

Abstract
Although the CNS is an established immune-privileged site, it is under surveillance by the immune system, particularly under pathological conditions. In the current study we examined the lymphocyte infiltration, a key component of this neuroimmune surveillance, into the axotomized facial motor nucleus and analyzed the changes in proinflammatory cytokines and the blood–brain barrier.
Peripheral nerve transection led to a rapid influx of CD3-, CD11a (αL, LFA1α)- and CD44-immunoreactive T-cells into the axotomized mouse facial motor nucleus, with a first, low-level plateau 2–4 d after injury, and a second, much stronger increase at 14 d. These T-cells frequently formed aggregates and exhibited typical cleaved lymphocyte nuclei at the EM level. Immunohistochemical colocalization with thrombospondin (TSP), a marker for phagocytotic microglia, revealed aggregation of the T-cells around microglia removing neuronal debris. The massive influx of lymphocytes at day 14 was also accompanied by the synthesis of mRNA encoding IL1β, TNFα, and IFN-γ. There was no infiltration by the neutrophil granulocytes, and the intravenous injection of horseradish peroxidase also showed an intact blood–brain barrier. However, mice with severe combined immunodeficiency (SCID), which lack differentiated T- and B-cells, still exhibited infiltration with CD11a-positive cells. These CD11a-positive cells also aggregated around phagocytotic microglial nodules.
In summary, there is a site-selective infiltration of activated T-cells into the mouse CNS during the retrograde reaction to axotomy. The striking aggregation of these lymphocytes around neuronal debris and phagocytotic microglia suggests an important role for the immune surveillance during neuronal cell death in the injured nervous system.
Keywords: CD3, chemotaxis, microglia, cytokines, NK cells, scid
The CNS has long been seen as an established, immune-privileged site, as shown, for example, by the much longer survival of heterologous tissue transplanted into the brain than that transplanted into the periphery (Medawar, 1948; Barker and Billingham, 1977). This protection of the neural tissue is apparently attributable to the presence of several barriers against attack from the immune system. Normal CNS shows extremely low levels of lymphocytes that enter neural parenchyma (Wekerle et al., 1986; Hickey et al., 1991). Unstimulated microglia, the resident, macrophage-related cells, express only low levels of the major histocompatibility complex (MHC) molecules (Wong et al., 1984; Vass et al., 1986; Streit et al., 1989a,b; Raivich et al., 1993), which are essential for antigen presentation to T-cells (Ford et al., 1996; Dangond et al., 1997). Finally, the normal blood–brain barrier, well developed in the mature CNS (Brightman et al., 1970; Kniesel et al., 1997), leads to an almost complete block of the influx of immunoglobulins and complement (Scolding et al., 1989; Poduslo et al., 1994), the molecular mediators of humoral immunity. Despite this multiple immune-privilege, viral, bacterial, or parasitic infection of the CNS frequently leads to a rapid activation of the immune system, influx of lymphocytes, monocytes, and immunoglobulin into the affected tissue and the inactivation of the pathogenic agent (Griffin et al., 1992;Dietzschold, 1993; Schluter et al., 1996; Rodriguez et al., 1996;Deckert-Schluter et al., 1997).
Although this influx of immune cells and molecules into the CNS is a well studied phenomenon in both infectious and autoimmune disease, the initial stages of this process are not well understood. At present, there are two major concepts to explain the initiation of the immune attack in the neural tissue, based on accidental encounter and on chemotaxis by the lesioned neural parenchyma. The first concept is based on the fact that there is a low level of infiltrating lymphocytes even in the normal CNS (Wekerle et al., 1986). After a specific peripheral activation, a small proportion of reactive lymphocytes will also enter the CNS (Hickey et al., 1991; Zeine and Owens, 1992). When presented with the right antigen, together with MHC (Maehlen et al., 1989; Konno et al., 1990; Molleston et al., 1993), these lymphocytes can initiate the immune response, which will then be followed by a secondary recruitment of further circulating lymphocytes (Cross et al., 1990; Olsson et al., 1992; Kawai et al., 1993; Schnell et al., 1997). In the second hypothesis, a primary, selective injury to the neural parenchyma, for example during an infection or a neurodegenerative process, can lead to a local production of proinflammatory cytokines and chemotactic molecules (Wesselingh et al., 1994; Calvo et al., 1996;McGeer and McGeer, 1996; Schluesener et al., 1996; Klein et al., 1997), followed by secondary changes in the adhesion properties of the surrounding vascular endothelium and a site-specific chemotaxis of circulating lymphocytes. Interestingly, recent studies have shown a site-specific lymphocyte infiltration in human neurodegenerative diseases such as Alzheimer’s dementia (McGeer et al., 1993) and amyotrophic lateral sclerosis (Kawamata et al., 1992; Engelhardt et al., 1993), providing indirect evidence for such a parenchymal recruitment.
In the current study we explored this possible interaction between injured brain parenchyma and lymphocytes in the adult mouse facial motor nucleus after a peripheral nerve transection. Interestingly, this model shows considerable species-specific differences in the extent of post-traumatic neuronal cell death. Facial motoneurons in the adult rat exhibit very little degeneration after a simple axotomy (Streit and Kreutzberg, 1988). In the adult mouse, however, this model leads to an easily visible, late degeneration of ∼20–35% of the axotomized motoneurons (Sendtner et al., 1996; Ferri et al., 1998) and their removal by phagocytotic microglia, with a maximum 14 d after injury (Torvik and Skjörten, 1971; Möller et al., 1996). As shown in this study, axotomy of the mouse facial nerve is accompanied by a significant influx of T-cells to the sites of neuronal degeneration and production of proinflammatory cytokines, but also by the maintenance of an intact blood–brain barrier.
MATERIALS AND METHODS
Animals and surgical procedures. Three different groups of experimental animals (2- to 3-month-old mice) were used in this study. C57BL/6 mice were imported from Jackson Laboratory (Bar Harbor, ME; BL6/JL) and Charles River (Hannover, Germany; BL6/CR). Normal BALB/c mice and homozygous animals with severe combined immunodeficiency (SCID) on a BALB/c background were bred in our animal facility. In BL6/JL mice, the right facial nerve was cut under ether anesthesia, and the animals survived for 1–66 d after axotomy. Axotomized BL6/CR, BALB/c, and SCID-BALB/c mice were used for the day 14 time point. The animal experiments and care protocols were approved by the Regierung von Oberbayern (AZ 211-2531-10/93 and AZ 211-2531-37/97).
Light microscopic immunohistochemistry. After the animals were killed with ether, they were first perfused intracardially (30 ml/min) with 200 ml of PBS (10 mmNa2HPO4, 0.84% NaCl, pH 7.4), followed by 200 ml of 4% formaldehyde (FA) in PBS (4% FA/PBS), and the brain stem was removed and post-fixed by a 2 hr immersion in 1% FA/PBS at 4°C on a rotator (8 rpm). The tissue was cryoprotected by an overnight rotating immersion in sucrose (30% sucrose, 10 mm Na2HPO4, pH 7.4, 4°C), frozen on dry ice, and then cut at −15°C in a cryostat at the level of the facial motor nucleus. Sections (20 μm) were collected on warm, 0.5% gelatin-dipped slides (Merck, Darmstadt, Germany), refrozen on dry ice, and stored at −80°C before use. For immunohistochemistry, the tissue sections were stained as described by Möller et al. (1996), with overnight incubation with the primary antibodies (see Table 1), followed by biotinylated secondary antibodies (goat anti-rat and goat anti-rabbit, respectively; Vector, Wiesbaden, Germany) and avidin–biotin peroxidase complex (ABC; Vector), and then visualized with diaminobenzidine/H2O2 (DAB; Sigma, St. Louis, MO), with Co/Ni intensification (Adams, 1981) (see Figs.1, 2, 6). Statistical analysis on the number of CD3- and CD11a-immunoreactive cells per tissue was performed using Jandel Sigmaplot 3.02 software (Erkrath) using a two-tailed Student’st test.
Table 1.
Summary of primary antibodies
| Antigen | Antibody | Dilution | Cellular IR | Source |
|---|---|---|---|---|
| CD3 | α-CD3, RtM | 1:500 | T | Chemicon (Palo Alto, CA) |
| CD11a/αL | MCA819, RtM | 1:6000 | T, NK | Camon (Wiesbaden, Germany) |
| CD11b/αM | 5C6, RtM | 1:6000 | MG | Camon |
| CD44 | α-CD44, RtM | 1:3000 | T, NK, N1-a | Chemicon |
| MHC1 | ER-HR52, RtM | 1:50 | pMG, MG1-a | Camon |
| MHC2 | 1199293, RtM | 1:1000 | PVM | Boehringer, Mannheim (Mannheim, Germany) |
| NG | MCA771, RtM | 1:400 | NG | Camon |
| TCR αβ | H57-597, RtM | 1:800 | T1-a | Pharmingen (Hamburg, Germany) |
| TSP | α-TSP, RbP | 1:6000 | pMG, MG,1-a N1-a | Alexis (Gruenberg, Germany) |
| IBA1 | α-IBA1, RbP | 1:6000 | MG | Dr. Y. Imai, Department of Neurochemistry, National Institute of Neuroscience (Tokyo, Japan) |
αL, αL integrin subunit; αM, αM integrin subunit; IR, immunoreactivity; MHC1, MHC class I; MHC2, MHC class II; TCRαβ, T-cell receptor αβ; TSP, thrombospondin; NG, neutrophil granulocytes; T, T-cells; NK, natural killer cells; N, neurons; MG, microglia, pMG, phagocytotic microglia (microglial nodules); PVM, perivascular macrophages; RtM, rat monoclonal; RbP, rabbit polyclonal.
