

Abstract
Ciliary neurotrophic factor (CNTF) acts instructively to switch multipotent stem cells of the CNS to an astrocytic fate. Here we show that CNTF causes activation of janus kinase–signal transducers and activators of transcription and mitogen-activated protein kinase (MAPK) pathways with differential kinetics in these cells. Inhibition studies indicate that activation of the MAPK pathway is required early in the differentiation process, whereas activation of signal transducer and activator of transcription (STAT) proteins is required for commitment to an astrocytic fate. Bone morphogenetic proteins have also been shown to cause astrocytic differentiation but do not cause STAT activation or astrocytic differentiation in fibroblast growth factor 2-expanded fetal stem cells used here. These results show that there are two distinct routes to initiate astrocytic commitment in multipotent CNS precursors.
Keywords: astrocytic differentiation, CNS stem cells, mammalian CNS development, intracellular signaling, CNTF, BMP4, EGF
Multipotent cells that give rise to neurons, astrocytes, and oligodendrocytes have been defined using both monolayer and aggregate culture systems. We have used stem cells isolated from rat embryos to study the molecular correlates of differentiation leading to the development of astrocytes. When maintained in a proliferative state in fibroblast growth factor (FGF), these cells remain multipotent and can differentiate into the three major cell types of the CNS: neurons, astrocytes, and oligodendrocytes. Ciliary neurotrophic factor (CNTF) causes 98% of these cells to differentiate into astrocytes, as specified by glial fibrillary acidic protein (GFAP) expression (Johe et al., 1996). Epidermal growth factor (EGF) also promotes astrocytic differentiation as EGF-expanded CNS stem cells yield a higher number of astrocytes on mitogen withdrawal (Johe et al., 1996). Bone morphogenetic proteins (BMPs) have been shown to induce astrocytic differentiation in cells that are isolated from the fetal subventricular zone and grown as aggregates in the presence of EGF (Gross et al., 1996). Both CNTF and BMP are thought to act instructively on multipotent cells, committing them to an astrocytic fate. The robust response to CNTF provides a technically accessible experimental paradigm for a biochemical dissection of the mechanism of lineage determination by CNTF.
CNTF signaling is accomplished through several pathways. The best-defined are the signal cascades that involve the janus kinases (JAKs) and the mitogen-activated protein kinase (MAPK) pathway. Signaling is initiated by members of the JAK family of tyrosine kinases, specifically Jak1, Jak2, and Tyk2 (Lutticken et al., 1994;Stahl et al., 1994). On activation, Src homology 2 (SH2) domain proteins bind to the receptor. These proteins include the signal transducers and activators of transcription (STAT) proteins (Stahl et al., 1995), activation of which leads to direct transcriptional activation (Symes et al., 1994). Activated Jak2 can also cause the activation of MAPK possibly through a ras–raf pathway that activates transcription factors including cAMP response element-binding protein (CREB) and TCF/Elk (Winston and Hunter, 1996). STATs have also been shown to be activated by EGF (Fu and Zhang, 1993; Schindler and Darnell, 1995), and this activation could be involved in astrocytic differentiation. In contrast, BMPs signal by activating Sma-MAD homolog (SMAD) proteins, a distinct family of transcriptional regulators (Lagna et al., 1996; Liu et al., 1996).
Effects of CNTF are mirrored by leukemia inhibitory factor (LIF) in CNTF-responsive cells, because the signaling moieties of the two receptors are identical. Mice lacking LIF receptor β (LIFR-β) show a phenotype of decreased numbers of astrocytes (Ware et al., 1995). In this report we show that activation of both the MAPK and JAK–STAT pathways are positively coupled to astrocytic differentiation in vitro. Inhibition of activation of the MAPK pathway delays the initiation of differentiation, whereas inhibiting Stat3 function causes a total block in astrocytic differentiation. However, there is no activation of the JAK–STAT system by either EGF or BMP4 in the cell system studied here. These results show that distinct molecular mechanisms initiate astrocytic commitment in response to BMP4 and CNTF in multipotent CNS precursors.
MATERIALS AND METHODS
Materials
DMEM/F-12 was obtained from Life Technologies (Gaithersburg, MD). Insulin, transferrin, and fibronectin were from Intergen (Purchase, NY), and progesterone, selenium, putrescine, polyornithine, 1,4-diazabicyclo-(2.2.2)-octane (DABCO), Tween 20, bovine serum albumin, and dimethyl sulfoxide (DMSO) were from Sigma (St. Louis, MO). Horseradish peroxidase-tagged secondary antibodies for enhanced chemiluminescence (ECL) and poly(dI-dC) were from Boehringer Mannheim (Indianapolis, IN). CNTF, EGF, and FGF were obtained from R & D Systems (Minneapolis, MN), and BMP4 was from Genetics Institute (Cambridge, MA). PD098059 was obtained from Research Biochemicals (Natick, MA). The ECL kit was obtained from Pierce (Rockford, IL). The following were the antibodies used in this study: GFAP monoclonal from ICN (Irvine, CA), GFAP polyclonal from Chemicon (Temecula, CA), antibody against the FLAG epitope from Eastman Kodak (Rochester, NY), Stat1 monoclonal α-ISGF3 (anti-ISGF3) and α-pan-ERK antibody from Transduction Laboratories (Lexington, KY), and Stat3 polyclonal from Santa Cruz Biotechnology (Santa Cruz, CA). The appropriate fluorescence-tagged secondary antibodies for immunofluorescence were obtained from Jackson ImmunoResearch (West Grove, PA).
Culture of neuroepithelial stem cells and differentiation protocol
Cultures of stem cells were prepared according to the protocol of Johe et al. (1996). Briefly, cerebral cortices were dissected from embryonic day 14 (E14) rat embryos; cells were mechanically dissociated by trituration and were plated on dishes coated with 15 mg/ml polyornithine and 1 mg/ml fibronectin at a concentration of 1 million per 10 cm dish. Cells were maintained in 10 ng/ml FGF. Cultures were passaged on approximately the fourth day. Experiments were performed on cultures either in the first or second passage.
Cultures were differentiated into astrocytes by treating with 10 ng/ml CNTF.
Electrophoretic mobility shift assay
The preparation of nuclear extracts from treated and untreated cells was prepared according to the protocol of Symes et al. (1994). All treatments were performed at 10 ng/ml except for EGF, which was performed at 20 ng/ml. Cells were treated with CNTF every 24 hr for time points that were >1 d. Electrophoretic mobility shift assay (EMSA) was also performed according to the protocol of Symes et al. (1994). The sequence of the SIE probe used was 5′-AGCTTCATTTCCCGTAAATCCCTA-3′ and 3′-AGTAAAGGGCATTTAGGGATTGA-5′. For supershift assays, the relevant antibody was included in the binding reaction.
