide phosphohydrolase polyclonal antibody). Compared with that seen with sham controls, in the injured cortex, significant apoptosis occurred at 24 hr (65 ± 19 cells; p < 0.05) with a second, more pronounced response at 1 week after injury (91 ± 24 cells;p < 0.05). The number of apoptotic cells in the white matter was increased as early as 12 hr after injury and peaked by 1 week (33 ± 6 cells; p < 0.05). An increase in apoptotic cells was observed in the hippocampus at 48 hr (13 ± 8), whereas in the thalamus, the apoptotic response was delayed, peaking at 2 weeks after injury (151 ± 71 cells;p < 0.05). By 2 months, the number of apoptotic cells in most regions had returned to uninjured levels. At 24 hr after injury, TUNEL-labeled neurons and oligodendrocytes were localized primarily to injured cortex. By 1 week after injury, populations of TUNEL-labeled astrocytes and oligodendrocytes were present in the injured cortex, while double-labeled neurons were present predominantly in injured cortex and thalamus, with a few scattered in the hippocampus. DNA agarose gels confirmed morphological identification of apoptosis. These data suggest that the apoptotic response to trauma is regionally distinct and may be involved in both acute and delayed patterns of cell death.
Keywords: apoptosis, traumatic brain injury, delayed cell death, DNA fragmentation, neurodegeneration, programmed cell death, rat, TUNEL
Various animal models have been used to characterize the behavioral and pathophysiological effects of traumatic brain injury (TBI). The rat model of lateral fluid-percussion (FP) brain injury simulates many clinically relevant sequelae of human closed-head injury, including acute compromise of the blood–brain barrier (Cortez et al., 1989; Soares et al., 1992; Schmidt and Grady, 1993; Dietrich et al., 1994) and alterations in cerebral blood flow and metabolism (Vink et al., 1987, 1988; Hovda et al., 1990; Yamakami and McIntosh, 1991; Yoshino et al., 1991), as well as chronic neurological motor dysfunction and cognitive impairment (McIntosh et al., 1989;Smith et al., 1991, 1994; Hamm et al., 1992; Bramlett et al., 1995;Pierce et al., 1998). Recent studies attempting to evaluate the temporal pattern of regionally selective post-traumatic cell death have shown that neuronal degeneration after lateral FP brain injury can be detected as early as 10 min after injury in the neocortex, thalamus, and hippocampus ipsilateral to the injury site (Hicks et al., 1996). In addition, a progressive, diffuse pattern of cell death and axonal injury has been observed throughout the brain after experimental brain injury (Povlishock et al., 1992; Dietrich et al., 1994; Gennarelli, 1994; Soares et al., 1995; Hicks et al., 1996; Bramlett et al., 1997), which persists for up to 1 year after injury (Smith et al., 1997). However, the pathophysiological mechanisms underlying acute and delayed cell death after brain trauma remain poorly understood.
Traumatically induced cell death has been suggested to be primarily necrotic in nature (Dietrich et al., 1994; Hicks et al., 1996) and is characterized by swelling of the nucleus and cytoplasmic organelles as well as by an early loss of plasma-membrane integrity and cell lysis (Dietrich et al., 1994). Apoptosis, the morphological manifestation of programmed cell death (PCD), is associated with normal CNS development (Oppenheim, 1991). In contrast to necrosis, a cell undergoing apoptosis is characterized by uniform internucleosomal DNA fragmentation, nuclear shrinkage, chromatin compaction as well as by cytoplasmic condensation and disintegration. In the later stages of apoptosis, the cell surface membrane undergoes blebbing and breaks down into spherical apoptotic bodies, which appear to be quickly absorbed via phagocytosis by surrounding cells (Kerr et al., 1972; Wyllie et al., 1980). In addition to its role in normal physiological cell death, apoptosis has been associated with specific pathological conditions in the CNS, including Alzheimer’s and Huntington’s disease (Portera-Cailliau et al., 1995;Smale et al., 1995), cerebral ischemia (MacManus et al., 1993, 1995; Li et al., 1995; Nitatori et al., 1995; Du et al., 1996), and spinal cord injury (Li et al., 1996; Crowe et al., 1997).
We have demonstrated previously that neuronal apoptosis occurs acutely within the first 12–72 hr after lateral FP brain injury using terminal deoxynucleotidyl-transferase-mediated biotin-dUTP nick end labeling (TUNEL) histochemistry and electron microscopy (Rink et al., 1995). These observations have been more recently replicated by others using both cortical contusion and lateral FP models of brain injury (Colicos and Dash, 1996; Clark et al., 1997; Yakovlev et al., 1997). Virtually nothing is known, however, concerning the precise temporal and spatial patterns of apoptotic cell death and its association with delayed cell death in the chronic post-traumatic period. Because delayed and progressive cell death appear to be a hallmark of both experimental (Bramlett et al., 1997; Smith et al., 1997) and clinical closed-head injury (Groswasser-Reider et al., 1993; Gale et al., 1995), the present study was undertaken to characterize the temporal and regional patterns of apoptotic cell death in multiple ipsilateral and contralateral structures up to 2 months after injury.
MATERIALS AND METHODS
Fluid-percussion injury and tissue preparation. Adult male Sprague Dawley rats (350–400 gm; n = 45) were anesthetized with sodium pentobarbital (60 mg/kg, i.p.) 5 min after an injection of atropine (0.06 mg, i.m.). After placement in a stereotactic frame, the scalp and temporal muscle were reflected, and a 5.0 mm craniotomy was performed over the left parietal cortex, midway between the lambda and bregma sutures. Rats in group I (n = 21) were subjected to experimental lateral FP injury of moderate severity (2.4–2.6 atm) as described previously (McIntosh et al., 1989). Briefly, a female Leur-Lok fitting is cemented to the craniotomy site, enabling the animal to be attached to the fluid-percussion device. The device introduces a rapid (21–23 msec) bolus of saline into the closed cranial cavity, causing mechanical deformation of the brain. Animals in group II (n = 6) were subjected to either anesthesia (naive controls; n = 3) or anesthesia and surgery without injury (sham controls; n = 3). Animals in group III (n = 15) were subjected to lateral FP brain injury of identical severity (2.4–2.6 atm) and used for DNA agarose gel electrophoresis with a separate group of sham controls (group IV;n = 3) used as uninjured controls.
At 12 hr, 24 hr, 48 hr, 1 week, 2 weeks, 1 month, and 2 months after injury, group I animals (n = 3/time point) were reanesthetized with sodium pentobarbital (200 mg/kg, i.p.) and perfused with heparinized saline followed by 4% paraformaldehyde in phosphate buffer, pH 7.6. Animals in group II were likewise killed at 24 hr after anesthesia and/or surgery. All brains were post-fixed within the skull in 4% paraformaldehyde at 4°C for 3–4 hr after perfusion. At that time, the brains were removed from the skull and immersed in fixative for 24 hr at 4°C. After fixation, all brains were paraffin embedded and cut into 6 μm sections. Brain-injured animals in group III were reanesthetized with sodium pentobarbital (200 mg/kg, i.p.) and decapitated at 30 min, 2 hr, 15 hr, 24 hr, and 1 week after injury (n = 3/time point) for DNA agarose gel electrophoresis. Sham control animals in group IV were similarly killed at 24 hr after surgery. Brains from animals in groups III and IV were quickly removed, and the injured cortex and hippocampus were dissected on a cold plate and snap frozen in liquid nitrogen. Tissue was stored at −80°C until used. All protocols were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania, and in all studies we adhered to the animal welfare guidelines set forth in the Guide for the Care and Use of Laboratory Animals (United States Department of Health and Human Services, Publication No. 85-23, 1985).
