

Abstract
We have previously shown that estradiol causes a twofold rise in dendritic spine density in cultured rat hippocampal neurons, as it doesin vivo. More recently, estrogen receptors have been localized to aspiny inhibitory hippocampal interneurons, indicating that their effect on spiny pyramidal neurons may be indirect. We therefore examined the possibility that estradiol affects spine density by regulating inhibition in cultured hippocampal interneurons. Immunocytochemically, estrogen receptors were found to be co-localized with glutamate decarboxylase (GAD)-positive neurons (∼21% of total neurons in the culture). Exposure of cultures to estradiol for 1 d caused a marked decrease (up to 80%) in the GAD content of the interneurons, measured both by immunohistochemistry and Western blotting. Also, the number of GAD-positive neurons in the cultures decreased to 12% of the total cell population. Moreover, GABAergic miniature IPSCs were reduced in both size and frequency by estradiol, whereas miniature EPSCs increased in frequency. We then mimicked the proposed effects of estradiol by blocking GABA synthesis with mercaptopropionic acid (MA). Cultures treated with MA expressed a dose-dependent decrease in GABA immunostaining that mimicked that seen with estradiol. MA-treated cultures displayed a significant 50% increase in dendritic spine density over controls, similar to that produced by estradiol. These results indicate that estradiol decreases GABAergic inhibition in the hippocampus, which appears to effectively increase the excitatory drive on pyramidal cells, and thus may provide a mechanism for formation of new dendritic spines.
Keywords: hippocampus, dendritic spines, interneuron, estrogen receptors, GABA, cultures
Dendritic spines are the primary loci of excitatory synapses in central neurons and have long been associated with neuronal plasticity (for review, see Harris and Kater, 1994). Consequently, studies have attempted to identify memory-related changes in dendritic spine morphology. The type and magnitude of reported spine changes after various training protocols are as yet unsettled. Recently we found that dissociated hippocampal neurons grown in culture can double their dendritic spine density in response to estradiol (Murphy and Segal, 1996), confirming earlier in vivo experiments (Woolley and McEwen, 1992). We extended these observations to propose that the phosphorylation of cAMP response element binding protein (CREB) is a necessary condition for this estradiol-induced increase in spine density (Murphy and Segal, 1997).
The classical role of estrogen in other areas of the brain related to reproductive function has been well characterized. Briefly, estrogen binds to its receptor, and the complex then binds the estrogen response element gene to regulate gene expression. In the hippocampus, an area unassociated with reproduction, the localization of estrogen receptors has been disputed. However, it appears that the originally defined estrogen receptors are not found in pyramidal neurons; rather, they are found on nonspiny interneurons (Weiland et al., 1996). How then does estradiol affect spine formation in spiny neurons? In the present experiments, we examined the hypothesis that estradiol indirectly causes formation of dendritic spines by reducing GABA inhibition, which would, in turn, enhance excitatory activity. To this end, we measured glutamic acid decarboxylase (GAD) immunoreactivity after treatment of cultured neurons with estradiol and recorded inhibitory activity in these cells. We also mimicked the effect of estradiol with an agent that reduces GABA synthesis. We propose that estradiol causes a reduction in GABAergic inhibition, leading to an increase in excitatory tone that may provide a mechanism for formation of new dendritic spines.
MATERIALS AND METHODS
Hippocampal cultures
Hippocampal cultures were prepared as described previously (Papa et al., 1995; Murphy and Segal, 1996). Briefly, 19- to 20-d-old embryos were taken out of anesthetized Sprague Dawley rats. Brains were removed and placed in ice-cold L15 medium supplemented with 0.6% glucose and 15 μg/ml gentamycin. The hippocampus was dissected and mechanically disaggregated by gentle trituration using a Pasteur pipette. Dissociated cells were plated onto 12 mm glass coverslips for immunofluorescence (500,000 cells per well) or onto tissue culture plastic for Western blotting (12 well plates at a density of 1.3 × 106 cells per well). These were coated with poly-l-lysine and UV-sterilized. Cells were also prepared on collagen-coated (50 μg/ml) tissue culture inserts (Millicell) for electrophysiology. The plating medium was Eagle’s minimum essential medium (MEM) containing 10% heat-inactivated horse serum, 5% fetal calf serum, 2 mm glutamine, 0.6% glucose, and 15 μg/ml gentamycin. Cells were incubated at 37°C with 8% CO2. The first change of medium, ∼4–6 d after plating, included 50 μg/ml uridine and 20 μg/ml deoxyuridine to prevent glial cell overgrowth. The cultures were fed thereafter one or two times a week with Eagle’s MEM containing 10% horse serum.
Drug treatments
17β-Estradiol. Cells were grown for 2.5–3 weeks to ensure maturity of GABAergic activity (Berninger et al., 1995; Marty et al., 1996), at which time they were dosed with 0.1 μg/ml water-soluble estradiol (Sigma, St. Louis, MO) for the following dosing regimens. For Western blots, cells were sampled after 12, 24, 36, and 48 hr of estradiol treatment. For GAD and GABA immunolabeling, cells were fixed at 24 hr. For spine counts, cells were incubated for at least 48 hr with estradiol, the time at which the changes in spine density become significant (Murphy and Segal, 1996). For electrophysiological experiments, cells were dosed for 24 and 48 hr. Controls received ordinary media.
Mercaptopropionic acid. Cultures were treated for 24 hr with graded concentrations of mercaptopropionic acid (MA), a drug known to reduce GAD activity (Netopilova et al., 1995, 1997). Cells were fixed and stained for GABA immunoreactivity or for detection of dendritic spines with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) (see below).
Tetrodotoxin. Tetrodotoxin (TTX, 2 μm; Sigma) was prepared daily from a freshly thawed aliquot and added to the cultures once a day for 3 d of estradiol treatment. The effects of TTX on estradiol-induced spine formation were measured using DiI (see below).
