

Abstract
In an attempt to elucidate the pathological implications of intracellular accumulation of the amyloid precursor protein (APP) in postmitotic neurons in vivo, we transferred APP695 cDNA into rat hippocampal neurons by using a replication-defective adenovirus vector. We first improved the efficiency of adenovirus-mediated gene transfer into neurons in vivoby using hypertonic mannitol. When a β-galactosidase-expressing recombinant adenovirus suspended in 1 m mannitol was injected into a dorsal hippocampal region, a number of neurons in remote areas were positively stained, presumably owing to increased retrograde transport of the virus. When an APP695-expressing adenovirus was injected into the same site, part of the infected neurons in the hippocampal formation underwent severe degeneration in a few days, whereas astrocytes near the injection site showed no apparent degeneration. These degenerating neurons accumulated different epitopes of APP, and β/A4 protein (Aβ)-immunoreactive materials were undetected in the extracellular space. A small number of degenerating neurons showed nuclear DNA fragmentation. Electron microscopic examinations demonstrated that degenerating neurons had shrunken perikarya along with synaptic abnormalities. Microglial cells/macrophages were often found in close proximity to degenerating neurons, and in some cases they phagocytosed these neurons. These results suggest that intracellular accumulation of wild-type APP695 causes a specific type of neuronal degeneration in vivoin the absence of extracellular Aβ deposition.
Keywords: Alzheimer’s disease, amyloid precursor protein, neurodegeneration, apoptosis, microglia, synapse, hippocampus, hypertonic mannitol, adenovirus vector
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by massive amounts of neuronal death associated with prominent histopathological features such as extracellular deposition of amyloid fibrils and accumulation of intracellular neurofibrillary tangles. The principal component of the extracellular amyloid fibrils is β/A4 protein (Aβ) (Glenner and Wong, 1984), which is derived from the amyloid precursor protein (APP) (Kang et al., 1987). APP is thought to be a membrane-associated protein, but the extracellular domain is secreted after proteolytic cleavage in the interior of Aβ (Esch et al., 1990). Differentiated postmitotic neurons express abundant APP mRNA, especially APP695 mRNA (Yoshikawa et al., 1990), but do not process significant amounts by secretory cleavage (Hung et al., 1992). APP is transported to synaptic sites by the fast anterograde component (Koo et al., 1990). Although the physiological implications of APP in neurons have not yet been fully elucidated, it is inferred that pathological accumulations of APP within neurons cause physiological functions such as synaptic transmission and signal transduction to deteriorate.
Previous histopathological studies have demonstrated that APP-immunoreactive materials are accumulated within neurons in the brain affected by AD (Benowitz et al., 1989; Cole et al., 1991;Cummings et al., 1992). These observations suggest that intracellular accumulation of APP is related to the pathogenesis of AD. However, it remains unclear whether degenerating neurons in AD brain accumulate APP abnormally or conversely whether intracellular APP accumulations cause neuronal degeneration. We have previously demonstrated that overexpression of full-length APP induces degeneration in vitro of postmitotic neurons derived from embryonal carcinoma cells (Yoshikawa et al., 1992). These neurons intracellularly accumulate APP C-terminal fragments, which are also toxic to glioma-derived cells (Hayashi et al., 1992). Moreover, an APP C-terminal 100 amino acid residue fragment, which includes the entire Aβ domain, forms amyloid fibril-like structures within transfected cells (Maruyama et al., 1990). These observations prompted us to examine whether overexpression of wild-type APP induces cellular degeneration in vivo in the brain of experimental animal models.
Replication-defective adenovirus vectors have been used to transfer foreign genes directly into the brain parenchyma (Akli et al., 1993;Bajocchi et al., 1993; Davidson et al., 1993; Le Gal La Salle et al., 1993). In these experiments, various cell types such as vascular endothelial cells, glial cells, and neurons are infected with recombinant adenoviruses. Here we demonstrate, using an improved method for in vivo adenovirus-mediated gene transfer, that overexpression of APP695 induces rapid degeneration of neurons in vivo. APP-immunoreactive materials were accumulated within the degenerating neurons, and Aβ immunoreactivity was undetected in the extracellular space, suggesting that neurons in vivo are vulnerable to intracellular accumulation of APP in the absence of extracellular Aβ deposition.
MATERIALS AND METHODS
Cosmid construction. Recombinant adenoviruses for expression of LacZ and APP 695 were constructed using cassette cosmids pAxcw and pAxCAwt, respectively (Miyake et al., 1996). LacZ transcription unit driven by the CAG promoter (Niwa et al., 1991) was inserted into pAxcw at the SwaI site (designated pAxCALacZ). Full-length cDNA of human APP 695 (Kang et al., 1987; Yoshikawa et al., 1992) was blunt-ended and inserted into pAxCAwt at the SwaI site (designated pAxCAYAP), so that the inserted cDNA is transcribed under the control of CAG promoter. The recombinant viruses were prepared according to the method described previously (Miyake et al., 1996). Briefly, cosmid DNA was co-transfected with the EcoT221-digested DNA-terminal protein complex of Ad5-dlX into 293 cells to generate the recombinant viruses by homologous recombination. The recombinant viruses, designated AxCALacZ (for LacZ expression) and AxCAYAP (for APP expression), were propagated in 293 cells. After the third propagation, virions were extracted from 293 cells, purified by double cesium step-gradient purification (Kanegae et al., 1994), dialyzed against a vehicle solution containing 10% glycerol in PBS, pH 7.4, and stored at −80°C. The titers of recombinant viruses were determined by the modified end-point cytopathic effect assay on 293 cells (Kanegae et al., 1994) and expressed in plaque-forming units (pfu). Positive expression of the inserted gene product was confirmed by immunohistochemical detection using COS-1 cells or NIH 3T3 cells. Experiments using recombinant adenovirus were approved by the Recombinant DNA Committee of the Osaka University and performed according to institutional guidelines.
Gene delivery to the hippocampus and tissue preparation. One hundred forty-four male Wistar rats (Nippon SLC, Shizuoka, Japan) weighing 220–250 gm were used: 46 rats for β-galactosidase (β-gal) expression and 98 rats for APP expression. The rats were anesthetized with sodium pentobarbital and secured on a stereotactic platform (Narishige, Tokyo). Using sterile techniques, we exposed the skull and made a 2 mm burr hole. Through the burr hole, a fine glass micropipette attached to a 5 μl Hamilton microsyringe was unilaterally introduced into the dorsal region of the left hippocampus according to the brain atlas of Paxinos and Watson (1986) (stereotactic coordinates: anterior, 4.5 mm caudal to bregma; lateral, 2.7 mm left lateral to midline; ventral, 3.2 mm ventral to dural surface at toothbar setting at ∼1–2 mm below the interaural line). Five microliters of AxCALacZ or AxCAYAP suspended in 1 m mannitol solution diluted in PBS were administered over a 10 min period. The adenovirus-injected rats showed neither apparent abnormal behaviors nor seizures.