Histological staining and quantitation of TUNEL-positive cells. TUNEL staining was performed using previously described methods (Gavrieli et al., 1992; Rink et al., 1995). In brief, 6 μm coronal sections at 3.6–3.8 mm posterior to bregma were adhered to poly-l-lysine–coated slides by heating them at 60°C for 15–20 min. After deparaffinization and rehydration, the tissue was digested for 15 min in proteinase K (20 μg/ml; Sigma, St. Louis, MO). The reaction was terminated with tap water, and the tissue was treated with buffer A (25 mmol/l Tris, pH 6.6, containing 200 mmol/l potassium cacodylate and 0.25 mg/ml bovine serum albumin) for at least 5 min. Sections were incubated at 37°C with labeling solution containing TdT (0.3 U/ml; Boehringer Mannheim, Indianapolis, IN), biotinylated-16-dUTP (20 mmol/l, Boehringer Mannheim), and 1.5 mmol/l cobalt chloride in buffer A for 1 hr in a humid chamber. The reaction was terminated with 2× SSC (300 mm sodium chloride and 30 mmsodium citrate, pH 7.4). After washing with 0.1 m Tris, pH 7.4, the sections were blocked with 10% goat serum in 0.1m Tris for 15 min. DNA was visualized by treating the tissue with a 1:40 dilution of streptavidin-conjugated alkaline phosphatase (BioGenex, San Ramon, CA) and was stained with fast red (Sigma Fast Red Kit; Sigma). The tissue was washed, counterstained with hematoxylin, and mounted in an aqueous medium. Brain tissue from an 8-d-old rat pup was used as a positive control as described previously (Fung et al., 1995). TUNEL-positive cells present in the cortex; the white matter, including the fimbria, cingulum, internal and external capsules, and corpus callosum; the hippocampus, including CA1–CA3 and the dentate gyrus and hilus; and the thalamus of the ipsilateral and contralateral hemispheres were examined and counted using light microscopy at 40× and under oil immersion at 63×. Cells were differentiated as being apoptotic and nonapoptotic based on TUNEL-positive staining, together with specific histological verification of two or more of the classic morphological hallmarks of apoptosis including membrane blebbing, chromatin condensation, nuclear shrinkage, and cytoplasm condensation and disintegration. Consequently, apoptotic cells exhibit intense nuclear staining with blebbing and breakdown of the cell surface into spherical apoptotic bodies. Swollen, diffusely TUNEL-stained cells not exhibiting apoptotic morphology were considered to be nonapoptotic. Three slides per brain, each separated by at least 42 μm, were examined in detail for the presence of apoptotic and nonapoptotic TUNEL-positive cells. Cell counts from the three slides were summed together according to region, giving a representative total number of cells within each brain. An average total cell count was then calculated from three brains at each time point. To evaluate gross cell loss, we performed toluidine blue staining on tissue sections adjacent to those stained with TUNEL (data not shown).
Immunohistochemistry and TUNEL double-label studies.To identify the type(s) of cells undergoing DNA fragmentation, we subjected 6μ coronal sections from brain-injured animals killed at 24 hr and 1 week (n = 3 animals/time point) to TUNEL-labeling and to subsequent immunohistochemical analysis for neurons [anti-neurofilament (RMO44); monoclonal; 1:50 (Pleasure et al., 1989)], astrocytes (anti-glial fibrillary acidic protein; polyclonal; 1:100; Sigma), or oligodendrocytes [anti-cyclic nucleotide 3′-phosphohydrolase (CNP); polyclonal; 1:100 (Raible and McMorris, 1989)]. After the TUNEL procedure, sections were rinsed in 0.1 m Tris, pH 7.0, incubated with 2% normal horse serum to block nonspecific binding of antibodies, and incubated overnight (18–20 hr) at 4°C with the primary antibodies described above. Sections were washed in 0.1 m Tris, pH 7.0, and incubated at ambient temperature for 2 hr with fluorescein isothiocyanate (FITC)-tagged donkey anti-mouse IgG (1:100; to detect RMO44) or FITC-donkey anti-rabbit IgG (1:200; to detect anti-GFAP and anti-CNP). Sections were washed in 0.1 m Tris, pH 7.0, coverslipped using 1,4-diazabicyclo[2,2,2]octane. A Nikon FXA photomicroscope equipped with epifluorescence and a FITC filter cube was used for photomicrography. Bright-field as well as fluorescent photomicrographs were taken using a Nikon 40× plan apochromat lens that can detect both fluorescence and Fast Red histochemistry (Schmidt et al., 1997). A distinct population of TUNEL-labeled cells did not stain with any of the antibodies used, suggesting that the cell death process affected the expression of these surface antigens. Thus, quantitation of TUNEL-positive cells, rather than of double-labeled cells, was performed.
DNA agarose gel electrophoresis. DNA-end labeling and DNA agarose gel electrophoresis were performed on cortical and hippocampal tissue according to the methods described previously (Tilly and Hsueh, 1993). Frozen tissue was homogenized in 0.3 mol/l Tris, pH 8.0, containing 0.1 mol/l NaCl, 0.01 mol/l EDTA, 0.2 mol/l sucrose, and 10% SDS and was incubated with agitation at 68°C for 1 hr. Protein precipitation was initiated with the addition of 8 mpotassium acetate and incubation on ice for 1 hr. The suspensions were centrifuged at 14,000 rpm at 4°C, and the supernatants were digested with DNase-free RNase (2 mg/ml) for 1 hr at 37°C. DNA was extracted with phenol and chloroform and precipitated with isopropanol, and the pellet was washed, dried, and dissolved in distilled water. Purified DNA was treated with 30 U of TdT (Life Technologies, Gaithersburg, MD), 25 μCi of [α-32P]ddATP (Amersham, Arlington Heights, IL), and 10 μl of 5× concentrated buffer (Life Technologies) in a reaction volume of 50 μl and was incubated at 37°C for 1 hr. The reaction was stopped with 2.5 μl of 0.5 mol/l EDTA, pH 8.0. Precipitation of the labeled DNA was achieved with three volumes of 100% ethanol and 10 mol/l ammonium acetate at −80°C for 1 hr, using 50 μg of yeast tRNA as a carrier. The suspension was centrifuged, and the resulting pellet was dried, redissolved in 50 μl of buffer (10 mmol/l Tris, pH 8.0, containing 1 mmol/l EDTA) twice, and stored at −20°C. The DNA was fractionated on a 2% agarose gel in 40 mmol/l Tris-acetate, pH 8.0, containing 1 mmol/l EDTA and was exposed to autoradiographic film (Kodak XAR5, Rochester, NY) for 24 hr.
Statistical analysis. Mean values of apoptotic and nonapoptotic TUNEL-positive cells in injured brains were compared with sham mean values using one-way ANOVA followed by a post hoc Dunnett’s test. A p value of <0.05 was considered to be statistically significant. All mean values are reported with SEM.