Immunofluorescence
Double-immunofluorescent staining of FLAG-Stat3 and GFAP. Cells were fixed with freshly mixed methanol and acetone in a 1:1 ratio for 2 min at room temperature. They were washed four times in Tris-buffered saline (TBS) and calcium (0.05 m Tris HCl, pH 7.4, 0.15 m NaCl, and 1 mmCaCl2). Double immunofluorescence was performed with a biotinylated anti-FLAG monoclonal antibody at 30 μg/ml and anti-GFAP polyclonal antibody at a 100-fold dilution. Primary antibodies were left on the cells for 1 hr at room temperature or overnight at 4°C. The cells were then washed with TBS and calcium for 1 hr with four changes and incubated with streptavidin–FITC- and lissamine rhodamine-coupled goat anti-rabbit antibody (100-fold dilution) for 30 min to 1 hr at room temperature. After four more washes cells were mounted in 70% glycerol in PBS with 2% DABCO.
GFAP immunofluorescent staining. Cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature. After three washes in PBS they were incubated in blocking solution (PBS with 0.1% Triton X-100 and 5% normal goat serum) for 1 hr. Cells were then incubated with monoclonal GFAP antibody at a 200-fold dilution for 2 hr at room temperature. After four washes in PBS with 0.1% Triton X-100 (PBST), cells were incubated with a 100-fold diluted solution of FITC-coupled donkey anti-mouse antibody for 1 hr at room temperature. After four more washes in PBST and one wash in PBS, cells were mounted in 70% glycerol and 2% DABCO. Dilution of all antibodies was performed in the blocking solution.
Nestin immunofluorescent staining. The protocol used was identical to the GFAP staining. Cells fixed in 4% paraformaldehyde were stained with a nestin antibody at a 500-fold dilution. Appropriate secondary antibodies were used at a 100-fold dilution.
Immunoblotting
Cells were washed once in PBS and lysed in NP-40 lysis buffer (50 mm Tris HCl, pH 8.0, 170 mm NaCl, and 0.5% NP-40). After a 20 min incubation, extracts were cleared by centrifuging at 14,000 rpm in a Microfuge for 20 min. All operations were performed at 4°C. Extracts were fractionated on 12.5% SDS-PAGE (acrylamide/bisacrylamide ratio of 30:0.2), and the separated proteins were transferred onto nitrocellulose filters. After blocking in TBS-Tween (TBST; 20 mm Tris, pH 7.6, 137 mmNaCl, and 0.1% Tween 20) with 2% bovine serum albumin, filters were incubated with primary antibody at a 1000-fold dilution for 3 hr at room temperature. After three washes with TBST, the appropriate secondary antibody conjugated to horseradish peroxidase was used at a 20,000-fold dilution, and ECL was performed according to the manufacturer’s instructions. All antibody dilutions were performed in the blocking solution.
Transfection, infection, and PD098059 experiments
Transfections were performed on cells in first passage by the calcium phosphate method. The plasmids used were the wild-type (WT) Stat3 and a DNA-binding mutant with a three-amino acid mutation at residues 462–464 (VVV→AAA). Both plasmids had an N-terminal FLAG and were described by Horvath et al. (1995). The precipitate was left on the cells for 2 hr; the cells were washed and left in FGF for 48 hr. Cells were then treated for 48 hr with 10 ng/ml CNTF.
Infections and PD098059 experiments were performed on cells in first or second passage. An adenovirus vector expressing a constitutively activated form of MAPK kinase (MAPKK) was used. The activated form of the protein has a two-amino acid substitution (S218E and S222E) and its nuclear localization signal is deleted (deletion 32–51). Cells were infected at the indicated multiplicity of infection and fixed 24 or 48 hr after infection.
A 20 mm stock of PD098059 was made in DMSO. Cells were treated at a final concentration of 10 or 20 μm for 24 or 48 hr before fixing in paraformaldehyde. For the 48 hr time points, cells were retreated with CNTF and PD098059 at 24 hr. The CNTF-treated cells that serve as control were treated with the relevant amount of DMSO. There is no appreciable difference in differentiation of DMSO-treated cells compared with untreated cells (data not shown).
RESULTS
CNTF causes differentiation of stem cells into astrocytes
Stem cells maintained in FGF were homogeneous in morphology and in their expression of nestin, a marker of neuroepithelial stem cells (Lendahl et al., 1990). A transient treatment with CNTF is sufficient to commit stem cells to an astrocytic fate in vitro (Johe et al., 1996). Exposure to CNTF causes these cells to express GFAP (Fig.1). Consistent with in vivoobservations, the intensity of nestin staining decreased with an increase in GFAP expression (Hockfield et al., 1985; Frederiksen et al., 1988).
Fig. 1.

CNTF causes differentiation of stem cells into astrocytes. Cultures of FGF-expanded stem cells were prepared as described previously (Johe et al., 1996) and treated for 2 d with 10 ng/ml CNTF. After fixation cells were stained with a nestin antibody (a stem cell marker; A, C) and α-GFAP antibody (an astrocytic marker; B, D). Scale bar: D, 100 μm. There is no GFAP expression when cells are maintained in FGF. These cells are uniformly positive for nestin. GFAP is expressed after CNTF treatment. Several of the differentiated cells also express nestin, but nestin is downregulated in cells intensely fluorescent for GFAP.
STAT proteins are activated by CNTF in neuroepithelial stem cells
One of the major cytoplasmic signaling pathways activated by CNTF is JAK–STAT. Activation of STAT proteins involves phosphorylation of a specific tyrosine residue (Shuai et al., 1993, 1994). Activated STAT proteins can be detected by their ability to dimerize and bind specific DNA target sites. This interaction can be detected by an EMSA. Specific nucleoprotein complexes were formed when a STAT-specific DNA-binding sequence (SIE) was incubated with nuclear proteins prepared from stem cells treated with CNTF. Complexes labeled “a” and “b” were formed when the cells were treated with CNTF for times ranging from 15 min to 8 d (Fig. 2). The amount of complex b formed decreased with time, whereas complex a was activated for prolonged periods. The constitutive complex that migrates just below complex b is present even in FGF-treated cells and is not induced by CNTF treatment. Although there is some Stat3 complex in the control conditions, indicating basal levels of activation of this protein in the cell, these data clearly show that CNTF rapidly activates proteins that bind to the SIE probe.
Fig. 2.

STAT proteins are activated by CNTF in neuroepithelial stem cells. Stem cell cultures were treated with FGF and CNTF either separately or in combination and nuclear proteins were prepared from these cells at the time points mentioned. Cells were treated every 24 hr for time points >1 d. Cells were mechanically removed from the culture dish in PBS, and nuclear proteins were prepared. EMSA assays were then performed to determine the extent of activation of the STAT proteins in these extracts. The complexes formed were resolved on a 6% nondenaturing polyacrylamide gel with 0.5× TBE buffer at 4°C. The positions of the complexes formed as a result of CNTF treatment are marked a and b. There is prolonged activation of STAT proteins as seen by the continued appearance of complex a and, to a lesser extent, complexb.
