

Abstract
Soman (pinacolymethylphosphonofluoridate), a highly potent irreversible inhibitor of acetylcholinesterase (AChE), causes seizures and rapidly increases Fos and glial fibrillary acidic protein (GFAP) staining in piriform cortex (PC). This suggests that the inhibition of AChE by soman leads to increased acetylcholine (ACh) and neuronal excitability in PC. The sole source of cholinergic input to PC is from the nucleus of the diagonal band (NDB). To investigate the role of ACh in soman-induced seizures, we lesioned cholinergic neurons in NDB unilaterally with 192-IgG-saporin. By 10 d, saporin eliminated staining for choline acetyltransferase (ChAT), the synthetic enzyme for ACh, in NDB ipsilateral to the lesion. Staining for AChE, the degradative enzyme for ACh, was eliminated in PC ipsilateral to the lesioned NDB. By 45–60 min after soman, increased Fos and GFAP staining in PC was evident only ipsilateral to the unlesioned NDB. By 90–120 min after soman, Fos and GFAP staining increased bilaterally in PC.
In a second experiment, electrical stimulation electrodes were implanted unilaterally in the NDB to activate focally the projections to PC in unanesthetized rats. Within 5 min of NDB stimulation, there were clear behavioral and EEG signs of convulsions. After 45–60 min of NDB stimulation, there was increased Fos and GFAP staining in layer II of PC ipsilateral to the stimulation site. Pretreatment with the selective muscarinic receptor antagonist scopolamine blocked the convulsions and prevented increased Fos and GFAP staining in PC. These results suggest that ACh release in PC triggers the initiation of seizures and gliosis after soman administration, predominantly by the activation of muscarinic receptors.
Keywords: soman, Fos, GFAP, muscarinic receptors, nucleus of the diagonal band (NDB), 192-IgG-saporin, choline acetyltransferase (ChAT)
Soman is a “nerve gas.” It is a highly potent irreversible inhibitor of acetylcholinesterase (AChE), the enzyme that hydrolyzes acetylcholine (ACh). Exposure to soman causes intense convulsions, neuropathology, and, ultimately, death. We recently reported that a single, systemic, convulsive dose of soman causes a rapid (30–45 min) induction of the immediate early gene protein product, Fos, in layer II of the piriform cortex (PC) (Zimmer et al., 1997a). PC is one of the first CNS regions to show intense neuropathology after soman poisoning (Petras, 1990; Switzer et al., 1990; Wall et al., 1990). Soman also causes rapid gliosis, including hypertrophy of astrocytic cell bodies and processes concomitant with increased glial fibrillary acidic protein (GFAP) staining in the same layers of PC exhibiting Fos staining (Zimmer et al., 1997b).
Similar to soman, systemic injections of the potent muscarinic agonist pilocarpine also induce seizures in PC (Millan et al., 1986). Microinjections of muscarinic receptor agonists in PC trigger focal seizures, which then spread throughout the brain (Turski et al., 1989). PC receives cholinergic innervation that arises exclusively from basal forebrain neurons in the nucleus of the diagonal band (NDB) (Rye et al., 1984). Taken together, these findings suggest that soman, by inhibiting acetylcholinesterase, causes increased levels of endogenously released ACh in PC; this, in turn, activates muscarinic receptors on PC neurons, leading to seizures. If this hypothesis is correct, then unilateral lesions of NDB cholinergic neurons may prevent seizures and attenuate increased Fos and GFAP staining in PC after soman. Further, focal activation of NDB, the sole source of ACh input to PC, should lead to seizures and elevated Fos and GFAP staining in PC similar to those caused by soman. The goal of the present study was to test these two predictions.
MATERIALS AND METHODS
NDB lesion study
192-IgG-saporin injections. Adult male Sprague Dawley rats (270–290 gm; Harlan Laboratories, Indianapolis, IN) were housed in pairs with free access to food and water for at least 1 week before use. Rats were anesthetized deeply with chloral hydrate (400 mg/kg, i.p.) and placed in a stereotaxic apparatus; a hole was drilled through the cranium at the coordinates for NDB (−0.6–0.3 mm to bregma, 1.4–2.2 mm lateral to midline). Microinjections of the specific cholinergic toxin 192-IgG-saporin (50–100 nl each; 0.5 mg/ml) were made through glass pipettes (tip diameter, 40 μm) with a hydraulic microinjection syringe. Because cholinergic neurons in NDB are distributed over a considerable area, microinjections were made at four sites in the NDB (−0.4 mm bregma, 1.6 mm lateral, 8.8 mm ventral; −0.4 mm bregma, 2.0 mm lateral, 8.6 mm ventral; 0.1 mm bregma, 1.6 mm lateral, 8.8 mm ventral; 0.1 mm bregma, 2.0 mm lateral, 8.6 mm ventral).
Soman administration. Ten days after saporin injections, rats were subjected to a convulsive dose (77.7 μg/kg, i.m.) of the irreversible AChE inhibitor, soman. Rats developed convulsions within 20 min of injection and maintained convulsions throughout the designated survival period. Behavioral signs of convulsions were scored as previously described (El-Etri et al., 1991; Zimmer et al., 1997a). Control animals were injected with proportional volumes of vehicle, saline (0.9%).
Fos, choline acetyltransferase (ChAT), and GFAP immunocytochemistry. At the end of the specified survival times, animals were anesthetized deeply with sodium pentobarbital (80 mg/kg) and perfused transcardially with 100–200 ml of 0.9% saline for 1 min, followed by 900–1000 ml of 4.0% paraformaldehyde in 0.1 mphosphate buffer (PB), pH 7.4, at 4°C for 20 min. Brains were removed rapidly from the skull and post-fixed in the same solution (4°C) for 1.5 hr and then placed in 30% sucrose in 0.1 m PB for 24 hr. Serial 40-μm-thick frozen sections were cut in the coronal plane and alternately placed in separate trays containing 0.1 mPB for immunohistochemical visualization of Fos, GFAP, and ChAT. To limit variability in staining intensity, we simultaneously processed free-floating sections from experimental and control animals as follows: (1) rinsed (for 30 min) in 0.1 m PBS; (2) placed in 2.0% normal rabbit serum (NRS, Fos, and ChAT) or 2.0% normal goat serum (NGS, GFAP) in PBS containing 0.2% Triton X-100 (TX) overnight; (3) incubated in anti-sheep primary antibody to Fos (Cambridge Biochemicals, Cambridge, MA) at 1:5000, anti-rat primary antibody to ChAT (Boehringer Mannheim, Indianapolis, IN) at 1:5000, or anti-rabbit primary antibody to GFAP (Dako, Carpinteria, CA) at 1:25,000 overnight in PBS-TX and 2% NRS for 24–48 hr at 4°C with gentle agitation; (4) rinsed in PBS-TX (1 hr) and then incubated in biotinylated rabbit anti-sheep IgG (Fos), rabbit anti-rat (ChAT), or goat anti-rabbit in PBS-TX (for 1.5 hr at room temperature); (5) rinsed in PBS-TX (1 hr) and incubated for 1 hr in avidin–biotin–peroxidase complex in 0.1m PBS-TX and then rinsed in PBS-TX (30 min); (6) incubated in 0.05% diaminobenzidine (DAB) with 0.1% hydrogen peroxide in PBS-TX for 10 min and rinsed in PB (30 min); (7) finally, sections were mounted on subbed slides, air-dried, dehydrated in graded alcohol and xylene, and coverslipped with DPX.
AChE histochemistry. Animals were perfused as above, and tissue sections were processed according to a modification of the Koelle–Friedenwald (Van Ooteghem and Shipley, 1984) AChE histochemical reaction. Sections were rinsed (seven times for 1 min each) in distilled water to remove excess phosphates and then incubated with gentle agitation for 2 hr (37°C) in the following solution containing (in mm): 2 copper (II) sulfate, 10 glycine, 50 sodium acetate, 4.2 acetylthiolcholine iodide, and 0.21 ethopropazine; this solution was freshly prepared and adjusted to a final pH of 5.25 with glacial acetic acid. After incubation, sections were rinsed (seven times for 1 min each) in distilled water and then reacted for 1 min (with agitation) in a freshly prepared solution of 1.0% sodium sulfide, adjusted to pH 7.8 with concentrated hydrochloric acid; this was followed by water rinses (seven times for 1 min each). Then the sections were reacted in a freshly prepared solution of 1.0% silver nitrate (1 min, constant agitation), followed by water rinses (three times for 1 min each).