RESULTS
Morphology of TUNEL-stained cells
Both naive and sham control brains exhibited few, if any, TUNEL-stained cells demonstrating apoptotic morphology, scattered randomly throughout regions of the cortex, white matter, hippocampus, and thalamus in both hemispheres of control brains. Lateral FP brain injury resulted in markedly increased numbers of TUNEL-stained cells in all regions of the injured hemisphere examined, with little to no change in the hemisphere contralateral to the injured side. Analysis of TUNEL histochemistry revealed two populations of stained cells in the traumatically injured brain. One population of TUNEL-positive cells (type II) were intensely stained, round, and shrunken and exhibited morphological features of apoptosis cells such as condensed nuclei (Fig.1A,B). These cells appeared to be morphologically similar to those seen in developing (8-d-old) rat brain, suggestive of the apoptotic nature of traumatic cell death. However, the other population (nonapoptotic or type I) of TUNEL-labeled cells exhibited diffuse, uneven cytoplasmic staining, lacked nuclear condensation and fragmentation, and on occasion appeared neuron-like (Fig. 1C,D). Interestingly, no differences were observed in the morphology of apoptotic cells seen in the different regions at the various time points studied after injury. Thus, TUNEL-stained, apoptotic cells in the injured cortex at 24 hr (Fig. 1A) exhibited similar morphological features to those observed in the injured thalamus at 2 weeks after injury (Fig. 1B). Figure2 shows the temporal progression of apoptotic cell death from injured cortex through deeper structures such as the hippocampus and white matter and into thalamic nuclei over time.
Fig. 1.

Photomicrographs illustrating type II apoptotic cells (A, B) and type I nonapoptotic cells (C, D) in the rat brain after lateral FP brain injury of moderate severity. Sections were counterstained with hematoxylin. A, B, Apoptotic cells were identified based on the rounded, shrunken nature of the cytoplasm and nucleus and on the intense staining of the nucleus. Note the similar intense TUNEL reactivity and morphological characteristics in apoptotic cells in the injured cortex at 24 hr after injury (A) and in injured thalamus at 2 weeks after injury (B). C,D, Type I nonapoptotic cells were characterized by diffuse TUNEL reactivity, uneven staining in the cytoplasm, and a lack of shrunken cytoplasm. C illustrates a nonapoptotic cell in the cortex at 12 hr after injury, whereas Dillustrates a typical type I TUNEL-stained cell (white arrow) in the thalamus at 1 week after injury. Note the neuron-like appearance and cytoplasmic TUNEL stain of cells inD and a relatively normal-appearing neuron stained with hematoxylin (D, black arrow). Scale bar, 50 μm.
Fig. 2.
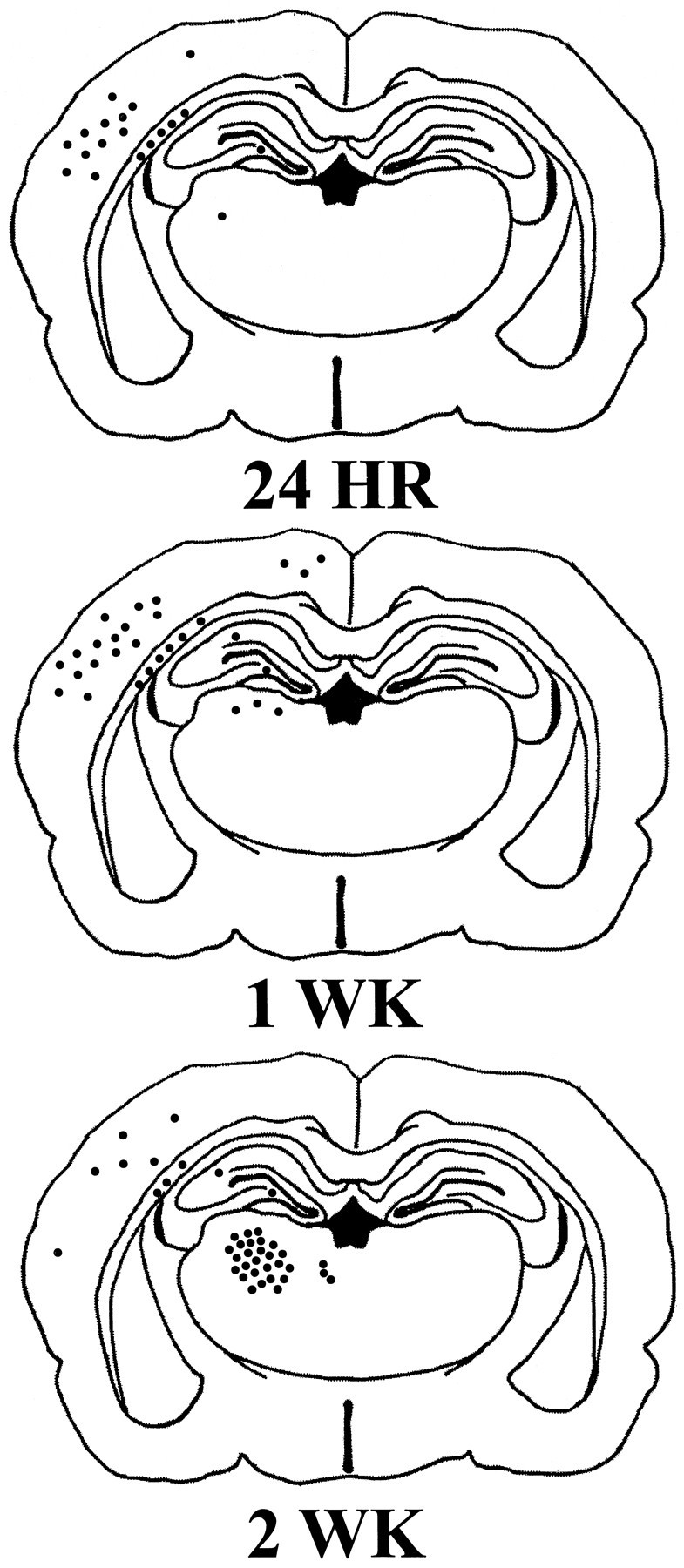
Schematic of TUNEL-stained sections of the rat brain 24 hr, 1 week, and 2 weeks after injury illustrating the regional distribution of apoptotic cells. Initially, apoptotic cells were observed primarily in the injured cortex at 24 hr after injury and over time were detected in internal structures, such as the thalamus and hippocampus. Each filled circle (•) represents five TUNEL-positive cells exhibiting apoptotic morphology.
Cortex
Analysis of the cortical hemisphere ipsilateral to the impact site revealed intense TUNEL-labeled apoptotic cells primarily in the injured parietal and temporal cortices (Figs. 2,3A; p < 0.001 using ANOVA). By 24 hr after injury, the number of TUNEL-positive apoptotic cells was found to be significantly increased when compared with those in sham animals (p < 0.05). Although the number of apoptotic cells appeared to decline by 48 hr after injury, a second increase in apoptotic cells was observed in the injured cortex, peaking by 1 week after injury (p < 0.05 compared with sham). TUNEL-positive apoptotic cells were observed even at 2 weeks and 1 month after injury, but by 2 months the number of apoptotic cells had reached sham levels in all cortical regions.
Fig. 3.

Quantitation of TUNEL-positive cells exhibiting apoptotic morphology in the rat brain after lateral FP injury of moderate severity (2.4–2.6 atm) or sham surgery (n= 3/time point). A, Cortex. Average number of apoptotic cells in the cortex peaked at 24 hr, with a second, delayed peak at 1 week after injury. B, White matter. Average number of apoptotic cells in the subcortical white matter, comprising the internal and external capsule, fimbria, corpus callosum, and cingulum, peaked at 1 week after injury. C, Hippocampus. Average number of apoptotic cells in the hippocampus, comprising the CA1–CA3, dentate hilus, and gyrus regions, peaked at 48 hr after injury.D, Thalamus. Average number of apoptotic cells in the thalamus peaked at 2 weeks after injury. By 2 months, apoptotic cells in all regions was similar to that found in sham control brains.Open bars represent the ipsilateral hemisphere;shaded bars represent the contralateral hemisphere; *p < 0.05 when compared with sham. Average number of cells was determined as described in Materials and Methods. Error bars represent SEM.