Electrophysiology
Cover glasses were removed from the incubator and washed with the recording medium containing (in mm): NaCl 130, KCl 5, glucose 30, HEPES 25, CaCl 2, and MgCl 1. Osmolarity was adjusted to 320 mOsm with sucrose, pH 7.4. Recording was made from medium to large (15–20 μm in diameter) pyramidal-shaped neurons, with patch pipettes having access resistance of 4–6 MΩ. In cases in which evoked synaptic currents were recorded, the patch-recording pipette contained (in mm): K-acetate 120, MgATP 2, HEPES 20, KCl 10, and Na2-phosphocreatine 10. Where miniature inhibitory currents were measured, the extracellular recording medium contained 1 μm TTX, 20 μm6,7-dinitroquinoxaline-2,3-dione (DNQX), and 20–50 μmaminophosphonobutyrate (APV). When miniature excitatory synaptic currents were recorded, APV and DNQX were replaced by 50 μm bicuculline. The recording pipettes in these experiments contained 120 mm CsCl, 2 mm MgATP, 10 mm Na2-phosphocreatine, 10 mmHEPES, 20 mm KCl, 25 mm sucrose, 200 μm EGTA, and 300 μm GTP-Tris. In experiments using cell-attached patch configuration, the pipette was filled with extracellular recording medium, so as not to affect ionic gradients across the membrane and to verify that GABA is indeed inhibitory in these neurons. Hyperosmotic medium, GABA, or glutamate was injected through a pressure pipette with brief pulses near the cell somata. The hyperosmotic medium contained normal recording medium supplemented with 300 mm sucrose and was used for evoking miniature IPSCs (mIPSCs). Experiments were conducted at room temperature. Signals were amplified with Axopatch-200 and stored on an IBM computer for off-line analysis that included counting of synaptic currents and measurements of their peaks using Axon Instruments software.
Immunolabeling and imaging
Cells were fixed in 4% paraformaldehyde in PBS for 1 hr at room temperature and then washed and permeabilized with 0.1% saponin in blocking buffer (5% goat serum in PBS) for 30 min. Coverslips were then drained and directly inverted over microdrops of primary antibodies to GAD (Boehringer Mannheim, Indianapolis, IN), estrogen receptor (ERα, Novocastra Labs) (Stressgen antibodies to the originally classified estrogen receptor; Kuiper and Gustaffson, 1997), or GABA (Sigma). Cells were incubated overnight at 4°C, after which they were washed in PBS and incubated in fluorescein isothiocyanate- or rhodamine-labeled secondary antibodies for 1 hr at room temperature. Cells were then mounted in Vectashield (Vector Laboratories, Burlingame, CA), sealed, and imaged on a confocal laser scanning microscope (CLSM) using a 100× 1.4 numerical aperture (NA) oil immersion lens. Random fields were selected from control and estradiol-treated cells, and a fluorescence and differential interference contrast (DIC) image was taken for each field. Identical confocal settings were used for each treatment group.
Intensity analysis and cell counts
Neuronal cell bodies were counted from each DIC field and summed for each treatment group. At the magnification used, the neurons were at a different focal point in the depth of field; thus, glial cells were out of focus and not counted. Cells that were GAD-positive, indicating interneurons, were counted for each field and summed as a percentage of the total for each group. Statistical significance was determined using nonparametric χ2 analysis. The same fluorescence images were then sent to the collaborating laboratory for the fluorescence intensity analysis. The analysis was performed in a double-blind procedure. Intensity of intracellular labeling in interneurons as well as the number, size, and intensity of the GAD-positive terminals impinging on GAD-negative cells were examined by thresholding and automatic counting of terminals using NIH Image software. Groups were compared using unpaired two-tailed ttests.
Western blots
Cultures were harvested in 75 μl of boiling (95°C) SDS sample buffer containing 10% glycerol, 2% SDS, 5% 2-mercaptoethanol, and 0.05% bromphenol blue. Samples were centrifuged and boiled for 3 min before loading on a 10% SDS-PAGE gel. Equal protein amounts were loaded in each lane. Gels were run for 45 min at room temperature. A 1 hr transfer of proteins to nitrocellulose membranes was performed in transfer buffer (25 mm Tris, 192 mm glycine, and 20% methanol, pH 8.3). Membranes were blocked in low-fat milk (10% in Tris buffer and 0.05% Tween) and washed. They were then incubated overnight at 4°C with GAD antibody, (rabbit polyclonal; Chemicon, Temecula, CA; 1:4000 for identification of GAD67, 1:1500 for both GAD65 and GAD67). The blots were washed, incubated for 1 hr with protein-A–horseradish peroxidase (Amersham, Arlington Heights, IL), and developed by a standard enhanced chemiluminescence routine. The density of the bands was recorded.
Morphological analysis of dendritic spines
Cells were fixed in 4% paraformaldehyde for 1 hr at room temperature. Individual cells were stained by pressurized microinjection of DiI dissolved in oil, as detailed elsewhere (Papa et al., 1995). Cells were imaged at a 2× zoom on a CLSM with a 100× 1.4 NA oil immersion objective. Z sections were collected and serially reconstructed at the time of imaging using Zeiss confocal software. Single sections and reconstructions were used jointly to count the number of spines for measured lengths of dendrite, ensuring that each spine was counted only once. Spines were counted if they were <3–4 μm in length and possessed a mature “head.” Spine density in 50 μm dendritic segments was counted as before (Murphy and Segal, 1996). For each treatment, at least 12–15 cells were reconstructed, and 48–60 dendritic segments were measured.