Weak immunoreactivity.
Fig. 1.

CD3 immunohistochemistry in the normal and axotomized mouse facial motor nucleus. CD3-immunoreactive T-lymphocytes are absent in the normal facial nucleus (0d), but appear 1 d after axotomy (1d, arrows), reach a maximum at day 14, and disappear almost completely at 66 d (66d) after injury. The extent of the facial motor nucleus is indicated by thedotted lines in this and in the following figure (Fig.2). All magnifications 49×. Bottom right, Quantitative time course of CD3-positive cells in the axotomized and contralateral facial nuclei (mean ± SEM, n = 3 animals per time point). Note the early plateau of two to three labeled cells per section 1–4 d after axotomy, and a further 10-fold increase at day 14. No statistically significant increase on the contralateral side.
Fig. 2.

Distribution of CD3-immunoreactive T-lymphocytes in the axotomized facial motor nucleus 14 d after injury. A, Diffuse distribution. B, A rare perivascular infiltrate (thin arrow) surrounding a large vessel (v). C, D, Focal aggregates of CD3-immunoreactive T-lymphocytes (arrows). Magnification, 49×.
Fig. 6.

Immunohistochemical distribution of MHC class I (A, B), CD3 (C, D), and CD11a (E, F) immunoreactivity in normal (A, C, E) and SCID mice (B, D, F), 14 d after facial axotomy. A, B, Strong, focal increase of MHC class I immunoreactivity in the axotomized facial motor nuclei (right side). No specific immunoreactivity on the contralateral, unoperated side. Note the similar increase in MHC class I in normal and SCID animals. Magnification, 15×. C, D, CD3 immunoreactivity. Complete absence of specific staining in the SCID animal. E, F, CD11a immunoreactivity. Note the reduction in the number of CD11a-positive cells in the immunodeficient mouse. Magnification: C–F, 110×.
Immunofluorescence/confocal laser microscopy. The fixed/spread tissue sections from 14 d axotomized facial motor nuclei were pretreated as above, with the only modification a preincubation in PB/5% donkey serum (DS; Dianova, Hamburg, Germany). The sections were incubated overnight with a combination of two primary antibodies: a rat monoclonal antibody against CD3, CD11a, CD11b, CD44, or MHC class I and a rabbit polyclonal antibody against thrombospondin (Möller et al., 1996) or IBA1 (Imai et al., 1996). This was followed by a combination of FITC-conjugated goat anti-rat (1:100, Sigma) and biotinylated donkey anti-rabbit IgG (1:100 in PB/BSA; Dianova) secondary antibodies for 1 hr at room temperature and 1 hr incubation with FITC-conjugated donkey anti-goat IgG tertiary antibody (1:100 in PB/BSA; Dianova), and then by a 1 hr incubation with Texas Red-avidin (1:100 in PB; Vector). The sections were covered with Vectashield (Vector), and digital micrographs (1024 × 1024 pixels) of FITC and Texas Red immunofluorescence were taken with a 100× objective in a Leica TCS 4D confocal laser microscope. Ten consecutive equidistant levels were taken per section (total vertical span 12 μm) and condensed to a 1 megabyte TIFF file for each fluorescence wavelength using the MaxIntens condensation algorithm. The algorithm picks the maximum intensity value for each pixel from 10 available levels.
Immunoelectron microscopy. For electron microscopy, 14 d axotomized animals were killed in ether and then first perfused slowly (8 ml/min) with 40 ml of MgPBS (10 mmMgCl2, 0.75% NaCl, 10 mmNa2HPO4, pH 7.4) to wash out red blood cells, followed by 80 ml of 0.5% glutaraldehyde/4% FA/MgPBS to achieve rapid cross-linking and then by 80 ml of 4% FA/MgPBS to wash out glutaraldehyde. MgCl2 was added to avoid vascular spasms during glutaraldehyde perfusion. The brainstems were rotation post-fixed for 2 hr at 4°C in 1% FA/PBS; 80 μm vibratome sections were cut at the level of the facial motor nucleus, followed by pre-embedding immunohistochemistry with a CD3 rat monoclonal antibody using a slightly modified immunohistochemistry (IHC) procedure on floating sections: treatment with acetone was omitted, the sections were preincubated for 4 hr in PBS/5% goat serum containing 0.01% Triton X-100, the secondary antibody was applied for 8 hr, and incubation with the ABC reagent was performed overnight (4°C). For DAB staining with Co/Ni intensification, vibratome sections were first preincubated for 20 min in DAB/CoNi without H2O2, followed by a 15 min DAB/H2O2/CoNi reaction at room temperature. After the DAB reaction, sections were fixed for 1 week in 2% glutaraldehyde in PBS and then processed for electron microscopy (araldite embedding) as described in Möller et al. (1996). For high resolution light microscopy (LM) (see Fig. 3A–D), 1 μm semithin araldite sections were scanned with a 100× objective and Practica Color Scanner (Dresden, Germany) with 24-bit RGB and 2700 × 3590 pixel resolution.
Fig. 3.
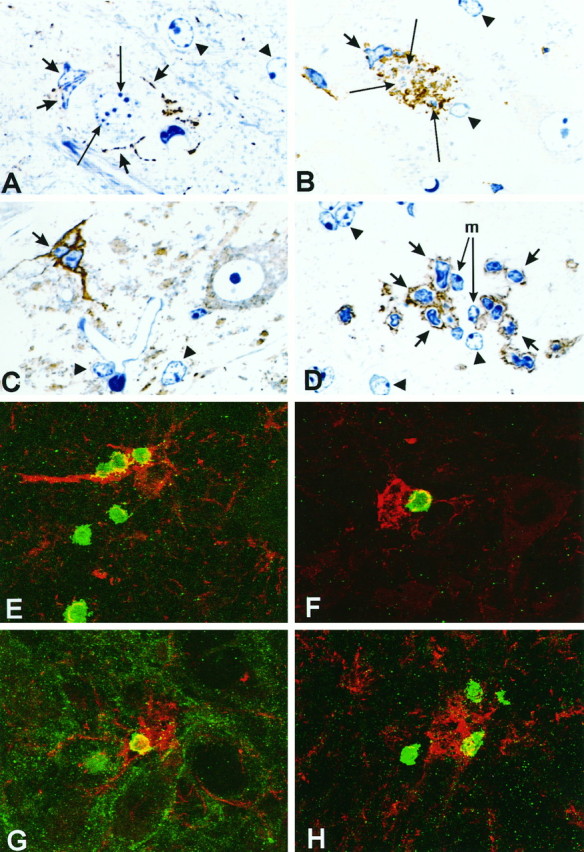
A–D, Different stages of microglial nodules in the mouse facial motor nucleus 14 d after injury in normal B6C3 mice; immunohistochemistry (brown staining) for TSP (A–C) and CD11a (D), 1 μm semithin araldite sections, methylene blue counterstain. A, Two activated microglia with slender TSP-immunoreactive processes (short arrows) adhere to an apoptotic neuron with nuclear chromatin condensation (long arrows). The arrowheads point to the TSP-negative astrocytes with clear and regular oval nuclei (also inB–D). B, Microglial phagocytosis of neuronal debris; strongly TSP-immunoreactive microglial nodule (short arrow) containing fragmented, methylene blue-counterstained cellular remnants (long arrows).C, Late stage TSP-immunoreactive microglial nodule (short arrow) consisting of three microglial cells after removal of the neuronal debris. The cellular structure of the TSP-immunoreactive nodules in this and the preceding micrograph (Fig.3B) is similar to that in E–H and Figure7C–F. D, Two microglial cells at the center of the nodule (m, long arrows) surrounded by CD11a-immunoreactive lymphocytes (short arrows).E–H, Colocalization of infiltrating lymphocytes and phagocytotic microglial nodules in the axotomized facial motor nucleus.E–G, Normal B6C3 mice, double immunofluorescence for thrombospondin and the T-lymphocyte markers CD3 (E), CD11a (F), and CD44 (G) 14 d after injury. Note the direct contact of T-lymphocytes (green) with the TSP-immunoreactive microglia (red). The CD44 immunoreactivity (G) is also present on the surface of axotomized motoneurons (Jones et al., 1997).H, SCID mouse facial motor nucleus, 14 d after injury. Apposition of CD11a-immunoreactive cells (green) on an IBA1-labeled microglial nodule (red). Magnification: A, 1140×;B–D, 900×; E–H, 950×.