White matter
Analysis of regional white matter tracts, including the internal and external capsules, fimbria, corpus callosum, and cingulum, demonstrated a marked increase in TUNEL-positive apoptotic cells as early as 12 hr after injury (p < 0.05 using ANOVA). Fewer TUNEL-positive apoptotic cells were observed in white matter than in the overlying injured cortex in the acute period after injury. At all time points, the majority of apoptotic cells were observed in the internal and external capsules and fimbria ipsilateral to the injury site (Fig. 2). Although the number of apoptotic cells appeared not to change between 12 hr and 1 month after injury, a significant peak was noted at 1 week (Fig. 3B;p < 0.05 compared with sham). By 2 months after injury, the number of apoptotic cells had returned to sham (uninjured) values.
Hippocampus
Compared with the cell counts in other brain structures, quantitation of apoptotic cells in the hippocampal CA1–CA3 regions and dentate hilus and gyrus revealed that these regions showed the least marked apoptotic response to injury with respect to the absolute number of TUNEL-positive cells (Figs. 2, 3C). A prominent apoptotic response to injury in hippocampus was observed at 12 hr after injury. Although a secondary peak of apoptosis was observed by 48 hr, these differences were not statistically significant when compared with sham controls. The number of apoptotic cells returned to sham levels by 1 month after injury (Fig. 3C). Interestingly, apoptotic cells were not localized to a particular region but were randomly scattered throughout the hippocampal structures.
Thalamus
TUNEL-positive cells exhibiting apoptotic morphology remained at sham levels in the thalamus until 1 week after injury, when a slight and gradual increase was observed, which continued up to 4 weeks after injury (Fig. 3D). However, a dramatic and delayed increase in the number of apoptotic cells was observed by 2 weeks after injury in the ventroposterior medial (VPM) and lateral (VPL) thalamic nuclei (Figs. 2, 3D; p < 0.05 compared with sham controls). The number of apoptotic cells began to decline by 1 month after injury, reaching sham levels by 2 months.
Immunohistochemical analysis of TUNEL-labeled cells
Double-labeling experiments combining TUNEL and immunohistochemistry revealed a mixture of TUNEL-labeled neurons (Fig.4A) and oligodendrocytes (Fig. 4D) primarily localized to the injured cortex at 24 hr after injury. By 1 week after injury, a population of TUNEL-positive astrocytes were first observed in the injured cortex (Fig. 4C) together with double-labeled neurons (Fig. 4B) and oligodendrocytes (Fig.4D). Double-labeled neurons were also present in the thalamus by 1 week after injury. The few double-labeled cells observed in the hippocampus at 24 hr and 1 week after injury were immunopositive for neurofilament (RM044) and TUNEL and were classified as neurons. Immunohistochemical labeling in the white matter was predominantly restricted to oligodendrocytes staining positive for CNP, but staining was diffuse and not localized to nuclei or processes; neuronal staining was not detected in white matter. No apparent changes occurred in the proportion of the specific populations of cells showing in situ nick end labeling during the first 7 d after injury. Chromatin condensation, consistent with apoptosis, was detected in neurons (Fig. 4B) and astrocytes (Fig.4C). Adjacent sections not subjected to the TUNEL procedure exhibited immunoreactive profiles identical to that of sections that were TUNEL-stained previously, suggesting that the TUNEL protocol did not affect the immunoreactivity of the cellular antigens used.
Fig. 4.

Representative photomicrographs illustrating TUNEL-labeled neurons (A, B), astrocytes (C), and oligodendrocytes (D) in the injured cortex. Neurons were observed at 24 hr (A) and 1 week (B) after injury, whereas GFAP-positive astrocytes were observed predominantly at 1 week (C) after injury. CNPase-labeled oligodendrocytes (D) were observed at 24 hr after injury. Arrows indicate TUNEL-positive nuclei, and arrowheads indicate staining in cell bodies. Tissue sections were subjected to the TUNEL procedure followed by immunohistochemistry as described in Materials and Methods. Scale bar, 50 μm.
DNA agarose gel electrophoresis
To validate analysis of TUNEL histochemistry, we fractionated DNA from cortical and hippocampal tissue at 30 min, 2 hr, 15 hr, 24 hr, and 1 week after injury on a 2% agarose gel (Fig.5). Uniform DNA fragmentation [180–200 base pairs (bp)] associated with apoptosis was observed as early as 30 min in the injured cortex (Fig. 5A). These bands appeared to intensify with time, reaching a maximum at 24 hr after injury, with evidence of uniform DNA fragmentation persisting until 1 week after injury. The injured hippocampus showed slight evidence of DNA banding at 2 hr after injury, with maximal DNA banding at 15 hr after injury; however, uniform DNA fragmentation was not observed beyond 24 hr after injury (Fig. 5B). No DNA banding was detected in naive control brains or in the hemisphere contralateral to the injury (data not shown).
Fig. 5.

Representation of internucleosomal DNA fragmentation at 30 min (lane 3), 2 hr (lane 4), 15 hr (lane 5), 24 hr (lane 6), and 1 week (lane 7) after injury in the cortex (A) and hippocampus (B). DNA fragments in 180–200 bp intervals were detected in injured tissue but not in sham tissue (lane 2). Molecular weight standards are shown in lane 1 of each gel with molecular weight markers illustrated.
Quantitation of nonapoptotic cells
In the parietal and temporal cortices, TUNEL-positive cells not exhibiting apoptotic morphology (type I) were observed at 12 hr after brain injury with the maximal number of cells at 24 hr after injury (p < 0.05 compared with sham), declining gradually to sham levels by 1 month after injury (Fig.6A). An increase in TUNEL-positive, nonapoptotic cells was observed in white matter structures between 24 and 48 hr after injury (Fig.6B), particularly in the internal and external capsules and fimbria. By 1 week, however, few nonapoptotic, TUNEL-positive cells were detected, with the number of cells reducing to sham levels by 2 months after injury. Nonsignificant increases in the number of TUNEL-positive, nonapoptotic cells were observed in injured hippocampus at both 12 and 48 hr after injury (Fig.6C). In the injured thalamus (Fig. 6D), a peak in the number of nonapoptotic, TUNEL-stained cells was observed at 12 hr (p < 0.05 compared with sham), with a second broader, nonsignificant peak between 48 hr and 1 month after injury. The number of nonapoptotic, TUNEL-positive cells eventually reached sham levels by 2 months after injury.
Fig. 6.

Quantitation of TUNEL-positive, nonapoptotic cells in the rat brain after lateral FP injury of moderate severity. Nonapoptotic cells were identified based on diffuse, uneven TUNEL staining in the nucleus and cytoplasm with little to no cellular shrinkage. A, Cortex. Average number of nonapoptotic cells peaked at 24 hr after injury. B, White matter. Average number of nonapoptotic cells peaked between 24 and 48 hr after injury. C, Hippocampus. Average number of TUNEL-positive, nonapoptotic cells in the injured hippocampus increased slightly (nonsignificantly) at 12 and 48 hr after injury.D, Thalamus. Average number of TUNEL-positive, nonapoptotic cells in the injured thalamus revealed a biphasic response, with a significant peak at 12 hr and a second broader, nonsignificant peak between 48 hr and 1 month after injury. By 2 months after injury, the number of nonapoptotic TUNEL-positive cells had reached sham levels in all regions. * p < 0.05 when compared with sham. Average number of cells was determined as described in Materials and Methods. Error bars represent SEM.