The GFAP promoter may be used as a paradigm to study transcriptional regulation during astrocytic differentiation, because it is robustly and specifically expressed in these cells. Promoter analysis of the GFAP promoter in CG4 cells showed that regions −1857 to −1546 and −384 to −106 are important for CNTF-induced GFAP expression (Kahn et al., 1997). Another report of promoter analysis of the GFAP promoter in U251 cells showed the following regions to be necessary and sufficient for GFAP expression: −1757 to −1604, −1612 to −1489, and −132 to −57 (Besnard et al., 1991). On examination of the GFAP promoter we identified three putative STAT sites: −1512 to −1504, TTCCGAGAA; −1292 to −1284, TTCCCAGAA; and 277 to 285, TTCCTGGAA; one of which (−1512 to −1504) is stronger in STAT complex formation by EMSA than the other two (data not shown). These observations suggest that GFAP induction by CNTF may be mediated by direct interaction of STAT complexes with the GFAP promoter.
The activated STAT proteins are Stat1 and Stat3
Supershifting of nucleoprotein complexes with antibodies against specific transcription factors indicate the involvement of these proteins in the formation of that complex. The identity of the STAT proteins activated by CNTF was established by supershift assays (Fig.3). Complex b was supershifted by the Stat1 antibody, forming complex c, indicating that Stat1 is a part of complex b (Fig. 3a, lane 4). An α-Stat3 antibody caused the disappearanceof both complexes a and b, forming the supershifted complex d (Fig. 3b, lane 8). Thus, complex a comprises homodimers of Stat3 complexed with DNA, whereas complex b comprises heterodimers of Stat1 and Stat3 with DNA.
Fig. 3.

The STAT proteins activated are Stat1 and Stat3.a, Extracts prepared from cells treated with CNTF for 15 min were used here. EMSA was performed as described in the legend to Figure 2, except for the inclusion of either a relevant antibody or unlabeled SIE oligomer DNA. a, lanes 3, 4, An antibody against Stat1 (α-Stat1) was included in the incubation during complex formation. The resultant supershifted complexes are indicated by c. The efficiency of formation of complex b is simultaneously reduced (compare with control, lane 2). b, Similarly, complex formation in lanes 7 and8 was performed in the presence of Stat3 antibody (αStat3). A supershifted band d forms, with a simultaneous reduction in complexes a andb (compare lanes 6, 8). c, EMSA was performed with a 20-fold excess of unlabeled SIE oligomer. There is a decrease in the intensity of complexes a andb and the constitutive complex formed in the presence of the competitor DNA.
The specificity of the complexes formed was determined by competition with 20-fold excess of unlabeled SIE oligo (Fig. 3c,lanes 11, 12). Both the induced (complexes a and b) and the constitutive complexes were competed with the unlabeled oligo, indicating that both are specific. However, the constitutive complex does not contain either Stat1 or Stat3, as shown by the supershift assays. There was no competition with an unrelated probe of similar length (data not shown).
CNTF induces the formation of three complexes in several other systems studied (Bonni et al., 1993; Symes et al., 1994; Rajan et al., 1995,1996). The largest is composed of Stat3 homodimers, the middle of Stat1 and Stat3 heterodimers, and the fastest-migrating one of Stat1 homodimers. From these previous observations it is probable that a third complex comprising homodimers of Stat1 migrates at the same position as the constitutive complex that forms just below complex b. It is of interest to note that there are two distinct complexes supershifted by the Stat1 antibody, indicating that there are two species of Stat1 activated by CNTF treatment in these cells. These results show that both Stat1 and Stat3 are activated by CNTF in the stem cells.
Blocking of Stat3 function prevents astrocytic differentiation by CNTF
The requirement of STAT activation in CNTF-mediated astrocytic differentiation was determined by assaying the effect of blocking Stat3 transcriptional function. Stat3 was targeted because EMSA indicated that it was the major protein that was activated. Plasmids expressing FLAG-tagged WT and mutant forms of the Stat3 protein (Horvath et al., 1995) were transfected into cells. The mutant Stat3 has a three-amino acid substitution in the DNA-binding domain, thus functioning as a dominant negative because of loss of DNA-binding function. The effect of the overexpressed transfected proteins on astrocytic differentiation was determined by double immunofluorescence with antibodies against GFAP and the FLAG epitope (Fig.4a). Transfection of WT Stat3 allows astrocytic differentiation in 80% of the cells, whereas differentiation takes place in only ∼25% of cells transfected with the mutant Stat3 (Fig. 4b), indicating that Stat3 function is required for astrocytic differentiation. The fact that differentiation is not 100% in the WT transfectants and 0% in the mutant one could be attributable to the fact that perturbations in this important signaling pathway cause responses of the cells to be skewed or, in the case of the mutant transfections, that the levels of dominant negative protein expressed are not sufficient to override the effect of the endogenous WT protein.
Fig. 4.
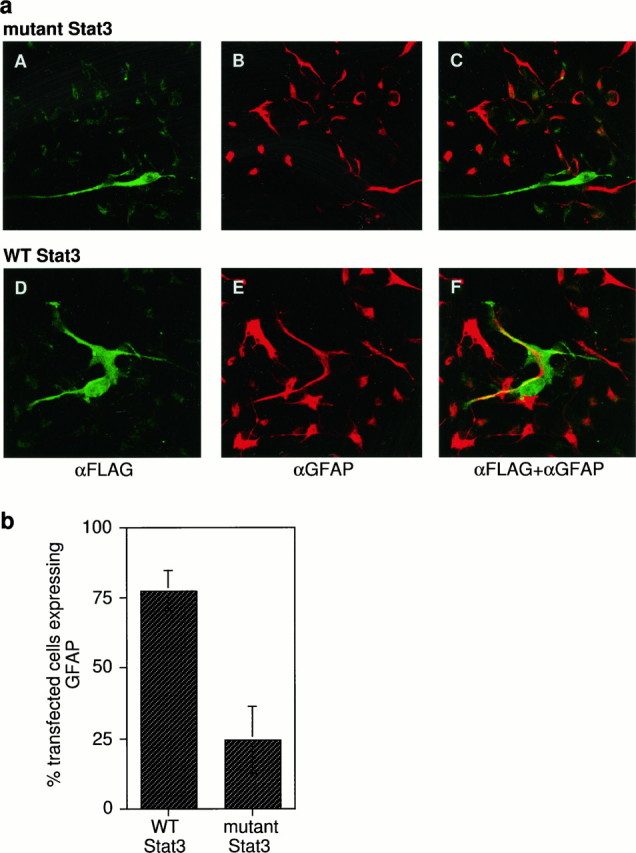
Effect of blocking Stat3 function on astrocytic differentiation. a, Cells were transiently transfected by the calcium phosphate method with plasmids expressing either wild-type (WT) or dominant negative mutant of Stat3 defective for DNA binding (mutant), both proteins expressing an N-terminal FLAG epitope. Forty-eight hours after transfection cells were treated with CNTF for 48 hr and fixed with freshly mixed methanol and acetone in a 1:1 ratio for 2 min at room temperature, and double-immunofluorescent staining was performed for FLAG and GFAP. A–C, Representative experiment of cells transfected with the mutant Stat3. D–F, Representative experiment of cells transfected with WT Stat3. A, D show staining specific for the FLAG epitope on the transfected proteins,B, E for GFAP, and C, F for both antigens. b, Based on experiments performed as described in Figure 3a, the percentage of transfected cells expressing GFAP was plotted for each of the two plasmids. Blocking of Stat3 function with a dominant negative protein inhibits CNTF-mediated astrocytic differentiation.