RESULTS
Immunohistochemistry
Cultures were double-immunostained for GAD and ERα to test for co-localization. In untreated cultures, estrogen receptor staining was confined to interneurons expressing GAD. Many of these cells displayed a typical bipolar morphology that was distinguishable from the pyramidal cell shape. ERα was localized in the cytoplasm as well as the nucleoplasm of the GAD-stained cells (Fig.1A,B). GAD staining was seen in the cytoplasm (Golgi), as well as in discrete puncta corresponding to GABAergic axon terminals that clustered around pyramidal cells and other GAD-positive neurons (Fig. 1C). ERα staining was also observed in the cytoplasm and nuclei of underlying glial cells (data not shown).
Fig. 1.

Estrogen receptors are found in GAD-positive neurons of rat hippocampal cultures. A,B, Double staining of GAD immunoreactivity (red) and estrogen receptor (green) produce orange fluorescing cells.C, An estrogen receptor-negative pyramidal neuron surrounded by GAD-positive puncta, staining GABAergic terminals on the soma. Scale bar, 20 μm. Note GAD-positive terminals on the somata of the estrogen receptor-positive neurons, indicating that ERαs are confined to the somata and do not travel to the GAD-positive terminals.
Estradiol-treated cultures displayed a marked decrease in the number of GAD-positive neurons, and a marked decrease in intensity of GAD staining (Fig.2A,B). This effect was most prominent within 24 hr of exposure to estradiol, but a decrease was also noticed after 48 hr of estradiol exposure. Although most of the experiments were conducted with a standard dose of 0.1 μg/ml, similar effects on GAD were observed also with 0.01 μg/ml estradiol (data not shown). The following analysis was done with cells exposed to estradiol for 24 hr. In control cultures, the percentage of GAD-positive neurons was 20.1% of the total neurons in the culture (data from three experiments, 110 GAD-positive cells of 529 cells counted in a total of 44 random fields), analogous to the percentage commonly seen in this type of hippocampal culture preparation (Segal, 1983). This number dropped to 12% after exposure to estradiol (three experiments, a total of 78 GAD-positive cells of 646 cells counted in 49 random fields; χ2 analysis,p < 0.01) (Fig. 2C).
Fig. 2.
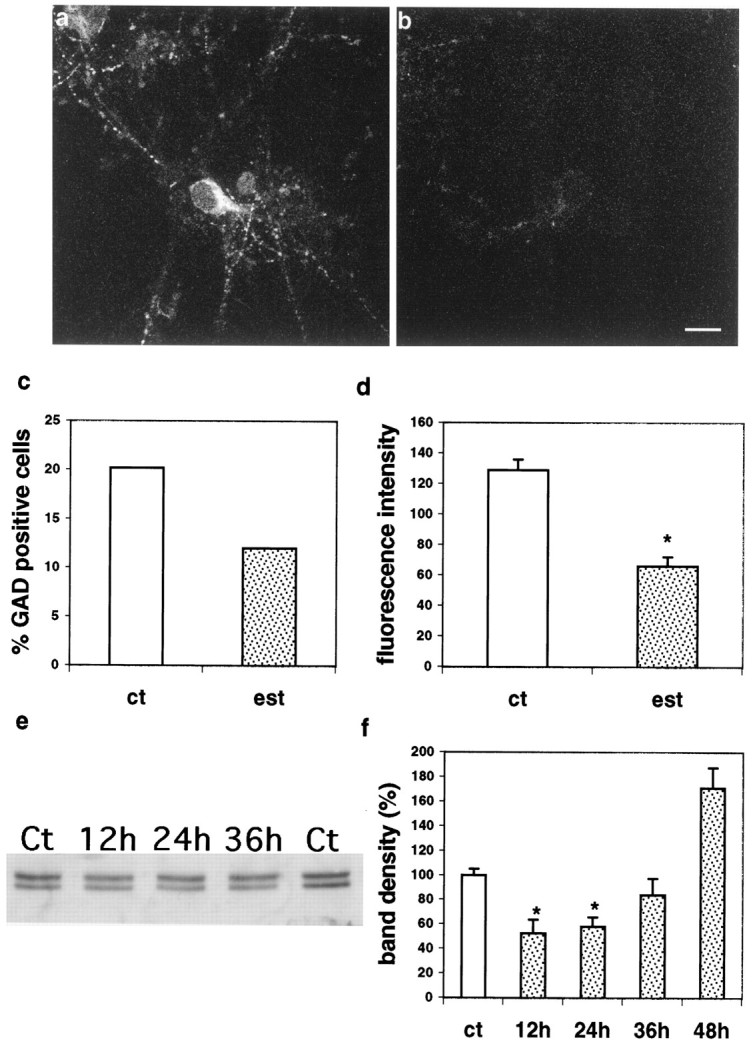
Estradiol suppresses GAD expression in cultured hippocampal neurons. A, Control. B, Twenty-four-hour exposure to estradiol. Scale bar, 20 μm.C, Percentage of GAD-positive neurons of the total cells in all of the randomly sampled fields from control (ct) and estradiol (est)-treated cultures; estradiol cultures are different from controls using χ2 analysis;p < 0.01. D, Estradiol reduces the fluorescence intensity of GAD staining (scale in arbitrary fluorescence units); mean ± SEM. E, Western blotting of GAD-stained gels, detecting both GAD65 (bottom band) and GAD67 (top band). F, Summary of densitometry analysis of effects of estradiol on total GAD immunoreactivity shown in E. Scale is percentage of matched untreated control lanes. In D andF, asterisks denote difference from controls using t tests; p < 0.01.
The intensity of GAD staining was measured in 33 control and 30 estradiol- treated cells. GAD fluorescence intensity in the estradiol-treated cultures decreased to ∼51% of control (control, 129 ± 6.8 arbitrary fluorescence units; estradiol-treated cells, 66 ± 6.1, in an 8 bit scale) (Fig. 2D). In two separate experiments, the overall number of fluorescent puncta dropped from 5363 particles in control cultures to 2797 in estradiol-treated cultures (10 random fields analyzed in each group) and in a second experiment from 5792 to 2037 (13 fields analyzed in each group; data not shown). Additionally, there was a modest decrease in the size of the remaining particles from 13.4 ± 0.096 pixels in controls to 10.8 ± 0.089 pixels (data not shown).