DISCUSSION
Experimental brain injury induced regionally distinct patterns of apoptotic cell death in the brain during both the acute and chronic period after injury. In the injured cortex, a biphasic pattern of apoptosis was observed, which included both an acute and a second, delayed increase in TUNEL-positive apoptotic cells at 24 hr and 1 week, respectively. The peak in apoptotic cell death in subcortical white matter was observed at 1 week after injury. Few apoptotic cells were observed in the thalamus during the acute post-traumatic period, although a massive and delayed increase in apoptosis was observed by 2 weeks after injury. By 2 months after injury, the number of apoptotic cells in all structures returned to sham levels. DNA agarose gel electrophoresis revealed that acute and delayed uniform DNA fragmentation into 180–200 bp fragments occurred in the injured cortex and hippocampus, confirming the presence of apoptosis. Because DNA laddering represented DNA fragmentation in the entire injured cortex whereas TUNEL-labeling was performed in specific coronal sections, the sample size used for the gels was larger, and therefore discrepancies may occur in the intensity of DNA fragmentation as characterized by agarose gel electrophoresis versus TUNEL immunohistochemistry. Moreover, although TUNEL staining in situ identified local appearances of DNA fragmentation, agarose gels provided information regarding the presence of DNA damage associated with apoptosis. To our knowledge, these data are the first to document both biphasic and prolonged apoptosis after experimental brain injury, suggesting delayed apoptosis can occur during the chronic post-traumatic period in selected brain regions. These data also suggest that DNA fragmentation may occur initially at the injury site in the cortex, with progression into deeper subcortical structures over time.
Compared with the number of apoptotic cells, a markedly higher number of TUNEL-positive, nonapoptotic cells was observed in the injured brain. In the majority of brain regions (cortex, hippocampus, and thalamus), the increase in the numbers of TUNEL-positive nonapoptotic cells appeared to precede those of TUNEL-positive apoptotic cells, suggesting that TUNEL-labeled cells with fragmented DNA may eventually progress into morphologically distinct apoptotic cells. It has been suggested that the different morphologies may reflect different stages of the same death process (Wilcox et al., 1995). However, it is equally possible that the range of morphologies observed here may be indicative of simultaneously activated apoptotic and necrotic processes (Portera-Cailliau et al., 1997).
The pattern of DNA fragmentation and apoptotic cell death as illustrated in Figure 2 correlates both regionally and temporally with patterns of neuronal injury after experimental brain injury in the rat (Dietrich et al., 1994; Rink et al., 1995; Hicks et al., 1996). During the first 10 min to 2 hr after injury, both acid fuchsin-stained and silver degeneration-stained neurons have been observed in structures in the injured hemisphere (Dietrich et al., 1994; Hicks et al., 1996). By 24 hr, these neurons are shrunken and show evidence of advanced degradation (Povlishock et al., 1992; Dietrich et al., 1994). Because our data demonstrate abundant DNA fragmentation occurring in the injured cortex by 24 hr, it is likely that a subpopulation of cells is experiencing irregular DNA fragmentation associated with necrosis. However, the prominent DNA “laddering” observed using agarose gel electrophoresis at 2 and 15 hr after injury suggests that a component of this acute cell death may also be apoptotic.
Using a contusion model of spinal cord injury in the rat, Crowe et al. (1997) recently reported the presence of apoptotic neurons and oligodendrocytes in the spinal cord within 6 hr after injury. Interestingly, the number of apoptotic cells was significantly increased in areas radiating progressively outward in both directions from the site of spinal cord contusion for up to 8 d after injury, suggesting that apoptotic cell death occurred over a delayed period in regions increasingly distal to the site of injury. Likewise, in another model of spinal cord compression injury, marked apoptosis of oligodendrocytes and astrocytes was observed at 4 and 9 d after contusion in the region both cranial and caudal to the injury site, suggesting that the selective vulnerability of glial cells was related to delayed apoptotic mechanisms (Li et al., 1996). These results closely parallel those observed in the present study after traumatic brain injury. By 24 hr after injury, in addition to oligodendrocytes, we observed DNA fragmentation (TUNEL-labeling) in neurons localized primarily in the injured cortex. Interestingly, TUNEL-positive, nonapoptotic cells observed in the chronic post-traumatic period (up to 1 month) exhibited distinct neuronal morphology, suggestive of an ongoing degenerative process. By 1 week after injury, populations of TUNEL-labeled astrocytes began to appear in the injured cortex, together with double-labeled neurons and oligodendrocytes with a few scattered neurons in the hippocampus. TUNEL-positive neurons were also observed in the thalamus beginning 1 week after injury. Our results replicate and extend the findings of earlier reports indicating that both DNA fragmentation and dystrophic/apoptotic neurons can be detected in injured cortex and hippocampus after experimental weight drop (Pravdenkova et al., 1996), cortical contusion (Colicos and Dash, 1996), and lateral FP brain injury (Rink et al., 1995) and support the recent observations of Yakovlev et al. (1997) that TUNEL-positive neurons (labeled with the NeuN antibody) can be observed in injured cortex at 24 and 72 hr after lateral FP brain injury. Our results further extend these observations regionally and temporally and suggest also that populations of astrocytes and oligodendrocytes also show DNA fragmentation after brain trauma.
In the present study, a distinct population of TUNEL-positive cells did not stain with any of the antibodies used, suggesting that the cell death process affected the expression of various surface antigens. Alternatively, these cells may represent a population of macrophage or microglial cells undergoing apoptosis. However, a recent study byShuman et al. (1997) has reported that many cells exhibiting apoptotic profiles after traumatic spinal cord injury did not label with immunoreactive markers for microglia/macrophages but rather double-labeled with markers for oligodendrocytes. However, these authors also observed a small population of OX42-immunopositive cells containing apoptotic bodies and suggested that some microglial cells may undergo apoptosis or may simply be in the process of phagocytizing apoptotic oligodendrocytes.
The robust increase in apoptotic cells in the thalamus beginning 2 weeks after injury and extending for up to 1 month after injury suggests that this region undergoes delayed and persistent apoptosis after brain injury. Furthermore, TUNEL labeling was observed at the chronic time points in cells that appeared neuron-like, suggestive of an ongoing degenerative process. The thalamus appears to be particularly vulnerable to focal injury induced in the cerebral cortex, as axonal projections from the thalamus may undergo retrograde damage (Iizuka et al., 1990; Pierce et al., 1995b; Hicks et al., 1996). Importantly, specific thalamocortical projections exist between the VPL/VPM thalamic nuclei (Paxinos, 1985) (where delayed apoptosis was observed) and the regions of the temporoparietal cortex that show characteristic and reproducible neurodegeneration in this and other models of experimental brain injury. This delayed post-traumatic apoptosis observed in the thalamus may be associated with activation of calpain (Squire et al., 1994), and, in fact, evidence of post-traumatic delayed calpain activation in the medial geniculate nucleus and the ventroposterior and laterodorsal thalamic nuclei has been shown to occur only after 1 week after injury (Saatman et al., 1996).