CNTF treatment causes activation of MAPK in neuroepithelial stem cells
Activated Jak2 can cause the activation of MAPK, possibly through a ras–raf pathway (Winston and Hunter, 1996). To determine whether CNTF activates the MAPK pathway in the stem cells, immunoblots were performed on cell extracts treated with CNTF for time points ranging from 15 min to 24 hr. CNTF causes activation of p44 MAPK in the neuroepithelial stem cells (Fig. 5). A time course shows that this activation is transient and peaks at 15–30 min. The levels of activation return to baseline by 4 hr after CNTF treatment (Fig. 5, compare lanes 9, 6). FGF-treated cells were used as a positive control because FGF is a known activator of MAPK.
Fig. 5.

CNTF activates MAP kinase in the neuroepithelial stem cells. NP-40 extracts of cells treated with FGF (lanes 1–5) or CNTF (lanes 6–9) for the mentioned time points were resolved on 12.5% SDS-PAGE, proteins were blotted on nitrocellulose, and filters were probed with an anti-pan-ERK antibody. After incubation with the appropriate horseradish peroxidase, coupled secondary antibody protein bands were visualized with ECL. The major protein seen is the p44 MAP kinase, the phosphorylated form moving as the heavier band at time points in which activation is elicited. Activation of MAP kinase is slightly prolonged after FGF treatment when compared with CNTF treatment. Activation of MAP kinase is completely back to control levels (Mock, lane 6) at 4 hr after CNTF treatment (lane 9).
Inhibition of activation of MAPKK causes a delay in astrocytic differentiation by CNTF
To determine the relevance of MAPK activation in astrocytic differentiation, activation of MAPK was blocked by inhibiting the activation of its upstream activator, MAPKK. PD098059 is a specific inhibitor of MAPKK at concentrations of ≤50 μm (Alessi et al., 1995). The effect of adding PD098059 to cells differentiating in response to CNTF was determined, as shown in Figure6, a and b. Addition of CNTF to cells in which activation of MAPKK has been inhibited leads to a complete lack of differentiation at 24 hr after the addition of CNTF (Fig. 6a, compare C,D with B). However, by 48 hr the extent of differentiation in CNTF-treated cells is comparable both in the presence and absence of PD098059 (Fig. 6a, compareK, L with J). Thus there is a delay in the onset of differentiation by CNTF in the absence of normal levels of MAPK activation.
Fig. 6.
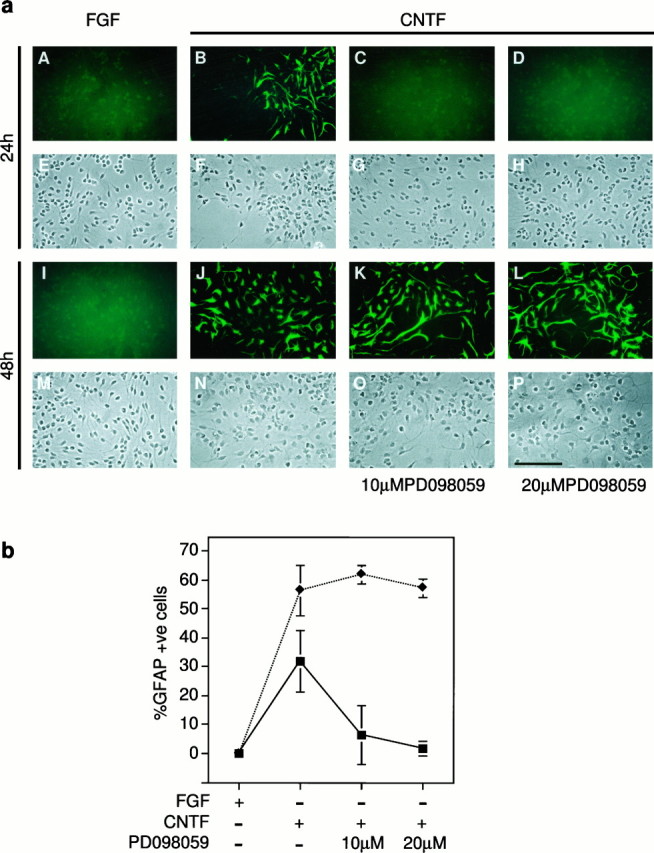
Blocking MAPK activation causes a delay in astrocytic differentiation. a, MAPKK inhibitor PD098059 was used to block MAPK activation. Cells were treated with CNTF for 24 or 48 hr in the presence or absence of 10 or 20 μmPD098059, fixed with 4% paraformaldehyde, and stained for GFAP. For the 48 hr time points, cells were retreated with CNTF and PD098059 at 24 hr. A representative experiment of the data plotted inb is shown. A–D, I–L, Immunofluorescent staining of GFAP. E–H,M–P, Corresponding bright-field (phase) images of these fields, E being the same field as A and so on. A–H, Images of cells fixed 24 hr after treatment and I–P at 48 hr after treatment. Scale bar:P, 100 μm. b, Average percentage of GFAP-positive cells was plotted for each condition.Squares, 24 hr time points; diamonds, 48 hr time points. Astrocytic differentiation is prevented by PD098059 only at the 24 hr time point.
The CNTF-mediated activation of MAPK in these cells peaks at 15 min, whereas the effect of its inhibition is seen at 24 hr, indicating that activation of MAPK leads to events downstream that regulate differentiation. The eventual differentiation at 48 hr may be a consequence of the residual activation of MAPK in the cell despite the inhibitor (see Fig. 7a), which is sufficient for differentiation to occur in a delayed manner. These results indicate that one of the earliest steps in CNTF-mediated astrocytic differentiation includes the activation of MAPK.
Fig. 7.