NDB stimulation study
Surgery. Adult male Sprague Dawley rats (270–290 gm; Harlan Laboratories, Indianapolis, IN) were housed in pairs with free access to food and water for at least 1 week before use. Animals were anesthetized deeply with chloral hydrate (400 mg/kg, i.p.) and placed in a stereotaxic apparatus. A hole was drilled at the coordinates for NDB (−0.5–0.5 mm to bregma, 1.9–2.1 mm lateral to midline; Paxinos and Watson, 1986). Insulated bipolar microwire electrodes (125 μm), exposed at the tips, were chronically implanted unilaterally in NDB (8.4–8.6 mm ventral to the skull surface); electrodes were secured to adjacent skull screws with dental cement. Four animals had epidural skull screws implanted over the parietal cortex (2.0 mm posterior to lambda, 2.0 mm lateral to midline) ipsilateral to the stimulation electrode for EEG recordings throughout the experiment.
Animals were allowed 1 week to recover from surgery to eliminate Fos staining in PC induced by surgical procedures (Dragunow and Robertson, 1988). Animals were placed in a transparent plastic receptacle 60 min before stimulation, and flexible leads were connected to the EEG and stimulation electrodes. Sham controls were treated in an identical manner but received no stimulation. EEG recordings (Grass Instruments, Polygraph, Quincy, MA) were sampled at 5 min intervals before, during, and after focal stimulation of the NDB. Animals received 45 min of focal stimulation (12 Hz, 5 sec on/3 sec off, 30–45 min, 400–500 μA). Animals were observed during the entire stimulation period for behavioral signs of convulsions, including unilateral eye blinking, head tremors, and occasional rearing with forepaw grasping. At the end of specified survival times (between 45 and 60 min), rats were anesthetized deeply (see above) and processed for Fos and GFAP immunocytochemistry.
Pharmacological agents. Some animals were given vehicle (0.9% saline, i.p.), the selective muscarinic receptor antagonist scopolamine (2 mg/kg, i.p.), or the selective nicotinic receptor antagonist mecamylamine (5 mg/kg, i.p.) 30 and 5 min before NDB stimulation.
Cell counts. Sections were visualized with a Leitz microscope (Wetzlar, Germany), and the number of Fos-positive cells per section was counted independently by two lab personnel who used a computer-based image analysis system (Neurolucida, MicroBrightfield, Baltimore, MD). To be considered Fos-positive, the nucleus of cells had to have a brown stain characteristic of DAB. To examine Fos expression along the rostral–caudal axis of PC, we made Fos counts at three evenly spaced rostral–caudal levels of PC corresponding to +2.0, 0.0, and −2.0 mm with respect to bregma. Specific anatomical landmarks (i.e., anterior commissure crossing at 0.0 mm bregma) were used to match sections across experiments. At each level of PC, a 6 × 90 μm2 rectangular grid was superimposed on each region to be counted. In each tissue section the Fos-positive cells were counted in three separate dorsal/ventral regions of PC perpendicular to the brain surface. Each grid included cells from layers I, II, and III of PC. Identical methods were used to evaluate Fos-positive neurons in other cortical regions (i.e., frontal cortex). In each case the same procedures were performed on the contralateral side of the brain to compare changes in Fos on stimulated/lesioned versus control hemispheres. The average number of Fos-positive cells in each experiment did not differ significantly under any experimental conditions on the ipsilateral rostral–caudal plane (p > 0.2); thus, these data were combined to represent the average number of Fos-positive cells throughout PC.
RESULTS
Lesions of the nucleus of the diagonal band
The working hypothesis of this research is that soman-induced inhibition of AChE leads to a buildup of unhydrolyzed ACh in PC, which activates muscarinic receptors and causes seizures. If correct, then elimination of the cholinergic innervation to PC should provide protection from the seizurogenic actions of soman. To test this prediction, we initially made electrolytic lesions of the basal forebrain, including the NDB, and then investigated the ability of soman to induce seizures and Fos. Large lesions that completely destroyed the NDB significantly reduced the sequelae of soman in the ipsilateral PC (data not shown). However, the large and neurochemically indiscriminate nature of these lesions made the findings difficult to interpret. To lesion NDB cholinergic neurons selectively, we made unilateral injections of the immunotoxin 192-IgG-saporininto the NDB (Wiley et al., 1991; Book et al., 1992, 1994; Wenk et al., 1994). Ten days after 192-IgG-saporin, ChAT-positive neurons were eliminated from the NDB around the injection sites. However, Nissl-stained cells were present around the injection site. ChAT staining was still present in the intact contralateral NDB (Fig.1). To confirm further that saporin injections were located discretely in the NDB and to assess the degree of cholinergic denervation of PC and other structures targeted by NDB cholinergic axons, we stained alternate sections for histochemical detection of AChE. AChE staining was reduced markedly in the NDB, medial septum, PC, main olfactory bulb, and anterior cingulate cortex ipsilateral to the injection site (Fig.2). Moderate decreases in AChE were present in the entorhinal cortex. Normal AChE staining was present in all of these sites in the intact hemisphere.
Fig. 1.

Choline acetyltransferase (ChAT) staining in the nucleus of the diagonal band (NDB). A, ChAT staining in NDB 10 d after microinjections of192-IgG-saporin unilaterally in NDB. Note the lack of ChAT-positive cells in NDB ipsilateral to the saporin lesion.B, ChAT staining in the contralateral control NDB. Scale bar, 700 μm.
Fig. 2.

Acetylcholinesterase (AChE) staining in piriform cortex. A, AChE staining in PC 10 d after microinjections of 192-IgG-saporin unilaterally in NDB. Note the lack of AChE staining in regions of PC corresponding to NDB cholinergic terminals in PC, ipsilateral to the saporin lesion.B, AChE staining in the contralateral control PC. Scale bar, 750 μm. I, II, III, Layers of PC;LOT, lateral olfactory tract.
Fos staining
In the next set of experiments the animals received a convulsive dose of soman 10 d after unilateral 192-IgG-saporininjections in the NDB. Control animals that received unilateral injections of saline in the NDB instead of 192-IgG-saporin(n = 7) had robust Fos staining after soman administration in PC ipsilateral (234.7 ± 70.6 cells/6 × 90 μm2) and contralateral to the injection site (243.7 ± 68.7 cells/6 × 90 μm2) (Figs.3A,B,4, columns 1, 2). Animals that received unilateral injections of192-IgG-saporin had few Fos-positive neurons in PC ipsilateral to the 192-IgG-saporin injection site (6.7 ± 1.6 cells/6 × 90 μm2) (Figs. 4,column 3, 5A). This number did not differ significantly from the contralateral PC (8.3 ± 2.4 cells/6 × 90 μm2) (Figs. 4,column 4, 5B). By 45 min after soman administration, Fos-positive neurons were present in layer II and some layer III neurons throughout the rostral–caudal extent of PC contralateral to the lesioned NDB (115.3 ± 23.0 cells/6 × 90 μm2) (Figs. 4, column 6,5D; n = 7). In striking contrast, there were far fewer Fos-positive cells in PC ipsilateral to the lesioned NDB (15.3 ± 6.1 cells/6 × 90 μm2) (Figs.4, column 5, 5C; p < 0.02), and the number did not differ from control animals (p > 0.07). The number of Fos-positive cells bilaterally in the frontal cortex (data not shown) did not differ between control and soman cases (p > 0.3).
Fig. 3.

Fos staining in the piriform cortex. Sections were stained for Fos immunohistochemistry 45 min after an intramuscular injection of soman 10 d after unilateral injections of vehicle, saline ipsilaterally in the NDB. A, Fos staining in PC ipsilateral to the saline-injected NDB. B, Fos staining in the contralateral control PC. Scale bar, 750 μm. I, II, III, Layers of PC; LOT, lateral olfactory tract.
