

Abstract
Glucocorticoids (GCs) act via intracellular mineralocorticoid (MR) and glucocorticoid receptors (GR). However, it has recently been recognized that GC access to receptors is determined by the presence of tissue-specific 11β-hydroxysteroid dehydrogenases (11β-HSDs) that catalyze the interconversion of active corticosterone and inert 11-dehydrocorticosterone. 11β-HSD type 1 (11β-HSD1) is a bidirectional enzyme in vitro that acts predominantly as a reductase (regenerating corticosterone) in intact neurons. In contrast, 11β-HSD type 2 (11β-HSD2) is a higher affinity exclusive dehydrogenase that excludes GCs from MR in the kidney, producing aldosterone-selectivity in vivo. We have examined the ontogeny of 11β-HSD mRNAs and enzyme activity during prenatal brain development and correlated this with GR and MR mRNA development. These data reveal that (1) 11β-HSD2 mRNA is highly expressed in all CNS regions during midgestation, but expression is dramatically reduced during the third trimester except in the thalamus and cerebellum; (2) 11β-HSD2-like activity parallels closely the pattern of mRNA expression; (3) 11β-HSD1 mRNA is absent from the CNS until the the third trimester, and activity is low or undectectable; and (4) GR mRNA is highly expressed throughout the brain from midgestation, but MR gene expression is absent until the last few days of gestation. High 11β-HSD2 at midgestation may protect the developing brain from activation of GR by GCs. Late in gestation, repression of 11β-HSD2 gene expression may allow increasing GC activation of GR and MR, permitting key GC-dependent neuronal and glial maturational events.
Keywords: 11β-hydroxysteroid dehydrogenases, glucocorticoid receptor, mineralocorticoid receptor, ontogeny, fetal brain, rats
A wealth of studies suggest that early life environment influences brain development and subsequent adult CNS function. In particular, maternal stress during pregnancy produces persisting neuroendocrine and behavioral abnormalities in the offspring, altering motor development, enhancing emotionality, amplifying amphetamine self-administration, and increasing locomotor responses to novelty (Ader and Blackman, 1971; Barlow et al., 1978;Demeniere et al., 1992). Prenatal stress also modifies the offspring hypothalamic–pituitary–adrenal (HPA) axis, a key neuroendocrine effector of the stress response (Meaney et al., 1996; Weinstock et al., 1997). Clinically, HPA axis dysregulation is associated with psychiatric syndromes including depression, eating disorders, anxiety, and age-related cognitive dysfunction (Holsboer et al., 1994; Seckl and Olsson, 1995), and neurodevelopmental factors may be etiologically important in several of these syndromes. Indeed, HPA axis activity in elderly men correlates with birth weight, which is a measure of the prenatal environment (Phillips et al., 1997). Together, these data suggest that maternal stress during pregnancy could increase the vulnerability of the offspring to behavioral brain-related pathologies. These changes could be mediated by in utero exposure of the developing brain to elevated levels of maternal glucocorticoids (GCs) secreted during HPA axis activation by stress that can cross the placenta barrier and reach the developing fetal brain (Zarrow et al., 1970). Prenatal GC exposure permanently “programs” several central functions, including dopamine sensitivity (Diaz et al., 1997), serotonergic activity (Slotkin et al., 1996), and HPA axis parameters (Levitt et al., 1996).
GC actions are primarily mediated via intracellular glucocorticoid receptor (GR) and mineralocorticoid receptor (MR) (de Kloet, 1991). MRs bind both physiological GCs (cortisol, corticosterone) and mineralocorticoids (aldosterone) with an equally high affinity (Kd, ≃0.5 nm), whereas GR preferentially binds cortcosterone but with a lower affinity (2.5–5.0 nm). However, it has recently been recognized that the effects of GCs on their target cells are not only regulated by plasma steroid levels, corticosteroid-binding globulin (CBG), and receptor density, but also by the presence of 11β-hydroxysteroid dehydrogenases (11β-HSDs) (Seckl, 1997a). 11β-HSDs catalyze the interconversion of physiological GCs (cortisol and corticosterone) and inert 11-keto metabolites (cortisone and 11-dehydrocorticosterone). Recently two isozymes have been characterized, encoded by distinct genes (White et al., 1997). 11β-HSD type 1 (11β-HSD1) is a widely expressed NADPH-dependent oxidoreductase (Agarwal et al., 1989). In intact cells, including neurons, 11β-HSD1 is a predominant 11β-reductase that regenerates active corticosterone from inert 11-dehydrocorticosterone (Hundertmark et al., 1995; Jamieson et al., 1995; Rajan et al., 1996). In contrast, 11β-HSD type 2 (11β-HSD2) is a nicotinamide adenine dinucleotide (NAD)-dependent high-affinity isozyme and is an exclusive 11β-dehydrogenase that rapidly inactivates GCs, thus protecting otherwise nonselective MR from GCs in aldosterone target tissues (e.g., distal nephron) (White et al., 1997). Administration of 11β-HSD inhibitors during rat pregnancy adversely programs peripheral systems, producing hypertension and hyperglycemia in adult offspring (Lindsay et al., 1996a,b). Although 11β-HSD bioactivity has been previously demonstrated in adult and perinatal brain (Moisan et al., 1990; Lakshmi et al., 1991; Moisan et al., 1992), these studies predated the identification of the two isozymes and their distinct reaction directions. We therefore investigated the detailed ontogeny of 11β-HSD mRNAs and enzyme activity and correlated this with GR and MR expression during rat brain prenatal development to identify potential time- and locus-dependent “windows” for GC action.
MATERIALS AND METHODS
Subjects. Female Sprague Dawley rats were put into the home cages of individually housed male rats at 5:00 P.M. and were removed the next morning. Coitus was determined by vaginal smear. The morning of sperm detection was designated embryonic day 0.5 (E0.5). At least two dams were decapitated at each of successive gestational days between 11:00 A.M. and 12:00 P.M., and the embryos were removed by cesarean section. Embryos aged E11.5–E14.5 were immediately frozenin toto into a −20°C isopentane bath. From E15.5, embryos were decapitated before freezing. All tissues were stored at −80°C.
11β-HSDs and GR and MR probes. The MR cRNA was prepared from a 513 bp EcoRI fragment of rat mineralocorticoid receptor cDNA (representing parts of the steroid-binding domain and 3′ untranslated region) and was subcloned into the vector pGEM4 (Arriza et al., 1988). The plasmid was linearized with either HindIII or EcoRI and used as a template for SP6 or T7 RNA polymerase to generate the antisense and sense, respectively. The GR cRNA was prepared from a 674 bp PstI–EcoRI fragment of the rat glucocorticoid receptor cDNA corresponding to the 3′ portion of the coding region (steroid-binding domain) and was subcloned into the vector pGEM3 (Seckl et al., 1990). The antisense and sense probes were generated using AvaI (T7) and EcoRI (SP6), respectively. The 11β-HSD2 was prepared from a 613 bp fragment obtained by reverse transcription-PCR from rat kidney RNA using primers to conserved regions of the 11β-HSD2 and subcloned into a pGEM-T vector (Leckie et al., 1995). The antisense and sense probes were generated using NcoI (SP6) and NotI (T7), respectively. The 11β-HSD1 was prepared from a 1.2 kbEcoRI fragment of the rat 11β-HSD1 cDNA and subcloned into the vector pBluescript SKII (Agarwal et al., 1989). The antisense and sense probes were generated using StyI (T3) andPstI (T7), respectively. 35S-Uridine 5′-triphospate (UTP)-labeled RNA antisense and sense probes were transcribed in vitro using the appropriate template and phage RNA polymerase, purified using Nensorb Columns (DuPont, Billerica, MA), and checked on denaturing acrylamide gels.