Histochemistry and immunohistochemistry. Virus-infected rats were anesthetized deeply and fixed by intracardiac perfusion with 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4. The brains were then removed and post-fixed with the same fixative overnight. After cryoprotection with 20% sucrose in 0.1 mPB, horizontal 30-μm-thick sections were prepared and used for staining. For histochemistry for β-gal, cryosections were washed with PBS and stained by immersion in 5 mmK3Fe(CN)6, 5 mmK4Fe(CN)6, 2 mmMgCl2, 0.01% sodium deoxycholate, 0.02% Nonidet P-40, and 2 mg/ml 5-bromo-4-chloro-3-indolyl-β-galactoside (X-gal) in PBS at 37°C overnight. Sections were then washed with PBS and placed onto gelatin-coated slides. The sections were counterstained with eosin and mounted with mounting media (Mount-Quick; Daido Sangyo, Tokyo) after dehydration through a graded series of ethanol. For immunohistochemistry, cryosections were incubated at 4°C for 2 d with the following antibodies in PBS containing 0.1% Triton-X: rabbit anti-β-gal serum (1:1000) (5 Prime→3 Prime, West Chester, PA), rabbit antiserum against the C-terminal 25 amino acid residues of APP (671–695) (AC-1) (Yoshikawa et al., 1992) (1:500), rabbit antiserum against the Aβ (1–24) amino acid residues (Rb758) (Ishii et al., 1989) (1:500), mouse monoclonal anti-NeuN antibody (A60) (Mullen et al., 1992) (1:50), mouse monoclonal antibody against glial fibrillary acidic protein (GFAP) (1:5000), and mouse monoclonal antibody against the amino terminus of APP (P2-1) (Van Nostrand et al., 1989) (1:500). After the sections were rinsed thoroughly, they were incubated at 4°C overnight with rhodamine B- or fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit IgG (1:200) (Tago, Burlingame, CA) for rabbit antibodies and with FITC- or rhodamine B-conjugated goat anti-mouse IgG (1:200) (Tago) for mouse antibodies. Immunoreactivities were visualized with a fluorescence microscope (BX 50– 34-FLAD 1, Olympus, Tokyo) or with a confocal laser scanning microscope (LSM-GB200, Olympus). To identify microglial cells, the cryosections were stained with 20 μg/ml Griffonia simplicifolialectin-FITC conjugate (Sigma, St. Louis, MO) in PBS containing 0.1% Triton X-100 at room temperature for 2 hr (Streit, 1990). To examine the association of microglia and APP (or β-gal)-overexpressing neurons, the sections were doubly stained for Griffonialectin and APP (or β-gal) immunoreactivity.
Western blot analysis. Four days after viral inoculation, the bilateral hippocampi were removed from the rat. The gross regions (∼10 mg wet weight) were dissected from the injection site, and two control areas: rostral areas of the ipsilateral (distant from the injection site) and contralateral hippocampi. The tissues were homogenized, sonicated for 30 sec in a lysis buffer (0.5% Nonidet P-40, 0.1% sodium lauryl sulfate, 10 μmphenylmethanesulfonyl fluoride), and centrifuged at 20,000 ×g for 10 min. The samples (20 μg protein) were separated by 10% SDS polyacrylamide gels and immunoblotted with the antibodies AC-1 and P2-1. For detection of Aβ peptides, the tissue samples (20 μg protein) and synthetic Aβ1–40 (100 ng) (Sigma) were separated by a 14% Tris-Tricine SDS polyacrylamide gel and immunoblotted with the antibody Rb758 (Hayashi et al., 1992).
Quantification of the degenerating neurons. AxCALacZ or AxCAYAP (each 3.7 × 107 pfu/5 μl) was injected into the dorsal hippocampus. Intrahippocampal regions (the subiculum, CA3 pyramidal cell layer, hilus of the dentate gyrus, and granule cell layer of the dentate gyrus) distant from the injection site were selected for the analysis. Four days after viral inoculation, degenerating neurons among β-gal- and APP-immunopositive neurons (>20 immunopositive cells) in each region were counted. The degenerating neurons were identified with those showing at least one of the following morphological abnormalities: irregular contour of soma, distortion or dilatation of processes, and disorganized intracellular membrane. Statistical significance of the results was assessed using Student’s t test.
DNA nick-end labeling. The nuclei of the APP-overexpressing cells were labeled by the terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick-end labeling (TUNEL) reaction according to the modified method of Gavrieli et al. (1992). Briefly, after treatment with 0.3% H2O2 in methanol for 30 min, they were incubated with 100 U/ml TdT (Takara, Tokyo) and 10 μm biotin-16-dUTP (Boehringer Mannheim, Mannheim, Germany) in TdT buffer (100 mm sodium cacodylate, pH 7.0, 1 mm cobalt chloride, 50 μg/ml gelatin) at 37°C for 2 hr. DNA fragmentation was detected by the peroxidase-conjugated avidin–biotin complex method (Vector Laboratories, Burlingame, CA) using 3, 3′-diaminobenzidine tetrahydrochloride (DAB), and the reaction was enhanced with sulfate nickel ammonium. After TUNEL reaction, the sections infected with AxCAYAP and AxCALacZ were immunostained with the antibody AC-1 and anti-β-gal serum, respectively, as described above. The FITC fluorescence for APP (or β-gal) immunoreactivity and the peroxidase-stained DNA fragmentation in the same fields were visualized with the fluorescence microscope.
Electron microscopy. Virus-infected rats were anesthetized deeply and fixed by intracardiac perfusion with 2% paraformaldehyde, 2% glutaraldehyde in 0.1 m PB. The brain tissues were cut into ∼1-mm-thick blocks, including the left hippocampal formation. The specimens were then post-fixed with 2% osmium tetroxide in 0.1m PB, dehydrated with a graded series of ethanol, and embedded in Epon 812. Degenerating neurons were identified on semithin sections stained with 1% toluidine blue, and then ultrathin sections containing these neurons were cut with an ultramicrotome (Ultracut N, Reichert-Nissei, Tokyo) and mounted on copper grids. After they were stained with a saturated aqueous solution of uranyl acetate and lead citrate, the sections were observed with an electron microscope (H-7100, Hitachi, Tokyo).
RESULTS
Efficient adenovirus-mediated gene transfer into in vivo neurons
We injected the recombinant adenovirus containing the LacZ cDNA (AxCALacZ) (6.3 × 107 pfu/5 μl) suspended in PBS into the parenchyma of the rat cerebral cortex. We found that vascular endothelial cells and glia-like cells were efficiently infected, but only a few neurons near the injection site were labeled (data not shown). Thus, we inferred that these non-neuronal cells, like those consisting of the blood–brain barrier (BBB), prevent the virus from entering neurons. In an attempt to increase the viral accessibility to neurons, we used hypertonic mannitol, which is often applied to the transient opening of the BBB by intracarotid administration (Muldoon et al., 1995). For determination of infected neurons, the hippocampal formation was used as a model system, because neuronal types and their connections in this structure are well defined. Moreover, the hippocampal formation is one of the regions in which neurons are severely affected by AD (Esiri et al., 1997).