Fig. 4.

Fos-positive cell counts in PC. All NDB injections were made into the left hemisphere. Animals receiving unilateral injections of vehicle, saline in NDB (left hemisphere) had large increases in Fos-positive cells bilaterally in PC 45 min after an intramuscular injection of soman (columns 1, 2). Animals receiving a unilateral injection of 192-IgG-saporin in the NDB (left hemisphere) had few Fos-positive cells 45 min after an intramuscular injection of vehicle, saline (columns 3, 4). Unilateral injections of192-IgG-saporin in NDB (left hemisphere) inhibited the large increases in Fos-positive cells in PC 45 min after soman administration (column 5). Note that the number of Fos-positive cells in PC ipsilateral to the lesioned NDB 45 min after soman (column 5) was not significantly different from animals receiving 192-IgG-saporin and no soman (columns 3, 4). Also, the number of Fos-positive cells in PC ipsilateral to the unlesioned NDB (column 6) was significantly fewer than in control animals 45 min after soman administration (columns 1, 2). By 90 min after soman the number of Fos-positive cells in PC ipsilateral to the lesioned NDB (column 7) was significantly fewer than the number of Fos-positive cells 45 min after soman administration in animals receiving vehicle, saline (columns 1, 2). The number of Fos-positive cells in PC ipsilateral to the unlesioned NDB (column 8) was similar to the number in control animals 45 min after soman administration (columns 1, 2).
Fig. 5.
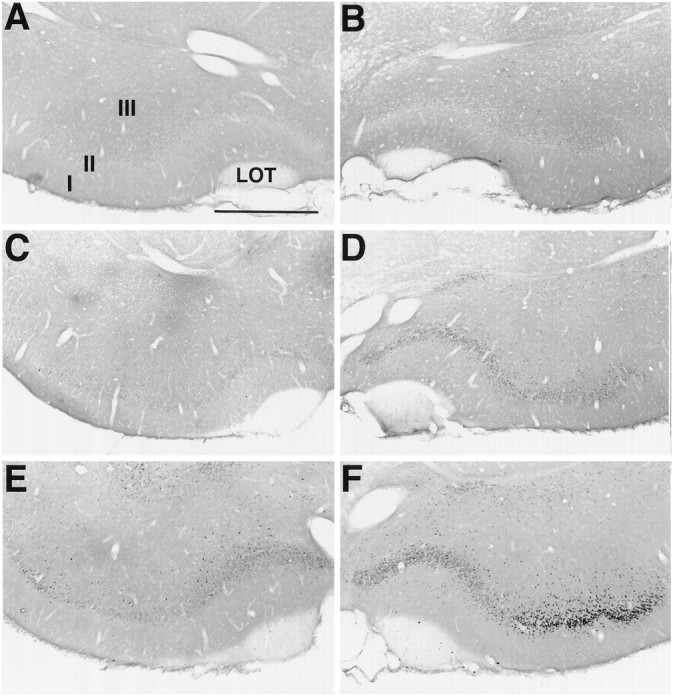
Fos staining in the piriform cortex. Sections were stained for Fos immunohistochemistry 10 d after unilateral lesions with 192-IgG-saporin ipsilaterally in the NDB. Fos staining in control (no soman) animals is absent in the PC ipsilateral (A) and contralateral (B) to the lesion site. By 45 min after an intramuscular injection of soman, Fos staining is absent in the PC ipsilateral to the lesion injection site (C). However, robust Fos staining is present in the contralateral PC (D). By 90 min after soman, increased Fos staining is observed in the PC ipsilateral to the lesioned NDB (E). Further increases in Fos staining are observed in the contralateral PC (F). Scale bar, 750 μm. I, II, III, Layers of PC; LOT, lateral olfactory tract.
The number of Fos-positive neurons increased significantly in PC contralateral to the lesioned NDB 90 min after soman administration (279.5 ± 36.4 cells/6 × 90 μm2) (Figs.4, column 8, 5F). Significantly fewer Fos-positive neurons were evident in PC ipsilateral to the lesioned NDB (95.7 ± 22.5 cells/6 × 90 μm2) (Figs.4, column 7, 5E; p < 0.002), but the number had increased dramatically over controls. In addition, Fos staining was present bilaterally in the hippocampus, entorhinal cortex, and the anterior cingulate cortex. Fos also was evident bilaterally in layers IV, V, and VI of other cortical regions. Fos staining was absent in all CNS regions examined in control animals receiving vehicle (0.9% saline, i.p.).
GFAP staining
By 1 hr after soman, GFAP staining in PC ipsilateral to the lesioned NDB was similar to staining in control animals receiving vehicle, saline (0.9%) (Fig. 6;n = 5). By contrast, GFAP staining was increased markedly in layer II of the contralateral PC by 1 hr. By 2 hr after soman, increased GFAP staining was present bilaterally in layer II of PC. Elevated GFAP staining also was evident in the hippocampus and layers I, IV, and VI of the neocortex bilaterally.
Fig. 6.
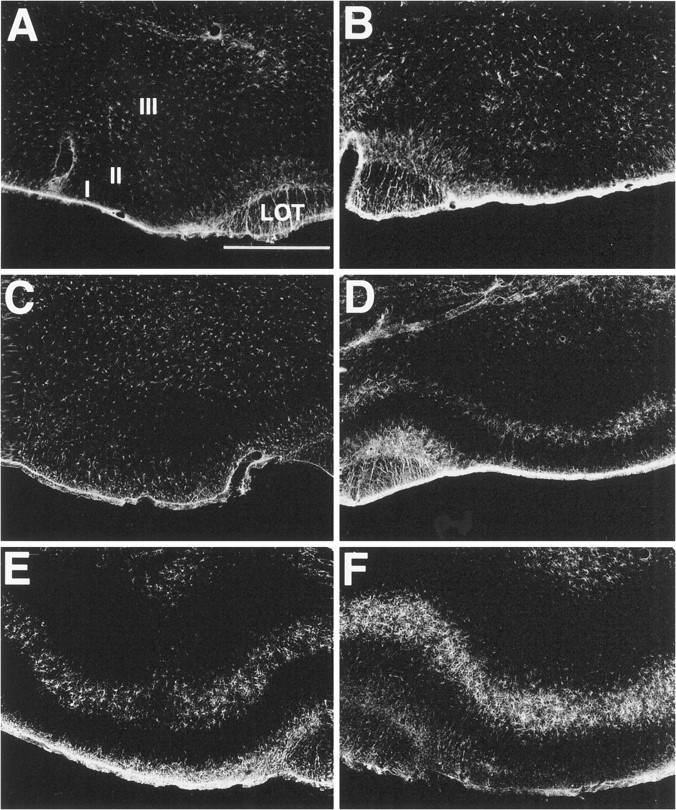
Glial fibrillary acidic acid (GFAP) staining in the piriform cortex. Sections were stained for GFAP histochemistry 10 d after unilateral lesions with 192-IgG-saporinipsilaterally in the NDB. Low levels of GFAP staining in control (no soman) animals are present in the PC ipsilateral (A) and contralateral (B) to the lesion site. By 45 min after an intramuscular injection of soman, GFAP staining is indistinguishable from controls in the PC ipsilateral to the lesion injection site (C). However, discrete layer-specific GFAP staining is present in layer II of the contralateral PC (D). By 90 min after soman, increased layer-specific increases in GFAP staining are observed in the PC ipsilateral to the lesioned NDB (E). Further increases in GFAP staining in layer II are observed in the contralateral PC (F). Scale bar, 750 μm. I, II, III, Layers of PC;LOT, lateral olfactory tract.
Taken together, the results of these experiments showed that lesions of NDB cholinergic neurons caused a significant delay in soman-induced Fos and GFAP staining in PC ipsilateral to the lesion. The finding that these seizure-associated changes were only delayed and not prevented altogether is probably attributable to the spread of seizures from other parts of the brain, including the contralateral PC (see below).