Both apoptotic and necrotic cell death has been reported in brains of Huntington’s disease patients (Portera-Cailliau et al., 1995) and in the hippocampal region of Alzheimer’s disease patients (Smale et al., 1995). Regional apoptotic cell death has been observed in the acute and chronic period in models of permanent or transient cerebral ischemia (MacManus et al., 1993; Li et al., 1995). After focal cerebral ischemia in mice, selectively vulnerable neurons within the inner boundary zone of the infarct exhibited DNA fragmentation with necrotic and apoptotic morphologies (Li et al., 1995). Likewise, by the use of gel electrophoresis and in situ end labeling, apoptosis was observed in hippocampal CA1–CA2 and striatal neurons after global ischemia in the rat (MacManus et al., 1993). Recently, an investigation of the progression of apoptotic cell death 3 d after mild focal ischemia in rats suggested a role for delayed apoptosis in the postischemic period (Du et al., 1996).
The molecular and cellular mechanism(s) underlying the induction of apoptotic cell death remains to be elucidated. Calcium ion influx has been shown to occur in the injured CNS (Young, 1992; Fineman et al., 1993). This ion influx has been suggested to activate Ca+2- and Mg+2-dependent endonucleases responsible for DNA fragmentation preceding apoptosis (Giannakis et al., 1991). Acute activation of both non-NMDA and NMDA receptors has been associated with neuronal vulnerability after brain trauma (McIntosh et al., 1990; Hayes et al., 1992; Toulmond et al., 1993; Hicks et al., 1994) and may participate in the progression of apoptosis in the injured hemisphere.
Expression of known genetic regulators of apoptosis, such as bcl-2 and bax, has been correlated with the temporal distribution of postischemic cell loss or survival (Gillardon et al., 1996). After global ischemia in the rat, immunostaining revealed an increase in regional levels of bax with a concurrent decrease in bcl-2 in the CA1 region of the hippocampus (Krajewski et al., 1996), whereas expression of bcl-2 has been shown to increase in the cortex after focal ischemia (Chen et al., 1995). Lateral FP brain injury in the rat has been shown to induce a loss of bcl-2 immunoreactivity in the injured parietotemporal cortex and hippocampus, which precedes neuronal cell loss (Raghupathi and McIntosh, 1995), whereas Clark et al. (1997) reported that bcl-2 immunoreactivity was increased in surviving neurons but was not present in TUNEL-positive cells after cortical impact injury in the rat.Yakovlev et al. (1997) have recently shown that gene expression for both caspase-1 and caspase-3 were increased in injured cortex at 24 hr after FP brain injury, implicating these proteases in neuronal apoptosis induced by TBI. Interestingly, the increase in caspase-3 expression in the injured cortex is temporally correlated with the peak of apoptotic cells observed in the cortex in the present study. In addition, inducible transcription factors, such as c-jun, which are upregulated after traumatic CNS injury (Dragunow et al., 1993;Raghupathi and McIntosh, 1996), may contribute to the progression of apoptosis. These data suggest that the post-traumatic altered expression of specific genes may stimulate a programmed cell death cascade resulting in apoptosis.
Disruption of the trophic factor milieu may also contribute to the delayed post-traumatic cell death observed in the rat brain. After weight drop and controlled cortical contusion models of brain injury, at 3 and 7 d after injury, respectively, levels of NGF protein increased markedly in the injured cortical region (DeKosky et al., 1994). Despite the potentially regional protective effects of increased NGF levels, a concurrent decrease in NGF-receptor immunoreactivity has been documented after brain injury (Leonard et al., 1994), suggesting that endogenous levels of NGF after trauma may be insufficient to halt cellular suicide pathways induced by withdrawal of trophic factors. To this end, previous studies have shown that treatment with NGF after a traumatic brain injury can attenuate regional apoptotic cell death in the septum (Sinson et al., 1997). Future studies are needed to determine whether apoptosis plays a deleterious or, in fact, an adaptive role in the histopathological events after brain injury. It is hoped that with this knowledge, more focused therapies could be developed and implemented in the treatment of neuronal cell death associated with traumatic brain injury.
Footnotes
This work was supported, in part, by National Institutes of Health Grants P01-NS08803 (T.K.M.) and RO1-NS26818 (T.K.M.) from the National Institute of Neurological Disorders and Stroke and RO1-GM34690 (T.K.M.) from the National Institute of General Medical Sciences and by a Veterans Administration Merit Review grant (T.K.M.) and AG-09215 and AG-10124 (J.Q.T.). We thank O. Becher, J. Kwon, Dr. Chen Zhang, Adrienne Brown, and Seamus Fernandez for their invaluable technical assistance, Dr. F. Arthur McMorris for the kind donation of the anti-CNP antibody, Sam Chiu for assistance with photography, and Laura Meehan and Jeanne Marks for careful preparation of this manuscript.
Correspondence should be addressed to Dr. Tracy K. McIntosh, 3320 Smith Walk, Room 102B, Philadelphia, PA 19104-6316.
REFERENCES
- 1.Bramlett HM, Green EJ, Dietrich WD, Busto R, Globus MY-T, Ginsberg MD. Posttraumatic brain hypothermia provides protection from sensorimotor and cognitive behavioral deficits. J Neurotrauma. 1995;12:289–298. doi: 10.1089/neu.1995.12.289. [DOI] [PubMed] [Google Scholar]
- 2.Bramlett HM, Dietrich WD, Green EJ, Busto R. Chronic histopathological consequences of fluid-percussion brain injury in rats: effects of post-traumatic hypothermia. Acta Neuropathol (Berl) 1997;93:190–199. doi: 10.1007/s004010050602. [DOI] [PubMed] [Google Scholar]
- 3.Colicos MA, Dash PK. Apoptotic morphology of dentate gyrus granule cells following experimental cortical impact injury in rats: possible role in spatial memory deficits. Brain Res. 1996;739:120–131. doi: 10.1016/s0006-8993(96)00824-4. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Graham SH, Chan PH, Lan H, Zhou RL, Simon RP. bcl-2 is expressed in neurons that survive focal ischemia in the rat. NeuroReport. 1995;6:394–398. doi: 10.1097/00001756-199501000-00040. [DOI] [PubMed] [Google Scholar]
- 5.Clark RSB, Chen J, Watkins SC, Kochanek PM, Chen M, Stetler RA, Loeffert JE, Graham SH. Apoptosis-suppressor gene bcl-2 expression after traumatic brain injury in rats. J Neurosci. 1997;17:9172–9182. doi: 10.1523/JNEUROSCI.17-23-09172.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cortez SC, McIntosh TK, Noble L. Experimental fluid percussion brain injury: vascular disruption and neuronal and glial alterations. Brain Res. 1989;482:271–282. doi: 10.1016/0006-8993(89)91190-6. [DOI] [PubMed] [Google Scholar]