In situ hybridization. Sagittal (in relation to the head) 20 μm cryostat sections were thaw-mounted on 3-aminopropyltriethoxysilane-coated slides and stored at −80°C. Tissue sections were fixed, prehybridized, hybridized (hybridization was made with ∼3 × 106 cpm/section35S-UTP-labeled RNA probe at 50°C for 12–14 hr), and washed essentially as described previously (Belluardo et al., 1997). After RNase A washing, treated slides were hydrated, dried, placed against β-Max (Hyperfilm β-film; Amersham, Arlington Heights, IL), and stored at room temperature for 1–2 weeks. Films were developed in D19 developer for 2 min and fixed in a 1:5 dilution of Amfix fixative for 2 min. Adjacent sections were stained with cresyl violet to allow neuroanatomical localization of the specifically labeled CNS regions (data not shown). The atlas of prenatal rat brain development by Altman and Bayer (1995) was consulted. Nonspecific hybridization was determined by incubating adjacent sections with the respective35S-UTP-labeled sense cRNA probe for the above cDNAs under identical conditions. Nonspecific background was very low in all sections analyzed from all fetal ages (data not shown).
11β-HSD activity. 11β-Reductase and 11β-dehydrogenase activity was determined from homogenates of rat brain tissue at ages E12.5, E14.5, E17.5, and E20.5. The assays were performed according to a standard protocol described elsewhere (Brown et al., 1996) and included 100 μm cofactor (NAD or NADP for dehydrogenase or NADPH for reductase) and 12 nm final3H-labeled steroid ([3H]corticosterone for dehydrogenase and [3H]11-dehydrocorticosterone for reductase). In addition, we assayed samples of liver (for reductase) and placenta (for dehydrogenase) from each fetus as an internal control to demonstrate that 11β-HSD1 and 11β-HSD2 were detectable in tissues outside the brain at each gestational age. Preliminary experiments showed that 0.25 mg/ml homogenate protein was the optimal concentration in the curve describing the relationship between protein concentration and enzyme activity. All assays used 0.25 mg/ml; two time points (10 and 40 min) were used for dehydrogenase and three time points were used for reductase (10, 40, and 90 min). Because the results for the dehydrogenase were similar at the two time points, only the 10 min data are presented.
Corticoterone levels. Plasma corticosterone levels were measured by radioimmunoassay at ages E12.5, E14.5, E17.5, and E20.5, as described previously (Holmes et al., 1997), but modified for microtiter scintillation plate assay (SPA, Amersham). Briefly, the incubation volume was 120 μl, consisting of 20 μl of 10× diluted plasma, 50 μl of antiserum (1:10,000)/[3H]corticosterone mix (12,000 cpm/tube 1,2,6,7-[3H]corticosterone, Amersham), and 50 μl of SPA beads. After an overnight incubation at room temperature, the microtiter plates were counted on a Wallac microbeta liquid scintillation counter.
RESULTS
Ontogeny of 11β-HSD2 mRNA
High 11β-HSD2 gene expression is observed throughout the CNS neuroepithelium, including the pituitary gland primordium at E11.5 (Fig. 1) (brain areas are labeled in Figs.1-5). 11β-HSD2 mRNA expression remains high until E13.5. By E14.5, the expression starts to decrease in the neocortex, pallidal area, and spinal cord (data not shown). By E15.5, 11β-HSD2 expression is restricted to several discrete areas that include thalamus (although Fig. 1 only shows anterior and posterior areas, its expression is more widely distributed), cerebellum, midbrain (superior colliculus differentiating field), various pontine regions (e.g., locus coeruleus and precerebellar area), septum, hypothalamus (preoptic area and anterior and ventral hypothalamus), medulla, pallidum (data not shown), and strianuclear complex. At E16.5, 11β-HSD2 mRNA expression is still high in several areas that include the straitum, midbrain (superior and inferior tectal neuroepithelium, inferior colliculus, and pretectum), thalamus, preoptic area, and ventral hypothalamus (e.g., arcuate nucleus), cerebellum (external granular layer), and low to moderate in septum (data not shown) (Fig. 3). 11β-HSD2 mRNA expression remains similar from E16.5 until E19.5, when expression ceases in the midbrain and is low in the septum. However, 11β-HSD2 mRNA is still present in preoptic area and ventral hypothalamus (e.g., arcuate nucleus). By E20.5, 11β-HSD2 gene expression is restricted to the external granular layer of the cerebellum, precerebellar area, and thalamus, and at E22.5 (the day of birth) 11β-HSD2 mRNA is only found in the external granular layer of the cerebellum and thalamus (Figs. 3,5).
Fig. 1.
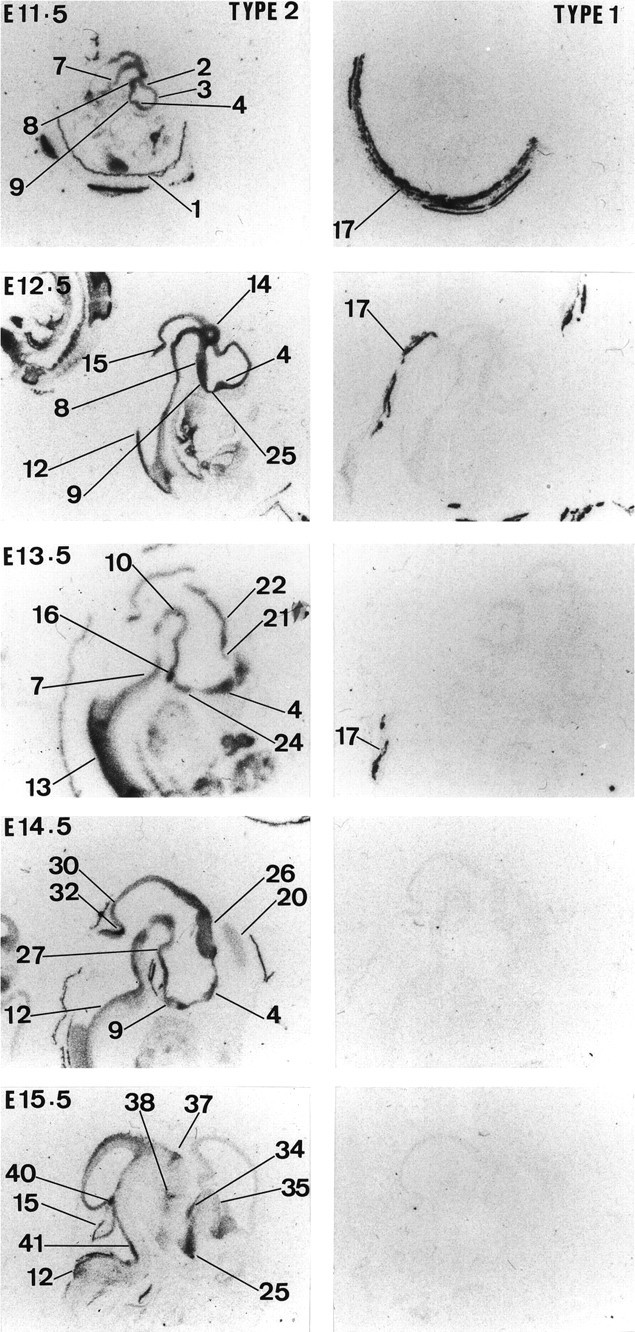
Expression of 11β-HSD type 2 and 11β-HSD type 1 in the rat brain from E11.5 to E15.5. Results of in situ hybridization for 11β-HSD TYPE 2 and 11β-HSD TYPE 1 are shown from left toright, respectively, for each gestation. The following are labeled in Figures 1-5: 1, extraembryonic membrane;2, hippocampal (Ammon’s horn) and subicular regions;3, neocortex; 4, septum;5, liver; 6, dorsal mesencephalon;7, pontine area; 8, intermediate, mammillary, and posterior hypothalamic areas; 9, hypothalamus and anterior hypothalamus; 10, tegmental area; 11, intermediate thalamus; 12, medulla; 13, spinal cord; 14, dorsal diencephalon; 15, cerebellum; 16, pituitary; 17, uterine wall; 18, taenia tecta; 19, indesium griseum; 20, cingulate cortex; 21, lamina terminalis;22, epithalamus; 23, pretectum;24, ventral hypothalamus; 25, preoptic area; 26, thalamus; 26a, posterior thalamus and epithalamus; 26b, thalamus and anterior-medial area; 27, mammillary area;28, posterior hypothalamus; 29, ventral isthmal area; 30, inferior colliculus;31, superior colliculus; 32, dorsal isthmal area; 33, precerebellar area; 34, basal ganglia and strionuclear area; 35, pallidum;36, arcuate nucleus and ventral medial hypothalamus;37, posterior thalamus; 38, anterior thalamus; 39, olfactory bulb; 40, locus coerulus; 41, anterior pontine area; 42, posterior pontine area; 43, dorsal raphe;44, pineal gland; 45, choroid plexus and lateral ventricle; 46, skin; 47, precerebellar area and above choroid plexus; and 48, dorsal periaqueductal gray and superior colliculus.