Detection of cytokine mRNA. For RNA studies, the brainstem was removed immediately after animals were killed, frozen on dry ice, and cut to the level of the facial motor nucleus. The facial motor nuclei were cut out on the operated and contralateral side, and the RNA was isolated and reverse-transcribed as described by Klein et al., (1997). PCR was performed in a volume of 50 μl containing 1 μl of the transcribed cDNA sample, dNTPs (0.2 mm; Pharmacia, Piscataway, NJ), 2.5 U of Ampli Taq(Perkin-Elmer/Cetus, Emeryville, CA), and PCR buffer (Perkin-Elmer/Cetus). The cDNA was first denaturated at 95°C for 3 min, and primers (100 pmol) were added at 80°C (hot start). The PCR to detect gene transcripts for IFN-γ, TNFα, and IL1β was performed by 35 cycles of the following regimen: 93°C, 1 min; 60°C, 1 min; 72°C, 1 min. The PCR to detect message for glucose 6-phosphate dehydrogenase (GAPDH) was performed in parallel using the same protocol with 30 cycles. Forward and reverse primers were each selected from two different exons with the program PRIMER (Whitehead Institute, Cambridge, MA). The respective primer sequences are as follows: GAPDH (GenBank-EMBL accession number M32599) 5′-TCCGCCCCTTCTGCCGATG-3′ (plus strand), 5′-CACGGAAGGCCATGCCAGTGA-3′ (minus strand); IFN-γ (GenBank-EMBL accession number K00083) 5′-CCACGGCACAGTCATTGAAAGCC-3′ (plus strand), 5′-TTTCCGCTTCCTGAGGCTGGATT-3′ (minus strand); TNFα (GenBank-EMBL accession number M13049) 5′-GGGGTGATCGGTCCCCAAAGG-3′ (plus strand), 5′-CGGGGCAGCCTTGTCCCTTG-3′ (minus strand); IL1β (GenBankEMBL accession number M15131) 5′-AAGCCTCGTGCTGTCGGACCC-3′ (plus strand), 5′-TCCAGCTGCAGGGTGGGTGTG-3′ (minus strand). PCR amplification was controlled with a water sample instead of cDNA. Ten microliters of the amplified fragments were run along with the molecular weight marker (φX 174, HaeIII-digested, Pharmacia) on a 1.7% agarose gel stained with ethidium bromide.
For Southern blotting, the PCR fragments subjected to electrophoresis were then blotted onto a nylon transfer membrane (Hybond-N+, Amersham, Arlington Heights, IL) and hybridized with a digoxigenin 3-end-labeled (DNA 3′-End Labeling, Boehringer Mannheim, Mannheim, Germany) internal oligonucleotide probe. The nucleotide sequences of the probes were designed with the program Oligo 5.0 (NBI, Plymouth, MN) from the published sequence data: GAPDH, 5′-CCCCCTGGCCAAGGTCATCCA-3′ (21-mer); IFN-γ, 5′-CCACAGGTCCAGCGCCAAGCA-3′ (21-mer); TNFα, 5′-TCCATGCCGTTGGCCAGGAG-3′ (20-mer); IL1β, 5′-AAAATACCTGTGTGCCTTGGGC-3′ (21-mer). The hybridized oligonucleotide was detected with alkaline phosphatase-conjugated antibodies directed against digoxigenin together with a chemiluminescent system (Dig Luminescent Detection, Boehringer Mannheim) and then exposed to autoradiography film.
Blood–brain barrier function. To assess possible changes in the blood–brain barrier, 13 d axotomized animals (BL6/JL) were injected intravenously with 8 mg of horseradish peroxidase (HRP; Sigma) in 400 μl of PBS, killed 24 hr after injection, and then processed in the same way as for light-microscopic IHC (perfusion fixation, immersion fixation, cryoprotection, etc.) and cut at the level of area postrema and facial motor nucleus. Fixed/spread tissue sections were first incubated for 10 min with 1% biotin tyramide (NEN DuPont, Dreieich, Wiesbaden) and 0.01% H2O2 at room temperature in PB to enhance the HRP signal by chemoconversion to biotin. The tissue sections were washed three times in PB, incubated for 1 hr with ABC reagent in PB, and then visualized with DAB/H2O2 with Co/Ni intensification.
To detect possible neutrophil granulocytes in the axotomized facial motor nuclei, tissue sections from normal, 14 d axotomized BL6/JL mice were stained for endogenous peroxidase for 10 min at room temperature with DAB/H2O2. Peroxidase-positive neutrophil granulocytes in spleen sections served as a positive control. Neutrophil granulocytes were also detected using conventional LM-IHC with the rat monoclonal antibody MCA771 (Camon).
RESULTS
CD3-immunoreactive cells in the normal and axotomized facial motor nucleus
All of the primary antibodies used in the current study are summarized in Table 1. Infiltrating T-lymphocytes were detected using CD3 immunoreactivity. These CD3-positive cells are very rare in the normal CNS, with a density of ∼0.3 cells per 20-μm-thick section of the facial motor nucleus (∼1 cell/mm2). Facial nerve transection led to a biphasic increase in the number of CD3-positive, round cells in the axotomized facial motor nucleus. The first increase was observed as soon as 1 d after injury (1 DAI) and reached a plateau of two to three cells per facial motor nucleus section 2–4 DAI (Fig.1). A small but statistically not significant increase was also observed on the contralateral unoperated side (Fig. 1, bottom right). A second, much stronger increase was observed 7–21 DAI, with a maximum of 27 ± 10 CD3-positive cells per section (mean ± SEM, n = 3) at day 14 (Fig. 1, bottom right), a 90-fold increase over the normal facial motor nucleus. This second increase was followed by a gradual decline to almost normal levels 66 DAI.
T-lymphocytes aggregate around neuronal debris and phagocytotic microglia
Figure 2 shows the distribution of the CD3-positive lymphocytes at their peak level at 14 DAI. Although some tissue sections showed a scattered distribution (Figs. 1,2A), CD3-positive cells frequently formed aggregates (one to two per section) consisting of 5–50 cells around focal points in neural parenchyma (Fig. 2C,D). Very rarely, some CD3-positive cells also aggregated around big vessels passing through the facial motor nucleus (Fig. 2B).
Interestingly, a similar distribution was also observed for TSP immunostaining on the cellular nodules in the axotomized mouse facial motor nucleus, with a maximum 14 d after transection (Möller et al., 1996). At high magnification, these nodules consisted of dying neurons or neuronal debris, surrounded by TSP-immunoreactive, phagocytotic microglia (Fig.3A–C). To define a possible correlation between both phenomena, infiltration of lymphocytes and phagocytotic microglia, we performed immunofluorescence double staining using polyclonal rabbit antibody against thrombospondin and rat monoclonal antibodies against CD3, 14 DAI. As shown in Figure3E, these TSP-immunoreactive microglial nodules were often surrounded by CD3-positive cells with a direct contact to the TSP-immunoreactive structures. Figure 3, D, F, and G, shows a similar contact of the microglial nodule with two further lymphocyte activation markers, CD11a (LFA-1α, αL-integrin subunit) and CD44 (Raine et al., 1990; Zeine and Owens, 1992). This direct contact was further confirmed at the electron microscopical level using the CD11a immunoreactivity. Figure4A shows a degenerating neuron surrounded by microglia, astrocytes, and numerous CD11a-immunoreactive cells. These CD11a-positive cells frequently demonstrated typical features of activated lymphocytes with extensive membrane ruffling, clear cytoplasm, and cleaved nuclei. Similar structural details were also observed on T-lymphocytes in the facial motor nucleus identified by the CD3 immunoreactivity (Fig.4B,C).
Fig. 4.

Ultrastructural localization of CD11a- and CD3-immunoreactivity in the 14 d axotomized facial motor nucleus.A, CD11a immunostaining of a cellular aggregate, consisting of a degenerating neuron at the center, surrounded by microglia (M), astrocytes (A), and the CD11a-positive lymphocytes (L). These CD11a-positive cells frequently showed a clear cytoplasm, deeply cleaved nuclei, and ruffled, CD11-immunoreactive cell membranes (short arrows). The curved arrow points to phagosomes in a CD11a-negative cell process adhering to a CD11a-immunoreactive cell. These phagosomes are a common, characteristic feature in the phagocytotic microglial cells. Magnification, 5400×. B, C, CD3 immunoreactivity on the cell membrane of infiltrating T-lymphocytes (T). Note the typical cleaved or deformed lymphocyte nuclei. Adjacent vessels (V) and astrocytes (A) are unlabeled. Magnification:B, 5800×; C, 6800×.