- 7.Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- 8.DeKosky ST, Goss JR, Miller PD, Styren SD, Kochanek PM, Marion D. Upregulation of nerve growth factor following cortical trauma. Exp Neurol. 1994;130:173–177. doi: 10.1006/exnr.1994.1196. [DOI] [PubMed] [Google Scholar]
- 9.Dietrich WD, Alonso O, Halley M. Early microvascular and neuronal consequences of traumatic brain injury: a light and electron microscopic study in rats. J Neurotrauma. 1994;11:289–301. doi: 10.1089/neu.1994.11.289. [DOI] [PubMed] [Google Scholar]
- 10.Dragunow M, Young D, MacGibbon G, Lawlor P, Singleton K, Sirimanne E, Beilharz E, Gluckman P. Is c-Jun involved in nerve cell death following status-epilepticus and hypoxic-ischemic brain injury? Mol Brain Res. 1993;18:347–352. doi: 10.1016/0169-328x(93)90101-t. [DOI] [PubMed] [Google Scholar]
- 11.Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischemia: a role for apoptosis? J Cereb Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Fineman I, Hovda DA, Smith M, Yoshino A, Becker DP. Concussive brain injury is associated with a prolonged accumulation of calcium: a 45Ca autoradiographic study. Brain Res. 1993;624:94–102. doi: 10.1016/0006-8993(93)90064-t. [DOI] [PubMed] [Google Scholar]
- 13.Fung K, Lee VM-Y, Trojanowski JQ. Dynamics of cell proliferation and cell death during the emergence of primitive neuroectodermal tumors of the immature central nervous system in transgenic mice. Am J Pathol. 1995;146:1376–1387. [PMC free article] [PubMed] [Google Scholar]
- 14.Gale SD, Johnson SC, Bigler ED, Blatter DD. Trauma-induced degenerative changes in brain injury: A morphometric analysis of three patients with preinjury and post-injury MR scans. J Neurotrauma. 1995;12:151–158. doi: 10.1089/neu.1995.12.151. [DOI] [PubMed] [Google Scholar]
- 15.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gennarelli TA. Animate models of human head injury. J Neurotrauma. 1994;11:357–368. doi: 10.1089/neu.1994.11.357. [DOI] [PubMed] [Google Scholar]
- 17.Giannakis C, Forbes IJ, Zalewski PD. Ca2+/Mg2+-dependent nuclease: tissue distribution, relationship to inter-nucleosomal DNA fragmentation and inhibition by Zn2+. Biochem Biophys Res Commun. 1991;181:915–920. doi: 10.1016/0006-291x(91)91278-k. [DOI] [PubMed] [Google Scholar]
- 18.Gillardon F, Lenz C, Waschke KF, Krajewski S, Reed JC, Zimmermann M, Kuschinsky W. Altered expression Bcl-2, Bcl-x, Bax, and c-Fos colocalizes with DNA fragmentation and ischemic cell damage following middle cerebral artery occlusion in rats. Mol Brain Res. 1996;40:254–260. doi: 10.1016/0169-328x(96)00059-9. [DOI] [PubMed] [Google Scholar]
- 19.Groswasser-Reider I, Cohen M, Costeff H, Groswasser Z. Late CT findings in brain trauma: relationship to cognitive and behavioral sequelae and to vocational outcome. Am J Roentgenol. 1993;160:147–152. doi: 10.2214/ajr.160.1.8416613. [DOI] [PubMed] [Google Scholar]
- 20.Hamm RJ, White-Gbadebo DM, Lyeth BG, Jenkins LW, Hayes RL. The effect of age on motor and cognitive deficits after traumatic brain injury in rats. Neurosurgery. 1992;31:1072–1078. doi: 10.1227/00006123-199212000-00013. [DOI] [PubMed] [Google Scholar]
- 21.Hayes RL, Jenkins LW, Lyeth BG. Neurotransmitter-mediated mechanisms of traumatic brain injury–acetylcholine and excitatory amino acids. J Neurotrauma. 1992;9:S173–S187. [PubMed] [Google Scholar]
- 22.Hicks RR, Smith DH, McIntosh TK. Kynurenate is neuroprotective following experimental brain injury in the rat. Brain Res. 1994;655:91–96. doi: 10.1016/0006-8993(94)91601-2. [DOI] [PubMed] [Google Scholar]
- 23.Hicks RR, Soares HD, Smith DH, McIntosh TK. Temporal and spatial characterization of neuronal injury following lateral fluid-percussion brain injury in the rat. Acta Neuropathol (Berl) 1996;91:236–246. doi: 10.1007/s004010050421. [DOI] [PubMed] [Google Scholar]
- 24.Hovda DA, Yoshino A, Kawamata T, Katayama Y, Fineman I, Becker DP. The increase in local cerebral glucose utilization following fluid percussion brain injury is prevented with kynurenic acid and is associated with an increase in calcium. Acta Neurochir [Suppl] (Wien) 1990;51:331–333. doi: 10.1007/978-3-7091-9115-6_112. [DOI] [PubMed] [Google Scholar]
- 25.Iizuka H, Sakatani K, Young W. Neural damage in the rat thalamus after cortical infarcts. Stroke. 1990;21:7990–7994. doi: 10.1161/01.str.21.5.790. [DOI] [PubMed] [Google Scholar]
- 26.Kerr JFR, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krajewski S, Mai JK, Krajewska M, Sikorska M, Mossakowski MJ, Reed JC. Upregulation of bax protein levels in neurons following cerebral ischemia. J Neurosci. 1995;15:6364–6376. doi: 10.1523/JNEUROSCI.15-10-06364.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leonard JR, Maris DO, Grady SM. Fluid percussion injury causes loss of forebrain choline acetyltransferase and nerve growth factor receptor immunoreactive cells in the rat. J Neurotrauma. 1994;11:379–392. doi: 10.1089/neu.1994.11.379. [DOI] [PubMed] [Google Scholar]
- 29.Li GL, Brodin G, Farooque M, Funa K, Holtz A, Wang WL, Olsson Y. Apoptosis and expression of Bcl-2 after compression trauma to rat spinal cord. J Neuropathol Exp Neurol. 1996;55:280–289. doi: 10.1097/00005072-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 30.Li Y, Chopp M, Jiang N, Yao F, Zaloga C. Temporal profile of in situ DNA fragmentation after transient middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1995;15:389–397. doi: 10.1038/jcbfm.1995.49. [DOI] [PubMed] [Google Scholar]
- 31.MacManus JP, Buchan AM, Hill IE, Rasquinha I, Preston E. Global ischemia can cause DNA fragmentation indicative of apoptosis in rat brain. Neurosci Lett. 1993;164:89–92. doi: 10.1016/0304-3940(93)90864-h. [DOI] [PubMed] [Google Scholar]
- 32.MacManus JP, Hill IE, Preston E, Rasquinha I, Walker T, Buchan AM. Differences in DNA fragmentation following transient cerebral or decapitation ischemia in rats. J Cereb Blood Flow Metab. 1995;15:728–737. doi: 10.1038/jcbfm.1995.93. [DOI] [PubMed] [Google Scholar]
- 33.McIntosh TK, Vink R, Noble L, Yamakami I, Fernyak S, Faden AI. Traumatic brain injury in the rat: characterization of a lateral fluid percussion model. Neuroscience. 1989;28:233–244. doi: 10.1016/0306-4522(89)90247-9. [DOI] [PubMed] [Google Scholar]
- 34.McIntosh TK, Vink R, Soares HD, Hayes RL, Simon R. Effects of non-competitive blockade of N-methyl-d-aspartate receptors on the neurochemical sequelae of experimental brain injury. J Neurochem. 1990;55:1170–1179. doi: 10.1111/j.1471-4159.1990.tb03122.x. [DOI] [PubMed] [Google Scholar]
- 35.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia in apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 37.Paxinos G. The rat nervous system. Vol 1, Forebrain and midbrain. Academic; Kensington, New South Wales, Australia: 1985. [Google Scholar]
- 38.Pierce JES, Smith DH, Trojanowski JQ, McIntosh TK (1998) Enduring cognitive, neurobehavioral and histopathological changes present for up to one year following severe experimental brain injury in rats. Neuroscience, in press [DOI] [PubMed]
