

Abstract
Loss of central glucocorticoid receptor (GR) function is thought to be involved in the development of neuroendocrine and psychiatric disorders associated with corticotropin-releasing hormone (CRH) hyperactivity. The possible causal relationship between defective GR function and altered activity of CRH neurons was studied in transgenic mice (TG) expressing antisense RNA against GR. Immunocytochemical studies showed significant reductions in CRH immunoreactive neurons in the paraventricular nucleus (PVN) and in CRH and vasopressin (AVP) stores in the external zone of the median eminence. Concomitantly, stimulus-evoked CRH secretion from mediobasal hypothalami of TG micein vitro was reduced significantly. However, CRH mRNA levels in the PVN of TG mice were marginally lower than those in wild-type (WT) mice. 125I-CRH binding autoradiography revealed no differences between WT and TG animals in any of the brain regions that were studied. Basal plasma corticosterone (cort) levels and 125I-CRH binding, CRH-R1 mRNA, POMC mRNA, and POMC hnRNA levels in the anterior pituitary gland were similar in WT and TG mice. Intraperitoneal injection of interleukin-1β (IL-1β) increased plasma cort levels, CRH mRNA in the PVN, and anterior pituitary POMC hnRNA similarly in WT and TG mice. The injection of saline significantly reduced anterior pituitary CRH-R1 mRNA levels in WT mice, but not in TG mice, whereas IL-1β produced a decrease in these mRNA levels in both strains.
The data show that long-term GR dysfunction can be associated with reduced activity of CRH neurons in the PVN and decreased sensitivity of pituitary CRH-R1 mRNA to stimulus-induced downregulation. Moreover, the hypothalamic changes observed in this model suggest that impaired GR function, at least if present since early embryonic life, does not necessarily result in CRH hyperexpression characteristics of disorders such as major depression.
Keywords: corticotropin-releasing hormone (CRH), glucocorticoid receptor (GR), transgenic (TG) mice, IL-1β, paraventricular nucleus, HPA axis, CRH receptor
Impaired glucocorticoid receptor (GR) function has been considered to be an important factor in the development and/or maintenance of various pathological conditions in man, including (familial) glucocorticoid resistance (Lamberts et al., 1992) and major depression (Barden et al., 1995). Patients suffering from major depression typically show increased circulating cortisol levels (Linkowski et al., 1985; Rubin et al., 1987; Deuschle et al., 1997), inadequate suppression of cortisol by dexamethasone (Carroll et al., 1968; Rubin et al., 1987), blunted adrenocorticotropic hormone (ACTH) responses to a corticotropin-releasing hormone (CRH) challenge (Holsboer et al., 1984; Gold et al., 1986), and exaggerated ACTH and cortisol responses to CRH after low-dose dexamethasone pretreatment (Heuser et al., 1994). These changes in hormonal responses may be the result of CRH-producing neurons in the hypothalamic paraventricular nucleus (PVN) (Owens and Nemeroff, 1991; Chrousos and Gold, 1992;Holsboer et al., 1994). In line with this notion is the observation that patients with major depression show increased CRH levels in the cerebrospinal fluid (Nemeroff et al., 1984; Banki et al., 1992), increased numbers of CRH and CRH/vasopressin (AVP)-expressing neurons in the PVN, (Raadsheer et al., 1994), and increased CRH mRNA levels in the PVN (Raadsheer et al., 1995). Decreased levels of central CRH receptors have been found in the frontal cortex of suicide victims, possibly as a result of chronic hyperactivity of brain CRH systems (Nemeroff et al., 1988). Recently, it was shown that chronically elevated brain CRH levels produce marked changes in baseline and stimulus-evoked physiological, neurochemical, and behavioral responses, suggesting that chronic CRH hypersecretion is an important factor in the etiology of stress-related disorders (Linthorst et al., 1997). However, until now, studies in humans could not resolve whether hypersecretion of CRH or GR dysfunction plays a primary role or any role in the physiological and behavioral anomalies associated with stress-related disorders.
To test whether long-term GR dysfunction is a determining factor for hyperactivity of brain CRH neurons, we studied the numbers of hypothalamic CRH neurons, CRH mRNA levels, CRH and AVP peptide stores, and CRH release from mediobasal hypothalami (MBH) in vitroin transgenic (TG) mice with impaired GR function. In addition, we studied putative changes in CRH receptor expression and regulation in several brain regions.