Stimulation of the nucleus of the diagonal band
The preceding experiments demonstrated that removal of the cholinergic innervation to PC provides significant attenuation of the seizurogenic actions of soman. This is consistent with the hypothesis that the persistence of unhydrolyzed ACh in PC after soman plays a causal role in the generation of seizures. To assess further this hypothesis, we next performed a set of experiments to determine whether focal activation of NDB projections to PC was sufficient to trigger seizures, induce Fos expression, and increase reactive gliosis.
Within 10 min of intermittent NDB stimulation (see Materials and Methods), the rats displayed behavioral signs of convulsions, including unilateral eye blinking, head tremors, and occasional rearing with forepaw grasping. All four animals with EEG records had large amplitude spike and wave EEG activity within 10–20 min of electrical stimulation (Fig. 7). This EEG activation and behavioral convulsions persisted for at least 20 min after the termination of stimulation.
Fig. 7.

Cortical EEG recordings before (Baseline, top panel), during NDB stimulation (middle panel), and 20 min after the termination of NDB stimulation (bottom panel). Note that focal NDB stimulation increases EEG spike and wave activity resembling cortical seizures. This increased activity persists as long as 20–40 min after the termination of NDB stimulation.
Fos staining
Sections from control (implanted, but not stimulated) and stimulated animals were paired and processed simultaneously to compare Fos staining before and after stimulation of NDB (Fig.8). Control animals that did not receive stimulation lacked Fos-positive cells [9.7 ± 1.2 cells/6 × 90 μm2 ipsilateral (Fig.9, column 1) and 8.2 ± 1.4 cells/6 × 90 μm2 contralateral (Fig. 9,column 2) to the NDB electrode (see Fig.8A,B)]. Stimulated animals displaying convulsions (12 of 13) had an increased number of Fos-positive cells (see Fig.8D) by 45 min throughout the entire rostral–caudal extent of PC ipsilateral to the stimulation site (222.6 ± 31.8 cells/6 × 90 μm2) (Fig. 9, column 4). There were significantly fewer Fos-positive cells in the contralateral PC (56.2 ± 15.4 cells/6 × 90 μm2) (Figs. 8C, 9, column 3;p < 0.0001). The number of Fos-positive cells in the frontal cortex did not differ between control and stimulation cases (p > 0.2). In the one animal lacking behavioral responses to NDB stimulation, there was no Fos staining in PC or other CNS regions.
Fig. 8.
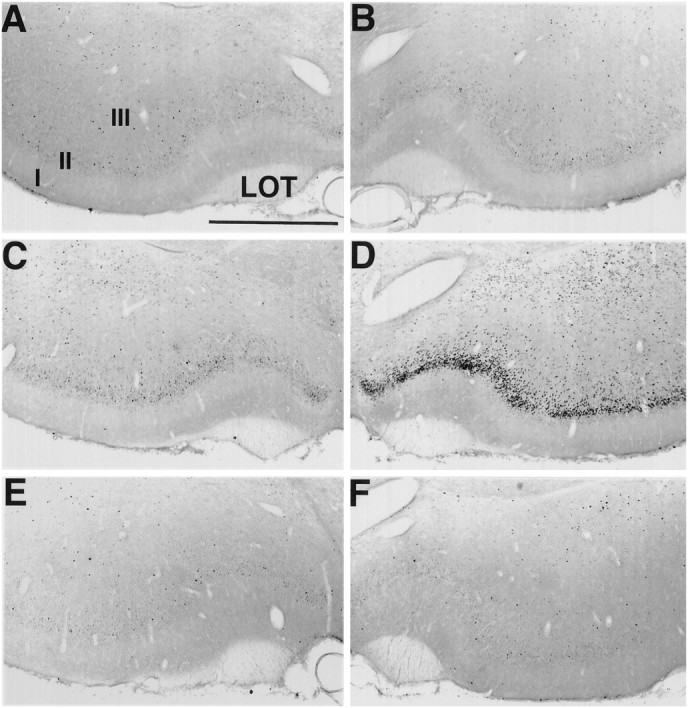
Fos staining in the piriform cortex. Sections were stained for Fos immunohistochemistry after focal ipsilateral stimulation of the NDB. Fos staining in control (no stimulation) animals is absent in the PC contralateral (A) and ipsilateral (B) to the stimulation electrode. By 45 min after focal stimulation of the NDB, Fos staining is similar to that in controls in the PC contralateral to the stimulation site (C). However, robust Fos staining is present in the PC ipsilateral to the NDB stimulation site (D). Pretreatment with the selective muscarinic receptor antagonist scopolamine inhibits increased Fos staining in the PC after 45 min of NDB stimulation (ipsilateral, in E; contralateral, in F). Scale bar, 750 μm.I, II, III, Layers of PC; LOT, lateral olfactory tract.
Fig. 9.

Fos-positive cell counts in PC. Stimulation electrodes were targeted to the right NDB in all cases. By 45 min after an intraperitoneal injection of vehicle, saline, few Fos-positive cells were present in PC bilaterally in the absence of NDB stimulation (columns 1, 2). A significant increase in Fos-positive cells was observed in PC (column 4) after 45 min of NDB stimulation, as compared with controls. Note that the increase in Fos-positive cells in the unstimulated contralateral PC (column 3), however, was significantly less than in PC ipsilateral to the stimulated NDB. Pretreatment with the selective muscarinic receptor antagonist scopolamine before NDB stimulation significantly decreased the number of Fos-positive cells bilaterally in PC (columns 5, 6), as compared with stimulated animals lacking scopolamine pretreatment (columns 3, 4).
GFAP staining
To determine whether astrocytes were activated after unilateral stimulation of NDB, we stained sections from control and stimulated animals for GFAP (Fig. 10). By 1 hr after electrical stimulation of NDB (n = 7), GFAP staining was increased in PC layers I, II, and III ipsilateral to the stimulation site. GFAP staining in PC caudal to the NDB was limited to PC layer II. In a few cases, light GFAP staining was evident bilaterally throughout the hippocampus, layer II of entorhinal cortex, and in layers I, IV, and VI of other cortical regions. Fos staining was not evident in astrocytes in any studies (data not shown).
Fig. 10.
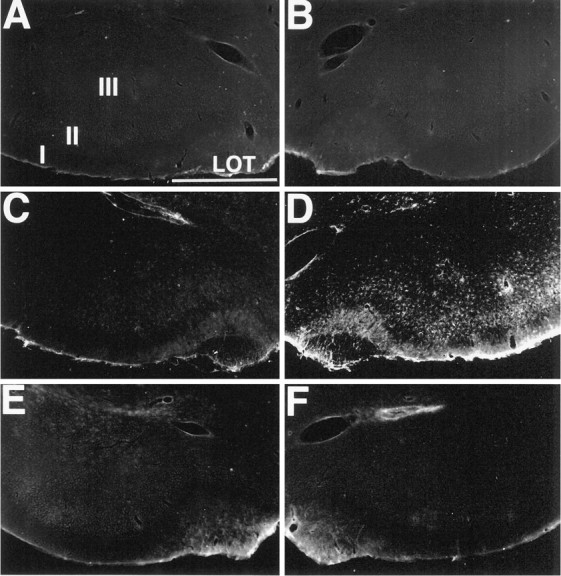
GFAP staining in the piriform cortex. Sections were stained for GFAP histochemistry after focal ipsilateral stimulation of the NDB. GFAP staining in control (no stimulation) animals is absent in the PC contralateral (A) and ipsilateral (B) to the stimulation electrode. By 45 min after focal stimulation of the NDB, GFAP staining is similar to that of controls in the PC contralateral to the stimulation site (C). However, robust GFAP staining is present in the PC ipsilateral to the NDB stimulation site (D). Pretreatment with the selective muscarinic receptor antagonist scopolamine inhibits increased GFAP staining in PC after 45 min of NDB stimulation (contralateral, in E; ipsilateral, in F). Scale bar, 750 μm.I, II, III, Layers of PC; LOT, lateral olfactory tract.