Fig. 2.
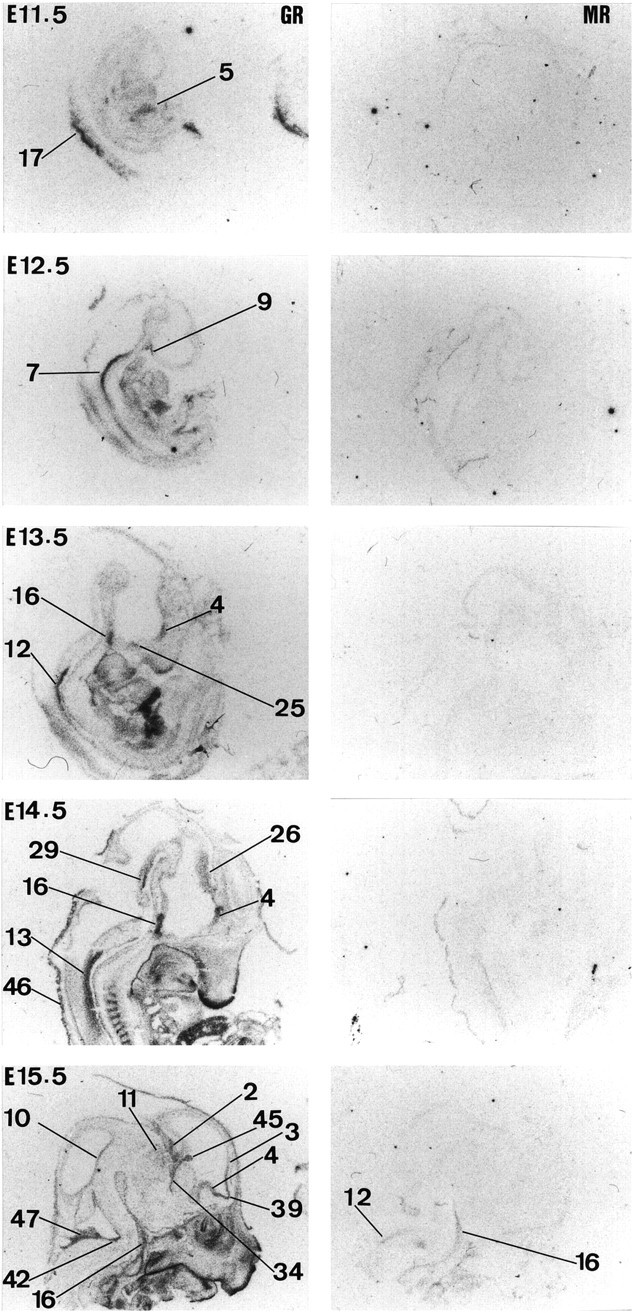
Expression of GR and MR in the rat brain from E11.5 to E15.5. Results of in situ hybridization forGR and MR are shown fromleft to right, respectively, for each gestation. For labeling of regions see text of Figure 1.
Fig. 3.
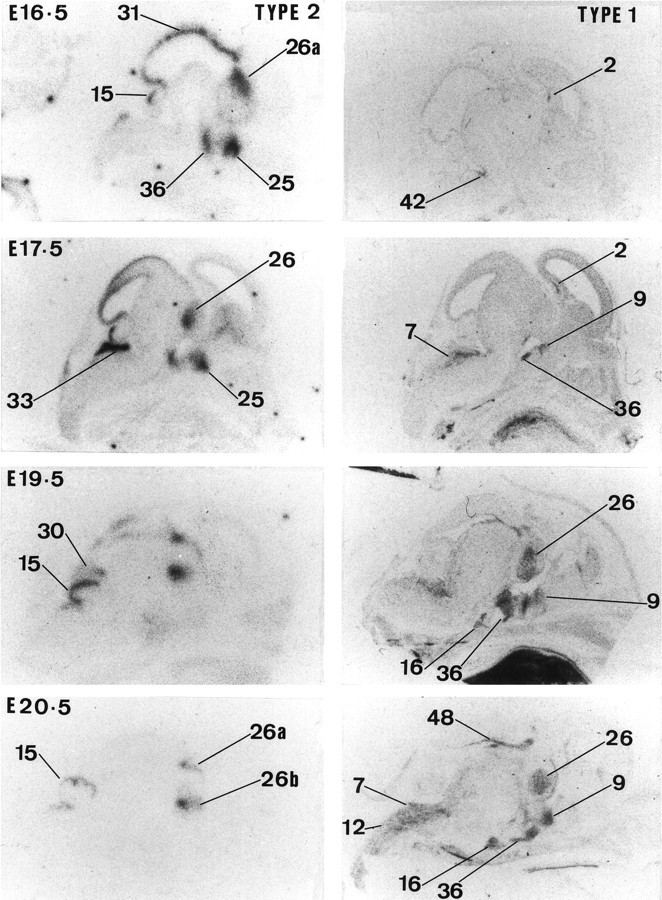
Expression of 11β-HSD type 2 and 11β-HSD type 1 in the rat brain from E16.5 to E20.5. Results of in situ hybridization for 11β-HSD TYPE 2 and 11β-HSD TYPE 1 are shown from left toright, respectively, for each gestation. For labeling of regions see text of Figure 1.
Fig. 4.