Effects of timing and SCID background on lymphocyte infiltration
The data presented in Figures 1-3 show a time-dependent infiltration of T-lymphocytes into the axotomized mouse facial motor nucleus, with a maximum at 14 DAI. This could be caused by an autoimmune process of lymphocyte activation, which reaches a peak 14 DAI and is selective for the axotomized facial nucleus. To explore this possible autoimmune-mediated infiltration, we examined the effects of timing and SCID phenotype.
The effects of timing were studied using a sequential approach, with a 14 d axotomy of the right and a 3 d axotomy of the left facial nucleus in the same animal, with the two operations 11 d apart. The reasoning was that if infiltration depended on peripheral lymphocyte activation, this sequential axotomy should lead to a similar influx on both sides. As shown in Figure5A, however, the technique caused a massive infiltration of CD3+ cells in the 14 d axotomized nucleus (22.5 ± 4.7, mean ± SD; n = 4), but only a 10-fold lower number on the 3 d injured side (2.3 ± 0.4).
Fig. 5.

Effects of timing and SCID-phenotype on lymphocyte infiltration. A, Effects of consecutive, bilateral axotomy. Bilateral infiltration of CD3 lymphocytes, 14 d after transection of the right and 3 d after transection of the left facial nerve. Note the ∼10-fold higher influx of lymphocytes on the 14 d axotomized side. *p < 0.001 in a paired, two-sided Student’s t test; mean ± SD (n = 4 animals). B, C, Infiltration of CD3- (B) and CD11a-immunoreactive cells (C) in normal and SCID mice in the BALB/c genetic background, 14 d after facial nerve transection (mean ± SEM,n = 5 animals). The SCID phenotype leads to a 98% decrease in the number of CD3-positive cells (p < 0.001) and a 60% decrease in the number of CD11a-positive cells (p < 0.01). Unpaired t test.
Mice with the homozygous SCID mutation exhibit an almost complete absence of differentiated T- and B-lymphocytes (Bosma et al., 1983;Dorshkind et al., 1984), which can be used to differentiate between autoimmune and nonautoimmune mechanisms. This defect is specific for T- and B-lymphocytes, and the animals still have a persistent population of the lymphocyte-related natural killer cells (Bancroft and Kelly, 1994), which carry the CD11a antigen (Nishimura and Itoh, 1988). As demonstrated in Figure6A,B, both normal (BALB/c wild type) and SCID mice (BALB/c, scid/scid) also show a strong, focal increase in MHC class I immunoreactivity in the axotomized facial motor nucleus, 14 DAI. There was no effect of the SCID phenotype on the axotomy-mediated increase in MHC class II (data not shown). Compared with normal animals, the SCID mice revealed an almost complete absence of the CD3+ cells in the axotomized facial motor nucleus (Figs. 5B, 6C,D). Of the five SCID mice examined, only one showed the presence of CD3+ cells, with two labeled cells in one of the two examined sections. Overall, this is a 98% reduction, compared with the average of 10 CD3+ cells per section in the wild-type mice (Fig. 5B). In contrast to the CD3+ lymphocytes, all SCID animals had round, CD11a+ cells in the axotomized facial nucleus 14 DAI, although their number was 60% lower compared with the control mice (Figs. 5C,6E,F).
To further define these CD11a+ cells, Figures 3H and6A–F show the results of a colocalization with a rabbit polyclonal antibody against IBA1, a cytoplasmic antigen expressed in cells of the monocyte/macrophage lineage (Imai et al., 1996). Interestingly, the CD11a+ cells were still able to aggregate around the IBA1+ microglial nodules, despite the SCID immunodeficiency (Fig. 3H). However, there was no direct colocalization of CD11a immunoreactivity on the IBA1-labeled microglia (Fig. 7A,B). In contrast to CD11a, the IBA1+ cells clearly exhibited the CD11b/αM-integrin immunoreactivity (Fig. 7C,D), a typical marker for both normal and activated brain microglia (Perry and Gordon, 1988;Raivich et al., 1994). As shown in Figure 7E,F, MHC1 immunoreactivity was present on both the IBA1+ microglia and the adjacent, round IBA1-negative cells. This MHC1 immunoreactivity was particularly prominent on the phagocytotic microglial nodules.
Fig. 7.

Facial motor nucleus, 14 d after axotomy, SCID mouse. A–F, Double immunofluorescence of microglial IBA1 immunoreactivity (red, A, C, E) with superimposed CD11a (B), CD11b (D), and MHC class I (F) labeling (green). Note the absence of colocalization of IBA1 with CD11a (B) and the colocalization with CD11b immunoreactivity (D, yellow). MHC class I immunoreactivity (F) is present both on IBA1-positive microglia (yellow) and on round, IBA1-negative cells (green, arrows). Thearrowheads point to the large microglial nodules. Magnification, 1050×.
Effects of axotomy on the blood–brain barrier, infiltration of neutrophil granulocytes, and the expression of proinflammatory cytokines
To assess possible changes in the blood–brain barrier, 13 d axotomized animals were injected intravenously with 8 mg of HRP and perfused after 24 hr with PBS to remove the intravenous enzyme. Sensitivity to HRP was further enhanced by the HRP-catalyzed reaction of H2O2 with biotinylated tyramide followed by a detection of the tissue-conjugated biotin residues with routine ABC histochemistry (see Materials and Methods). In most parts of the brain, intravenous injection of HRP only led to a strong labeling of the vascular endothelium, with very little staining in the adjacent neural parenchyma (Fig.8A–D). Brain regions with permeable vascular endothelium such as area postrema showed clear parenchymal staining (Fig. 8A). Interestingly, particularly strong staining was observed in the ∼500 μm region surrounding area postrema, which may be attributable to the outward diffusion of HRP in the 24 hr interval between the injection and perfusion with PBS. Transection of the facial nerve, however, did not lead to enhanced peroxidase staining in the parenchyma of the axotomized facial motor nucleus, 14 DAI (Fig. 8D). Similar, low staining intensity was also seen on the unoperated side (Fig. 8C).
Fig. 8.

Effects of axotomy on the blood–brain barrier (A–D) and the infiltration of neutrophil granulocytes, 14 d after facial nerve transection (E–G). A, Detection of HRP extravasation in area postrema and in the surrounding parenchyma.B, No gross HRP extravasation in the brain stem at the level of the facial motor nucleus. C, D, Higher magnification of the contralateral (C) and axotomized facial nucleus (D) only shows a specific HRP staining of the brain vasculature. E–H, Histochemical and immunohistochemical staining for neutrophil granulocytes in the spleen (E, G) and in the axotomized facial nucleus (F, H). E, F, Immunohistochemistry with a rat monoclonal antibody MCA771 against neutrophil granulocytes. G, H, Endogenous peroxidase. Both methods show the absence of granulocyte staining in the facial nucleus. Magnification: A, B, 13×;C–H, 53×.
The presence or absence of neutrophil granulocytes was determined using two different methods, by staining for the endogenous peroxidase and by immunohistochemistry with a monoclonal antibody MCA771 against neutrophil granulocytes (Table 1). In both cases, mouse spleen served as a positive control (Fig. 8F,H). As shown in Figure 8E,G, both methods revealed the absence of neutrophil granulocytes in the axotomized facial motor nucleus, 14 DAI.
Figure 9 shows the expression of mRNA coding for proinflammatory cytokines IL1β, TNFα, and IFN-γ using RT-PCR in the axotomized and contralateral facial nuclei, at the time point of the first plateau at day 3, and of the maximal lymphocyte infiltration, 14 d after transection. The PCR amplification of specific DNA fragments was confirmed by Southern blotting with digoxigenin end-labeled internal oligonucleotide probes. At day 3, a moderate increase was observed for IL1β and TNFα, but only in one (TNFα) or two (IL1β) of the four axotomized facial motor nuclei. IFN-γ mRNA was not detected. At day 14, all three animals showed a clear increase in IL1β, TNFα, and IFN-γ mRNA on the axotomized side. The constitutively expressed glucose 6-phosphate dehydrogenase mRNA served as a recovery standard for RNA extraction, reverse transcription, and amplification with PCR.
Fig. 9.

RT-PCR detection of mRNA for IL1β, TNFα, and IFN-γ, 3 and 14 d after facial nerve transection in the axotomized facial motor nuclei (A1–A4) and on the contralateral side (C1–C4). Southern blotting with digoxigenin-end-labeled internal oligonucleotide probes. Day 3 shows a moderate increase for IL1β and TNFα, but not for IFN-γ mRNA. All animals showed a clear increase in IL1β, TNFα, and IFN-γ mRNA on the axotomized side at day 14. The constitutively expressed glucose 6-phosphate dehydrogenase (GAPDH) mRNA served as a recovery standard for RNA extraction, reverse transcription, and amplification with PCR.PC, Positive control with added synthetic cytokine RNA.NC, Negative control, omission of added RNA. The results for days 3 and 14 are from two separate experiments and preclude a direct comparison of the absolute amount of mRNA on the contralateral side at these two time points.