The adenovirus AxCALacZ suspended in 1 m mannitol was stereotactically injected into a left dorsal hippocampus, and the tissues were examined 4 d after viral inoculation. When AxCALacZ-infected cells in the horizontal sections were visualized by X-gal histochemistry, the majority of intensely stained cells were neuron-like cells in the granule cell layer of the dentate gyrus (Fig.1A). Moreover, a number of cells in remote areas such as the Ammon’s horn (CA) 3 region (Fig.1C), perforant pathway (Fig. 1E), and ipsilateral entorhinal cortex (Fig. 1G) were intensely stained. On the other hand, only a few cells in these regions were positively stained when injected with isotonic PBS (Fig.1B,D,F,H). The infected cells were then characterized by double immunostaining for β-gal and NeuN, a neuronal nuclear marker (Mullen et al., 1992) (Fig.2). In the hilus of the dentate gyrus, all of the β-gal-immunopositive cells were neurons with NeuN-immunoreactive nuclei, and no β-gal-immunopositive glia-like cells were detected (Fig. 2A,B). Moreover, a group of the NeuN-immunoreactive neurons in the subiculum and CA3 regions of the ipsilateral hippocampus were also infected (data not shown). β-gal-immunopositive neurons in intrahippocampal regions were morphologically intact. In the ipsilateral entorhinal cortex, a group of medium- to large-sized NeuN-immunoreactive neurons in layers II and III were immunopositive for β-gal, whereas small neurons in deep layers were hardly stained (Fig. 2C,D). Neurons in the layer II project their axons into the dentate gyrus via the perforant pathway, which was also intensely stained (Fig.1E). Therefore, it is inferred that this specific group of neurons in the ipsilateral entorhinal cortex takes up the virions at the injection site and transports them to the somata in a retrograde manner.
Fig. 1.
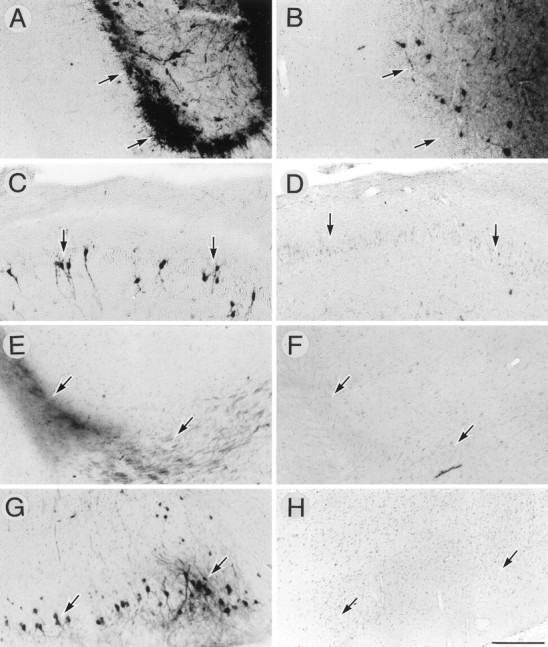
Enhancement of adenovirus infectivity in vivo by hypertonic mannitol. AxCALacZ (6.3 × 107 pfu/5 μl) suspended in 1 mmannitol (A, C, E, G) or in isotonic PBS (B, D, F, H) was stereotactically injected into the left dorsal hippocampus. Five days later, infected cells were histochemically detected for β-gal activity (X-gal staining). A, B, The dentate gyrus of the hippocampus; C, D, the pyramidal cell layer of the CA3 region; E, F, the perforant pathway; G, H, the ipsilateral entorhinal cortex (layer II). Arrows point to the regions above for orientation. Scale bar (shown in H forA–H): 200 μm.
Fig. 2.
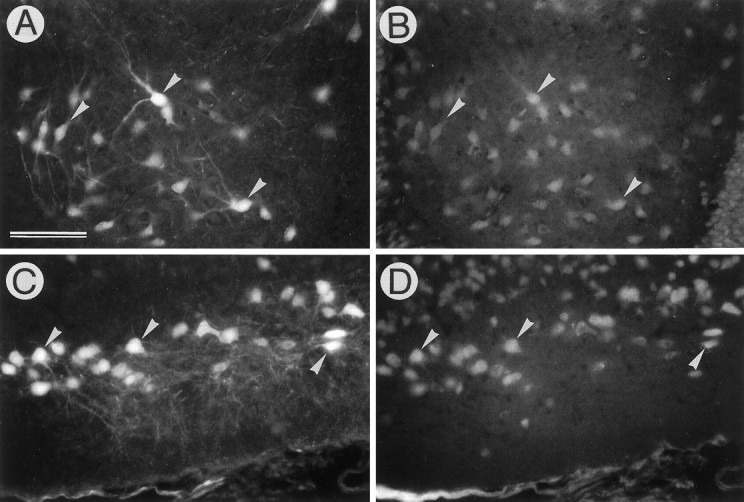
Identification of infected cells as neurons. AxCALacZ (6.3 × 107 pfu/5 μl) suspended in 1m mannitol was injected into the left dorsal hippocampus, and infected cells were stained by fluorescent immunohistochemistry for β-gal (A, C) and NeuN (B, D). A,B, The hilus of the dentate gyrus. All of the β-gal-immunopositive cells possess NeuN-immunoreactive nuclei;C, D, the ipsilateral entorhinal cortex. Note that many neurons in superficial layers (layers II and III) are infected. Arrowheads inA–D point to representative neurons expressing both exogenous β-gal (A, C) and endogenous NeuN (B, D). Scale bar (shown in A for A–D): 100 μm.
To examine whether the increased infectivity is dependent on the tonicity of the vehicle solution, we quantified the infected neurons in serial 30-μm-thick sections of the ipsilateral entorhinal cortex after injecting AxCALacZ with isotonic PBS, 0.2 m mannitol, 1 m mannitol, and 1 m sucrose. The maximal numbers of infected neurons were more than 70 per section in 1m mannitol-treated and 1 m sucrose-treated samples, whereas a few neurons (less than five per section) were infected with the virus suspended in isotonic PBS and 0.2 mmannitol (data not shown). These findings indicate that hypertonic solutions increase the efficiency of neuronal infection.
Adenovirus-mediated transfer of APP cDNA into the hippocampus
Using the modification with hypertonic mannitol, we transferred the recombinant adenovirus carrying cDNA encoding human APP 695 (AxCAYAP), which is abundantly expressed in postmitotic neurons (Yoshikawa et al., 1992), into hippocampal neurons in vivo. We first analyzed exogenous APP expression in the infected region by Western blotting (Fig. 3). A gross region, including the injection site, contained higher levels of ∼110 kDa APP-immunoreactive molecules than those of control regions, as detected with an antibody against APP C-terminus (Fig. 3A,APP-C). Antibody P2-1, a monoclonal antibody raised against native human APP (nexin-2) (Van Nostrand et al., 1989), reacted with exogenous human APP expressed at the injection site but not with endogenous rat APP (Fig. 3A, APP-N). APP C-terminus-immunoreactive degraded fragments, which were generated in APP-overexpressing P19 cells (Yoshikawa et al., 1992), were hardly detected in vivo 4 d after viral infection. In addition, ∼4 kDa Aβ immunoreactive materials were undetected in the AxCAYAP-infected region (Fig. 3B, Aβ). These results suggest that AxCAYAP-infected cells contain human APP695 as a full-length form without being processed further into smaller fragments.
Fig. 3.