Muscarinic receptors
Focal stimulation of NDB activates cholinergic as well as noncholinergic neurons. Thus, although behavioral seizures and patterns of Fos and GFAP mimicked those caused by soman and thus might be attributable to excess ACh release, it is possible that these responses potentially are mediated by the release of other neurotransmitters. To investigate this possibility, we performed another series of experiments in which NDB was stimulated as before, but the animals received systemic administration of the muscarinic receptor antagonist scopolamine (see Materials and Methods) before the onset of NDB stimulation (see Fig. 8E,F). Scopolamine completely blocked or significantly attenuated NDB stimulation-induced convulsions and Fos-positive cells (14.0 ± 1.8 cells/6 × 90 μm2 ipsilateral and 4.8 ± 0.8 cells/6 × 90 μm2contralateral to the stimulation site; p < 0.001;n = 10) (see Fig. 9, columns 5, 6) in PC and throughout the CNS, as compared with animals lacking scopolamine pretreatment. By contrast, pretreatment with the nicotinic receptor antagonist mecamylamine (n = 4) failed to attenuate NDB-evoked Fos staining in PC ipsilateral to the stimulation site. Scopolamine pretreatment (6 of 7) prevented increased GFAP (Fig. 10) staining in PC and other CNS regions after NDB stimulation, whereas pretreatment with mecamylamine did not attenuate NDB-evoked increases in GFAP staining.
DISCUSSION
The results of this study demonstrate the following. (1) Selective chemolytic lesions of cholinergic neurons in NDB prevented or delayed soman-induced Fos induction and increased GFAP in PC. (2) Focal unilateral activation of NDB produced a pattern of effects similar to those caused by soman: convulsions, robust induction of Fos, and increased staining for GFAP in PC. (3) The anatomical distribution and intensity of Fos and GFAP staining elicited by focal stimulation exhibited a consistent spatiotemporal pattern, as previously reported (Zimmer et al., 1997a,b). Fos staining was always observed in PC before the neocortex and hippocampus. (4) The selective muscarinic cholinergic receptor antagonist scopolamine prevented NDB-evoked seizures, Fos expression, and increased GFAP staining. Taken together with our recent findings (Zimmer et al., 1997a,b), the present results support the hypothesis that the “nerve” gas soman causes increased ACh buildup from NDB cholinergic terminals and protracted muscarinic receptor activation of PC neurons. This, in turn, triggers increased neuronal activity, seizures, and reactive gliosis in PC.
Extent and specificity of NDB lesions
PC receives a dense cholinergic input from the NDB in the basal forebrain (Rye et al., 1984; Woolf et al., 1986). The accumulation of endogenously released ACh in PC after soman may increase PC neuronal activity and lead to seizures. To determine whether cholinergic NDB terminals in PC play a role in the rapid induction of Fos after soman, we destroyed NDB cholinergic neurons with the selective neurotoxin192-IgG-saporin. 192-IgG-saporin binds to the low-affinity NGF receptor, is internalized by the cell, and inhibits protein synthesis, resulting in cell death (Wiley et al., 1991). Because only cholinergic neurons in NDB contain the low-affinity NGF receptor (Fisher et al., 1988; Gritti et al., 1993), microinjections of192-IgG-saporin into the NDB selectively destroy these neurons. In the present experiment the specificity of the192-IgG-saporin lesions was evaluated in two ways: (1) immunohistochemical staining for ChAT showed that cholinergic neurons were eliminated from the region of NDB, and (2) noncholinergic cells, as revealed by Nissl staining, were present throughout the lesion site.192-IgG-saporin lesions eliminated AChE staining in the NDB and in ipsilateral cortical regions receiving ACh input from the NDB, including PC, the olfactory bulb, and the anterior cingulate cortex (Lamour et al., 1984; Rye et al., 1984). Taken together, these findings indicate that there was a selective elimination of NDB-derived cholinergic terminals ipsilateral to the 192-IgG-saporinlesion.
NDB lesions markedly attenuate soman-induced Fos staining in PC
If soman-induced Fos in PC is caused by increased ACh, then Fos expression in PC should be attenuated or delayed on the side of the brain in animals with unilateral NDB lesions. Large increases in Fos were present only in PC contralateral to the lesioned NDB by 45 min after soman. This indicates that intact cholinergic terminals in PC are necessary for “rapid” increases in neuronal activity and Fos staining after soman. By 90 min, Fos staining was present in PC ipsilateral to the lesion. Thus, the effect of removing the cholinergic input to PC is mainly to delay the onset of Fos expression. The most reasonable explanation for this is that cholinergic input is required for the initiation of seizures, but, in their absence, seizures initiated in other brain structures may spread secondarily into PC. In our previous study of the time course of expression of Fos after soman (Zimmer et al., 1997a), it was found that PC is the earliest cortical area to express Fos, but with time, nearly all cortical areas will have increased Fos. In the present experiments, Fos was expressed rapidly (<45 min) in PC in the hemisphere where the cholinergic innervation was intact. Association neurons in PC project extensively to other cortical regions. There is a strong projection, via the anterior commissure, to the contralateral PC (Haberly and Price, 1978). Thus, seizures initiated in PC of the intact hemisphere could spread to the contralateral PC. In agreement with this, seizures triggered unilaterally in PC have been shown to spread throughout the brain bilaterally (Piredda and Gale, 1985, 1986; Halonen et al., 1994). In this regard, it would be interesting to determine whether transection of the anterior commissure would prevent or further delay the induction of Fos in the hemisphere ipsilateral to the NDB lesion.
Electrical stimulation of the NDB causes seizures and rapid Fos staining in PC
The NDB lesion experiments are consistent with the hypothesis that the NDB–PC cholinergic circuit is necessary for rapid increases in neuronal activity and seizures after soman. If this is true, then it is reasonable to predict that unilateral focal stimulation of NDB neurons would lead to seizures and increased Fos expression, as does soman. This prediction was supported by the finding that unilateral NDB stimulation caused increased Fos staining in layer II neurons in the ipsilateral PC by 45 min.
There are several potential limitations to these results, however. Stimulation of the NDB may activate neurons and/or fiber tracts in the NDB that are not cholinergic. Thus, noncholinergic inputs to PC could cause seizures and/or Fos induction (Haberly and Price, 1978; Bobillier et al., 1979; Fisher et al., 1988; Vertes, 1988; Gritti et al., 1993). Also, activated cholinergic neurons may release other neurotransmitters or neuropeptides colocalized with ACh, and these transmitters could excite PC neurons and trigger seizures. Neither of these possibilities appeared to play a significant role in the present experiments, however, because muscarinic receptor antagonism prevented behavioral convulsion, changes in EEG activity, and increased Fos and GFAP staining caused by NDB stimulation. This indicates that increased transmitter release from cholinergic terminals in PC activates layer II neurons. Although both muscarinic (mAChR) and nicotinic (nAChR) cholinergic receptors are present in PC (Wamsley et al., 1980; Buckley et al., 1988; van der Zee et al., 1992; Hill et al., 1993), the nAChR antagonist mecamylamine did not prevent seizures or increased Fos and GFAP staining.
The cellular mechanism by which muscarinic receptor stimulation in PC triggers seizures and Fos expression is not known. ACh and muscarinic receptor agonists are known to increase the excitability of neurons throughout the brain (Dodd et al., 1981; Halliwell and Adams, 1982;Cole and Nicoll, 1984; McCormick and Prince, 1985; Nicoll, 1988). Furthermore, muscarinic receptor activation may inhibit GABA release from GABAergic interneurons, causing disinhibition of excitatory neurons (Ben-Ari et al., 1981,Krnjevic, 1981; Krnjevic and Ropert, 1981). Taken together, these results support the hypothesis that rapid increases in ACh release cause increased neuronal excitability via the activation of muscarinic receptors in PC.