Western blotting was conducted in three experiments, with two or three replications in each. Cultures were grown in 12 well plates and treated with 0.5 μm estradiol for 12–48 hr. The analysis of the Western blots indicated that estradiol produced a large ∼50% reduction in GAD immunoreactivity within 12–24 hr of exposure, a partial recovery within 36 hr, and an overshoot within 48 hr of exposure to estradiol (Fig.2E,F). Two bands are seen in the gel (GAD65 and GAD67, Fig. 2E). The reduction was larger in the GAD65 band but also evident in the GAD67 band.
Taken together, these results indicate that GAD production in cultured hippocampal interneurons is markedly reduced by exposure to estradiol. Although it is not clear whether some interneurons actually disappear, the heterogenous effect on both GAD and GABA immunoreactivity (see below) indicates that the interneurons are actually alive but producing less GABA. The reduction in the number of GAD-positive neurons is most likely a consequence of reduced GAD levels below the detection threshold of the antibody.
Electrophysiology
The immunohistochemical findings indicate that there is a marked decrease in the production of GAD, the enzyme critical for the synthesis of GABA in interneurons, after exposure to estradiol. We tested the physiological consequences of estradiol treatment, focusing on possible effects on GABAergic inhibition. Spontaneous action potentials were recorded in both control and estradiol-treated cells recorded in a cell-attached configuration. There were no apparent differences between the two groups in spontaneous firing rates attributable to large variabilities within each group. In both groups of cells, spontaneous activity was reversibly suppressed by exposure to 10 μm GABA applied by pressure from an adjacent pipette (Fig. 3A). There was no difference in the inhibitory effect of GABA on action potential discharges in estradiol-treated (n = 10) and control (n = 8) cells (two experiments). GABA totally suppressed the spontaneous action potential discharges in all cells examined, as expected for an inhibitory neurotransmitter (in younger cells GABA can be excitatory, as seen elsewhere).
Fig. 3.

GABA evokes inhibitory responses in cultured hippocampal neurons. A, Cell-attached patch recording of action potential discharges in a cultured neuron. Spontaneous activity (top 4 traces) is inhibited by exposure of the cell to 10 μm GABA applied via an adjacent pressure pipette (middle 4 traces). Spontaneous activity recovers thereafter. Bottom traces, Culture treated with estradiol for 24 hr. Patch pipette contained extracellular recording medium. B, C, Similar postsynaptic responses to GABA in control and estradiol-treated cultures.B, GABA-induced currents in control (top) and estradiol-treated (bottom) neurons. Cells were held at −60, −30, 0, and +30 mV. Patch pipette contained CsCl.C, Averaged IV curves showing similar responses to GABA of eight control (Ct) and six estradiol (Est)-treated cells for 24 hr.
In another series of experiments, cells were recorded with CsCl-containing pipettes in the whole-cell configuration, and the effects of GABA were assessed. There was no effect of estradiol on either the magnitude or reversal potential of the response to pulse application of GABA (Fig. 3B,C; six control cells and eight estradiol-treated cells).
In a third series of experiments, pipettes containing K-acetate were used for recording, and synaptic responses to stimulation of afferent cells with short puffs of glutamate were measured. Evoked synaptic currents were recorded in most neurons (Fig.4). These synaptic currents could be easily divided into excitatory or inhibitory, based on the difference in reversal potential. On average, estradiol-treated cells responded to afferent stimulation with significantly smaller IPSCs than control cells (control mean IPSC at −60 mV, −102.8 ± 16.6 pA;n = 8 cells; estradiol-treated at −60 mV, −56 ± 12 pA; n = 6 cells; F = 5.11;p < 0.02).
Fig. 4.

Inhibitory synaptic currents evoked by glutamate application on an adjacent neuron. A brief pulse application of glutamate (top trace) evoked a series of IPSCs, responding to a series of action potentials generated in the afferent neuron. Recording was made with a patch pipette containing K-acetate. The cell was held at different potentials, and the inhibitory currents reversed near resting potential (−50 mV).
In the presence of TTX, DNQX, and 2-APV, six control cells responded to a 10 msec pressure application of hyperosmotic medium with a barrage of miniature inhibitory synaptic currents, lasting ∼3 sec. Reproducible barrages could be obtained from repetitive applications. Acute exposure to estradiol (0.1 μg/ml) did not cause a systematic change in cellular responses to application of hyperosmotic medium (mean IPSC amplitude in control, −47.8 ± 2.3 pA;n = 172 events; estradiol-treated cells, −50.4 ± 1.81 pA; n = 205 events for the same duration of analysis). Analysis of the synaptic currents evoked by hyperosmotic medium in cultures treated for 24 hr of estradiol indicated that both their magnitude (control, −45.6 ± 1.41 pA; estradiol,−22.7 ± 1.11 pA; t = 10.83;p < 0.001) and frequency (control, 53.5 ± 4.5 events/2.5 sec; estradiol, 35.6 ± 6.0 events/2.5 sec;t = 2.2; p < 0.04) were significantly reduced (Fig. 5). A similar but smaller effect on mIPSCs was seen also with cells exposed to estradiol for 48 hr. For example, in one experiment (seven control cells and five estradiol-treated cells for 48 hr), there was no difference in the magnitude of the mIPSCs (−45 pA in controls and −50 pA in estradiol-treated cells), but there were ∼30% more events in the control group (data not shown).
Fig. 5.