Western blot analysis of APP and Aβ expressed in AxCAYAP-infected hippocampal regions. AxCAYAP suspended in 1m mannitol was stereotactically injected into the dorsal hippocampus. Four days later, gross regions including the injection site and distant regions (as controls) were dissected. APP was detected by Western blotting with the antibodies AC-1 (APP-C), P2-1 (APP-N), and Rb758 (Aβ).R, A rostral region of the ipsilateral hippocampus;I, a dorsal hippocampal region including the injection site; C, a contralateral dorsal hippocampal region;S, synthetic Aβ1–40 standard (100 ng). Molecular weight markers are on the left. Arrowsindicate predicted positions of full-length APP and Aβ1–40.
We then infected equal amounts of AxCAYAP and AxCALacZ (each 2.4 × 107 pfu/5 μl) into the dorsal hippocampus and examined the infected tissues by immunohistochemistry. Some neurons in the hilus of the dentate gyrus showed β-gal immunoreactivity (Fig.4A), whereas very few neurons possessed APP immunoreactivity (Fig. 4B). On the other hand, many glia-like cells near the injection site contained large amounts of both β-gal and APP-immunoreactive materials (Fig.4A,B). This discrepancy may be because exogenous APP in the infected neurons is metabolized more intensively than β-gal, whereas both APP and β-gal are slowly metabolized in the infected glial cells. When a larger amount of AxCAYAP (3.7 × 107 pfu/5 μl) was injected, intensely APP-immunoreactive cells were detected at the stratum radiatum (Fig.4C). In this region, APP-immunopositive neuron-like cells appeared to be degenerating, whereas APP-immunopositive glia-like cells were apparently intact. A number of APP-immunoreactive neurons were found in the ipsilateral entorhinal cortex, but they showed little or no degeneration (Fig. 4D). The subiculum of the hippocampus contained some degenerating neurons, as identified by double immunostaining for APP and NeuN (Fig.4E,F). Degenerating neurons were consistently found in intrahippocampal regions, including the stratum radiatum, stratum lucidum, hilus and granule cell layer of the dentate gyrus, subiculum, and CA3 when infected with AxCAYAP (3.7 × 107 pfu/5 μl) (data not shown). On the other hand, APP-accumulating astrocytes, identified as GFAP-immunopositive cells, near the injection site showed no apparent degenerative changes (Fig. 4G,H). In the following experiments, the amount of 3.7 × 107 pfu/5 μl of AxCAYAP was used.
Fig. 4.
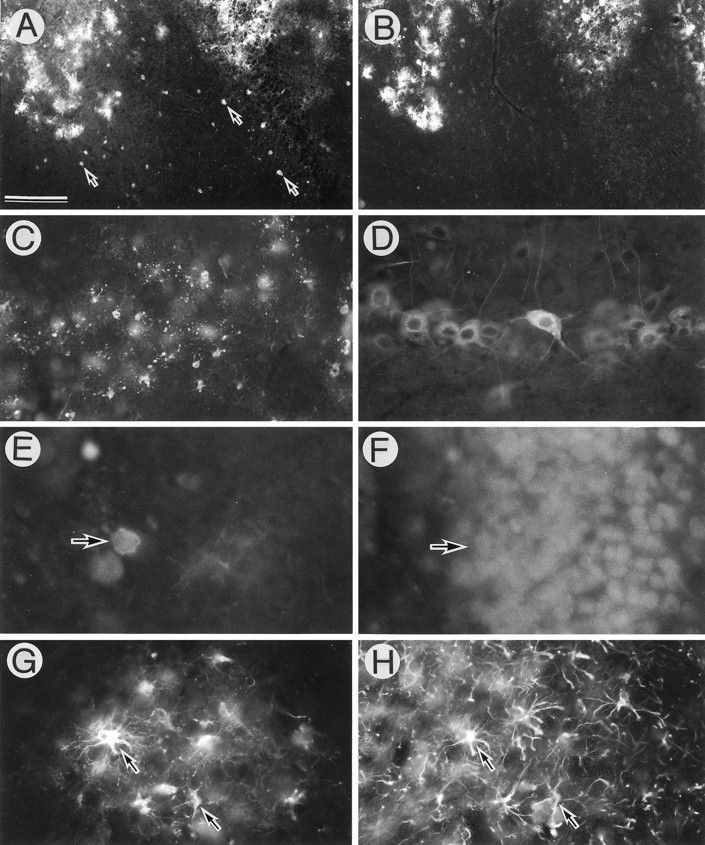
APP-accumulating cells in adenovirus-infected brain regions. AxCAYAP plus AxCALacZ (each 2.4 × 107 pfu/5 μl) (A, B), or AxCAYAP (3.7 × 107 pfu/5 μl) (C–H) suspended in 1 m mannitol was stereotactically injected into the dorsal hippocampus. Four days later, immunoreactivities of β-gal (A), APP C terminus (B–E, G), NeuN (F), and GFAP(H) were examined.A (β-gal) and B (APP), the hilus of the dentate gyrus (adjacent sections); C (APP), the stratum radiatum; D (APP), the ipsilateral entorhinal cortex;E (APP) and F (NeuN), the subiculum of the hippocampus; G (APP) and H (GFAP), the dentate gyrus. Arrows in A point to representative β-gal-immunopositive neurons. Arrows inE and F indicate the APP-immunoreactive degenerating neuron. Arrows in G andH point to APP-accumulating astrocytes. Scale bar (shown in A): A, B, 200 μm;C, 100 μm; D–H, 50 μm.
When morphological changes of APP-accumulating neurons in the hippocampus were examined 5 d after AxCAYAP injection, many neurons contained varying amounts of intracellular APP-immunoreactive materials. Some degenerating neurons with irregular contours had APP-immunoreactive granules in both the perikarya and the dilated processes (Fig. 5A,B). Moreover, disorganized APP-immunoreactive membranes (Fig.5C) and “ghost-like” depositions of APP-immunoreactive granules were detected (Fig. 5D). Such severely degenerating neurons were undetected in the tissues infected with the same amount of LacZ-carrying adenovirus (Fig. 5F). When the infected tissue samples were examined on days 5, 10, 15, 20, and 30 after viral infection, APP-accumulating neurons in the hilus disappeared on day 15 or later (data not shown), and only weakly APP-immunoreactive cells, presumably glial cells, were found in the dentate gyrus on day 15 (Fig.5E). These findings raise the possibility that intracellular accumulations of APP induce rapid neuronal death without leaving APP-immunoreactive debris in vivo. To examine the specificity of APP-induced neurodegeneration, we quantified the degenerating neurons in the hippocampus infected with AxCAYAP or AxCALacZ (Table 1). The hippocampal regions distant from the injection site were selected to avoid nonspecific neurodegeneration caused by tissue damage. In each region tested, the number of degenerating APP-immunoreactive neurons was significantly larger than that of degenerating β-gal-immunoreactive neurons. In the dentate gyrus near the injection site, a larger number of β-gal-immunopositive neurons showed degenerative changes, but the APP-induced neurodegeneration was significantly frequent. In this analysis, degrees of degeneration of AxCAYAP-infected neurons were much greater than those of AxCALacZ-infected neurons (data not shown). These results suggest that AxCAYAP-induced neurodegeneration is caused by overexpression of APP and not by nonspecific neurotoxicity of adenovirus.
Fig. 5.