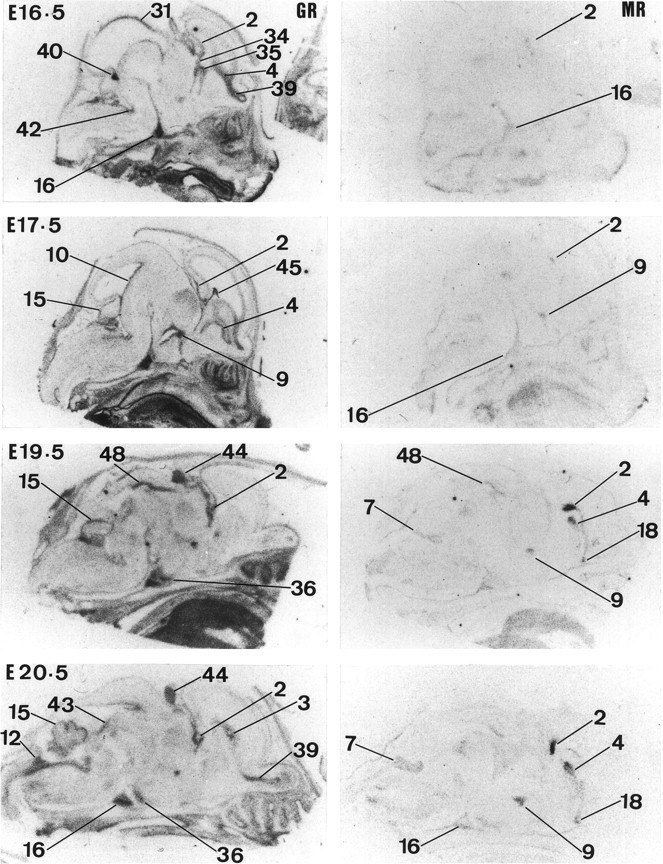
Expression of GR and MR in the rat brain from E16.5 to E20.5. Results of in situ hybridization forGR and MR are shown fromleft to right, respectively, for each gestation. For labeling of regions see text of Figure 1.
Fig. 5.
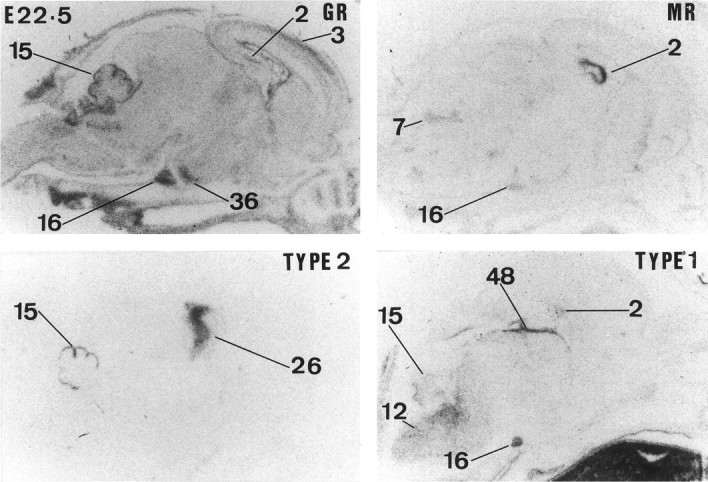
Expression of GR,MR, 11β-HSD TYPE 2, and 11β-HSDTYPE 1 mRNAs in the rat brain at E22.5. For labeling of regions see text of Figure 1.
Ontogeny of 11β-HSD1 mRNA
11β-HSD1 mRNA expression is clearly very low or absent before E15.5 (Figs. 1, 3). Moderate 11β-HSD1 mRNA expression is first seen at E16.5 in hippocampus, precerebellar area, and medulla (data not shown). From E17.5 to E22.5, there is a gradual increase in expression throughout the CNS, including the pituitary gland primordium. At E17.5, there is also expression in the tegmental area (data not shown). The pattern of 11β-HSD1 gene expression is similar between E18.5 and E22.5 (date of birth) (Figs. 3, 5). High 11β-HSD1 mRNA expression is observed in thalamus, neocortex, hypothalamus (particularly in the anterior and ventral parts), pituitary gland (primordium), brainstem (both pons and medulla areas), dorsal periaqueductal gray area, tegmental area, spinal cord, and hippocampus (Fig.6).
Fig. 6.
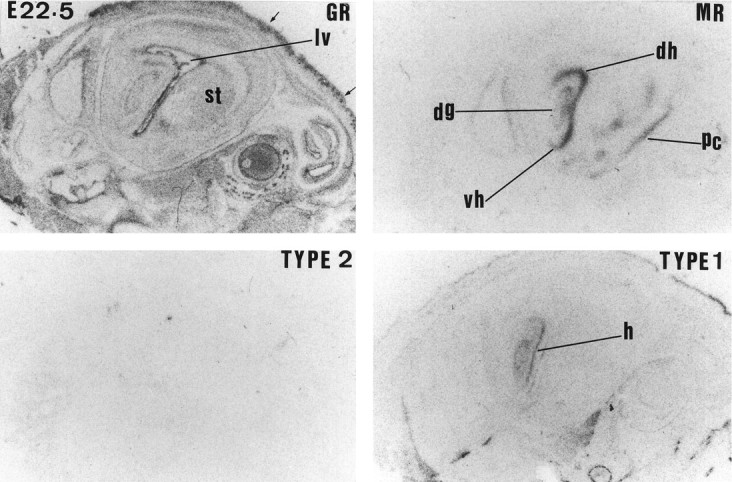
Expression of GR,MR, 11β-HSD TYPE 2, and 11β-HSDTYPE 1 mRNAs in the rat hippocampus at E22.5. The following regions are labeled in the figure: striatum (st); lateral ventricle (lv); hippocampus (hr); dorsal hippocampus (dh); ventral hippocampus (vh); and piriform cortex (pc). Arrows indicate skin tissue.
11β-HSD activity
NAD+-dependent 11β-dehydrogenase activity (11β-HSD2) is clearly detectable in whole brain and placental extracts at all gestational ages examined (Fig.7). 11β-HSD2 activity in the brain is high at E12.5 and E14.5 but by E17.5 is dramatically reduced to ∼24% of the levels observed during midgestation. 11β-HSD2 enzyme activity remains low at E20.5. The overall pattern of placental 11β-HSD2 activity was similar to that in the brain. In contrast, NADP+-dependent 11β-dehydrogenase (11β-HSD1) was very low in brain extracts at all gestational ages (Fig. 7). NADPH+-dependent 11β-reductase (11β-HSD1) was undetectable in brain extracts at all ages. However, in the liver there was an increase in 11β-reductase activity at E17.5 (10.65 ± 1.95%) and E20.5 (29.05 ± 0.75%) (90 min incubation).
Fig. 7.