Estradiol suppresses miniature IPSCs evoked by puff application of hyperosmotic medium near the recorded cell soma.A, Control cell responding to osmotic challenge with a typical barrage of miniature IPSCs. B, Estradiol-treated cell expresses only a small series of mIPSCs. C, Frequency histogram of the sizes of the mIPSCs (in picoamperes) recorded in six control cells (404 events counted in a subset of the data for each cell). D, Frequency histogram of the total mIPSCs evoked in six estradiol-treated cells (24 hr) (210 events counted in a subset of data for each cell). E, Cumulative histogram of all the events counted in C andD, illustrating that a large proportion of medium-sized mIPSCs are absent after estradiol treatment. Cells were recorded with a patch pipette containing CsCl, and the extracellular medium also contained TTX, DNQX, and 2-APV. Cells were held at resting membrane potential (−60 mV). Ct, Control; Est, estradiol.
Estradiol did not cause a marked change in the size of spontaneous (i.e., without the use of hyperosmotic medium) mEPSCs recorded in the presence of bicuculline and TTX (Fig.6; 12 cells recorded after 24 hr of exposure to estradiol, compared with 11 control cells, two experiments). Although the size of the mEPSCs was only slightly increased by estradiol (Fig. 6B), more events were found in the treated cells compared with controls (Fig.6C–E).
Fig. 6.

Estradiol effects on miniature EPSCs in cultured neurons. A, Spontaneous mEPSCs recorded from control and estradiol-treated neurons (24 hr) with a patch pipette containing CsCl. The recording medium contained TTX and 50 μm bicuculline. Cells were held at −80 mV. B, Averaged mEPSC of ∼20–30 such events in both control and estradiol-treated cells. The magnitude of synaptic current is about the same in the two cells, but its decay time constant is slightly slower in the estradiol-treated cell. C, D, Frequency distribution histograms of mEPSCs recorded in five control and six estradiol-treated cells, respectively. A sample of 20 sec was recorded from each cell. The histograms summarize a subset of 266 events recorded in the control and 506 events recorded in the treated group for an equal duration of time. The mean size of these events was −64.1 ± 2.69 pA in the control and −76.4 ± 2.95 pA in the estradiol-treated cells.E, Cumulative histograms of the events recorded in the control and estradiol-treated cells show an overall similar distribution of mEPSP sizes.
Morphology
We reasoned that if estradiol acts by decreasing GABA-mediated inhibitory activity of interneurons, then decreasing GABA synthesis by other means should produce a similar increase in spine density as that evoked by estradiol. We treated cultured neurons with MA (Netopilova et al., 1995, 1997) at various doses to block the production of GABA and subsequently stained for GABA immunoreactivity. Cells displayed a readily observable decrease in fluorescence intensity in MA-treated cultures that was analogous to that seen in estradiol-treated cultures (Fig. 7A–C). Random field images were then examined for fluorescence intensity as described above. MA treatment caused a significant decrease in the intensity of GABA fluorescence in a dose-dependent manner (controls, 188.6 ± 6.18 arbitrary fluorescence units in an 8 bit scale; 1 μmMA, 193.8 ± 7.3; 10 μm MA, 115.8 ± 11.3; 50 μm MA, 107.2 ± 7.8; and 100 μm MA, 60 ± 5.8). Estradiol also lowered GABA fluorescence intensity significantly compared with control (115.2 ± 7.2; Fig.8).
Fig. 7.

Effects of MA on GABA content and on dendritic spine formation in hippocampal cells. A–C, Control and estradiol- and MA-treated cultures, respectively, stained for GABA immunoreactivity. Scale bar, 20 μm. D–F, Control and estradiol- and MA-treated cultures, respectively, stained with DiI to reveal dendritic spines. Scale bar, 10 μm.
Fig. 8.

A, Effects of MA and estradiol (est) on GABA immunofluorescence measured in individual neurons at 24 hr. B, Effects of estradiol and MA on dendritic spine density in cultured hippocampal neurons after 48 hr. Error bars represent SE, and asterisks represent statistically significant difference from controls using Student’st tests; p < 0.01.
Having established that GABA was decreased in the presence of MA, cells were examined for spine density at the dose of MA in which virtually all of the puncta disappeared (100 μm). A significant increase in spine density was seen compared with controls (control, 8.63 ± 0.35 spines/50 μm dendritic segments; MA-treated cultures, 12.98 ± 0.528) that were slightly less than the increase in spine density evoked by estradiol alone (15.15 ± 0.379) (Figs. 7E–G, 8). MA in combination with estradiol did not cause an increase in spine density beyond that produced by estradiol alone, indicating the the two drugs may share a common mechanism.
Finally, if indeed estradiol produces spines by blocking inhibition and subsequently causing facilitation of excitation, then blockade of action potential discharges by tetrodotoxin should block the effects of estradiol. In a previous study (Murphy and Segal, 1996), we obtained only a partial blockade of estradiol action on spine formation by TTX. We repeated these experiments applying 2 μm TTX daily, as it may deteriorate over time in culture (see Materials and Methods). Under these conditions, TTX blocked the effects of estradiol (control, 8.29 ± 0.31 spines/50 μm dendritic segment; estradiol, 13.82 ± 0.58; TTX alone, 7.32 ± 0.31; and TTX and estradiol, 8.38 ± 0.35). This would indicate that the effects of estradiol may be mediated by action potential discharges in the cultured neurons.
DISCUSSION
It has now been demonstrated that estradiol acts to increase hippocampal CA1 dendritic spine density in vivo and in vitro (Woolley and McEwen, 1992; Murphy and Segal, 1996). However, it has also been demonstrated that pyramidal cells of the hippocampus do not express estrogen receptors (of the originally classified ERα)in vivo (Weiland et al. 1996), although they may express the ERβ type (Li et al., 1997). We have found that interneurons do express estrogen receptors in vitro of the ERα subtype, thereby suggesting that the spine-producing effects of estradiol in hippocampal pyramidal cells are possibly mediated by changes in inhibitory interneuronal synaptic efficacy.