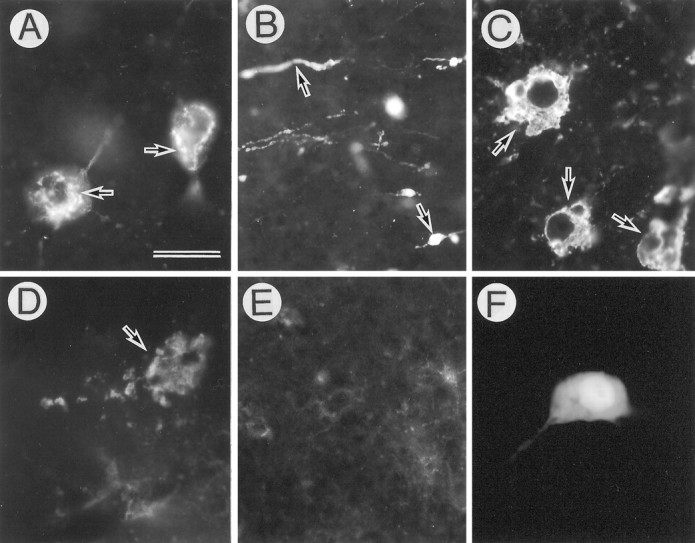
Degeneration of APP-accumulating hippocampal neurons. AxCAYAP was injected into the dorsal hippocampus. Tissues were stained with antibody AC-1 (A–E) and anti-β-gal antibody (F) on day 4 (A–D, F) and day 15 (E). The regions examined are A, C, D, F, the hilus of the dentate gyrus; B, the stratum lucidum; E, the granule cell layer of the dentate gyrus. Highly APP-immunoreactive materials are detected in the perikarya (arrows in A), dilated neurites (arrows in B), deformed intracellular membranes (arrows in C), and amorphous extracellular depositions (arrow inD). Very weak APP-immunoreactive materials are detected in the dentate gyrus on day 15 (E). Most of the β-gal-immunopositive neurons show no apparent degeneration (F). Scale bar (shown inA): A, C, D, F, 20 μm; B, E, 50 μm.
Table 1.
Quantification of degenerating neurons in the hippocampus
| Hippocampal region | Degenerating neurons (%) | |
|---|---|---|
| AxCALacZ (n = 4) | AxCAYAP (n = 3) | |
| Subiculum | 11.5 ± 0.9 | 51.4 ± 9.1* |
| CA3 | 4.8 ± 2.2 | 50.1 ± 5.1* |
| Hilus | 11.7 ± 0.7 | 59.8 ± 2.6* |
| Dentate gyrus | 17.6 ± 3.0 | 44.9 ± 2.2* |
AxCALacZ or AxCAYAP (each 3.7 × 107 pfu/5 μl) was injected into the dorsal hippocampus. Four days later, degenerating neurons among β-gal- and APP-immunopositive neurons (>20 immunopositive cells) in each region were counted. Degenerating neurons were assessed according to the criteria described in Materials and Methods. Each value represents the mean ± SEM.
* Significantly higher (p < 0.005) than the AxCALacZ value by Student’s t test.
We immunostained these degenerating neurons with antibodies against different epitopes of APP. Confocal laser microscopy revealed that the N- and C-terminal epitopes of APP showed closely similar distribution patterns (Fig.6A,B). This, together with the data of Western blot analysis (Fig. 3), suggests that these degenerating neurons contain a full-length form of APP695. Degenerating neurons were then doubly stained for APP N-terminal and Aβ1–24 epitopes. Aβ immunoreactivity was detected in degenerating neurons with intense APP N-terminus-immunoreactivity, but some APP-immunopositive cells contained no Aβ-immunoreactive materials (Fig. 6C,D). In the extracellular space adjacent to APP-immunopositive degenerating neurons, Aβ-immunoreactive materials were undetected.
Fig. 6.
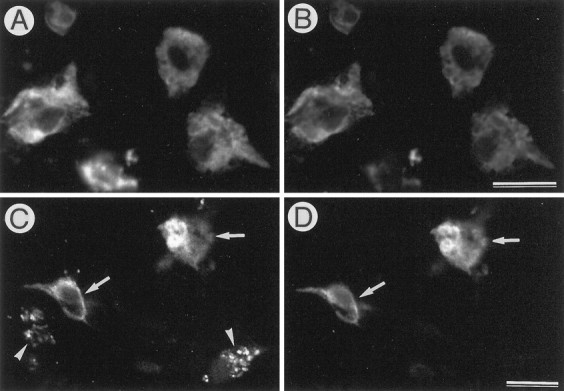
Distribution of different epitopes of APP in AxCAYAP-infected tissues. AxCAYAP was injected into the dorsal hippocampus. Four days later, the sections of the hilus of the dentate gyrus were immunostained for the N terminus (A,C), the C terminus (B) of APP, and Aβ 1–24 (D). Fluorescent images were visualized with a confocal laser microscope. APP N and C terminus show similar distribution patterns (A, B). Note the cells containing both APP N-terminal and Aβ immunoreactivities (arrowsin C, D) and the N-terminus-immunoreactive cells lacking Aβ immunoreactivity (arrowheads). Scale bars (shown inB and D for A–D): 20 μm.
To examine whether DNA fragmentation occurs during degeneration of APP-accumulating neurons, we doubly stained the cells by the APP immunohistochemistry and TUNEL method. We found that some APP-accumulating degenerating neurons (<20%) possessed TUNEL-positive nuclei (Fig.7A–D). In contrast, all β-gal-accumulating neurons (i.e., a negative control) had TUNEL-negative nuclei (Fig. 7E,F), whereas CA1 pyramidal neurons of the gerbil hippocampus after transient ischemia (Nitatori et al., 1995) (i.e., a positive control) showed numerous TUNEL-positive nuclei (Fig. 7G). To examine whether microglial cells/macrophages are involved in scavenging process for these degenerated neurons, we doubly stained the APP-accumulating cells with the antibody against the APP C terminus and Griffonialectin, a marker for microglia (Streit, 1990) (Fig.8). Microglial cells were often found in close proximity to the APP-accumulating degenerating neurons, and some of them appeared to phagocytose these degenerating neurons (Fig.8A,B). In the tissues infected with AxCALacZ, no microglial cells associated with β-gal-immunoreactive neurons were detected (Fig. 8C,D).
Fig. 7.