DISCUSSION
The current study describes a significant, site-selective influx of T-lymphocytes into the mouse CNS after transection of the facial nerve. These lymphocytes targeted the affected facial motor nucleus, aggregated around neuronal debris and phagocytotic microglia, and reached a maximum during the peak of delayed neuronal cell death 14 d after axotomy (Möller et al., 1996). The lymphocyte extravasation was also accompanied by the expression of proinflammatory cytokines IL1β, TNFα, and IFN-γ and a strong, focal increase in the MHC class I immunoreactivity. On the other hand, there was no disruption of the blood–brain barrier to intravenously injected horseradish peroxidase and no infiltration by neutrophil granulocytes. The scarcity of the perivascular lymphocytes and the site-specific infiltration of the CD11a-positive leukocytes even in animals with SCID also argue in favor of an initially not antigen-mediated, parenchymal recruitment of circulating lymphocytes into the axotomized mouse facial motor nucleus.
Lymphocyte recruitment into injured CNS: antigen-dependent versus antigen-independent mechanisms
Although the entry of lymphocytes into the CNS is known in both infectious and autoimmune disease, the initial stages of this process are not well understood. Recent studies provided evidence for a continuous patrol of the CNS by activated T-cells, which are able to enter the normal, uninjured brain (Wekerle et al., 1986; Hickey et al., 1991). Despite this presence of lymphocytes even in the normal brain, a critical requirement for the generation of the cellular immune response is the presentation of the antigen together with the appropriate MHC molecule. Although the levels of MHC class I and class II are very low in the normal CNS parenchyma, neural injury leads to a massive increase of these molecules on the activated and particularly the phagocytotic microglia (Akiyama and McGeer, 1989; Streit et al., 1989b; Kaur and Ling, 1992), which can serve as a competent antigen-presenting cell (Ford et al., 1996; Dangond et al., 1997). Interestingly, there is considerable delay between the passive transfer of encephalitogenic T-cells and the onset of neurological symptoms (Raine et al., 1990; Wekerle et al., 1994). The drastic reduction of the delay phase after direct or indirect CNS trauma coincides with the expression of microglial MHC molecules (Maehlen et al., 1989; Konno et al., 1990; Molleston et al., 1993) and strongly supports the immune-regulatory function of these brain-resident cells. When presented with the right antigen, the stimulated lymphocytes can then initiate the immune response, which may be followed by a secondary recruitment of additional leukocytes (lymphocytes, monocytes, granulocytes) and perivascular infiltrates and a disruption of the blood–brain barrier (Brosnan and Raine, 1996; Prineas and McDonald, 1997). In this conceptual framework, the initial CNS entry of activated T-cells is a constitutive phenomenon, and the secondary recruitment of lymphocytes is a specific, immune-mediated response based on the accidental encounter between the activated T-cell and the right, correctly presented antigen by the MHC-positive, microglial cell.
The data described in the current study strongly suggest the presence of a second, not antigen-mediated pathway for lymphocyte recruitment into the injured CNS. Despite the massive lymphocyte extravasation in the 14 d axotomized facial motor nucleus, there was no disruption of the blood–brain barrier or infiltration by neutrophil granulocytes or by rounded, IBA1-positive cells with macrophage morphology. Perivascular infiltrates, a key feature of the secondary lymphocyte recruitment (Brosnan and Raine, 1996; Prineas and McDonald, 1997), were very rare. Faced with the choice between day 3 and day 14 axotomized facial motor nucleus, the circulating CD3-positive lymphocytes showed a 10-fold higher influx to the longer-axotomized side. This argues against a general, time-dependent, peripheral activation of T-cells against the axotomized motoneurons with a maximum at day 14. Overall, these data support a site-specific chemotaxis by the degenerating neuron and the surrounding, phagocytotic microglia.
Morphological studies on lymphocyte recruitment, including the current work, are complicated by the ability of the T-cells to initiate an immune response, which could lead to a secondary lymphocyte influx. In the current study we examined this problem by looking at leukocyte infiltration in mice homozygous for SCID. As shown by previous studies, these SCID animals lack differentiated T- and B-lymphocytes (Bosma et al., 1983; Dorshkind et al., 1984), which can be used to differentiate between antigen-mediated and not antigen-mediated mechanisms (Nonoyama and Ochs, 1996). This defect is specific for T- and B-lymphocytes, and the animals still have a persistent population of the lymphocyte-related natural killer (NK) cells (Bancroft and Kelly, 1994), which carry the CD11a antigen, the α-subunit of the αLβ2 integrin (Nishimura and Itoh, 1988; Hynes, 1992),. This CD11a antigen is also expressed on circulating T-cells, granulocytes, and monocytes (Patarroyo et al., 1990). However, the absence of the CD3-positive T-cells, the absence of the endogenous peroxidase-positive granulocytes, and the failure to detect a colocalization of CD11a with the microglia/macrophage-marker IBA1–1 all suggest that the CD11a-positive cells in the SCID axotomized facial motor nucleus are NK cells. Here, the clear infiltration of these CD11a-positive cells around sites of neuronal degeneration and phagocytotic microglia in the SCID-immunodeficient animals argues in favor of the initially not antigen-mediated, parenchymal recruitment of the activated, circulating lymphocytes.
Entry of lymphocytes into the axotomized facial motor nucleus: species differences, blood–brain barrier function, and the induction of proinflammatory cytokines
The extensive lymphocyte infiltration into the mouse facial motor nucleus provides a noticeable contrast to previous results in the other commonly used experimental animal, the rat. With the exception of the study by Olsson et al. (1992), transection of the facial nerve did not lead to clearly observable entry of lymphocytes into the affected rat facial motor nucleus (Streit and Kreutzberg, 1988; Graeber et al., 1990). The extent of post-traumatic neuronal cell death could be an important reason for these species differences. Thus, facial motoneurons in the adult rat exhibit very little degeneration after a simple axotomy (Streit and Kreutzberg, 1988) but show pronounced, late neuronal cell death in the mouse model, affecting 20–35% of the axotomized neurons (Sendtner et al., 1996; Ferri et al., 1998). This notion is also supported by experiments with the retrograde axonal transport of ricin and adriamycin into the rat facial motor nucleus, which was followed by rapid neuronal cell death and lymphocyte infiltration (Graeber et al., 1990). However, the number of lymphocytes in these rat neurotoxic models was still just one to five T-cells per 20-μm-thick tissue section of the facial motor nucleus, and thus considerably lower than that observed in the current study in the mouse, with 10–30 CD3-positive T-cells per tissue section of the same thickness (Figs. 1F, 5A,B). These differences could point to the presence of additional, genetic factors that influence the extent of lymphocyte infiltration. For example, facial axotomy in the mouse is accompanied by a strong increase in the mRNA for three proinflammatory cytokines, IL1β, TNFα, and IFN-γ, which was not detected in the rat facial motor nucleus (Kiefer et al., 1993). Interestingly, these cytokines showed a similar increase in mRNA in the T- and B-cell-deficient scid mice, suggesting a local and T-cell-independent production in the injured mouse parenchyma (H. Neumann and G. Raivich, unpublished observations). Similar differences between rat and mouse were also observed for cell adhesion molecules such as ICAM−1, which were induced on activated mouse microglia in the axotomized facial motor nucleus (Werner et al., 1998) but not on the microglia in the rat model (Moneta et al., 1993). At present, the involvement of each of these molecules in the enhanced lymphocyte recruitment in the mouse facial motor nucleus remains to be shown. However, the current data do suggest important species differences in the intensity of immune surveillance in the injured nervous system that need to be considered when neuroimmunological studies performed in different species are compared.
One important functional parameter that was not affected by lymphocyte infiltration was the blood–brain barrier (BBB). Although there are a number of methods for examining the BBB at light microscopic level, such as immunohistochemistry for serum proteins or intravenous injection of HRP, there may be technical limits in detecting a low-level dysfunction. Primary antibodies show a certain level of nonspecific binding, which sets a lower limit for antigen detection (Raivich et al., 1993), and dilution of HRP during circulation and adsorption to endothelia strongly reduces the intensity of its enzymatic staining. In the present study, we therefore amplified the intensity of HRP staining by the HRP-catalyzed deposition of biotinylated tyramide in the presence of H2O2followed by a visualization of the tissue-conjugated biotin residues with ABC histochemistry. In addition to the strong labeling in the BBB-free brain regions such as area postrema, this enhanced technique allowed the detection of the enzyme diffusing for ∼500 μm into the surrounding parenchyma with an intact endothelial barrier. It also allowed the detection of the minute amounts of HRP adsorbed to brain vascular endothelia. However, there was no increased detection of HRP in the facial motor nucleus at the peak of lymphocyte infiltration 14 d after transection of the facial nerve. Although we cannot exclude a subthreshold increase in permeability, the current data strongly suggest an intact BBB and argue against a 1:1 relationship between the presence of brain lymphocytes and permeability to serum proteins. Despite the rapid influx of activated lymphocytes into the normal brain (Wekerle et al., 1986) or in the adoptive transfer of encephalitogenic T-cells (Raine et al., 1990;Wekerle et al., 1994), a severe disruption of the BBB is a more delayed phenomenon that appears to occur after specific antigen recognition (Linington et al., 1988; Seeldrayers et al., 1993). The apparent absence of BBB disruption in the injured facial motor nucleus indicates that this antigen recognition is a phenomenon that does not always occur and that lymphocyte infiltration can also follow a benign course with little or no tissue damage.