Seizures persist in the absence of electrical stimulation
After electrical stimulation of NDB, behavioral convulsions and EEG spike wave activity persisted for the duration of the experiment. Thus, seizures can persist in the absence of continued electrical stimulation of the NDB. This suggests that neurons in PC that are activated by continuous cholinergic muscarinic stimulation can maintain high levels of activity after the cessation of cholinergic stimulation. Muscarinic receptor activation dramatically increases the level and duration of hippocampal pyramidal neuron responses to coactivation of excitatory amino acid (EAA) inputs (Biscoe and Straughan, 1966; Dodd et al., 1981; Benardo and Prince, 1982; Cole and Nicoll, 1984; McCormick and Prince, 1985; Madison et al., 1987; Nieto-Sampedro and Berman, 1987; Nicoll, 1988) by reducing a slow afterhyperpolarization (AHP) (Biscoe and Straughan, 1966; Dodd et al., 1981; Benardo and Prince, 1982; Halliwell and Adams, 1982; Cole and Nicoll, 1984; McCormick and Prince, 1985; Nicoll, 1988). Reduction of the AHP leaves cortical neurons susceptible to overexcitation by EAAs. Furthermore, it is known that EAA activation of NMDA receptors is involved in the spread and maintenance of seizures in the PC and the hippocampus (Millan et al., 1986; Braitman and Sparenborg, 1989). In the present study, increased synaptic ACh release during NDB stimulation may activate muscarinic receptors in PC and attenuate AHPs that normally limit the excitability of PC neurons to EAAs. These neurons would become progressively more excited by EAA inputs from the main olfactory bulb and associational synapses from other parts of the PC and, as a result, release more EAAs from their own synaptic terminals in the PC and other cortical regions. This would increase the intensity of neural activity in the PC and, via associational PC axons, cause the spread of seizures to the cerebral cortex. Intense neuronal activity in PC could activate NMDA receptors located in PC. It is known that EAA activation of NMDA receptors is involved in the spread and maintenance of seizures in PC and the hippocampus (Millan et al., 1986; Braitman and Sparenborg, 1989). Thus, NMDA receptor activation may maintain seizure activity after the cessation of electrical stimulation of ACh inputs to PC.
An alternate hypothesis is that muscarinic activation initiates long-lasting excitation of pyramidal cells in layer II/III of PC because of slow depolarization after potentials (sADP). Bath application of the muscarinic receptor agonist oxotremorine-M induces a strong and persistent membrane depolarization, an increase in input resistance, and a sustained neuronal discharge (>30 min) of pyramidal cells in PC in vitro (Constanti et al., 1993). The sADP-responsive neurons are located in deep layer II/III of PC (Libri et al., 1994). Muscarinic activation of sADP in layer II/III neurons after ACh release from NDB terminals in PC could trigger sustained neuronal activity and long-lasting seizure discharges that persist after the cessation of ACh release.
ACh release from cholinergic nerve terminals in PC also may increase pyramidal cell excitability in PC by suppressing inhibitory synaptic input. Pyramidal cells in the hippocampus, when depolarized by a brief train of action potentials, demonstrate decreased inhibitory (GABA-mediated) synaptic input (Pitler and Alger, 1992a, 1994). This phenomenon is known as depolarization-induced suppression of inhibition (DSI). Cholinergic receptor activation enhances DSI in the hippocampus (Pitler and Alger, 1992b; Martin and Alger, 1996). Although DSI has not been demonstrated in PC, increased ACh release in PC in the present experiments could suppress GABA-mediated inhibition in PC and increase pyramidal cell activity. Increased pyramidal cell activity with concomitant decreases in GABAergic activity may predispose pyramidal cells in PC toward seizure-like activity.
In the present study, ACh release in PC triggers the initiation of seizures after both NDB stimulation and soman administration, predominantly by the activation of muscarinic receptors. It is known that repeated activation of neural pathways (kindling) can induce seizures and an epileptic state. It has been demonstrated that kindling induces synaptic reorganization (plasticity) and axonal sprouting in limbic pathways (Sutula et al., 1988). Sutula and colleagues suggest that alterations in limbic connectivity may make hippocampal pathways more susceptible to epileptiform events. Increased neuronal activity as observed in PC after soman administration may lead to permanent alterations in PC connectivity. Changes in PC connectivity may make neurons more susceptible to epileptic events and generalized seizures. Exposure to anticholinesterases, such as soman, may lead to dysfunctional plasticity in the CNS and permanent neurological deficits. Changes in CNS connectivity may help to explain the neurological complications of soldiers (Gulf War Syndrome) potentially exposed to neurotoxins, such as soman, during the Gulf War.
Glia
Reactive gliosis occurs in response to injury, including soman-induced seizures, in the mature CNS (Zimmer et al., 1997a,b). A salient manifestation of reactive gliosis is an increase in GFAP, a protein subunit of glial intermediate filaments found exclusively in astrocytes in the CNS (Eng et al., 1971; Amaducci et al., 1981; Eng and Shiurba, 1987). By 1 hr after soman, increased GFAP is present in astrocytes in precisely the same layers of PC that contain neurons that express Fos (Zimmer et al., 1997a,b). We previously suggested that this rapid increase in GFAP is associated with increased neuronal activity in neighboring PC neurons. If this is correct, then destruction of NDB cholinergic neurons, which prevents or delays increased excitability of PC neurons, should prevent or delay increased GFAP staining in PC.
Consistent with this hypothesis, 192-IgG-saporin lesions of the NDB prevented increased GFAP staining in layer II astrocytes of the ipsilateral PC within 1 hr after soman. GFAP staining in the contralateral intact PC increased similarly to control intact animals. This indicates that an intact cholinergic input is also necessary for rapid reactivity of PC astrocytes after soman and supports the idea that astrocytes respond to increased activity in neighboring neurons. Consistent with this, by 2 hr after soman, increased GFAP staining was observed bilaterally in PC and the hippocampus, consistent with the observation that Fos expression is delayed from 45 to 90 min in the NDB-lesioned animals.
NDB stimulation induced GFAP staining primarily in layer II of PC ipsilateral to the stimulation site. Pretreatment with scopolamine, but not mecamylamine, prevented increased GFAP staining after NDB stimulation. Astrocytes have receptors for and physiologically respond to ACh in culture (Murphy et al., 1986; Hosli et al., 1988; Salm and McCarthy, 1989, 1992; Cornell-Bell et al., 1990; Van Der Zee et al., 1993). Thus, ACh could have a direct action on PC astrocytes via muscarinic receptor activation. However, glial reactivity lags Fos expression by approximately the same amount of time either when the cholinergic input is intact or when it is destroyed and Fos expression in PC neurons is delayed. This suggests that PC astrocytes respond to signals from highly active adjacent neurons and not from direct cholinergic stimulation. Neuronal activation, such as occurs during seizures, increases extracellular levels of lactate and decreases extracellular pH (Meric et al., 1994). A reduction in extracellular pH in serum-free astrocyte cultures increased levels of GFAP staining with a time course similar to that seen in the present experiments (Oh et al., 1995). Thus, during seizures, hyperactive neurons may produce excess lactate, causing local decreases in pH. This could activate adjacent astrocytes, leading to increased GFAP expression.
Footnotes
This work was supported by the U.S. Army Medical Research and Development under contracts DAMD-17-91-C-1071 and DAMD-17-95-C-5031. The opinions and assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the U.S. Army or the Department of Defense.
Correspondence should be addressed to Dr. Michael T. Shipley, Department of Anatomy, University of Maryland School of Medicine, 685 West Baltimore Street, Baltimore, MD 21201.