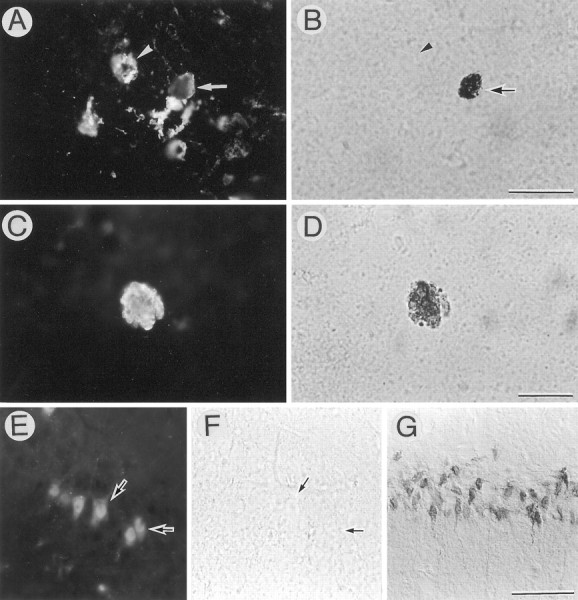
Nuclear DNA fragmentation in APP-accumulating neurons. AxCAYAP was injected into the dorsal hippocampus. Four days later, the sections of the stratum lucidum were doubly stained for APP C-terminus (A, C) and TUNEL reactivity (B, D). Note the APP-accumulating neurons with (arrow) and without (arrowhead) TUNEL-positive nuclei (A, B), and TUNEL-reactive granular materials spread throughout APP-accumulating soma (C, D). E, F (negative control), the pyramidal cell layer of CA3 region infected with AxCALacZ, doubly stained for β-gal (E) and TUNEL (F).Arrows in E and F point to β-gal-immunopositive neurons with TUNEL-negative nuclei.G (positive control), the pyramidal cell layer of CA1 region in the gerbil hippocampus after transient ischemia. The neurons possess numerous TUNEL-positive nuclei. Scale bars (shown inB for A and B): 50 μm; (shown in D for C and D), 20 μm; (shown in G for E–G), 100 μm.
Fig. 8.
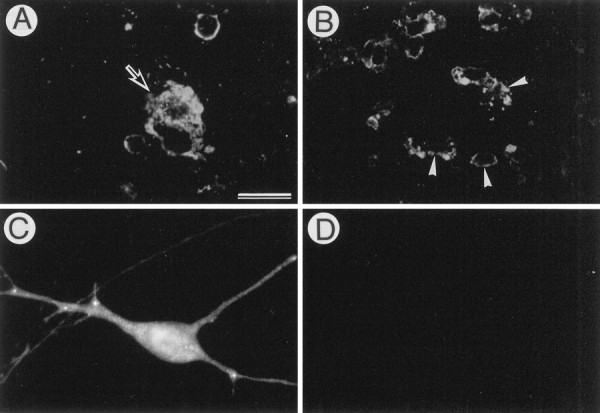
Association of microglia with APP-accumulating degenerating neurons. AxCAYAP (A, B) or AxCALacZ (C, D) was injected into the dorsal hippocampus. Four days later, cryosections were doubly labeled by fluorescent immunohistochemistry with antibody AC-1 (A) or anti-β-gal antibody (C) and the microglial marker Griffonia lectin (B, D).A–D, The hilus of the dentate gyrus.Griffonia lectin-positive microglial cells (arrowheads in B) are adjacent to the APP-accumulating degenerating neuron (arrow inA) but are absent from the vicinity of a β-gal-accumulating neuron (C, D). Scale bars (shown inA for A–D): 50 μm.
Several atrophic neurons in the CA3 region of AxCAYAP-infected hippocampus were intensely labeled by toluidine blue staining. These neurons had shrunken somata with irregular contours (Fig.9A). Electron microscopic examinations revealed that the atrophic neurons had electron-dense perikarya and deformed nuclei with a slight chromatin condensation (Fig. 9B). These neurons had moderately dilated endoplasmic reticulum (Fig. 9B–E). Some degenerating neurons had numerous clear vacuoles, multivesicular bodies, and dense bodies (Fig.9D,E), suggesting that autophasic processes are operative in these neurons (Nitatori et al., 1995). Moreover, microglial cells/phagocytes were detected in proximity to degenerating neurons (Fig. 9B) and a soma-like structure filled with numerous autophagic vacuoles (Fig. 9E). A swollen presynaptic ending (Fig. 9C) and a postsynaptic structure lacking the presynaptic element (Fig. 9D) were identified, suggesting that these synaptic abnormalities occur concomitantly with perikaryal shrinkage. These pathological features were observed in the areas containing APP-accumulating degenerating neurons (data not shown). Thus, it is likely that intracellular accumulation of wild-type APP695 causes “shrinkage-type” neuronal death along with autophagic processes and synaptic abnormalities, and that microglial cells are involved in scavenging processes of these affected neurons.
Fig. 9.
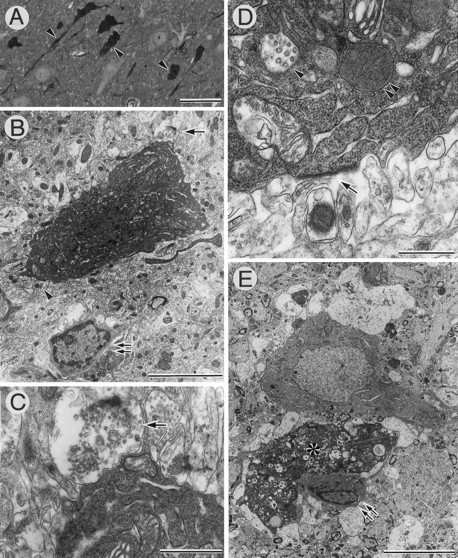
Electron micrographs of degenerating CA3 neurons of AxCAYAP-infected hippocampus. The hippocampal tissues were prepared 4 d after AxCAYAP infection and examined by electron microscopy.A, Toluidine blue staining of the CA3 region.Arrowheads point to atrophic degenerating neurons.B–E, Electron micrographs of CA3 neurons. C and D are enlarged images of the areas shown by the arrow andarrowhead in B, respectively. Note abnormalities such as electron-dense cytoplasm (B,E), numerous clear vacuoles (asterisk inE), multivesicular bodies (arrowhead inD), dense bodies (double arrowhead inD), swollen presynaptic ending (arrow inC), and postsynaptic density lacking presynaptic element (arrow in D). Microglial cells are adjacent to degenerating neurons (double arrows inB, E). Note the microglial cell extending the processes along the degenerated neuron (double arrows and asterisk in E). Scale bars: A, 50 μm; B, 5 μm;C, 1 μm; D, 500 nm; E, 10 μm.
DISCUSSION
This study has shown that hypertonic mannitol for the direct viral delivery into the hippocampal parenchyma markedly increases the number of infected neurons. The disruption of BBB with hypertonic mannitol has been previously applied to the adenovirus-mediated gene transfer via intracarotid administration, but only glia-like cells were frequently infected (Muldoon et al., 1995). After intracarotid mannitol administration, capillary endothelial cells that form BBB may shrink, and the tight junctions are temporarily opened, allowing the recombinant adenovirus to enter the perivascular space. Similarly, the direct adenovirus transfer into the brain parenchyma with hypertonic mannitol may shrink non-neuronal cells that block the viral accessibility to neurons. The retrograde transport of adenovirus has been demonstrated previously by injecting the virus into the striatum and detecting the labeled neurons in the substantia nigra (Ridoux et al., 1994). Therefore, intrahippocampal neurons in this study may be retrogradely infected, and hypertonic mannitol increases the viral accessibility to the nerve terminals, resulting in the increased retrograde transport to neuronal nuclei in which infected genes are transcribed. Because adenovirus has potential nonspecific cytotoxicities, we used the adenovirus vector carrying one of the strongest promoters currently available (Niwa et al., 1991; Miyake et al., 1996) to attain an equal effectiveness with less viral quantity. Using this vector and the hyperosmotic modification in combination, we have succeeded in demonstrating the APP-induced neurodegeneration.