Functional consequences of lymphocyte entry
As shown in the current study, a neurodegenerative process can lead to a highly selective, nonaccidental encounter between the phagocytotic microglia and activated T-cells in the mouse CNS. The role of microglia as a professional brain phagocytotic cell (Kreutzberg, 1996), the production of proteolytic enzymes (Banati et al., 1993) and proinflammatory cytokines (Seilhean et al., 1997; Uno et al., 1997; Williams et al., 1997), and the expression of MHC molecules (Akiyama and McGeer, 1989; Streit et al., 1989a,b;Kaur and Ling, 1992) all point to this cell as a competent antigen-presenting cell and a key counterpart of the immune system in the brain. Activated microglia produce several chemokines, such as MCP-1 (Calvo et al., 1996) and IL16 (Schluesener et al., 1996), which together with the proinflammatory cytokines (IL1, TNFα, IFN-γ) could change the adhesion properties of the vascular endothelium (Tang et al., 1996; Henninger et al., 1997) and induce lymphocyte extravasation and chemotaxis. Moreover, phagocytosis also leads to a strong upregulation of microglial cell adhesion molecules such as intercellular adhesion molecule 1/ICAM1 and the αMβ2-integrin (Möller et al., 1996; Werner et al., 1998). The presence of appropriate counter-receptors αLβ2-integrin and ICAM1, respectively, on the infiltrating lymphocytes [Raine et al. (1990); Werner et al. (1998); this study] could promote their adhesion to microglial nodules, enhancing the effect of antigen presentation.
Overall, the site-specific parenchymal recruitment of T-cells could play an important role as a protective mechanism that allows early contact of the immune system with cellular debris and then leads to a differentiation between unspecific degeneration and cell death caused by an infectious pathogen. In the latter case, the entry of lymphocytes and their specific activation will normally lead to the destruction of infected cells and the removal of pathogens from the CNS (Griffin et al., 1992; Dietzschold, 1993; Kreutzberg et al., 1996; Schluter et al., 1996; Deckert-Schlueter et al., 1997). The intensity of the first step of this neuroimmune surveillance, the initial lymphocyte entry, appears to vary even in closely related species such as mouse and rat and could have been subject to different evolutionary constraints. Interestingly, lymphocyte infiltration has also been described in noninfectious human neurodegenerative diseases such as Alzheimer’s dementia (McGeer et al., 1993) and amyotrophic lateral sclerosis (Kawamata et al., 1992; Engelhardt et al., 1993). In light of the current findings, this entry of lymphocytes could be a physiological phenomenon in response to a neurodegenerative process. However, the long-term presence of lymphocytes and the presentation of neural antigens by the surrounding phagocytotic microglia may lead to a secondary, antigen-mediated neurotoxicity (Shalit et al., 1995; McGeer and McGeer, 1996). This hypothesis is supported by the higher risk and/or the earlier onset of Alzheimer’s disease associated with specific MHC class 1 (Payami et al., 1997) and MHC class 2 (Frecker et al., 1994; Curran et al., 1997) alleles. Here, interference with this putative immune response (Aisen, 1996; McGeer and McGeer, 1996), and specifically with the initial lymphocyte recruitment into the affected CNS, could be of benefit for the long-term progression of this neurodegenerative disease.
In summary, neuronal cell death can lead to a significant influx of activated T-cells, which home on the neuronal debris and the neighboring phagocytotic microglia. Interestingly, this site-specific recruitment may serve as an important protective mechanism that permits early contact of the immune system with cellular debris and then allows the differentiation between unspecific degeneration and cell death attributable to an infectious pathogen. Errors during this process could be detrimental in two ways: by inducing an autoimmune reaction against the injured nervous system or by causing tolerance to a neural infection. The identification of the molecular signals that regulate this early influx of lymphocytes after brain injury could therefore be of clinical interest.
Footnotes
This work was supported by BMBF Grant 01KO9401/3 and DFG Grant Ra 486/3–1 to G.R. We thank Dietmute Büringer, Irmtraud Milojevic, Karin Brückner, Theresa Baethmann, and Marion Bohatschek for their expert technical assistance; Dr. James Chalcroft for help with digital micrography; Dr. Yoshinuri Imai (Department of Neurochemistry, National Institute of Neuroscience, Tokyo, Japan) for providing the IBA1 antibody; and Dr. Manuel Graeber (Department of Neuromorphology, Max-Planck-Institute) and Dr. Hartmut Wekerle (Department of Neuroimmunology, Max-Planck-Institute) for reading this manuscript.
Correspondence should be addressed to Dr. Gennadij Raivich, Department of Neuromorphology, Max-Planck-Institute for Psychiatry, Am Klopferspitz 18A, D-85152 Martinsried, Germany.
REFERENCES
- 1.Adams JC. Heavy metal intensification of DAB-based HRP reaction product. J Histochem Cytochem. 1981;29:775. doi: 10.1177/29.6.7252134. [DOI] [PubMed] [Google Scholar]
- 2.Aisen PS. Inflammation and Alzheimer disease. Mol Chem Neuropathol. 1996;28:83–88. doi: 10.1007/BF02815208. [DOI] [PubMed] [Google Scholar]
- 3.Akiyama H, McGeer PL. Microglial response to 6-hydroxydopamine-induced substantia nigra lesions. Brain Res. 1989;489:247–253. doi: 10.1016/0006-8993(89)90857-3. [DOI] [PubMed] [Google Scholar]
- 4.Banati RB, Rothe G, Valet G, Kreutzberg GW. Detection of lysosomal cysteine proteinases in microglia: flow cytometric measurement and histochemical localization of cathepsin B and L. Glia. 1993;7:183–191. doi: 10.1002/glia.440070208. [DOI] [PubMed] [Google Scholar]
- 5.Bancroft GJ, Kelly JP. Macrophage activation and innate resistance to infection in SCID mice. Immunobiology. 1994;191:424–431. doi: 10.1016/S0171-2985(11)80448-1. [DOI] [PubMed] [Google Scholar]
- 6.Barker CF, Billingham RE. Immunologically privileged sites. Adv Immunol. 1977;25:1–54. [PubMed] [Google Scholar]
- 7.Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301:527–530. doi: 10.1038/301527a0. [DOI] [PubMed] [Google Scholar]
- 8.Brightman MW, Klatzo I, Olsson Y, Reese TS. The blood-brain barrier to proteins under normal and pathological conditions. J Neurol Sci. 1970;10:215–239. doi: 10.1016/0022-510x(70)90151-6. [DOI] [PubMed] [Google Scholar]
- 9.Brosnan CF, Raine CS. Mechanisms of immune injury in multiple sclerosis. Brain Pathol. 1996;6:243–257. doi: 10.1111/j.1750-3639.1996.tb00853.x. [DOI] [PubMed] [Google Scholar]
- 10.Calvo CF, Yoshimura T, Gelman M, Mallat M. Production of monocyte chemotactic protein-1 by rat brain macrophages. Eur J Neurosci. 1996;8:1725–1734. doi: 10.1111/j.1460-9568.1996.tb01316.x. [DOI] [PubMed] [Google Scholar]
- 11.Cross AH, Cannella B, Brosnan CF, Raine CS. Homing to central nervous system vasculature by antigen-specific lymphocytes. I. Localization of [14C]-labeled cells during acute, chronic, and relapsing experimental allergic encephalomyelitis. Lab Invest. 1990;63:162–170. [PubMed] [Google Scholar]
- 12.Curran M, Middleton D, Edwardson J, Perry R, McKeith I, Morris C, Neill D. HLA-DR antigens associated with major genetic risk for late-onset Alzheimer’s disease. NeuroReport. 1997;8:1467–1469. doi: 10.1097/00001756-199704140-00028. [DOI] [PubMed] [Google Scholar]
- 13.Dangond F, Windhagen A, Groves CJ, Hafler DA. Constitutive expression of costimulatory molecules by human microglia and its relevance to CNS autoimmunity. J Neuroimmunol. 1997;76:132–138. doi: 10.1016/s0165-5728(97)00043-x. [DOI] [PubMed] [Google Scholar]
- 14.Deckert-Schluter M, Buck C, Weiner D, Kaefer N, Rang A, Hof H, Wiestler OD, Schluter D. Interleukin-10 downregulates the intracerebral immune response in chronic Toxoplasma encephalitis. J Neuroimmunol. 1997;76:167–176. doi: 10.1016/s0165-5728(97)00047-7. [DOI] [PubMed] [Google Scholar]