REFERENCES
- 1.Amaducci L, Forno KI, Eng L. Glial fibrillary acidic protein in cryogenic lesions of the rat brain. Neurosci Lett. 1981;21:27–32. doi: 10.1016/0304-3940(81)90052-5. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Ari Y, Krnjevic K, Reinhardt W, Ropert N. Intracellular observations of the disinhibitory action of acetylcholine in the hippocampus. Neuroscience. 1981;6:2475–2484. doi: 10.1016/0306-4522(81)90093-2. [DOI] [PubMed] [Google Scholar]
- 3.Benardo LS, Prince DA. Ionic mechanisms of cholinergic excitation in mammalian hippocampal pyramidal cells. Brain Res. 1982;249:333–334. doi: 10.1016/0006-8993(82)90067-1. [DOI] [PubMed] [Google Scholar]
- 4.Biscoe TJ, Straughan DW. Micro-electrophoretic studies of neurones in the cat hippocampus. J Physiol (Lond) 1966;183:341–359. doi: 10.1113/jphysiol.1966.sp007869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bobillier P, Seguin S, Degueurce A, Lewis BD, Pujol JF. The efferent connections of the nucleus raphe centralis superior in the rat as revealed by autoradiography. Brain Res. 1979;166:1–8. doi: 10.1016/0006-8993(79)90644-9. [DOI] [PubMed] [Google Scholar]
- 6.Book AA, Wiley RG, Schweitzer JB. Specificity of 192-IgG-saporin for NGF receptor-positive cholinergic basal forebrain neurons in the rat. Brain Res. 1992;590:350–355. doi: 10.1016/0006-8993(92)91121-t. [DOI] [PubMed] [Google Scholar]
- 7.Book AA, Wiley RG, Schweitzer JB. 192-IgG-saporin. I. Specific lethality for cholinergic neurons in the basal forebrain of the rat. J Neuropathol Exp Neurol. 1994;53:95–102. doi: 10.1097/00005072-199401000-00012. [DOI] [PubMed] [Google Scholar]
- 8.Braitman DJ, Sparenborg S. MK-801 protects against seizures induced by the cholinesterase inhibitor soman. Brain Res Bull. 1989;23:145–148. doi: 10.1016/0361-9230(89)90173-1. [DOI] [PubMed] [Google Scholar]
- 9.Buckley NJ, Bonner TI, Brann MR. Localization of a family of muscarinic receptor mRNAs in rat brain. J Neurosci. 1988;8:4646–4652. doi: 10.1523/JNEUROSCI.08-12-04646.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cole AE, Nicoll RA. Characterization of a slow cholinergic postsynaptic potential recorded in vitro from rat hippocampal pyramidal cells. J Physiol (Lond) 1984;352:173–188. doi: 10.1113/jphysiol.1984.sp015285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Constanti A, Bagetta G, Libri V. Persistent muscarinic excitation in guinea-pig olfactory cortex neurons: involvement of a slow poststimulus afterdepolarizing current. Neuroscience. 1993;56:887–904. doi: 10.1016/0306-4522(93)90135-3. [DOI] [PubMed] [Google Scholar]
- 12.Cornell-Bell AH, Thomas PG, Smith SJ. The excitatory neurotransmitter glutamate causes filopodia formation in cultured hippocampal astrocytes. Glia. 1990;3:322–334. doi: 10.1002/glia.440030503. [DOI] [PubMed] [Google Scholar]
- 13.Dodd J, Dingledine R, Kelly SJ. The excitatory action of acetylcholine on hippocampal neurones of the guinea pig and rat maintained in vitro. Brain Res. 1981;207:109–127. doi: 10.1016/0006-8993(81)90682-x. [DOI] [PubMed] [Google Scholar]
- 14.Dragunow M, Robertson HA. Localization and induction of c-fos protein-like immunoreactive material in the nuclei of adult mammalian neurons. Brain Res. 1988;440:252–260. doi: 10.1016/0006-8993(88)90993-6. [DOI] [PubMed] [Google Scholar]
- 15.El-Etri M, Shipley MT, Ennis M, Nickell WT. Cholinolytic seizures: possible role of nucleus locus coeruleus. Soc Neurosci Abstr. 1991;17:529. [Google Scholar]
- 16.Eng LF, Shiurba RA. Glial fibrillary acidic protein: a review of structure, function, and clinical application. In: Marangos PJ, Campbell I, Cohen RM, editors. Neurobiological research, Vol 2. Academic; New York: 1987. pp. 1–42. [Google Scholar]
- 17.Eng LF, Vanderhaeghen JJ, Bignami A, Gerstl B. An acidic protein isolated from fibrous astrocytes. Brain Res. 1971;28:351–354. doi: 10.1016/0006-8993(71)90668-8. [DOI] [PubMed] [Google Scholar]
- 18.Fisher RS, Buchwald NA, Hull CD, Levine MS. GABAergic basal forebrain neurons project to the neocortex: the localization of glutamic acid decarboxylase and choline acetyltransferase in feline corticopetal neurons. J Comp Neurol. 1988;272:489–502. doi: 10.1002/cne.902720404. [DOI] [PubMed] [Google Scholar]
- 19.Gritti I, Mainville L, Jones BE. Codistribution of GABA with acetylcholine-synthesizing neurons in the basal forebrain of the rat. J Comp Neurol. 1993;329:438–457. doi: 10.1002/cne.903290403. [DOI] [PubMed] [Google Scholar]
- 20.Haberly LB, Price JL. Association and commissural fiber systems of the olfactory cortex of the rat. I. Systems originating in the piriform cortex and adjacent areas. J Comp Neurol. 1978;178:711–740. doi: 10.1002/cne.901780408. [DOI] [PubMed] [Google Scholar]
- 21.Halliwell JV, Adams PR. Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res. 1982;250:71–92. doi: 10.1016/0006-8993(82)90954-4. [DOI] [PubMed] [Google Scholar]
- 22.Halonen T, Tortorella A, Zrebeet H, Gale K. Posterior piriform and entorhinal cortex relay seizures evoked from the area tempestas: role of excitatory and inhibitory amino acid receptors. Brain Res. 1994;548:248–255. doi: 10.1016/0006-8993(94)90328-x. [DOI] [PubMed] [Google Scholar]
- 23.Hill JA, Zoli M, Bourgeois J, Changeux J. Immunocytochemical localization of a neuronal nicotinic receptor: the β2-subunit. J Neurosci. 1993;13:1551–1568. doi: 10.1523/JNEUROSCI.13-04-01551.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hosli L, Hosli E, Briotta GD, Quadri L, Heuss L. Action of acetylcholine, muscarine, nicotine, and antagonists on the membrane potential of astrocytes in cultured rat brainstem and spinal cord. Neurosci Lett. 1988;92:165–170. doi: 10.1016/0304-3940(88)90054-7. [DOI] [PubMed] [Google Scholar]
- 25.Krnjevic K, Ropert N. Septo-hippocampal pathway modulates hippocampal activity by a cholinergic mechanism. Can J Physiol Pharmacol. 1981;59:911–914. doi: 10.1139/y81-140. [DOI] [PubMed] [Google Scholar]
- 26.Krnjevic K. Acetylcholine as modulator of amino-acid-mediated synaptic transmission. In: Kenny AD, Lombardini BJ, editors. The role of peptides and amino acids as neurotransmitters, Vol 68. Liss; New York: 1981. pp. 127–141. [PubMed] [Google Scholar]
- 27.Lamour Y, Dutar P, Jobert A. Cortical projections of the nucleus of the diagonal band of Broca and of the substantia innominata in the rat: an anatomical study using the anterograde transport of a conjugate of wheat germ agglutinin and horseradish peroxidase. Neuroscience. 1984;12:395–408. doi: 10.1016/0306-4522(84)90061-7. [DOI] [PubMed] [Google Scholar]
- 28.Libri V, Constanti A, Calaminici M, Nistico G. A comparison of the muscarinic response and morphological properties of identified cells in the guinea-pig olfactory cortex in vitro. Neuroscience. 1994;59:331–347. doi: 10.1016/0306-4522(94)90600-9. [DOI] [PubMed] [Google Scholar]
- 29.Madison DV, Lancaster B, Nicoll RA. Voltage clamp analysis of cholinergic action in the hippocampus. J Neurosci. 1987;7:733–741. doi: 10.1523/JNEUROSCI.07-03-00733.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin LA, Alger BE. Muscarinic receptor activation of interneurons susceptible to depolarization-induced suppression of inhibition (DSI). Soc Neurosci Abstr. 1996;22:1989. [Google Scholar]
- 31.McCormick DA, Prince DA. Two types of muscarinic response to acetylcholine in mammalian cortical neurons. Proc Natl Acad Sci USA. 1985;82:6344–6348. doi: 10.1073/pnas.82.18.6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meric PH, Barrere B, Peres M, Gillet B, Berenger G, Beloeil JC, Seylaz J. Effects of kainate-induced seizures on cerebral metabolism. A. Combined 1H and 31P NMR study in the rat. Brain Res. 1994;638:53–60. doi: 10.1016/0006-8993(94)90632-7. [DOI] [PubMed] [Google Scholar]
- 33.Millan MH, Patel S, Mello LM, Meldrum BS. Focal injection of 2-amino-7-phosphonoheptanoic acid into prepiriform cortex protects against pilocarpine-induced limbic seizures in rats. Neurosci Lett. 1986;70:69–74. doi: 10.1016/0304-3940(86)90439-8. [DOI] [PubMed] [Google Scholar]
- 34.Murphy S, Pearce B, Morrow C. Astrocytes have both M1 and M2 muscarinic receptor subtypes. Brain Res. 1986;364:177–180. doi: 10.1016/0006-8993(86)91000-0. [DOI] [PubMed] [Google Scholar]
- 35.Nicoll RA. The coupling of neurotransmitter receptors to ion channels in the brain. Science. 1988;241:545–551. doi: 10.1126/science.2456612. [DOI] [PubMed] [Google Scholar]
- 36.Nieto-Sampedro M, Berman MA. Interleulin-1-like activity in rat brain: sources, targets, and effect of injury. J Neurosci Res. 1987;17:214–219. doi: 10.1002/jnr.490170303. [DOI] [PubMed] [Google Scholar]
- 37.Oh TH, Markelonis J, Von Visger JR, Baik B, Shipley MT. Acidic pH rapidly increases immunoreactivity of glial fibrillary acidic protein in cultured astrocytes. Glia. 1995;13:319–322. doi: 10.1002/glia.440130408. [DOI] [PubMed] [Google Scholar]
- 38.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1986. [DOI] [PubMed] [Google Scholar]
- 39.Petras JM (1990) Brain damage induced by soman intoxication. Proceedings of the Workshop on Convulsions and Related Brain Damage Induced by Organophosphorus Agents, May, p 19.