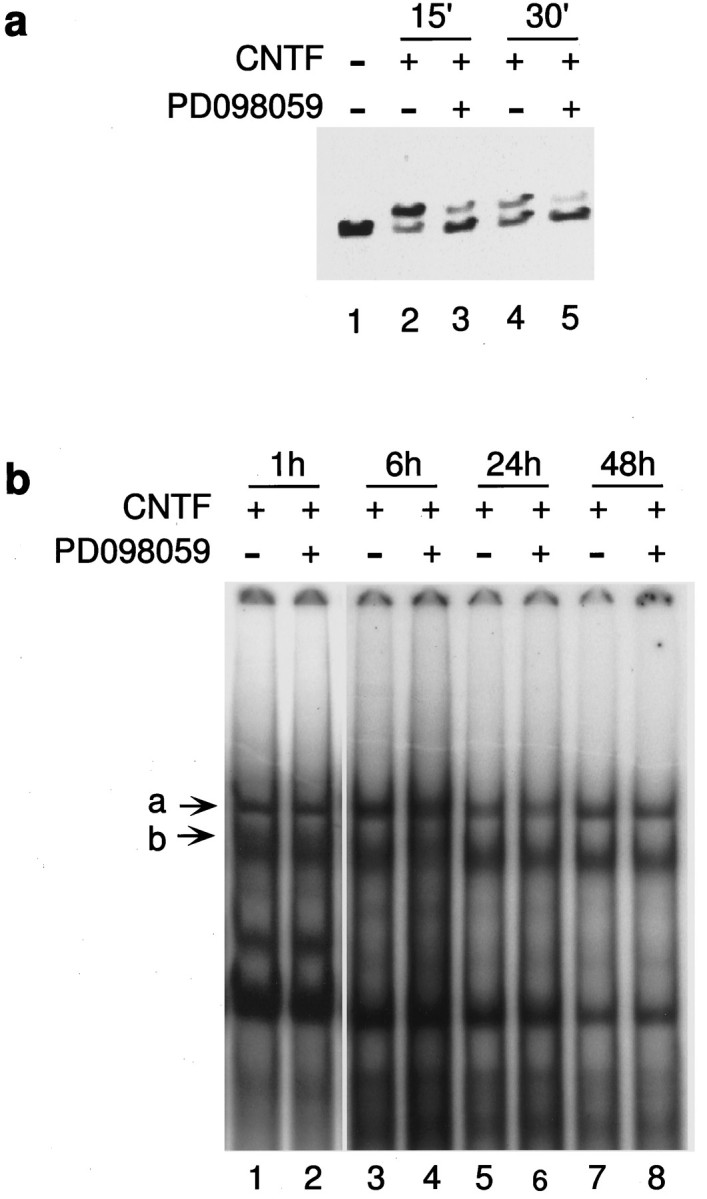
PD098059 inhibits MAPK activation but does not affect STAT activation. a, Cells were treated with CNTF in the presence or absence of 20 μm PD098059 for the indicated time points, and activation of MAPK was analyzed as described in Figure 5. At 20 μm, PD098059 blocks most of the MAPK activation caused by CNTF (compare lanes 3, 2 and5, 4). b, Nuclear extracts prepared from stem cells treated with CNTF for the time points indicated, in the presence or absence of PD098059. EMSA was performed on these extracts as described in Figure 2. The specific complexes formed as a result of CNTF treatment are marked a andb. There is no inhibition of STAT complex formation seen at early (1h, lanes 1, 2) or late (48h, lanes 7, 8) time points in the presence of 20 μm PD098059.
Overexpression of constitutively active MAPKK causes transient GFAP expression
Since the previous set of experiments established that MAPK is required in the initiation of astrocytic differentiation, we investigated the effects of aberrant activation of MAPKK in the absence of CNTF treatment. FGF-expanded stem cells were infected with an adenovirus vector expressing a constitutively activated form of MAPKK. A representative set of experiments is shown in Table1. The result is the inverse of that seen in Figure 6. There was a transient expression of GFAP at 24 hr after infection that was lost at 48 hr. Thus MAPK activation in the absence of other CNTF-activated pathways leads to a transient appearance of a differentiated astrocytic phenotype.
Table 1.
Activated MAPKK causes a transient expression of GFAP in the absence of CNTF treatment
| Condition | MOI | No. of GFAP-positive cells/40 × field | |
|---|---|---|---|
| 24 hr | 48 hr | ||
| Experiment 1 | |||
| Mock | 0.67 | 0.1 | |
| WT | 50 | 0.97 | 5.5 |
| MAPKK | 50 | 11.0 | 1.66 |
| Experiment 2 | |||
| Mock | 2.5 | 0 | |
| WT | 50 | 2.3 | 0 |
| MAPKK | 50 | 8.75 | 0.05 |
Cells were infected with either WT adenovirus or an adenovirus vector expressing a constitutively active form of MAPKK at a multiplicity of infection (MOI) of ∼50. Cells were fixed with 4% paraformaldehyde 24 or 48 hr after infection and stained for GFAP. The number of GFAP-positive cells per 40× field is shown. There is an 11.3-fold increase in GFAP expression at 24 hr after infection in experiment 1, and a 3.8-fold increase in experiment 2. When compared with a WT infection, there is no increase in GFAP expression at 48 hr after infection in an MAPKK infection.
Treatment of stem cells with PD098059 blocks activation of MAPK but does not affect STAT activation
The activation of MAPK by CNTF was appreciably inhibited by PD098059, as seen in Figure 7a. The intensity of the heavier bands denoting activation are reduced in lanes 3 and5 (+PD098059) when compared with lanes 2 and4 (control). Because MAPK and STAT activation both lead to astrocytic differentiation, it is possible that there is an interaction between the two pathways, and the inhibition of differentiation seen in Figure 6 could be attributed to a block of STAT activation by PD098059, via the inhibition of MAPK activity. As detected by EMSA, PD098059 did not affect STAT activation in response to CNTF (Fig. 7b). The amounts of complexes a and b formed are unchanged in the presence and absence of PD098059 at early (1 hr, lanes 1, 2) and late (48 hr, lanes 7, 8) time points. It appears that there is no interaction between the MAPK and JAK–STAT pathways at the level of STAT activation in the FGF-expanded stem cells.
EGF and BMP4 do not activate STAT proteins
EGF treatment of FGF-expanded stem cells leads to astrocytic differentiation (Johe et al., 1996). In addition, EGF-treated aggregates from the fetal subventricular zone show enhanced astrocytic differentiation in response to BMPs 2, 4, and 7 (Gross et al., 1996). These observations raise the possibility that EGF and BMPs might activate the STAT signaling system in CNS stem cells. Treatment of cells with EGF and BMP4 does not cause activation of STAT proteins (Fig. 8, compare lanes 2, 3with lane 1). Because BMP4 causes astrocytic differentiation in EGF-expanded stem cells, activation of STATs by combinations of these factors was determined. There was no enhancement of STAT activation caused by BMP4 or CNTF in the presence of EGF (Fig. 8). These data suggest that the astrocytic differentiation effects of EGF and BMP4 are not mediated through JAK–STAT activation.