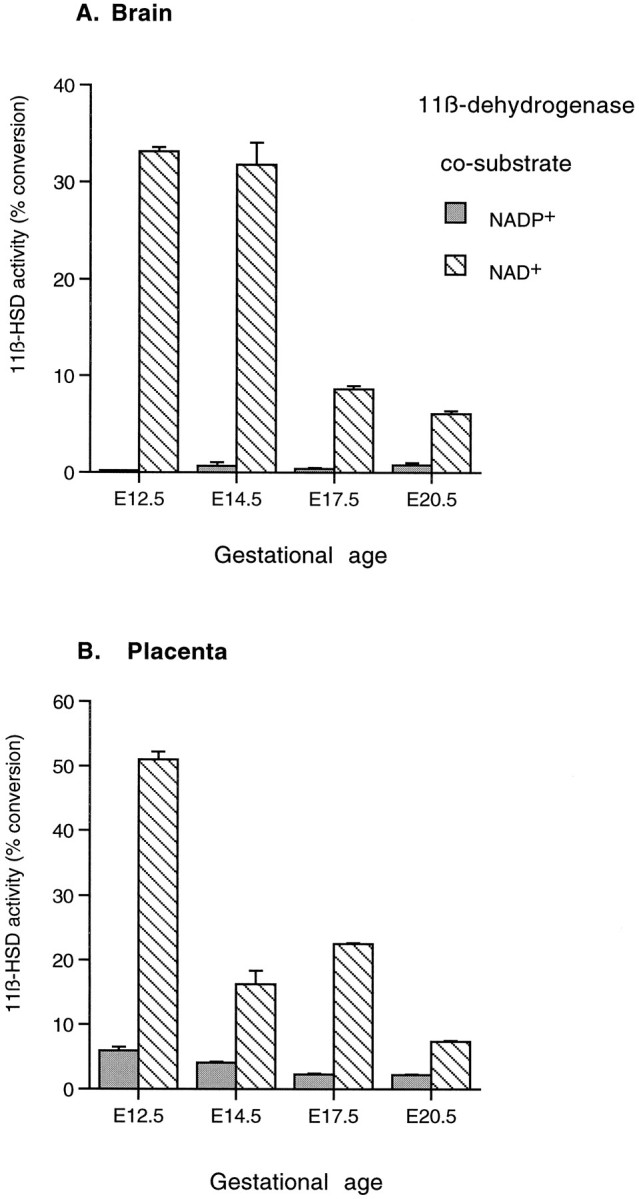
11β-Dehydrogenase activity from homogenates of rat brain tissue (A) and placenta (B) at ages E12.5, E14.5, E17.5, and E20.5. Cosubstrates were added at 100 μm. Note the predominant NAD+-dependent reaction, typical of 11β-HSD2.
Ontogeny of GR mRNA
GR mRNA expression is first seen at E12.5 in the neuroepithelium (high levels are already present in the periphery, e.g., liver 1 d earlier) (Fig. 2). At this stage, there is an overall increase in GR mRNA expression throughout the body. In the CNS, high expression is only seen in the pons (mainly posterior area), medulla (both ventral and dorsal parts), anterior hypothalamus, spinal cord, and pituitary gland (primordium). At E13.5, high expression of GR mRNA is also detected in septum, preoptic area, and hippocampus (data not shown). By E14.5, GR mRNA expression dramatically increases in the ventral part of the spinal cord and to a lesser extent in the thalamus. GR mRNA is also seen in the ventral isthmal area. By E15.5, there is increased GR mRNA expression in neocortex, olfactory bulb, precerebellar area, cerebellum, choroid plexus, tegmentum, and basal glanglia (striatal and pallidal neuroepithelia). In contrast, GR mRNA expression dramatically decreases in spinal cord and moderately decreases in the thalamus and septum. At E16.5, GR gene expression appears in the locus coeruleus and superior colliculus and continues to increase in septum, olfactory bulb, neocortex, and hippocampus (Fig. 4). Expression of GR mRNA is similar at E17.5. At E19.5, GR mRNA expression is also seen in the pineal gland and periaqueductal gray area. Moreover, there is an increase of GR mRNA in cerebellum. This pattern of expression persists until E22.5. However, during this period there is a clear increase in the level of GR expression throughout all the CNS-differentiating fields (Figs. 4, 5).
Ontogeny of MR mRNA
MR mRNA expression is clearly absent in the CNS during midgestation (Fig. 2). Moderate MR mRNA expression is first seen at E15.5 in pituitary gland (primordium), brainstem (medulla area), facial motor nucleus, tegmentum, and neuroepithelium of the septum and pallidum (data not shown). At E16.5, low expression can also be detected in hippocampal neuroepithelium. MR mRNA expression moderately increases by E17.5 and is first seen in the anterior hypothalamus (Fig.4). At E19.5, MR mRNA expression dramatically increases in hippocampus, septum, rhinencephalon (data not shown), anterior hypothalamus, periaqueductal gray area, and brainstem (pontine and medulla) neuroepithelium. There is also clear MR mRNA expression in the amygdala and piriform cortex. This pattern of expression persists until E22.5 (see Fig. 6).
Plasma corticosterone levels
It was not possible for plasma corticosterone levels to be assayed at E12.5 because of insufficient blood volume. At E14.5, corticosterone levels were <3 nm. On E17.5 there was a clear increase in corticosterone levels (161 nm). By E20.5 corticosterone levels increased further to double the levels observed on E17.5 (333 nmol). The intra-assay variation was well <10%, and the figures were consistently indicated by different dilutions of the plasma that was pooled from at least 5 fetuses (12 for E14.5).
DISCUSSION
This study represents the first description of the ontogeny of 11β-HSD isozymes during prenatal rat brain development. The results clearly demonstrate a distinct pattern of expression for each enzyme. 11β-HSD2, which potently inactivates GCs, is widely expressed in the neuroepithelium until the beginning of the third trimester, when the expression falls dramatically. In contrast, 11β-HSD1 mRNA (encoding an enzyme that largely reactivates GCs in vivo) is low or absent until late gestation. There are also precise temporal and spatial changes in expression of GR, and later MR, during prenatal rat brain development. The data clearly show potential time- and locus-dependent windows for adrenal steroid hormone action on fetal brain development via changes in both receptor expression and enzymatic control of GC access to receptors.
GR and MR expression in fetal brain
The data confirm and extend previous findings on corticosteroid receptor gene expression in fetal brain (Cintra et al., 1993; Rene et al., 1994; Yi et al., 1994; Kitraki et al., 1996). In agreement with Cintra et al. (1993), GR mRNA was found highly expressed in the neuroepithelium of multiple brain areas (including hippocampus, cerebellum, hypothalamus, raphe nuclei, locus coeruleus, and olfactory bulb) during the last trimester of gestation. In addition, we found GR mRNA expression within the septal, pallidal, and strionuclear complex neuroepithelia during this period. The present data also extend to earlier stages of development to reveal high GR mRNA expression during midgestation (starting at E12.5). Previous findings have demonstrated that GR mRNA is translated to an adult-like protein in fetal tissue, suggesting that GR is functional and participates in developmental processes such as cell birth, migration, differentiation, and maturation (Meaney et al., 1985). Another important observation is that toward the end of the third trimester GR mRNA expression increases throughout the brain, including within the differentiating fields, which coincides with the activation of the fetal HPA axis (Milkovic et al., 1973). These observations support the notion that endogenous GCs acting via GR may play a role in differentiation and maturational events during late fetal brain development. GCs may promote the transition between a proliferative and a differentiating stage by directly inhibiting cell division as well as activating the expression of specific genes characteristic of the differentiated mature phenotype. Indeed, GCs have been reported in vitro to decrease the rate of cell proliferation by preventing progression through the cell cycle and extending the G1 phase or G2 and M phases in non-neuronal cells (Fanger et al., 1987;Vintermyr et al., 1989; Hatakeyama et al., 1991; Sanchez et al., 1993).
In contrast to widespread GR gene expression, MR mRNA expression was confined to the neuroepithelium of the septal–hippocampal system and a few discrete areas of the anterior hypothalamus, pituitary, deep layers of the superior colliculus, piriform cortex, and brainstem. Also in contrast to GR mRNA expression, MR mRNA expression was very low or absent until the last 3 d of gestation, when it became more prominent in the hippocampus and lateral septum. Indeed, at the end of gestation, MR was higher than GR mRNA expression in the hippocampus and septum, confirming previous observations (Bohn et al., 1994). MR binding sites have been reported in both neurons and glia in fetal hippocampal cell cultures (Bohn et al., 1991), suggesting that MR mRNA is likely to be translated. Physiological MR agonists, such as aldosterone, affect neuronal and glial cell birth and death rates in early postnatal dentate gyrus (see Gould and Cameron, 1996), and MR, as well as GR, may regulate similar processes in the developing fetal brain. The mechanisms of MR induction late in gestation are unknown. Possible factors include (1) activation of the fetal HPA axis, although MR is slightly regulated by GCs during adult life (Seckl, 1997a), and dexamethasone in the last trimester reduces MR gene expression in adult life (Levitt et al., 1996); and (2) more likely, the contemporaneous development of neurotransmitter inputs (e.g., 5-hydroxytryptamine) that potently controls hippocampal GR and MR expression in perinatal and adult life (see Meaney et al., 1996).