- 40.Piredda S, Gale K. A crucial epileptogenic site in the deep piriform cortex. Nature. 1985;317:623–625. doi: 10.1038/317623a0. [DOI] [PubMed] [Google Scholar]
- 41.Piredda S, Gale K. Role of excitatory amino acid transmission in the genesis of seizures elicited from deep prepiriform cortex. Brain Res. 1986;377:205–210. doi: 10.1016/0006-8993(86)90859-0. [DOI] [PubMed] [Google Scholar]
- 42.Pitler TA, Alger BE. Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci. 1992a;12:4122–4132. doi: 10.1523/JNEUROSCI.12-10-04122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pitler TA, Alger BE. Cholinergic excitation of GABAergic interneurons in the rat hippocampal slice. J Physiol (Lond) 1992b;450:127–142. doi: 10.1113/jphysiol.1992.sp019119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pitler TA, Alger BE. Depolarization-induced suppression of GABAergic inhibition in rat hippocampal pyramidal cells: G-protein involvement in a presynaptic mechanism. Neuron. 1994;13:1447–1455. doi: 10.1016/0896-6273(94)90430-8. [DOI] [PubMed] [Google Scholar]
- 45.Rye DB, Wainer BH, Mesulam MM, Mufson EJ, Saper CB. Cortical projections arising from the basal forebrain: a study of cholinergic and non-cholinergic components employing combined retrograde tracing and immunohistochemical localization of choline acetyltransferase. Neuroscience. 1984;13:627–643. doi: 10.1016/0306-4522(84)90083-6. [DOI] [PubMed] [Google Scholar]
- 46.Salm AK, McCarthy KD. Expression of β-adrenergic receptors by astrocytes isolated from adult rat cortex. Glia. 1989;2:346–352. doi: 10.1002/glia.440020507. [DOI] [PubMed] [Google Scholar]
- 47.Salm AK, McCarthy KD. The evidence for astrocytes as a target for central noradrenergic activity: expression of adrenergic receptors. Brain Res Bull. 1992;29:265–275. doi: 10.1016/0361-9230(92)90056-4. [DOI] [PubMed] [Google Scholar]
- 48.Sutula T, He XX, Cavazos J, Scott G. Synaptic reorganization in the hippocampus induced by abnormal functional activity. Science. 1988;239:1147–1150. doi: 10.1126/science.2449733. [DOI] [PubMed] [Google Scholar]
- 49.Switzer III RC, Campbell SK, Murphy MR, Kerenyi SZ, Miller SA, Hartgraves SK (1990) Soman-induced convulsions and brain damage as a function of chronic and acute exposure in rats and diazepam therapy in rhesus monkeys. Proceedings of the Workshop on Convulsions and Related Brain Damage Induced by Organophosphorus Agents, May, pp 33–67.
- 50.Turski L, Ikonomidou C, Turski WA, Bortolotto ZA, Cavalheiro EA. Cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine [review]. Synapse. 1989;3:154–171. doi: 10.1002/syn.890030207. [DOI] [PubMed] [Google Scholar]
- 51.Van Der Zee EA, De Jong GI, Strosberg AD, Luiten PGM. Muscarinic acetylcholine receptor expression in astrocytes in the cortex of young and aged rats. Glia. 1993;8:42–50. doi: 10.1002/glia.440080106. [DOI] [PubMed] [Google Scholar]
- 52.van der Zee EA, Streefland C, Strosberg AD, Schroder H, Luiten PGM. Visualization of cholinoceptive neurons in the rat neocortex: colocalization of muscarinic and nicotinic acetylcholine receptors. Mol Brain Res. 1992;14:326–336. doi: 10.1016/0169-328x(92)90100-p. [DOI] [PubMed] [Google Scholar]
- 53.Van Ooteghem SA, Shipley MT. Factors affecting the sensitivity and consistence of the Koelle–Friedenwald histochemical method for localization of acetylcholinesterase. Brain Res Bull. 1984;12:543–553. doi: 10.1016/0361-9230(84)90170-9. [DOI] [PubMed] [Google Scholar]
- 54.Vertes RP. Brainstem afferents to the basal forebrain in the rat. Neuroscience. 1988;24:907–935. doi: 10.1016/0306-4522(88)90077-2. [DOI] [PubMed] [Google Scholar]
- 55.Wall HG, Jaax NK, Hayward IJ (1990) Motor activity and brain lesions in soman-intoxicated rhesus monkeys. Proceedings of the Workshop on Convulsions and Related Brain Damage Induced by Organophosphorus Agents, May, pp 21–29.
- 56.Wamsley JK, Zarbin MA, Birdsall JM, Kuhar MJ. Muscarinic cholinergic receptors: autoradiographic localization of high and low affinity agonist binding sites. Brain Res. 1980;200:1–12. doi: 10.1016/0006-8993(80)91089-6. [DOI] [PubMed] [Google Scholar]
- 57.Wenk GL, Stoehr JD, Quintana G, Mobley S, Wiley RG. Behavioral, biochemical, histological, and electrophysiological effects of 192-IgG-saporin injections into the basal forebrain of rats. J Neurosci. 1994;14:5986–5995. doi: 10.1523/JNEUROSCI.14-10-05986.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wiley R, Oeltmann T, Lappi D. Immunolesioning: selective destruction of neurons using immunotoxin to rat NGF receptor. Brain Res. 1991;562:149–153. doi: 10.1016/0006-8993(91)91199-b. [DOI] [PubMed] [Google Scholar]
- 59.Woolf NJ, Hernit MC, Butcher LL. Cholinergic and non-cholinergic projections form the rat basal forebrain revealed by combined choline acetyltransferase and Phaseolus vulgaris leukoagglutinin immunohistochemistry. Neurosci Lett. 1986;66:281–286. doi: 10.1016/0304-3940(86)90032-7. [DOI] [PubMed] [Google Scholar]
- 60.Zimmer LA, Ennis M, El-Etri M, Shipley MT. Anatomical localization and time course of Fos expression following soman-induced seizures. J Comp Neurol. 1997a;378:468–481. [PubMed] [Google Scholar]
- 61.Zimmer LA, Ennis M, Shipley MT. Soman-induced seizures rapidly activate astrocytes and microglia in discrete brain regions. J Comp Neurol. 1997b;378:482–492. doi: 10.1002/(sici)1096-9861(19970224)378:4<482::aid-cne4>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]