Many studies have linked estradiol to GABAergic activity in various regions of the brain. During development, steroid-induced changes in GAD and GABA neurotransmission may be responsible for sexual differentiation (Davis et al., 1996). Evidence also exists for changes in GAD activity in various parts of the brain during postdevelopmental hormonal cycling (Unda et al., 1995). In the hippocampus, estradiol appears to have no direct effects on expression of GABAAreceptor subunit mRNAs (Weiland and Orchinik, 1995) and similarly has no effect on the affinity or density of GABA binding sites (Jussofie et al., 1995). Our results on the lack of effect of estradiol on reactivity to GABA are consistent with these observations. However, estradiol has been found to differentially regulate the levels of mRNA for both forms of GAD (GAD65 and GAD67) in various regions of the rat brain (McCarthy et al., 1995). These findings would suggest the effects of estradiol on dendritic spines are indirect, probably mediated by a lowering of GAD and thus GABA production. A decrease in inhibitory activity is also consistent with the findings that estradiol facilitates kindled seizures in the hippocampus (Buterbaugh and Hudson, 1991) and that hippocampal seizure threshold is decreased during the estrous cycle in the rat (Terasawa and Timiras, 1968). Moreover, picrotoxin-induced convulsions are more readily seen in female than in male rats, and when males are primed with estradiol, they display the female-type sensitivity (Pericic et al., 1996). This estrogen-related reduction in inhibitory tone may also underlie catamenial epilepsy in women (Rodriguez Macias, 1996).
An increase in spine density during elevated levels of estradiol may thus be correlated with an increase in excitatory activity that will follow a decrease in inhibition. Picrotoxin and bicuculline, which block GABAergic activity, have been previously shown to cause an increase in dendritic spine density in a similar culture condition (Papa and Segal, 1996), much the same way as MA has done in the present study. The present results suggest that the estradiol-induced spine formation may therefore share a wider common mechanism with other treatments that cause spine formation; i.e., an increase in synaptic activity after blockade of inhibition is responsible for formation of new dendritic spines. The fact that estradiol and MA effects are not additive support this interpretation.
Additionally, the blockade of GABA inhibition may not be the sole reason for the formation of new dendritic spines. We observed here that estradiol produces a consistently larger effect on spine density than that produced by reduced inhibition by MA. The same was observed with bicuculline and picrotoxin in a previous study (Papa and Segal, 1996). The difference may reflect the presence of estrogen receptors on glial cells and our reported effect of estradiol on activation of CREB in glial cells (Murphy and Segal, 1997). Thus, it is possible that estradiol activates other processes, in addition to reducing GAD in interneurons, to facilitate formation of new spines. The nature of this action in glial cells remains to be determined.
Other effects of estradiol may also contribute to spine formation. It has been reported recently that 17β-estradiol enhances the outgrowth and survival of neocortical neurons in culture (Brinton et al., 1997, and references therein), an action that does not seem to involve the nuclear estrogen receptor. In addition, acute estradiol may affect AMPA (Wong and Moss, 1992) and NMDA (Xu et al., 1997) receptors. These effects may enhance excitability of the cells and may contribute to the ultimate effects of estradiol on spine formation. These actions, which are short-term and/or do not involve the nuclear estrogen receptor, are not likely to underlie the effects seen here, which develop over several days in vitro (Woolley and McEwen, 1992; Woolley et al., 1997) and in vivo. Interestingly, Wong and Moss (1992)found that in vivo estradiol priming causes a prolongation of EPSPs in hippocampal slices, an effect that can be attributed to a reduction in inhibition, as suggested here.
Our results are consistent with those of Weiland (1995), who found an increase in GAD mRNA in intact female rats after 2 d of exposure to estradiol. Such an increase may constitute a feedback response to the initial reduction in GAD, seen here but not in the results ofWeiland (1995). GAD mRNA increases after tetanic stimulation (Liang et al., 1996), and GAD activity is regulated by levels of internal calcium; i.e., calcium ionophores were found to increase GAD activity (Erecinska et al., 1996). The changes we observed are largest at 12–24 hr after onset of exposure to estradiol. This could increase excitatory activity and levels of internal calcium in pyramidal cells and interneurons, a feedback that increases GAD levels by 48 hr. Indeed, we see a slight overshoot in GAD protein at this time, analogous to the results of Weiland (1995) for GAD mRNA. It should be mentioned that message for GAD may not result in actual synthesis of GAD; GAD mRNA has been detected in hippocampal pyramidal neurons, known to be excitatory (Cao et al., 1996). Thus, the fact that mRNA for GAD is increased does not necessitate that GAD function increases as well. Additionally, detection of GAD enzyme on a Western gel does not differentiate its distribution in the cell body versus the terminals, where it can be seen with immunostaining methods used here. Thus, discrepancies between the two methods for quantifying GAD levels may be expected. The changes we observed were larger with the GAD65 isoform that supposedly undergoes short-term changes in relation to activity, whereas the other isoform, GAD67, is assumed to control the metabolic non-neurotransmitter-related form of GAD in a more phasic manner (Feldbaum et al., 1995). Changes in GAD65 would then be consistent with a short-term response to hormonal fluctuations.