11β-HSD expression in fetal brain
Previous studies suggested that the high 11β-HSD bioactivity in whole fetal brain extracts (Burton et al., 1968) is comparable to the levels in the adult kidney. Limited human studies suggest that high 11β-HSD is confined to early or midgestation and falls in the third trimester in many tissues, including brain (Murphy, 1981; Stewart et al., 1994). Our results demonstrate the predominance of the NAD-dependent 11β-HSD2 isozyme throughout the midgestation fetal rat brain, with 11β-dehydrogenase bioactivity paralleling the pattern of 11β-HSD2 mRNA expression. The high levels of 11β-HSD2 in the neuroepithelium during midgestation are likely to profoundly attenuate GC effects during the stage of CNS development. Similarly, abundant 11β-HSD2 in placenta during midgestation is thought to protect the fetus from the deleterious effects of high maternal GCs by ensuring that most maternal glucocorticoids are inactivated on crossing to the fetus (see Seckl, 1997b). Low plasma corticosterone levels in the fetus during midgestation may reflect rapid inactivation of GCs by widespread 11β-HSD2 in placenta and fetal tissues. However, there is no evidence available indicating that 11β-HSD2 is so effective as to abolish GR activation by GCs during this period. Thus, further studies are required to determine the relationship between brain corticosterone levels, GR occupancy, and 11β-HSD2 activity during early fetal development.
The present data do not support the notion that 11β-HSD2 protects MR during brain development, because MR mRNA is not expressed when 11β-HSD2 is most abundant. Given that GR gene expression is seen from early stages of development, and exposure of the developing fetus to excess GCs can result in fetal death (Yang et al., 1969), 11β-HSD2 may be protecting the developing CNS from the adverse effects of endogenous GCs acting via GR. Although severe brain abnormalities are not prominent in humans with the syndrome of apparent mineralocorticoid excess caused by deleterous mutations of the 11β-HSD2 gene (Mune et al., 1995; Wilson et al., 1995), more subtle CNS abnormalities have not been sought in these exceptional patients who rarely survive childhood. certainly, poor 11β-HSD2 substrate GCs, such as dexamethasone, have distinct programming effects on CNS maturation before birth (Levitt et al., 1996; Slotkin et al., 1996). It is also conceivable, although unlikely, that the function of 11β-HSD2 differs during development, perhaps catalyzing metabolism of putative alternative 11β-hydroxysteroid substrates. The mechanism downregulating 11β-HSD2 in fetal brain at midgestation is unclear, although widespread silencing may be related to C and G-rich sequences reported in the 5′ region of the 11β-HSD2 gene (Brown et al., 1996).
Recently, Rajan et al. (1996) demonstrated 11β-HSD1 gene expression and activity in fetal hippocampal cells in culture. The present work confirms this observation and also demonstrates 11β-HSD1 gene expression in other brain regions, including the thalamus, hypothalamus, pituitary, brainstem, and cortex. However, 11β-HSD1 gene expression was much lower than that of 11β-HSD2 mRNA and was restricted to late gestation, when 11β-HSD2 mRNA expression was declining. Interestingly, both 11β-HSD1 and MR expression increase after fetal adrenocortical activation has occurred and circulating fetal corticosterone levels are rising (Dupouy et al., 1975; Milkovic et al., 1973). Previous studies have demonstrated that dexamethasone administration, or stress, increase 11β-HSD1 mRNA expression and enzyme bioactivity in the adult rat hippocampus (Low et al., 1994). Similar GC induction of 11β-HSD1 occurs in primary rat hippocampal cultures from E18 (Rajan et al., 1996). Moreover, there is a putative glucocorticoid response element in the promoter of the rat 11β-HSD1 gene (Voice et al., 1996). It is thus possible that the rise in fetal corticosterone levels during the last part of the third trimester may induce 11β-HSD1 mRNA expression in fetal brain. 11β reduction is clearly the predominant reaction in intact cells from a variety of tissues, including brain, liver, and lung (Hundertmark et al., 1995;Jamieson et al., 1995; Rajan et al., 1996). However, any physiological role for the 11β-HSD1 in modulating exposure of the developing brain to GCs (either protecting against or increasing exposure to GCs) remains unclear, because 11β-HSD1 activity is barely, if at all, detected in the rat fetal brain. Because these studies were performed in homogenates of whole fetal brain, it is possible that regional differences in 11β-HSD1 activity could have been masked. Thus, at this early point in the investigation, the presence of 11β-HSD1 in the fetal brain seems likely, but requires further investigation.
The importance of the intrauterine environment has been emphasized by studies showing that prenatal stress exerts long-lasting effects on a variety of neuroendocrine and behavioral parameters (see Weinstock et al., 1997). It has been postulated that these changes could be mediated by direct effects on the developing brain after in uteroexposure to elevated levels of maternal or fetal GCs. Our data suggest exquisite time and region-specific windows for GC action within the developing CNS.
Footnotes
This work was supported by grants from the Wellcome Trust/Royal Society of Edinburgh, the Scottish Hospital Endowments Research Trust, and the Sir Stanley and Lady Davidson Research Fund (J.R.S.), a Medical Research Council Training Fellowship (R.W.B.), and a Karolinska Institute Training Fellowship (R.D.). We thank Dr. Megan Holmes for generously measuring the plasma corticosterone levels and Dr. Tommy Olsson for kindly providing R.D. with photographic facilities.
Correspondence should be addressed to Dr. Rochellys Diaz, Department of Medicine, Umeå University Hospital, S-901 85 Umeå, Sweden.