Fig. 8.
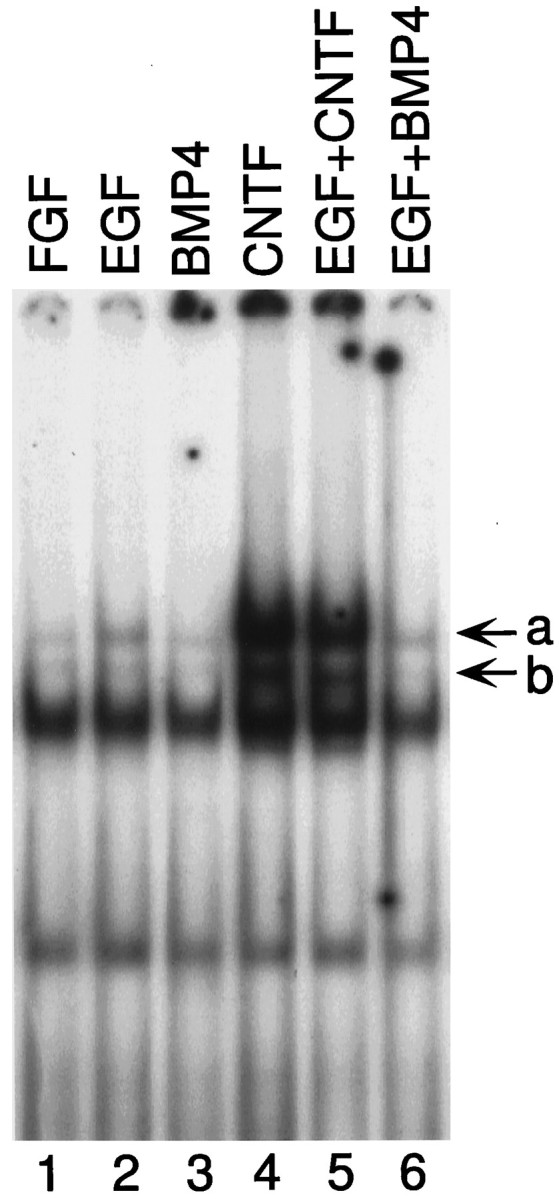
EGF and BMP4 do not activate STAT proteins. Stem cell cultures were treated with the mentioned factors for 15 min, and nuclear extracts were prepared. EMSA assays were performed to determine the extent of activation of STAT proteins as described in Materials and Methods and the legend to Figure 2. STAT complexes are markeda and b. EGF and BMP4 do not activate STAT proteins.
DISCUSSION
FGF-expanded stem cells maintained in monolayers are multipotent and give rise to astrocytes, oligodendrocytes, and neurons on withdrawal of the mitogen (McKay, 1997). All the experiments described here were performed on cultures that were in first or second passage. The cells in these cultures are homogeneous by morphological criteria and nestin immunohistochemistry. It is thus an excellent primary cell culture system in which to study the biochemical mechanisms of differentiation of CNS cells and, particularly, for defining the biochemical basis for the transition from a multipotent stem cell to a committed glial progenitor.
CNTF treatment leads to a prolonged activation of the JAK–STAT pathway and transient activation of the MAPK pathway. The requirement of these pathways in astrocytic differentiation was shown by blocking activation. A dominant negative Stat3 that prevents DNA binding of activated Stat3 homodimers and heterodimers completely blocks astrocytic differentiation, whereas blocking activation of MAPKK, the upstream activator of MAPK, causes a delay in astrocytic differentiation by CNTF. Overexpression of MAPKK causes the converse effect, a transient appearance of GFAP. Thus, the MAPK pathway functions in the initiation of astrocytic differentiation, whereas activation of Stat3 is required for complete differentiation. Stat3 activation as seen by EMSA occurs within 15 min of CNTF treatment. It is likely to be transcriptionally active at this time, thus indicating a function for this protein early in the differentiation process.
Our data suggest that there is interaction between the MAPK and JAK–STAT pathways during CNTF-mediated differentiation. The MAPK pathway has been shown to have an effect on STAT activation (Zhang et al., 1995; Chung et al., 1997) and on transactivation by STATs (Wen et al., 1995). The experiments shown here indicate that the MAPK pathway is not involved in CNTF-mediated STAT activation in FGF-expanded stem cells. Perhaps interaction between the two pathways occurs at the level of the GFAP promoter through an MAPK-sensitive transcriptional regulatory site. In the MAPKK overexpression experiment it appears that activation of MAPK causes a measure of astrocytic differentiation. FGF also causes MAPK activation but does not cause astrocytic differentiation. This may be a consequence of different levels of MAPK activation in FGF-treated and virus-infected cells. However, it may also be the case that FGF stimulates other pathways that inhibit astrocytic differentiation. The different response of the cells to FGF and CNTF is probably the result of the activation of distinct pathways in response to the two factors.
CNTF and LIF have been implicated in astrocytic differentiation in two experimental models. CNTF causes O2A progenitor cells to form astrocytes in vitro (Hughes et al., 1988, Lillien et al., 1990). The number of astrocytes was greatly reduced in E18.5 LIFRβ null animals when compared with a wild-type littermate, implicating LIF in either the survival or differentiation of astrocytes (Ware et al., 1995). In a related model, LIF may be involved in the appearance of astrocytes after CNS injury, because LIF expression is upregulated 30-fold within 24 hr after injury to the CNS (Banner et al., 1997). It is interesting to speculate that the mechanisms by which astrocytic differentiation occurs during embryonic development may be reactivated in an adult cell during injury.
Our results contrast with that of Bonni et al. (1997), who have recently suggested that the JAK–STAT pathway alone is instrumental in causing CNTF-mediated astrocytic differentiation. In a transfection–luciferase assay, inhibition of MAPK activation enhances activation from a transfected GFAP promoter. An explanation for this discrepancy is that the cultures being used in the two studies are different. The data presented here were obtained on a homogeneous population of cells that consistently gave 50–60% astrocytic differentiation after 2 d and ∼98% after 4–6 d of CNTF treatment. Because the cultures of Bonni et al. (1997) are 60% neuronal, the results of any biochemical assay in which the entire population of cells is used may reflect neuronal rather than stem cell responses.