Our findings indicate that estradiol acts indirectly on pyramidal cells by causing a transient lowering of GABA synthesis in interneurons. This effectively increases the relative level of excitatory activity on pyramidal cells, increasing intracellular calcium levels more frequently, which may stimulate the phosphorylation of CREB. Excessive stimulation leading to long-term potentiation has been shown to phosphorylate CREB (Deisseroth et al., 1996), and we have shown previously that CREB is indeed phosphorylated in response to estradiol (Murphy and Segal, 1997). It is through this sequence of events that we propose new spines to be generated, effectively countering increases in excitatory activity. Although we cannot directly relate the effects of estradiol on spine density to hippocampal functions, studies have shown estradiol to enhance spatial memory in rats (Packard et al., 1996) and to influence visual memory in adult males (Kampen and Sherwin, 1996). Estrogen may also aid in diseases such as Alzheimer’s disease; postmenopausal women receiving estrogen replacement therapy fare better than those who do not receive estrogen (Henderson, 1997). Indeed, spines are altered in the pathology of Alzheimer’s disease (Yamada et al., 1988). Thus, it is possible that the decrease in inhibitory activity and increase in spine density in response to estradiol have profound functional consequences in the hippocampus.
Footnotes
This study was supported in part while M.S. was a Scholar in Residence at the Fogarty International Center for Advanced Study in the Health Sciences, National Institutes of Health. We acknowledge Drs. T. S. Reese and S. B. Andrews for their support and thank the Light Imaging Facility at the National Institute of Neurological Diseases and Stroke for use of the confocal laser scanning microscope.
Correspondence should be addressed to Dr. D. D. Murphy, Laboratory of Neurobiology, National Institute of Neurological Diseases and Stroke, National Institutes of Health, Building 36, Room 2A25, Bethesda, MD 20892.
REFERENCES
- 1.Berninger B, Marty S, Zafra F, Berzaghi M, Thoenen H, Lindholm D. GABAergic stimulation switches from enhancing to repressing BDNF expression in rat hippocampal neurons during maturation in vitro. Development. 1995;121:2327–2335. doi: 10.1242/dev.121.8.2327. [DOI] [PubMed] [Google Scholar]
- 2.Brinton RD, Tran J, Proffitt P, Montoya M. 17-Beta-estradiol enhances the outgrowth and survival of neocortical neurons in culture. Neurochem Res. 1997;22:1339–1351. doi: 10.1023/a:1022015005508. [DOI] [PubMed] [Google Scholar]
- 3.Buterbaugh GG, Hudson GM. Estradiol replacement to female rats facilitates dorsal hippocampal but not ventral hippocampal kindled seizure acquisition. Exp Neurol. 1991;111:55–64. doi: 10.1016/0014-4886(91)90050-m. [DOI] [PubMed] [Google Scholar]
- 4.Cao Y, Wilcox KS, Martin CE, Rachinsky TL, Eberwine J, Dichter MA. Presence of mRNA for glutamic acid decarboxylase in both excitatory and inhibitory neurons. Proc Natl Acad Sci USA. 1996;93:9844–9849. doi: 10.1073/pnas.93.18.9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis AM, Grattan DR, Selmanoff M, McCarthy MM. Sex differences in glutamic acid decarboxylase mRNA in neonatal rat brain: implications for sexual differentiation. Horm Behav. 1996;30:538–552. doi: 10.1006/hbeh.1996.0057. [DOI] [PubMed] [Google Scholar]
- 6.Deisseroth K, Bito H, Tsien R. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 7.Erecinska M, Nelson D, Daikhin Y, Yudhoff M. Regulation of GABA level in rat brain synaptosomes: fluxes through enzymes of the GABA shunt and effects of glutamate, calcium, and ketone bodies. J Neurochem. 1996;67:2325–2334. doi: 10.1046/j.1471-4159.1996.67062325.x. [DOI] [PubMed] [Google Scholar]
- 8.Feldblum S, Dumoulin A, Anoal M, Sandillon F, Privat A. Comparative distribution of GAD65 and GAD67 mRNAs and proteins in the rat spinal cord supports a differential regulation of these two glutamate decarboxylases in vivo. J Neurosci Res. 1995;42:742–757. doi: 10.1002/jnr.490420603. [DOI] [PubMed] [Google Scholar]
- 9.Harris KM, Kater SB. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- 10.Henderson VW. The epidemiology of estrogen replacement therapy and Alzheimer’s disease. Neurology. 1997;48[Suppl 7]:S27–S35. doi: 10.1212/wnl.48.5_suppl_7.27s. [DOI] [PubMed] [Google Scholar]
- 11.Jussofie A, Korner I, Schell C, Hienke C. Time course of the effects of steroid hormone deprivation elicited by ovarietomy or ovariectomy plus adrenalectomy on the affinity and density of GABA binding sites in distinct rat brain areas. Exp Clin Endocrinol Diabetes. 1995;103:196–204. doi: 10.1055/s-0029-1211350. [DOI] [PubMed] [Google Scholar]
- 12.Kampen DL, Sherwin BB. Estradiol is related to visual memory in healthy young men. Behav Neurosci. 1996;110:613–617. doi: 10.1037//0735-7044.110.3.613. [DOI] [PubMed] [Google Scholar]
- 13.Kuiper G, Gustaffson J. The novel estrogen receptor-β subtype: potential role in the cell and promoter-specific actions of estrogens and anti-estrogens. FEBS Lett. 1997;410:87–90. doi: 10.1016/s0014-5793(97)00413-4. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Schwartz PE, Rissman EF. Distribution of estrogen receptor-β-like immunoreactivity in rat forebrain. Neuroendocrinology. 1997;66:63–67. doi: 10.1159/000127221. [DOI] [PubMed] [Google Scholar]