REFERENCES
- 1.Ader R, Blackman DE. Prenatal psychological stress and offspring behavior in rats and mice. Science. 1971;125:698–699. doi: 10.1002/dev.420040302. [DOI] [PubMed] [Google Scholar]
- 2.Agarwal A, Monder C, Eckstein B, White PC. Cloning and expression of rat cDNA encoding corticosteroid 11-dehydrogenase. J Biol Chem. 1989;264:18939–18943. [PubMed] [Google Scholar]
- 3.Altman J, Bayer SA. Atlas of prenatal rat brain development. CRC; London: 1995. [Google Scholar]
- 4.Arriza JL, Simerly RB, Swanson LW, Evans RM. The neuronal mineralocorticoid receptor as a mediator of glucocorticoid response. Neuron. 1988;1:887–900. doi: 10.1016/0896-6273(88)90136-5. [DOI] [PubMed] [Google Scholar]
- 5.Barlow SM, Knight AF, Sullivan FM. Delay in postnatal growth and development of offspring produced by maternal restraint stress during pregnancy in the rat. Teratology. 1978;18:211–218. doi: 10.1002/tera.1420180206. [DOI] [PubMed] [Google Scholar]
- 6.Belluardo N, Wu G, Mudo G, Hansson AC, Pettersson R, Fuxe K. Comparative localization of fibroblast growth factor receptor-1,-2, and -3 mRNAs in the rat brain: in situ hybridization analysis. J Comp Neurol. 1997;379:226–246. [PubMed] [Google Scholar]
- 7.Bohn MC, Howard E, Vielkind U, Krozowski Z. Glial cells express both mineralocorticoid and glucocorticoid receptors. J Steroid Biochem Mol Biol. 1991;40:105–111. doi: 10.1016/0960-0760(91)90173-3. [DOI] [PubMed] [Google Scholar]
- 8.Bohn MC, Dean D, Hussain S, Giuliano R. Development of mRNAs for glucocorticoid and mineralocorticoid receptors in rat hippocampus. Dev Brain Res. 1994;77:157–162. doi: 10.1016/0165-3806(94)90192-9. [DOI] [PubMed] [Google Scholar]
- 9.Brown RW, Kotelevtsev Y, Leckie C, Lindsay RS, Lyons V, Murad P, Mullins JJ, Chapman KE, Edwards CRW, Seckl JR. Isolation and cloning of human placental 11β-hydroxysteroid dehydrogenase-2 cDNA. Biochem J. 1996;313:1007–1017. doi: 10.1042/bj3131007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burton AF, Turnell RW. 11-dehydrocorticosteroids in tissues of mice. Can J Biochem. 1968;46:497–502. doi: 10.1139/o68-075. [DOI] [PubMed] [Google Scholar]
- 11.Cintra A, Solfrini V, Bunneman B, Okret S, Bortolotti F, Gustafsson J-Å, Fuxe K. Prenatal development of glucocorticoid receptor gene expression and immunoreactivity in the rat brain and pituitary gland: a combined in situ hybridization and immunocytochemistry analysis. Neuroendocrinology. 1993;57:1133–1147. doi: 10.1159/000126480. [DOI] [PubMed] [Google Scholar]
- 12.de Kloet ER. Brain corticosteroid receptor balance and homeostatic control. Front Neuroendocrinol. 1991;12:95–164. doi: 10.1016/j.yfrne.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 13.Demeniere JM, Piazza PV, Guegan G, Abrous N, Maccari S, Le Moal M, Simon H. Increased locomotor response to novelty and propensity to intravenous amphetamine self-administration in adult offspring of stressed mothers. Brain Res. 1992;586:135–139. doi: 10.1016/0006-8993(92)91383-p. [DOI] [PubMed] [Google Scholar]
- 14.Diaz R, Fuxe K, Ögren SV. Prenatal cortcosterone treatment induces long-term changes in spontaneous and apomorphine-mediated motor activity in male and female rats. Neuroscience. 1997;81:129–140. doi: 10.1016/s0306-4522(97)00141-3. [DOI] [PubMed] [Google Scholar]
- 15.Dupouy JP, Coffigny H, Magre S. Maternal and fetal corticosterone levels during late pregnancy in rats. J Endocrinol. 1975;65:347–352. doi: 10.1677/joe.0.0650347. [DOI] [PubMed] [Google Scholar]
- 16.Fanger BO, Schreifer J, Cidlowski JA. Glucocorticoids increase the length of the G2 and M phases of the HeLa S3 cell cycle. J Steroid Biochem Mol Biol. 1987;28:345–347. doi: 10.1016/0022-4731(87)91029-6. [DOI] [PubMed] [Google Scholar]
- 17.Gould E, Cameron HA. Regulation of neuronal birth, migration, and death in the rat dentate gyrus. Dev Neurosci. 1996;18:22–35. doi: 10.1159/000111392. [DOI] [PubMed] [Google Scholar]
- 18.Hatakeyama S, Susuki A, Yoshizumi N, Sato M, Nishiya I. Glucocorticoid-induced G1 arrest and the release effect of epidermal growth factor on human salivary gland adrenocarcinoma cell. Cell Biol Int Rep. 1991;15:55–65. doi: 10.1016/0309-1651(91)90082-t. [DOI] [PubMed] [Google Scholar]
- 19.Holmes Mg, French K, Seckl JR. Dysregulation of serotonin 5-HT2C and corticosteroid receptor gene expression in the hippocampus with food restriction and glucocorticoids. J Neurosci. 1997;17:4046–4065. doi: 10.1523/JNEUROSCI.17-11-04056.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holsboer F, Grasser A, Friess E, Wiedemann K. Steroid effects on central neurons and implications for psychiatric and neurological disorders. Ann NY Acad Sci. 1994;746:345–359. doi: 10.1111/j.1749-6632.1994.tb39255.x. [DOI] [PubMed] [Google Scholar]
- 21.Hundertmark S, Buhler H, Ragosch V, Dinkelborg L, Arabin B, Weitzel HK. Correlation of surfactant phosphatidylcholine synthesis and 11β-hydroxysteroid dehydrogenase in fetal lung. Endocrinology. 1995;136:2573–2578. doi: 10.1210/endo.136.6.7750479. [DOI] [PubMed] [Google Scholar]
- 22.Jamieson PM, Chapman KE, Edwards CRW, Seckl JR. 11β -Hydroxysteroid dehydrogenase is an exclusive 11-reductase in primary cultures of rat hepatocytes: effect of physicochemical and hormonal manipulations. Endocrinology. 1995;136:4754–4760. doi: 10.1210/endo.136.11.7588203. [DOI] [PubMed] [Google Scholar]
- 23.Kitraki E, Alexis MN, Stylianopoulou F. Glucocorticoid receptor gene expression in the embryonic rat brain. Neuroendocrinology. 1996;63:305–317. doi: 10.1159/000126971. [DOI] [PubMed] [Google Scholar]
- 24.Lakshmi V, Sakai RR, McEwen BS. Regional distribution of 11β-hydroxysteroid dehydrogenase in rat brain. Endocrinology. 1991;128:1741–1748. doi: 10.1210/endo-128-4-1741. [DOI] [PubMed] [Google Scholar]
- 25.Leckie C, Chapman KE, Edwards CRW, Seckl JR. LLC-PK1 cells model 11-hydroxysteroid dehydrogenase type 2 regulation of glucocorticoid access to renal mineralocorticoid receptors. Endocrinology. 1995;136:5561–5569. doi: 10.1210/endo.136.12.7588309. [DOI] [PubMed] [Google Scholar]
- 26.Levitt N, Lindsay RS, Holmes MC, Seckl JR. Dexamethasone in the last week of pregnancy attenuates hippocampal glucocorticoid receptor gene expression and elevates blood pressure in the adult offspring in the rat. Neuroendocrinology. 1996;64:412–418. doi: 10.1159/000127146. [DOI] [PubMed] [Google Scholar]
- 27.Lindsay RS, Lindsay RM, Edwards CRW, Seckl JR. Inhibition of 11β-hydroxysteroid dehydrogenase in pregnant rats and the programming of blood pressure in the offspring. Hypertension. 1996a;27:1200–1204. doi: 10.1161/01.hyp.27.6.1200. [DOI] [PubMed] [Google Scholar]