- 39.Pierce JES, Trojanowski JQ, Graham DI, Smith DH, McIntosh TK. Immunohistochemical characterization of alterations in the distribution of amyloid precursor proteins and amyloid β peptide following experimental brain injury in the rat. J Neurosci. 1996;16:1083–1090. doi: 10.1523/JNEUROSCI.16-03-01083.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pleasure SJ, Selzer ME, Lee VM-Y. Lamprey neurofilaments combine in one subunit the features of each mammalian NF triplet protein but are highly phosphorylated only in large axons. J Neurosci. 1989;9:698–709. doi: 10.1523/JNEUROSCI.09-02-00698.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos VE. Evidence of apoptotic cell death in Huntington disease and excitotoxic animal models. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Portera-Cailliau C, Price DL, Martin LJ. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: further evidence for an apoptosis-necrosis continuum. J Comp Neurol. 1997;378:88–104. [PubMed] [Google Scholar]
- 43.Povlishock JT, Erb DE, Astruc J. Axonal response to traumatic brain injury: reactive axonal change, differentiation, and neuroplasticity. J Neurotrauma. 1992;9:S189–S200. [PubMed] [Google Scholar]
- 44.Pravdenkova SV, Basnakian AG, James SJ, Anderson BJ. DNA fragmentation and nuclear endonuclease activity in rat brain after severe closed head injury. Brain Res. 1996;729:151–155. [PubMed] [Google Scholar]
- 45.Raghupathi R, McIntosh TK. Programmed cell death in traumatic brain injury: temporal alterations in bcl-2 immunoreactivity. J Neurotrauma. 1995;12:966. [Google Scholar]
- 46.Raghupathi R, McIntosh TK. Regionally and temporally distinct patterns of induction of c-fos, c-jun and junB mRNAs following experimental brain injury in the rat. Mol Brain Res. 1996;37:134–144. doi: 10.1016/0169-328x(95)00289-5. [DOI] [PubMed] [Google Scholar]
- 47.Raible DW, McMorris FA. Cyclic AMP regulates the rate of differentiation of oligodendrocytes without changing the lineage of commitment of their progenitors. Dev Biol. 1989;133:437–446. doi: 10.1016/0012-1606(89)90047-x. [DOI] [PubMed] [Google Scholar]
- 48.Rink AD, Fung K, Trojanowski JQ, Lee V, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- 49.Saatman KE, Bozyczko-Coyne D, Marcy VR, Siman R, McIntosh TK. Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996;55:850–860. doi: 10.1097/00005072-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 50.Schmidt ML, Lee VM, Forman M, Chiu TS, Trojanowski JQ. Monoclonal antibodies to a 100-kd protein reveal abundant A beta-negative plaques throughout gray matter of Alzheimer’s disease brains. Am J Pathol. 1997;151:69–80. [PMC free article] [PubMed] [Google Scholar]
- 51.Schmidt RH, Grady MS. Regional patterns of blood-brain barrier breakdown following central and lateral fluid percussion injury in rodents. J Neurotrauma. 1993;10:415–430. doi: 10.1089/neu.1993.10.415. [DOI] [PubMed] [Google Scholar]
- 52.Shuman SL, Bresnahan JC, Beattie MS. Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J Neurosci Res. 1997;50:798–808. doi: 10.1002/(SICI)1097-4547(19971201)50:5<798::AID-JNR16>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 53.Sinson G, Perri BR, Trojanowski JQ, Flamm ES, McIntosh TK. Neurotrophin infusion improves cognitive deficits and decreases cholinergic neuronal cell loss and apoptotic cell death following experimental traumatic brain injury. J Neurosurg. 1997;86:511–518. doi: 10.3171/jns.1997.86.3.0511. [DOI] [PubMed] [Google Scholar]
- 54.Smale G, Nichols NR, Brady DR, Finch CE, Horton WE., Jr Evidence for apoptotic cell death in Alzheimer’s disease. Exp Neurol. 1995;133:225–230. doi: 10.1006/exnr.1995.1025. [DOI] [PubMed] [Google Scholar]
- 55.Smith DH, Okiyama K, Thomas MJ, Claussen B, McIntosh TK. Evaluation of memory dysfunction following experimental brain injury using the Morris water maze. J Neurotrauma. 1991;8:259–269. doi: 10.1089/neu.1991.8.259. [DOI] [PubMed] [Google Scholar]
- 56.Smith DH, Lowenstein DH, Gennarelli TA, McIntosh TK. Persistent memory dysfunction is associated with bilateral hippocampal damage following experimental brain injury. Neurosci Lett. 1994;168:151–154. doi: 10.1016/0304-3940(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 57.Smith DH, Chen X-H, Pierce JES, Wolf JA, Trojanowski JQ, Graham DI, McIntosh TK. Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma. 1997;14:715–727. doi: 10.1089/neu.1997.14.715. [DOI] [PubMed] [Google Scholar]
- 58.Soares HD, Thomas MJ, Cloherty K, McIntosh TK. Development of prolonged focal cerebral edema and regional cation change following experimental brain injury in the rat. J Neurochem. 1992;58:1845–1852. doi: 10.1111/j.1471-4159.1992.tb10061.x. [DOI] [PubMed] [Google Scholar]
- 59.Soares HD, Hicks RR, Smith DH, McIntosh TK. Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J Neurosci. 1995;15:8223–8233. doi: 10.1523/JNEUROSCI.15-12-08223.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Squire MKT, Miller ACK, Malkinson AM, Cohen JJ. Calpain activation in apoptosis. J Cell Physiol. 1994;159:229–237. doi: 10.1002/jcp.1041590206. [DOI] [PubMed] [Google Scholar]
- 61.Tilly JL, Hsueh AJW. Microscale autoradiographic method for the qualitative and quantitative analysis of apoptotic DNA fragmentation. J Cell Physiol. 1993;154:519–526. doi: 10.1002/jcp.1041540310. [DOI] [PubMed] [Google Scholar]
- 62.Toulmond S, Duval D, Serrano A, Scatton B, Benavides J. Biochemical and histological alterations induced by fluid percussion brain injury in the rat. Brain Res. 1993;620:24–31. doi: 10.1016/0006-8993(93)90266-p. [DOI] [PubMed] [Google Scholar]
- 63.Vink R, McIntosh TK, Demediuk P, Faden AI. Decrease in total and free magnesium concentration following traumatic brain injury in rats. Biochem Biophys Res Commun. 1987;149:594–599. doi: 10.1016/0006-291x(87)90409-8. [DOI] [PubMed] [Google Scholar]
- 64.Vink R, McIntosh TK, Demediuk P, Weiner MW, Faden AI. Decline in intracellular free Mg2+ is associated with irreversible tissue injury following brain trauma. J Biol Chem. 1988;263:757–761. [PubMed] [Google Scholar]
- 65.Wilcox BJ, Applegate MD, Portera-Cailliau C, Koliatsos VE. Nerve growth factor prevents apoptotic cell death in injured central cholinergic neurons. J Comp Neurol. 1995;359:573–585. doi: 10.1002/cne.903590405. [DOI] [PubMed] [Google Scholar]
- 66.Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 67.Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yamakami I, McIntosh TK. Alterations in regional cerebral blood flow following brain injury in the rat. J Cereb Blood Flow Metab. 1991;11:655–660. doi: 10.1038/jcbfm.1991.117. [DOI] [PubMed] [Google Scholar]
- 69.Yoshino A, Hovda DA, Kawamata T, Katayama Y, Becker DP. Dynamic changes in local cerebral glucose utilization following cerebral concussion in rats: evidence of hyper and subsequent hypometabolic state. Brain Res. 1991;561:106–119. doi: 10.1016/0006-8993(91)90755-k. [DOI] [PubMed] [Google Scholar]
- 70.Young W. Role of calcium in central nervous system injuries. J Neurotrauma. 1992;9:S9–S25. [PubMed] [Google Scholar]