Since GFAP is robustly and specifically induced in astrocytes, it may be used as a paradigm for transcriptional regulation. It remains to be seen whether the STAT sites in the GFAP promoter are functional, and if so, whether they are equivalent. Also, it remains to be determined whether sites corresponding to transcription factors activated by MAPK-related pathways such as AP1 and CREB, which are present in this promoter, are functional (Besnard et al., 1991; Kahn et al., 1997). In other systems interaction between the MAPK-related and STAT pathways has been shown to occur through integrators such as the p300 and CREB-binding proteins (Horvai et al., 1997). It will also be important to determine whether SMAD proteins interact with the GFAP promoter either directly or indirectly. The prolonged activation of STAT for 8 d in the stem cells is in contrast to the brief activation usually seen in vitro (Symes et al., 1994; Rajan et al., 1996) but is reminiscent of LIF-induced in vivo STAT activation in injured peripheral neurons (Rajan et al., 1995). In the experiment shown in Figure 2, cells were treated every 24 hr for time points >24 hr. However, Stat1 and Stat3 activation was seen even 72 hr after one dose of CNTF (data not shown).
EGF promotes astrocytic differentiation, but it is not identical in its effects to CNTF. In a clonal assay done with FGF-expanded cells, mitogen withdrawal causes ∼10% of cells to differentiate into astrocytes (Johe et al., 1996). A similar experiment done with EGF-expanded cells causes the number of astrocytes to increase to 50% (Johe et al., 1996). In another experiment performed along the same lines, FGF-expanded cells were treated with EGF for the final passage, and both mitogens were withdrawn. This scenario also yields 50% astrocytes (K. Johe, T. Hazel, and R. D. G. McKay, unpublished results). Thus, exposure to EGF just before mitogen withdrawal causes the cells to have a greater propensity to differentiate into astrocytes. However, it is not instructive in its action like CNTF. Although EGF has been reported to activate STATs (Fu and Zhang, 1993; Schindler and Darnell, 1995), in the experiments reported here there is no evidence for STAT activation after EGF treatment. Although we have not investigated the possibility of STAT activation after EGF withdrawal, it seems highly unlikely. We conclude that EGF promotes astrocytic differentiation by mechanisms that do not include activation of STATs. BMP4 treatment causes astrocytic differentiation in EGF-expanded precursors isolated from the murine E17 subventricular zone grown in neurosphere cultures (Gross et al., 1996). This effect is not seen in our cells, which are isolated from E14 cortex and maintained in monolayers. Instead they respond to BMP4 by differentiating into neural crest progeny (Hazel et al., 1997). BMP4 also does not cause activation of STAT proteins in our system.
Thus, both EGF and BMP4 cause astrocytic differentiation in different paradigms. Both of these factors signal through SMAD proteins. BMP4 causes direct activation of the SMAD1 protein by receptor-mediated phosphorylation (Lagna et al., 1996; Liu et al., 1996), whereas EGF causes inhibition of transcriptional activation by the SMADs via the MAPK pathway (Kretzschmar et al., 1997). Astrocytic differentiation in both the EGF withdrawal and BMP4 treatment paradigms may be regulated positively by the SMAD proteins. In the case of BMP4 this would occur through direct activation, whereas withdrawal of EGF may cause an alleviation of SMAD inhibition, leading to astrocytic differentiation. These results are consistent with a model in which the involvement of either STAT or SMAD signaling is necessary for astrocytic differentiation depending on the type of stem cell undergoing differentiation and the ligand causing it. In the experimental models discussed here, CNTF causes astrocytic differentiation of FGF-expanded CNS stem cells by STAT activation, whereas EGF and BMP may cause astrocytic differentiation in their respective models via the SMAD pathway. It is possible that FGF primes the cells for the differentiation effects of CNTF in the model studied in this report, whereas EGF exposure is required in the model of Gross et al. (1996)for astrocytic differentiation in response to BMPs. Thus different growth factors may activate distinct groups of transcription factors, providing a method for plasticity in the timing, quantity, and quality of astrocytes generated in the adult brain.
Footnotes
We thank Drs. M. Molne, L. van Grunsven, and M. Brenner for helpful discussions, E. Saphier for technical assistance, Dr. J. E. Darnell Jr (Rockefeller University) for the Stat3 plasmids, and Dr. Y. Gotoh (Kyoto University, Kyoto, Japan) for the MAPKK virus vector.
Correspondence should be addressed to Ronald D. G. McKay, Laboratory of Molecular Biology, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Building 36, Room 5A29, Bethesda, MD 20892-4157.
REFERENCES
- 1.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 2.Banner L, Moayeri N, Patterson P. Leukemia inhibitory factor is expressed in astrocytes following cortical brain injury. Exp Neurol. 1997;147:1–9. doi: 10.1006/exnr.1997.6536. [DOI] [PubMed] [Google Scholar]
- 3.Besnard F, Brenner M, Nakatani Y, Chao R, Purohit HJ, Freese E. Multiple interacting sites regulate astrocyte-specific transcription of the human gene for glial fibrillary acidic protein. J Biol Chem. 1991;266:18877–18883. [PubMed] [Google Scholar]
- 4.Bonni A, Frank DA, Schindler C, Greenberg ME. Characterization of a pathway for ciliary neurotrophic factor signaling to the nucleus. Science. 1993;262:1575–1579. doi: 10.1126/science.7504325. [DOI] [PubMed] [Google Scholar]
- 5.Bonni A, Sun Y, Nadal-Vicens M, Bhatt A, Frank DA, Rozovsky I, Stahl N, Yancopoulos GD, Greenberg M. Regulation of gliogenesis in the central nervous system by the JAK-STAT signaling pathway. Science. 1997;278:477–483. doi: 10.1126/science.278.5337.477. [DOI] [PubMed] [Google Scholar]
- 6.Chung J, Uchida E, Grammar T, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997;17:6508–6516. doi: 10.1128/mcb.17.11.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frederiksen K, McKay R. Proliferation and differentiation of rat neuroepithelial precursor cells in vivo. J Neurosci. 1988;8:1144–1151. doi: 10.1523/JNEUROSCI.08-04-01144.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu X-Y, Zhang J-J. Transcription factor p91 interacts with the epidermal growth factor receptor and mediates activation of the c-fos promoter. Cell. 1993;74:1135–1145. doi: 10.1016/0092-8674(93)90734-8. [DOI] [PubMed] [Google Scholar]