- 28.Lindsay RS, Lindsay RM, Waddel B, Seckl JR. Programming of glucose tolerance in the rat: role of placental 11β-hydroxysteroid dehydrogenase. Diabetologia. 1996b;39:1299–1305. doi: 10.1007/s001250050573. [DOI] [PubMed] [Google Scholar]
- 29.Low SC, Moisan M-P, Edwards CRW, Seckl JR. Glucocorticoids and chronic stress upregulate 11β-hydroxysteroid dehydrogenase activity and gene expression in the hippocampus. J Neuroendocrinol. 1994;6:275–290. doi: 10.1111/j.1365-2826.1994.tb00584.x. [DOI] [PubMed] [Google Scholar]
- 30.Meaney Mj, Sapolsky RM, Aiteken DH, McEwen BS. 3H- Dexamethasone binding in the limbic brain of the fetal rat. Dev Brain Res. 1985;23:297–300. doi: 10.1016/0165-3806(85)90054-9. [DOI] [PubMed] [Google Scholar]
- 31.Meaney MJ, Diorio J, Francis D, Widdowson J, La Plante P, Caldji C, Sharma S, Seckl JR, Plotsky PM. Early environmental regulation of forebrain glucocorticoid receptor gene expression: implications for adrenocortical responses to stress. Dev Neurosci. 1996;18:49–72. doi: 10.1159/000111395. [DOI] [PubMed] [Google Scholar]
- 32.Milkovic S, Milkovic K, Paunovic J. The initiation of fetal adrenocortrophic activity in the rat. Endocrinology. 1973;92:380–384. doi: 10.1210/endo-92-2-380. [DOI] [PubMed] [Google Scholar]
- 33.Moisan M-P, Seckl JR, Edwards CRW. 11β-Hydroxysteroid dehydrogenase bioactivity and messenger RNA expression in rat forebrain: localization in hypothalamus, hippocampus, and cortex. Endocrinology. 1990;127:1450–1455. doi: 10.1210/endo-127-3-1450. [DOI] [PubMed] [Google Scholar]
- 34.Moisan M-P, Edwards CRW, Seckl JR. Ontogeny of 11β-hydroxysteroid dehydrogenase in rat brain and kidney. Endocrinology. 1992;130:400–444. doi: 10.1210/endo.130.1.1727713. [DOI] [PubMed] [Google Scholar]
- 35.Mune T, Rogerson FM, Nikkilä H, Agarwal AK, White PC. Human hypertension caused by mutations in the kidney isozyme of 11β-hydroxysteroid dehydrogenase. Nat Genet. 1995;10:394–399. doi: 10.1038/ng0895-394. [DOI] [PubMed] [Google Scholar]
- 36.Murphy BE. Ontogeny of cortisol-cortisone interconversion in humans tissues: a role for cortisone in human fetal development. J Steroid Biochem Mol Biol. 1981;14:811–817. doi: 10.1016/0022-4731(81)90226-0. [DOI] [PubMed] [Google Scholar]
- 37.Phillips DIW, Barker DJP, Fall CHD, Whorwood CB, Walker BR, Wood PJ. Low birth weight and raised plasma concentrations in adult life. Diabetologia. 1997;40:830. [Google Scholar]
- 38.Rajan V, Edwards CRW, Seckl JR. 11β-Hydroxysteroid dehydrogenase in cultured hippocampal cells reactivates inert 11-dehydrocorticosterone, potentiating neurotoxicity. J Neurosci. 1996;16:65–70. doi: 10.1523/JNEUROSCI.16-01-00065.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rene F, Hindelang C, Stoeckel ME, Felix JM. Ontogeny of glucocorticoid and D2 receptors in the rat pituitary: an in situ hybridization study. Mol Cell Endocrinol. 1994;105:65–75. doi: 10.1016/0303-7207(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez I, Goya L, Vallerga AK, Firestone GL. Glucocorticoids reversibly arrest rat hepatoma cell growth by inducing an early G1 block in cell cycle progression. Cell Growth Differ. 1993;4:215–225. [PubMed] [Google Scholar]
- 41.Seckl JR. 11β-hydroxysteroid dehydrogenase in the rat brain: a novel regulator of glucocorticoid action. Front Neuroendocrinol. 1997a;18:49–99. doi: 10.1006/frne.1996.0143. [DOI] [PubMed] [Google Scholar]
- 42.Seckl JR. Glucocorticoids, feto-placental 11β-hydroxysteroid dehydrogenase type 2, and the early life origins of adult diseases. Steroids. 1997b;62:89–94. doi: 10.1016/s0039-128x(96)00165-1. [DOI] [PubMed] [Google Scholar]
- 43.Seckl JR, Olsson T. Glucocorticoid hypersecretion and the age-impaired hippocampus: cause or effect? J Endocrinol. 1997;145:201–211. doi: 10.1677/joe.0.1450201. [DOI] [PubMed] [Google Scholar]
- 44.Seckl JR, Dickson KL, Fink G. Central 5,7-dihydroxytryptamine lesions decrease hippocampal glucocorticoid and mineralocorticoid receptor messenger ribonucleic acid expression. J Neuroendocrinol. 1990;2:911–916. doi: 10.1111/j.1365-2826.1990.tb00659.x. [DOI] [PubMed] [Google Scholar]
- 45.Slotkin TA, Barnes GA, McCook EC, Seidler FJ. Programming of brainstem serotonin transporter development by prenatal glucocorticoids. Dev Brain Res. 1996;93:155–161. doi: 10.1016/0165-3806(96)00027-2. [DOI] [PubMed] [Google Scholar]
- 46.Stewart PM, Murry BA, Mason JI. Type 2 11β-hydroxysteroid dehydrogenase in human tissues. J Clin Endocrinol Metab. 1994;78:1529–1532. doi: 10.1210/jcem.78.6.8200959. [DOI] [PubMed] [Google Scholar]
- 47.Vintermyr OK, Doskeland SO. Characterization of inhibitory effect of glucocorticoids on the DNA replication of adult rat hepatocytes growing at various cell densities. J Cell Physiol. 1989;138:29–37. doi: 10.1002/jcp.1041380106. [DOI] [PubMed] [Google Scholar]
- 48.Voice MW, Seckl JR, Edwards CRW, Chapman KE. 11β-Hydroxysteroid dehydrogenase Type I expression in 2S FAZA hepatoma cells is hormonally regulated; a model system for the study of hepatic glucocorticoid metabolism. Biochem J. 1996;317:621–625. doi: 10.1042/bj3170621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weinstock M. Does prenatal stress impair coping and regulation of hypothalamic-pituitary-adrenal axis? Neurosci Behav Rev. 1997;21:1–10. doi: 10.1016/s0149-7634(96)00014-0. [DOI] [PubMed] [Google Scholar]
- 50.White PC, Mune T, Agarwal AK. 11β-hydroxysteroid dehydrogenase and the syndrome of apparent mineralocorticoid excess. Endocr Rev. 1997;18:135–156. doi: 10.1210/edrv.18.1.0288. [DOI] [PubMed] [Google Scholar]
- 51.Wilson RC, Harbison MD, Krowski ZS, Funder JW, Shackleton CHL, HanauskeAbel HM, Wei JQ, Hertecant J, Moran A, Neiberger RE, Balfe JW, Fattah A, Daneman D, Licholai T, New MI. Several homozygous mutations in the gene for 11beta-hydroxysteroid dehydrogenase type 2 in patients with apparent mineralocorticoid excess. J Clin Endocrinol Metab. 1995;80:3145–3150. doi: 10.1210/jcem.80.11.7593417. [DOI] [PubMed] [Google Scholar]
- 52.Yang WH, Yang WP, Lin LL. Interruption of pregnancy in the rat by administration of ACTH. Endocrinology. 1969;84:1282–1285. doi: 10.1210/endo-84-5-1282. [DOI] [PubMed] [Google Scholar]
- 53.Yi SY, Masters JN, Baram TZ. Glucocorticoid receptor mRNA ontogeny in fetal and postnatal rat forebrain. Mol Cell Neurosci. 1994;5:385–393. doi: 10.1006/mcne.1994.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zarrow MX, Philpott JE, Denenberg VH. Passage of 14C-4-corticosterone from the rat mother to the fetus and neonate. Nature. 1970;226:1058–1059. doi: 10.1038/2261058a0. [DOI] [PubMed] [Google Scholar]