Intraneuronal accumulations of APP in AD brain have been demonstrated previously using various antibodies raised against different epitopes of APP: a large number of hippocampal CA1 neurons in AD contain abnormally dense APP C-terminal immunoreactive materials as compared with controls (Benowitz et al., 1989). Hippocampal pyramidal neurons in AD display an intense immunostaining with 10 different antibodies against subsequences of APP (Cole et al., 1991). Pyramidal neurons in hippocampal fields CA1–3 and entorhinal cortex in AD brain are strongly stained with an antibody against APP N terminus (P2-1) (Cummings et al., 1992). The areas containing these APP-accumulating neurons are consistent with those showing the most intense neuropathology in AD. However, it has been unclear whether intracellular accumulation of APP is a cause of neurodegeneration seen in AD brain. Using the adenovirus-mediated APP gene transfer, we were able to demonstrate that neurons in vivo are vulnerable to the intracellular accumulations of wild-type APP. The degenerating neurons had shrunken perikarya with deformed nuclei (Fig. 9). In the nucleus basalis of Meynert complex in AD brain, cholinergic neurons become smaller (Pearson et al., 1983), and the number of small neurons in this region is significantly increased (Vogels et al., 1990). Moreover, neuron shrinkage in the nucleus raphes dorsalis in AD has also been suggested (Aletrino et al., 1992). Therefore, neuron shrinkage may be a typical pathological feature of AD. Because APP-accumulating pyramidal neurons in the hippocampus show severe atrophy in AD brain (Benowitz et al., 1989), it is tempting to speculate that intracellular accumulation of APP is responsible for neuronal shrinkage seen in AD brain. Cell shrinkage is one of the typical features of apoptosis (Kerr et al., 1987). We found that nuclear DNA fragmentation, another feature of apoptosis, occurs in some APP-accumulating neurons (Fig. 7). Previous studies have revealed that nuclear DNA fragmentation is significantly increased in neurons in AD brain (Su et al., 1994; Lassmann et al., 1995). Moreover, degenerating APP-immunopositive neurons were often accompanied by reactive microglia (Fig. 8), which are prevalent in AD brain (McGeer et al., 1993). These findings together suggest that APP-accumulating neurons, at least in part, undergo degeneration in a manner similar to apoptosis, and that a specific type of neurodegeneration induced by APP occurs in AD brain.
Transgenic mice overexpressing APP mutants such as APP (Val717Phe) (Games et al., 1995) and APP695 (Lys670Asn/Met671Leu) (Hsiao et al., 1996) have been reported to show neuropathological changes accompanied by extracellular Aβ depositions. However, no overt neuronal loss is detected in the brain regions in which Aβ is extensively deposited in APP (Val717Phe) transgenic mice (Irizarry et al., 1997), suggesting that extracellular Aβ deposits per se are not toxic to neurons. The APP mutants used in the transgenic mice may not be accumulated to toxic levels within neurons, whereas adenovirus-mediated overexpression of wild-type APP695 induces rapid APP accumulations that exert toxic effects on neurons from inside. We infer that such rapid accumulations of APP are attainable only by strong overexpression systems such as those in vitro(Hayashi et al., 1992; Yoshikawa et al., 1992) and in vivo (present study). Molecular mechanisms underlying APP-induced neurodegeneration remains to be elucidated. We have recently demonstrated, by using the same APP695 cDNA-carrying adenovirus, that overexpression of full-length APP in cultured rat hippocampal neurons enhances the glutamate-induced rise of intracellular Ca2+ concentration (Tominaga et al., 1997). Such studies using the adenovirus-mediated APP gene transfer system might provide valuable information about molecular mechanisms whereby intracellular accumulation of wild-type APP causes neurodegeneration.
Footnotes
This work was supported by Grants-in-Aid (07557332/08458253) from the Ministry of Education, Science, and Culture of Japan (K.Y.). We thank Dr. Haruo Okado for the adenovirus system, Dr. Takayuki Gotoh for electron microscopy, and Dr. Tohru Nitatori for the TUNEL-positive tissue preparation. We appreciate the generous gifts of antibodies from Dr. Richard Mullen (A60), Dr. Tsuyoshi Ishii (Rb758), and Dr. William Van Nostrand (P2-1).
Correspondence should be addressed to Dr. Kazuaki Yoshikawa, Division of Regulation of Macromolecular Functions, Institute for Protein Research, Osaka University, 3–2 Yamadaoka, Suita, Osaka 565, Japan.
REFERENCES
- 1.Aletrino MA, Vogels OJM, Van Domburg PHMF, Ten Donkelaar HJ. Cell loss in the nucleus raphes dorsalis in Alzheimer’s disease. Neurobiol Aging. 1992;13:461–468. doi: 10.1016/0197-4580(92)90073-7. [DOI] [PubMed] [Google Scholar]
- 2.Akli S, Caillaud C, Vigne E, Stratford-Perricaudet LD, Poenaru L, Perricaudet M, Kahn A, Peschanski MR. Transfer of a foreign gene into the brain using adenovirus vectors. Nat Genet. 1993;3:224–228. doi: 10.1038/ng0393-224. [DOI] [PubMed] [Google Scholar]
- 3.Bajocchi G, Feldman SH, Crystal RG, Mastrangeli A. Direct in vivo gene transfer to ependymal cells in the central nervous system using recombinant adenovirus vectors. Nat Genet. 1993;3:229–234. doi: 10.1038/ng0393-229. [DOI] [PubMed] [Google Scholar]
- 4.Benowitz LI, Rodriguez W, Paskevich P, Mufson EJ, Schenk D, Neve RL. The amyloid precursor protein is concentrated in neuronal lysosomes in normal and Alzheimer disease subjects. Exp Neurol. 1989;106:237–250. doi: 10.1016/0014-4886(89)90156-8. [DOI] [PubMed] [Google Scholar]
- 5.Cole GM, Masliah E, Shelton ER, Chan HW, Terry RD, Saitoh T. Accumulation of amyloid precursor fragment in Alzheimer plaques. Neurobiol Aging. 1991;12:85–91. doi: 10.1016/0197-4580(91)90046-m. [DOI] [PubMed] [Google Scholar]
- 6.Cummings BJ, Su JH, Geddes JW, Van Nostrand WE, Wagner SL, Cunningham DD, Cotman CW. Aggregation of the amyloid precursor protein within degenerating neurons and dystrophic neurites in Alzheimer’s disease. Neuroscience. 1992;48:763–777. doi: 10.1016/0306-4522(92)90265-4. [DOI] [PubMed] [Google Scholar]
- 7.Davidson BL, Allen ED, Kozarsky KF, Wilson JM, Roessler BJ. A model system for in vivo gene transfer into the central nervous system using an adenoviral vector. Nat Genet. 1993;3:219–223. doi: 10.1038/ng0393-219. [DOI] [PubMed] [Google Scholar]
- 8.Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, McClure D, Ward PJ. Cleavage of amyloid B peptide during constitutive processing of its precursor. Science. 1990;248:1122–1124. doi: 10.1126/science.2111583. [DOI] [PubMed] [Google Scholar]