- 15.Liang F, Isackson PJ, Jones EG. Stimulus-dependent, reciprocal up- and downregulation of glutamic acid decarboxylase and Ca+2/calmodulin-dependent protein kinase II gene expression in rat cerebral cortex. Exp Brain Res. 1996;110:163–174. doi: 10.1007/BF00228548. [DOI] [PubMed] [Google Scholar]
- 16.McCarthy MM, Kaufman LC, Brooks PJ, Pfaff DW, Schwartz-Giblin S. Estrogen modulation of nRNA levels for the two forms of glutamic acid decarboxylase (GAD) in female rat brain. J Comp Neurol. 1995;360:685–697. doi: 10.1002/cne.903600412. [DOI] [PubMed] [Google Scholar]
- 17.Marty S, Berninger B, Carroll P, Thoenen H. GABAergic stimulation regulates the phenotype of hippocampal interneurons through the regulation of brain-derived neurotrophic factor. Neuron. 1996;16:565–570. doi: 10.1016/s0896-6273(00)80075-6. [DOI] [PubMed] [Google Scholar]
- 18.Murphy DD, Segal M. Regulation of dendritic spine density in cultured rat hippocampal neurons by steroid hormones. J Neurosci. 1996;16:4059–4068. doi: 10.1523/JNEUROSCI.16-13-04059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murphy DD, Segal M. Morphological plasticity of dendritic spines in central neurons is mediated by activation of cAMP response element binding protein. Proc Natl Acad Sci USA. 1997;94:1482–1487. doi: 10.1073/pnas.94.4.1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Netopilova M, Drsata J, Kubova H, Mares P. Differences between immature and adult rats in brain glutamate decarboxylase inhibition by 3-mercaptopropionic acid. Epilepsy Res. 1995;20:179–184. doi: 10.1016/0920-1211(94)00068-8. [DOI] [PubMed] [Google Scholar]
- 21.Netopilova M, Drsata J, Haugvicova R, Kubova H, Mares P. Inhibition of glutamate decarboxylase activity by 3-mercaptopropionic acid has different time course in the immature and adult rat brains. Neurosci Lett. 1997;226:68–70. doi: 10.1016/s0304-3940(97)00241-3. [DOI] [PubMed] [Google Scholar]
- 22.Packard MG, Kohlmaier JR, Alexander GM. Posttraining intrahippocampal estradiol injections enhance spatial memory in male rats: interaction with cholinergic systems. Behav Neurosci. 1996;110:626–632. doi: 10.1037//0735-7044.110.3.626. [DOI] [PubMed] [Google Scholar]
- 23.Papa M, Segal M. Morphological plasticity in dendritic spines of cultured hippocampal neurons. Neuroscience. 1996;71:1005–1011. doi: 10.1016/0306-4522(95)00490-4. [DOI] [PubMed] [Google Scholar]
- 24.Papa M, Bundman MC, Greenberger V, Segal M. Morphological analysis of the development of dendritic spines in primary cultures of hippocampal neurons. J Neurosci. 1995;15:1–11. doi: 10.1523/JNEUROSCI.15-01-00001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pericic D, Manev H, Bujas M. Gonadal hormones and picrotoxin-induced convulsions in male and female rats. Brain Res. 1996;736:174–179. doi: 10.1016/0006-8993(96)00677-4. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez Macias KA. Catamenial epilepsy: gynecological and hormonal implications. Five case reports. Gynecol Endocrinol. 1996;10:139–142. doi: 10.3109/09513599609097905. [DOI] [PubMed] [Google Scholar]
- 27.Segal M. Rat hippocampal neurons in culture: responses to electrical and chemical stimuli. J Neurophysiol. 1983;50:1249–1264. doi: 10.1152/jn.1983.50.6.1249. [DOI] [PubMed] [Google Scholar]
- 28.Terasawa E, Timiras PL. Electrical activity during the estrous cycle of the rat: cyclical changes in limbic structures. Endocrinology. 1968;83:207–216. doi: 10.1210/endo-83-2-207. [DOI] [PubMed] [Google Scholar]
- 29.Unda R, Brann DW, Mahesh VB. Progesterone suppression of glutamic acid decarboxylase (GAD67) levels in the preoptic area: correlation with luteinizing hormone surge. Neuroendocrinology. 1995;62:562–570. doi: 10.1159/000127064. [DOI] [PubMed] [Google Scholar]
- 30.Weiland NG, Orchinik M. Specific subunit mRNAs of the GABAa receptor are regulated by progesterone in subfields of the hippocampus. Mol Brain Res. 1995;32:271–278. doi: 10.1016/0169-328x(95)00087-9. [DOI] [PubMed] [Google Scholar]
- 31.Weiland NG, Orikasa C, Hayashi S, McEwen BS. Localization of estrogen receptors in the hippocampus of male and female rats. Soc Neurosci Abstr. 1996;22:618. [Google Scholar]
- 32.Wong M, Moss RL. Long-term and short-term electrophysiological effects of estrogen on the synaptic properties of hippocampal CA1 neurons. J Neurosci. 1992;12:3217–3225. doi: 10.1523/JNEUROSCI.12-08-03217.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woolley CS, McEwen BS. Estradiol mediates fluctuation in hippocampal synapse density during the estrous cycle in the adult rat. J Neurosci. 1992;12:2549–2554. doi: 10.1523/JNEUROSCI.12-07-02549.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Woolley CS, Weiland NG, McEwen BS, Schwartzkroin PA. Estradiol increases the sensitivity of hippocampal CA1 pyramidal cells to NMDA receptor mediated synaptic input: correlation with dendritic spine density. J Neurosci. 1997;17:1848–1859. doi: 10.1523/JNEUROSCI.17-05-01848.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu J, Xie X, Brinton RD, Berger TW. Direct enhancement of NMDA receptor-mediated EPSPs by 17β-estradiol in hippocampal CA1. Soc Neurosci Abstr. 1997;23:2104. [Google Scholar]
- 36.Yamada M, Wada Y, Tsukagoshi H, Otmo E, Hayakawa M. A quantitative Golgi study of basal dendrites of hipporampal C1 pyramidal cells in senile dementia of Alzheimer’s type. J Neurol Neurosurg Psychiatry. 1988;51:1088–1090. doi: 10.1136/jnnp.51.8.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]