- 9.Gross RE, Mehler MF, Mabie PC, Zang Z, Santschi L, Kessler JA. Bone morphogenetic proteins promote astroglial lineage commitment by mammalian subventricular zone progenitor cells. Neuron. 1996;17:595–606. doi: 10.1016/s0896-6273(00)80193-2. [DOI] [PubMed] [Google Scholar]
- 10.Hazel T, Panchision D, Warriner P, McKay R. Regional plasticity of multipotent precursors from the developing CNS. Soc Neurosci Abstr. 1997;23:319. [Google Scholar]
- 11.Hockfield S, McKay R. Identification of major cell classes in the developing mammalian central nervous system. J Neurosci. 1985;5:3310–3328. doi: 10.1523/JNEUROSCI.05-12-03310.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horvai AE, Xu L, Korzus E, Brard G, Kalafus D, Mullen T-M, Rose DW, Rosenfeld MG, Glass CK. Nuclear integration of JAK/STAT and Ras/AP1 signaling by CBP and p300. Proc Natl Acad Sci USA. 1997;94:1074–1079. doi: 10.1073/pnas.94.4.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horvath CM, Wen Z, Darnell JE., Jr A STAT protein domain that determines DNA sequence recognition suggests a novel DNA-binding domain. Genes Dev. 1995;9:984–994. doi: 10.1101/gad.9.8.984. [DOI] [PubMed] [Google Scholar]
- 14.Hughes SM, Lillien LE, Raff MC, Rohrer H, Sendtner M. Ciliary neurotrophic factor induces type-2 astrocyte differentiation in culture. Nature. 1988;335:70–72. doi: 10.1038/335070a0. [DOI] [PubMed] [Google Scholar]
- 15.Johe KK, Hazel TG, Muller T, Dugich-Djordjevic MM, McKay RDG. Single factors direct the differentiation of stem cells from fetal and adult central nervous system. Genes Dev. 1996;10:3129–3140. doi: 10.1101/gad.10.24.3129. [DOI] [PubMed] [Google Scholar]
- 16.Kahn MA, Huang CJ, Caruso A, Barresi V, Nazarian R, Condorelli DF, de Vellis J. Ciliary neurotrophic factor activates JAK/Stat signal transduction cascade and induces transcriptional expression of glial fibrillary acidic protein in glial cells. J Neurochem. 1997;68:1413–1423. doi: 10.1046/j.1471-4159.1997.68041413.x. [DOI] [PubMed] [Google Scholar]
- 17.Kretzschmar M, Doody J, Massague J. Opposing BMP and EGF signalling pathways converge on the TGF-β family mediator Smad1. Nature. 1997;389:618–622. doi: 10.1038/39348. [DOI] [PubMed] [Google Scholar]
- 18.Lagna G, Hata A, Hemmati-Brivanlou A, Massague J. Partnership between DPC4 and SMAD proteins in TGF-β signalling pathways. Nature. 1996;383:832–836. doi: 10.1038/383832a0. [DOI] [PubMed] [Google Scholar]
- 19.Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- 20.Lillien LE, Sendtner M, Raff MC. Extracellular matrix associated molecules collaborate with ciliary neurotrophic factor to induce type-2 astrocyte development. J Cell Biol. 1990;111:635–644. doi: 10.1083/jcb.111.2.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu F, Hata A, Baker J, Doody J, Carcamo J, Harland R, Massague J. A human Mad protein acting as a BMP-regulated transcriptional activator. Nature. 1996;381:620–623. doi: 10.1038/381620a0. [DOI] [PubMed] [Google Scholar]
- 22.Lutticken C, Wegenka UM, Yuan J, Buschmann J, Schindler C, Ziemiecki A, Harpur AJ, Wilks AF, Yasukawa K, Taga T, Kishimoto T, Barbieri G, Pellegrini S, Sendtner M, Heinrich PC, Horn F. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science. 1994;263:89–92. doi: 10.1126/science.8272872. [DOI] [PubMed] [Google Scholar]
- 23.McKay R. Stem cells in the central nervous system. Science. 1997;276:66–71. doi: 10.1126/science.276.5309.66. [DOI] [PubMed] [Google Scholar]
- 24.Rajan P, Stewart CL, Fink JS. LIF-mediated activation of STAT proteins after neuronal injury in vivo. NeuroReport. 1995;6:2240–2244. doi: 10.1097/00001756-199511000-00033. [DOI] [PubMed] [Google Scholar]
- 25.Rajan P, Symes AJ, Fink JS. STAT proteins are activated by ciliary neurotrophic factor in cells of central nervous system origin. J Neurosci Res. 1996;43:403–411. doi: 10.1002/(SICI)1097-4547(19960215)43:4<403::AID-JNR2>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 26.Schindler C, Darnell JE., Jr Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu Rev Biochem. 1995;64:621–651. doi: 10.1146/annurev.bi.64.070195.003201. [DOI] [PubMed] [Google Scholar]
- 27.Shuai K, Stark GR, Kerr IM, Darnell JE., Jr A single phosphotyrosine residue of Stat91 required for gene activation by interferon-γ. Science. 1993;261:1744–1746. doi: 10.1126/science.7690989. [DOI] [PubMed] [Google Scholar]
- 28.Shuai K, Horvath CM, Huang LHT, Qureshi SA, Cowburn D, Darnell JE., Jr Interferon activation of the transcription factor Stat91 involves dimerization through SH2-phosphotyrosyl peptide interactions. Cell. 1994;76:821–828. doi: 10.1016/0092-8674(94)90357-3. [DOI] [PubMed] [Google Scholar]
- 29.Stahl N, Boulton TG, Farruggella T, Ip NY, Davis S, Witthuhn BA, Quelle FW, Silvennoinen O, Barbieri G, Pellegrini S, Ihle JN, Yancopoulos GD. Association and activation of Jak/Tyk kinases by CNTF/LIF/OSM/IL6 β receptor components. Science. 1994;263:92–95. doi: 10.1126/science.8272873. [DOI] [PubMed] [Google Scholar]
- 30.Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE, Jr, Yancopoulos GD. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science. 1995;267:1349–1353. doi: 10.1126/science.7871433. [DOI] [PubMed] [Google Scholar]
- 31.Symes A, Lewis S, Corpus L, Rajan P, Hyman SE, Fink JS. STAT proteins participate in the regulation of the vasoactive intestinal peptide gene by the ciliary neurotrophic factor family of cytokines. Mol Endocrinol. 1994;8:1750–1763. doi: 10.1210/mend.8.12.7708062. [DOI] [PubMed] [Google Scholar]
- 32.Ware C, Horowitz MC, Renshaw BR, Hunt JS, Liggit D, Koblar SA, Gliniak BC, McKenna HJ, Papayannopoulou T, Thoma B, Cheng L, Donovan PJ, Peschon JJ, Bartlett PF, Willis CR, Wright BD, Carpenter MK, Davison BL, Gearing DP. Targeted disruption of the low-affinity leukemia inhibitory factor receptor gene causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development. 1995;121:1283–1299. doi: 10.1242/dev.121.5.1283. [DOI] [PubMed] [Google Scholar]
- 33.Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;62:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 34.Winston LA, Hunter T. Intracellular signalling: Putting JAKs on the kinase MAP. Curr Biol. 1996;6:668–671. doi: 10.1016/s0960-9822(09)00445-x. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X, Blenis J, Li H-C, Schindler C, Chen-Kiang S. Requirement of serine phosphorylation for formation of STAT-promoter complexes. Science. 1995;267:1990–1994. doi: 10.1126/science.7701321. [DOI] [PubMed] [Google Scholar]