- 15.Dietzschold B. Antibody-mediated clearance of viruses from the mammalian central nervous system. Trends Microbiol. 1993;1:63–66. doi: 10.1016/0966-842X(93)90035-P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorshkind K, Keller GM, Phillips RA, Miller RG, Bosma GC, O’Toole M, Bosma MJ. Functional status of cells from lymphoid and myeloid tissues in mice with severe combined immunodeficiency disease. J Immunol. 1984;132:1804–1808. [PubMed] [Google Scholar]
- 17.Engelhardt JI, Tajti J, Appel SH. Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Arch Neurol. 1993;50:30–36. doi: 10.1001/archneur.1993.00540010026013. [DOI] [PubMed] [Google Scholar]
- 18.Ferri CC, Moore FA, Bisby MA. Effects of facial nerve injury on mouse motoneurons lacking the p75 low-affinity neurotrophin receptor. J Neurobiol. 1998;34:1–9. [PubMed] [Google Scholar]
- 19.Ford AL, Foulcher E, Lemckert FA, Sedgwick JD. Microglia induce CD4 T lymphocyte final effector function and death. J Exp Med. 1996;184:1737–1745. doi: 10.1084/jem.184.5.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frecker MF, Pryse-Phillips WE, Strong HR. Immunological associations in familial and non-familial Alzheimer patients and their families. Can J Neurol Sci. 1994;21:112–119. doi: 10.1017/s0317167100049027. [DOI] [PubMed] [Google Scholar]
- 21.Graeber MB, Streit WJ, Kiefer R, Schoen S, Kreutzberg GW. New expression of myelomonocytic antigens by microglia and perivascular cells following lethal motor neuron injury. J Neuroimmunol. 1990;27:121–132. doi: 10.1016/0165-5728(90)90061-q. [DOI] [PubMed] [Google Scholar]
- 22.Griffin DE, Levine B, Tyor WR, Irani DN. The immune response in viral encephalitis. Semin Immunol. 1992;4:111–119. [PubMed] [Google Scholar]
- 23.Henninger DD, Panes J, Eppihimer M, Russell J, Gerritsen M, Anderson DC, Granger DN. Cytokine-induced VCAM-1 and ICAM-1 expression in different organs of the mouse. J Immunol. 1997;158:1825–1832. [PubMed] [Google Scholar]
- 24.Hickey WF, Hsu BL, Kimura H. T-lymphocyte entry into the central nervous system. J Neurosci Res. 1991;28:254–260. doi: 10.1002/jnr.490280213. [DOI] [PubMed] [Google Scholar]
- 25.Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- 26.Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun. 1996;224:855–862. doi: 10.1006/bbrc.1996.1112. [DOI] [PubMed] [Google Scholar]
- 27.Jones LL, Kreutzberg GW, Raivich G. Regulation of CD44 in the regenerating mouse facial motor nucleus. Eur J Neurosci. 1997;9:1854–1863. doi: 10.1111/j.1460-9568.1997.tb00752.x. [DOI] [PubMed] [Google Scholar]
- 28.Kaur C, Ling EA. Activation and re-expression of surface antigen in microglia following an epidural application of kainic acid in the rat brain. J Anat. 1992;180:333–342. [PMC free article] [PubMed] [Google Scholar]
- 29.Kawai K, Ito K, Imamura K, Hickey WF, Zweiman B, Takahashi A. Enhancing effects of irrelevant lymphocytes on adoptive transferred experimental allergic encephalomyelitis. J Neuroimmunol. 1993;42:39–45. doi: 10.1016/0165-5728(93)90210-p. [DOI] [PubMed] [Google Scholar]
- 30.Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am J Pathol. 1992;140:691–707. [PMC free article] [PubMed] [Google Scholar]
- 31.Kiefer R, Lindholm D, Kreutzberg GW. Interleukin-6 and transforming growth factor beta-1 mRNAs are induced in rat facial nucleus following motoneuron axotomy. Eur J Neurosci. 1993;5:775–781. doi: 10.1111/j.1460-9568.1993.tb00929.x. [DOI] [PubMed] [Google Scholar]
- 32.Klein MA, Möller JC, Jones LL, Bluethmann H, Kreutzberg GW, Raivich G. Impaired neuroglial activation in interleukin-6 deficient mice. Glia. 1997;19:227–233. doi: 10.1002/(sici)1098-1136(199703)19:3<227::aid-glia5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 33.Kniesel U, Risau W, Wolburg H. Development of blood-brain barrier tight junctions in the rat cortex. Dev Brain Res. 1997;96:229–240. doi: 10.1016/0165-3806(96)00117-4. [DOI] [PubMed] [Google Scholar]
- 34.Konno H, Yamamoto T, Suzuki H, Yamamoto H, Iwasaki Y, Ohara Y, Terunuma H, Harata N. Targeting of adoptively transferred experimental allergic encephalitis lesion at the sites of wallerian degeneration. Acta Neuropathol. 1990;80:521–526. doi: 10.1007/BF00294613. [DOI] [PubMed] [Google Scholar]
- 35.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 36.Linington C, Bradl M, Lassmann H, Brunner C, Vass K. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol. 1988;130:443–454. [PMC free article] [PubMed] [Google Scholar]
- 37.Maehlen J, Olsson T, Zachau A, Klareskog L, Kristensson K. Local enhancement of major histocompatibility complex (MHC) class I and II expression and cell infiltration in experimental allergic encephalomyelitis around axotomized motor neurons. J Neuroimmunol. 1989;23:125–132. doi: 10.1016/0165-5728(89)90031-3. [DOI] [PubMed] [Google Scholar]
- 38.McGeer PL, McGeer EG. Anti-inflammatory drugs in the fight against Alzheimer’s disease. Ann NY Acad Sci. 1996;777:213–220. doi: 10.1111/j.1749-6632.1996.tb34421.x. [DOI] [PubMed] [Google Scholar]
- 39.McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG. Microglia in degenerative neurological disease. Glia. 1993;7:84–92. doi: 10.1002/glia.440070114. [DOI] [PubMed] [Google Scholar]
- 40.Medawar PB. Immunity to homologous grafted skin: III. The fate of skin homografts transplanted to the brain, to subcutaneous tissue and to anterior chamber of the eye. Br J Exp Pathol. 1948;29:58–69. [PMC free article] [PubMed] [Google Scholar]
- 41.Möller JC, Klein MA, Haas S, Jones LL, Kreutzberg GW, Raivich G. Regulation of thrombospondin in the regenerating mouse facial motor nucleus. Glia. 1996;17:121–132. doi: 10.1002/(SICI)1098-1136(199606)17:2<121::AID-GLIA4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 42.Molleston MC, Thomas ML, Hickey WF. Novel major histocompatibility complex expression by microglia and site-specific experimental allergic encephalomyelitis lesions in the rat central nervous system after optic nerve transection. Adv Neurol. 1993;59:337–348. [PubMed] [Google Scholar]
- 43.Moneta ME, Gehrmann J, Toepper R, Banati RB, Kreutzberg GW. Cell adhesion molecule expression in the regenerating rat facial nucleus. J Neuroimmunol. 1993;45:203–206. doi: 10.1016/0165-5728(93)90181-w. [DOI] [PubMed] [Google Scholar]
- 44.Nishimura T, Itoh T. Higher level expression of lymphocyte function-associated antigen-1 (LFA-1) on in vivo natural killer cells. Eur J Immunol. 1988;18:2077–2080. doi: 10.1002/eji.1830181231. [DOI] [PubMed] [Google Scholar]
- 45.Nonoyama S, Ochs HD. Immune deficiency in SCID mice. Int Rev Immunol. 1996;13:289–300. doi: 10.3109/08830189609061753. [DOI] [PubMed] [Google Scholar]
- 46.Olsson T, Diener P, Ljungdahl A, Hojeberg B, van der Meide PH, Kristensson K. Facial nerve transection causes expansion of myelin autoreactive T cells in regional lymph nodes and T cell homing to the facial nucleus. Autoimmunity. 1992;13:117–126. doi: 10.3109/08916939209001912. [DOI] [PubMed] [Google Scholar]
- 47.Patarroyo M, Prieto J, Rincon J, Timonen T, Lundberg C, Lindbom L, Asjo B, Gahmberg CG. Leukocyte-cell adhesion: a molecular process fundamental in leukocyte physiology. Immunol Rev. 1990;114:67–108. doi: 10.1111/j.1600-065x.1990.tb00562.x. [DOI] [PubMed] [Google Scholar]
- 48.Payami H, Schellenberg GD, Zareparsi S, Kaye J, Sexton GJ, Head MA, Matsuyama SS, Jarvik LF, Miller B, McManus DQ, Bird TD, Katzman R, Heston L, Norman D, Small GW. Evidence for association of HLA-A2 allele with onset age of Alzheimer’s disease. Neurology. 1997;49:512–518. doi: 10.1212/wnl.49.2.512. [DOI] [PubMed] [Google Scholar]
- 49.Perry VH, Gordon S. Macrophages and microglia in the nervous system. Trends Neurosci. 1988;11:273–277. doi: 10.1016/0166-2236(88)90110-5. [DOI] [PubMed] [Google Scholar]
- 50.Poduslo JF, Curran GL, Berg CT. Macromolecular permeability across the blood-nerve and blood-brain barriers. Proc Natl Acad Sci USA. 1994;91:5705–5709. doi: 10.1073/pnas.91.12.5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prineas JW, McDonald WI (1997) Demyelinating diseases. In: Greenfield’s neuropathology, Ed 6 (Graham DI, Lantos PL, eds), pp 813–896. New York: Arnold/Oxford UP.