- 9.Esiri MM, Hyman BT, Beyreuther K, Masters CL. Ageing and dementia. In: Graham DI, Lantos PL, editors. Greenfield’s neuropathology, Ed 6. Arnold; London: 1997. pp. 153–233. [Google Scholar]
- 10.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagopian S, Johnson-Wood K, Khan K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 11.Gavrieli Y, Sherman Y, Ben-Sasson AJ. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 13.Hayashi Y, Kashiwagi K, Yoshikawa K. Protease inhibitors generate cytotoxic fragments from Alzheimer amyloid protein precursor in cDNA-transfected glioma cells. Biochem Biophys Res Commun. 1992;187:1249–1255. doi: 10.1016/0006-291x(92)90437-p. [DOI] [PubMed] [Google Scholar]
- 14.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 15.Hung AY, Koo EH, Haass C, Selkoe DJ. Increased expression of β-amyloid precursor protein during neuronal differentiation is not accompanied by secretory cleavage. Proc Natl Acad Sci USA. 1992;89:9439–9443. doi: 10.1073/pnas.89.20.9439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT. Aβ deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F(PDAPP) transgenic mouse. J Neurosci. 1997;17:7053–7059. doi: 10.1523/JNEUROSCI.17-18-07053.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishii T, Kametani F, Haga S, Sato M. The immunohistochemical demonstration of subsequences of the precursor of the amyloid A4 protein in senile plaques in Alzheimer’s disease. Neuropathol Appl Neurobiol. 1989;15:135–147. doi: 10.1111/j.1365-2990.1989.tb01216.x. [DOI] [PubMed] [Google Scholar]
- 18.Kanegae Y, Makimura M, Saito I. A simple and efficient method for purification of infectious recombinant adenovirus. Jpn J Med Sci Biol. 1994;47:157–166. doi: 10.7883/yoken1952.47.157. [DOI] [PubMed] [Google Scholar]
- 19.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Müller-Hill B. The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 20.Kerr JFR, Searle J, Harmon BV, Bishop CJ. Apoptosis. In: Potten CS, editor. Perspectives on mammalian cell death. Oxford UP; New York: 1987. pp. 93–128. [Google Scholar]
- 21.Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K, Fischer P, Masters CL, Price DL. Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA. 1990;87:1561–1565. doi: 10.1073/pnas.87.4.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lassmann H, Bancher C, Breitschopf H, Wegiel J, Bobinski M, Jellinger K, Wisniewski HM. Cell death in Alzheimer’s disease evaluated by DNA fragmentation in situ. Acta Neuropathol. 1995;89:35–41. doi: 10.1007/BF00294257. [DOI] [PubMed] [Google Scholar]
- 23.Le Gal La Salle G, Robert JJ, Berrard S, Ridoux V, Stratford-Perricaudet LD, Perricaudet M, Mallet J. An adenovirus vector for gene transfer into neurons and glia in the brain. Science. 1993;259:988–990. doi: 10.1126/science.8382374. [DOI] [PubMed] [Google Scholar]
- 24.Maruyama K, Terakado K, Usami M, Yoshikawa K. Formation of amyloid-like fibrils in COS cells overexpressing part of the Alzheimer amyloid protein precursor. Nature. 1990;347:566–569. doi: 10.1038/347566a0. [DOI] [PubMed] [Google Scholar]
- 25.McGeer PL, Kawamata T, Walker DG, Akiyama H, Tooyama I, McGeer EG. Microglia in degenerative neurological disease. Glia. 1993;7:84–92. doi: 10.1002/glia.440070114. [DOI] [PubMed] [Google Scholar]
- 26.Miyake S, Makimura M, Kanegae Y, Harada S, Sato Y, Takamori K, Tokuda C, Saito I. Efficient generation of recombinant adenovirus using adenovirus DNA-terminal protein complex and a cosmid bearing the full-length virus genome. Proc Natl Acad Sci USA. 1996;93:1320–1324. doi: 10.1073/pnas.93.3.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muldoon LL, Nilaver G, Kroll RA, Pagel MA, Breakefield XO, Chiocca EA, Davidson BL, Weissleder R, Neuwelt EA. Comparison of intracerebral inoculation and osmotic blood-brain barrier disruption for delivery of adenovirus, herpesvirus, and iron oxide particles to normal rat brain. Am J Pathol. 1995;147:1840–1851. [PMC free article] [PubMed] [Google Scholar]
- 28.Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- 29.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–200. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 31.Paxinos G, Watson C. The rat brain in stereotaxic coordinates, Ed 2. Academic; Sydney: 1986. [DOI] [PubMed] [Google Scholar]
- 32.Pearson RCA, Sofroniew MV, Cuello AC, Powell TPS, Eckenstein F, Esiri MM, Wilcock GK. Persistence of cholinergic neurons in the basal nucleus in a brain with senile dementia of the Alzheimer’s type demonstrated by immunohistochemical staining for choline acetyltransferase. Brain Res. 1983;289:375–379. doi: 10.1016/0006-8993(83)90046-x. [DOI] [PubMed] [Google Scholar]
- 33.Ridoux V, Robert JJ, Zhang X, Perricaudet M, Mallet J, Le Gal La Salle G. Adenoviral vectors as functional retrograde neuronal tracers. Brain Res. 1994;648:171–175. doi: 10.1016/0006-8993(94)91919-4. [DOI] [PubMed] [Google Scholar]
- 34.Streit WJ. An improved staining method for rat microglial cells using the lectin from Griffonia simplicifolia (GSAI-B4). J Histochem Cytochem. 1990;38:1683–1687. doi: 10.1177/38.11.2212623. [DOI] [PubMed] [Google Scholar]
- 35.Su JH, Anderson AJ, Cummings BJ, Cotman CW. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. NeuroReport. 1994;5:2529–2533. doi: 10.1097/00001756-199412000-00031. [DOI] [PubMed] [Google Scholar]
- 36.Tominaga K, Uetsuki T, Ogura A, Yoshikawa K. Glutamate responsiveness enhanced in neurones expressing amyloid precursor protein. NeuroReport. 1997;8:2067–2072. doi: 10.1097/00001756-199705260-00051. [DOI] [PubMed] [Google Scholar]
- 37.Van Nostrand WE, Wagner SL, Suzuki M, Choi BH, Farrow JS, Geddes JW, Cotman CW, Cunningham DD. Protease nexin-II, a potent antichymotrypsin, shows identity to amyloid β-protein precursor. Nature. 1989;341:546–549. doi: 10.1038/341546a0. [DOI] [PubMed] [Google Scholar]
- 38.Vogels OJM, Broere CAJ, Ter Laak HJ, Ten Donkelaar HJ, Nieuwenhuys R, Schulte BPM. Cell loss and shrinkage in the nucleus basalis Meynert complex in Alzheimer’s disease. Neurobiol Aging. 1990;11:3–13. doi: 10.1016/0197-4580(90)90056-6. [DOI] [PubMed] [Google Scholar]
- 39.Yoshikawa K, Aizawa T, Maruyama K. Neural differentiation increases expression of Alzheimer amyloid protein precursor gene in murine embryonal carcinoma cells. Biochem Biophys Res Commun. 1990;171:204–209. doi: 10.1016/0006-291x(90)91377-5. [DOI] [PubMed] [Google Scholar]
- 40.Yoshikawa K, Aizawa T, Hayashi Y. Degeneration in vitro of post-mitotic neurons overexpressing the Alzheimer amyloid protein precursor. Nature. 1992;359:64–67. doi: 10.1038/359064a0. [DOI] [PubMed] [Google Scholar]