- 52.Raine CS, Cannella B, Duijvestijn AM, Cross AH. Homing to central nervous system vasculature by antigen-specific lymphocytes. II. Lymphocyte/endothelial cell adhesion during the initial stages of autoimmune demyelination. Lab Invest. 1990;63:476–489. [PubMed] [Google Scholar]
- 53.Raivich G, Gehrmann J, Graeber MB, Kreutzberg GW. Quantitative immunohistochemistry in the rat facial nucleus with [125I]-iodinated secondary antibodies and in situ autoradiography: non-linear binding characteristics of primary monoclonal and polyclonal antibodies. J Histochem Cytochem. 1993;41:579–592. doi: 10.1177/41.4.8450197. [DOI] [PubMed] [Google Scholar]
- 54.Raivich G, Moreno-Flores MT, Möller JC, Kreutzberg GW. Inhibition of posttraumatic microglial proliferation in a genetic model of macrophage colony-stimulating factor deficiency in the mouse. Eur J Neurosci. 1994;6:1615–1618. doi: 10.1111/j.1460-9568.1994.tb00552.x. [DOI] [PubMed] [Google Scholar]
- 55.Rodriguez M, Pavelko KD, Njenga MK, Logan WC, Wettstein PJ. The balance between persistent virus infection and immune cells determines demyelination. J Immunol. 1996;157:5699–5709. [PubMed] [Google Scholar]
- 56.Schluesener HJ, Seid K, Kretzschmar J, Meyermann R. Leukocyte chemotactic factor, a natural ligand to CD4, is expressed by lymphocytes and microglial cells of the MS plaque. J Neurosci Res. 1996;44:606–611. doi: 10.1002/(SICI)1097-4547(19960615)44:6<606::AID-JNR11>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 57.Schluter D, Chahoud S, Lassmann H, Schumann A, Hof H, Deckert-Schluter M. Intracerebral targets and immunomodulation of murine Listeria monocytogenes meningoencephalitis. J Neuropathol Exp Neurol. 1996;55:14–24. doi: 10.1097/00005072-199601000-00002. [DOI] [PubMed] [Google Scholar]
- 58.Schnell L, Schneider R, Berman MA, Perry VH, Schwab ME. Lymphocyte recruitment following spinal cord injury in mice is altered by prior viral exposure. Eur J Neurosci. 1997;9:1000–1007. doi: 10.1111/j.1460-9568.1997.tb01450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scolding N, Linington C, Compston A. Immune mechanisms in the pathogenesis of demyelinating diseases. Autoimmunity. 1989;4:131–142. doi: 10.3109/08916938909034368. [DOI] [PubMed] [Google Scholar]
- 60.Seeldrayers PA, Syha J, Morrissey SP, Stodal H, Vass K, Jung S, Gneiting T, Lassmann H, Haase A, Hartung HP. Magnetic resonance imaging investigation of blood-brain barrier damage in adoptive transfer experimental autoimmune encephalomyelitis. J Neuroimmunol. 1993;46:199–206. doi: 10.1016/0165-5728(93)90250-3. [DOI] [PubMed] [Google Scholar]
- 61.Seilhean D, Kobayashi K, He Y, Uchihara T, Rosenblum O, Katlama C, Bricaire F, Duyckaerts C, Hauw JJ. Tumor necrosis factor-alpha, microglia and astrocytes in aids dementia complex. Acta Neuropathol. 1997;93:508–517. doi: 10.1007/s004010050646. [DOI] [PubMed] [Google Scholar]
- 62.Sendtner M, Gotz R, Holtmann B, Escary JL, Masu Y, Carroll P, Wolf E, Brem G, Brulet P, Thoenen H. Cryptic physiological trophic support of motoneurons by LIF revealed by double gene targeting of CNTF and LIF. Curr Biol. 1996;6:686–694. doi: 10.1016/s0960-9822(09)00450-3. [DOI] [PubMed] [Google Scholar]
- 63.Shalit F, Sredni B, Brodie C, Kott E, Huberman M. T lymphocyte subpopulations and activation markers correlate with severity of Alzheimer’s disease. Clin Immunol Immunopathol. 1995;75:246–250. doi: 10.1006/clin.1995.1078. [DOI] [PubMed] [Google Scholar]
- 64.Streit WJ, Kreutzberg GW. Response of endogenous glial cells to motor neuron degeneration induced by toxic ricin. J Comp Neurol. 1988;268:248–263. doi: 10.1002/cne.902680209. [DOI] [PubMed] [Google Scholar]
- 65.Streit WJ, Graeber MB, Kreutzberg GW. Functional plasticity of microglia: a review. Glia. 1988;1:301–307. doi: 10.1002/glia.440010502. [DOI] [PubMed] [Google Scholar]
- 66.Streit WJ, Graeber MB, Kreutzberg GW. Peripheral nerve lesion produces increased levels of major histocompatibility complex antigens in the central nervous system. J Neuroimmunol. 1989a;21:117–123. doi: 10.1016/0165-5728(89)90167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Streit WJ, Graeber MB, Kreutzberg GW. Expression of Ia antigen on perivascular and microglial cells after sublethal and lethal motor neuron injury. Exp Neurol. 1989b;105:115–116. doi: 10.1016/0014-4886(89)90111-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang T, Frenette PS, Hynes RO, Wagner DD, Mayadas TN. Cytokine-induced meningitis is dramatically attenuated in mice deficient in endothelial selectins. J Clin Invest. 1996;97:2485–2490. doi: 10.1172/JCI118695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Torvik A, Skjörten F. Electron microscopic observations on nerve cell regeneration and degeneration after axon lesions. II. Changes in the glial cells. Acta Neuropathol. 1971;17:265–282. doi: 10.1007/BF00685059. [DOI] [PubMed] [Google Scholar]
- 70.Uno H, Matsuyama T, Akita H, Nishimura H, Sugita M. Induction of tumor necrosis factor-alpha in the mouse hippocampus following transient forebrain ischemia. J Cereb Blood Flow Metab. 1997;17:491–499. doi: 10.1097/00004647-199705000-00002. [DOI] [PubMed] [Google Scholar]
- 71.Vass K, Lassmann H, Wekerle H, Wisniewski HM. The distribution of Ia antigen in the lesions of rat acute experimental allergic encephalomyelitis. Acta Neuropathol. 1986;70:149–160. doi: 10.1007/BF00691433. [DOI] [PubMed] [Google Scholar]
- 72.Wekerle H, Linington C, Lassmann H, Meyermann R. Cellular immune reactivity within the CNS. Trends Neurosci. 1986;9:271–277. [Google Scholar]
- 73.Wekerle H, Kojima K, Lannes-Vieira J, Lassmann H, Linington C. Animal models. Ann Neurol. 1994;36[Suppl]:S47–53. doi: 10.1002/ana.410360714. [DOI] [PubMed] [Google Scholar]
- 74.Werner A, Kloss CUA, Walter J, Kreutzberg GW, Raivich G (1998) Intercellular adhesion molecule-1 (ICAM1) in the mouse facial motor nucleus following axotomy and during nerve regeneration. J Neurocytol, in press. [DOI] [PubMed]
- 75.Wesselingh SL, Levine B, Fox RJ, Choi S, Griffin DE. Intracerebral cytokine mRNA expression during fatal and nonfatal alpha virus encephalitis suggests a predominant type 2 T cell response. J Immunol. 1994;152:1289–1297. [PubMed] [Google Scholar]
- 76.Williams A, Vandam AM, Ritchie D, Eikelenboom P, Fraser H. Immunocytochemical appearance of cytokines, prostaglandin e-2 and lipocortin-1 in the cns during the incubation period of murine scrapie correlates with progressive prp accumulations. Brain Res. 1997;754:171–180. doi: 10.1016/s0006-8993(97)00067-x. [DOI] [PubMed] [Google Scholar]
- 77.Wong GH, Bartlett PF, Clark-Lewis I, Battye F, Schrader JW. Inducible expression of H-2 and Ia antigens on brain cells. Nature. 1984;310:688–691. doi: 10.1038/310688a0. [DOI] [PubMed] [Google Scholar]
- 78.Zeine R, Owens T. Direct demonstration of the infiltration of murine central nervous system by Pgp-1/CD44high CD45RB(low) CD4+ T cells that induce experimental allergic encephalomyelitis. J Neuroimmunol. 1992;40:57–69. doi: 10.1016/0165-5728(92)90213-5. [DOI] [PubMed] [Google Scholar]