The TG mouse used as a model for impaired GR function was constructed by the insertion of a transgene expressing constitutively antisense RNA against GR (Pepin et al., 1992a). These TG mice show reduced GR mRNA levels in brain (Pepin et al., 1992a; Marchetti et al., 1994), pituitary (Morale et al., 1995), and some peripheral tissues (Pepin et al., 1992a), reduced brain glucocorticoid receptor binding (Pepin et al., 1992a), and reduced hypothalamus–pituitary–adrenal (HPA axis) sensitivity to glucocorticoids (Stec et al., 1994; Barden et al., 1997;Karanth et al., 1997). As a consequence of the impaired GR function, the regulation of the HPA axis in these animals is disturbed. Transgenic animals display exaggerated ACTH responses to stress and to exogenously administered CRH, whereas the responses in corticosterone (cort) are normal (Montkowski et al., 1995; Barden et al., 1997;Karanth et al., 1997) because of the hyposensitivity of the adrenal gland to ACTH (Barden et al., 1997). Hence, these TG mice show reduced GR capacity, which is not compensated for by elevations in circulating glucocorticoid levels.
In marked contrast to the predicted results, we found a substantial reduction in the number of CRH neurons, CRH mRNA levels, CRH and AVP stores, and in vitro CRH release despite augmented CRH- and stressor-induced ACTH responses, as previously reported in these TG mice (Montkowski et al., 1995; Barden et al., 1997; Karanth et al., 1997). To determine whether these responses in TG mice were attributable to adaptation at the level of the anterior pituitary gland, we studied 125I-CRH binding and POMC mRNA, POMC heteronuclear RNA (hnRNA), and CRH receptor mRNA levels under resting conditions and after intraperitoneal injection of interleukin-1β (IL-1β), a known activator of central CRH neurons (Berkenbosch et al., 1987; Sapolsky et al., 1987).
MATERIALS AND METHODS
Animals. Adult male mice of the B6C3F1 strain (wild-type, WT) were purchased from Charles River Wiga (Sulzfeld, Germany). Male B6C3F1 transgenic mice (TG; line 1.3) were used, in which a transgene was inserted in the genome constitutively expressing antisense RNA against the GR (Pepin et al., 1992a). The transgene was driven by a neurofilament promotor. In the cohort of animals used, GR mRNA levels in the brain and pituitary of TG mice were decreased by ∼50% as compared with the levels in WT mice. Mice were bred in the animal unit of the Clinical Institute of the Max Planck Institute of Psychiatry, Munich, Germany. The animals were housed five per cage in a temperature (23°C), humidity (60%), and light (lights on from 6:00 A.M. until 6:00 P.M.) controlled room. Mice used for quantitative immunofluorescence microscopy were housed two per cage. Food and tap water were availablead libitum. The animals were kept in the animal room for at least 2 weeks before the experiments.
Experimental procedures. In a first experiment, groups (n = 6) of WT and TG mice (60 d old) were anesthetized quickly with halothane (Hoechst, Frankfurt am Main, Germany) and decapitated. Their brains were removed from the skull and subsequently were processed for quantitative immunocytochemistry of CRH and AVP, as described elsewhere (De Goeij et al., 1991; Schmidt et al., 1995). In addition, we quantified the relative numbers of CRH immunoreactive neurons in the PVN of WT and TG mice.
In a second experiment, we studied the release of CRH from MBH in vitro. Groups of mice (n = 10–11) were anesthetized quickly with halothane and decapitated. The MBHs were dissected by sagittal cuts along the lateral hypothalamic sulci, and frontal cuts were made through the optic chiasma and anterior margin of the mamillary bodies. A horizontal cut 1 mm from the base of the brain was conducted to separate the island of MBH tissue from the rest of the brain. Subsequently, the dissected tissue was used for in vitro stimulation of CRH release (see below).
In a third series of experiments, groups (n = 10) of WT and TG animals were injected intraperitoneally with 0.9% saline (SAL) or human recombinant IL-1β (1 μg/mouse) (provided by Dr. E. B. De Souza and the DuPont-Merck Pharmaceutical Company, Wilmington, DE) or were left undisturbed (UNT). The animals were decapitated 4 hr later between 1:00 P.M. and 3:00 P.M. Trunk blood was collected within 45 sec after disturbance of the animals in ice-chilled EDTA-coated (1.5 ml) tubes containing 25 μg of aprotinin. This allowed for the measurement of undisturbed cort, but not of resting ACTH concentrations in plasma. Plasma was stored at −20°C until it was assayed for cort. Brains and pituitaries were dissected and rapidly frozen in isopentane at −40°C or in liquid nitrogen, respectively, and thereafter stored at −70°C until further processing for in situ hybridization histochemistry (for CRH mRNA and CRH-R1 mRNA in brain and POMC mRNA, POMC hnRNA, and CRH-R1 mRNA in the anterior pituitary) and 125I-CRH binding in brain and anterior pituitary.
Tissue processing for immunocytochemistry of CRH and AVP.After decapitation, brains were removed from the skull and immersion-fixed in 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.6, for 4 hr at 4°C. Subsequently, the brains were incubated in 5% sucrose in 0.1 m phosphate buffer for 48 hr at 4°C. Thereafter, the hypothalami were dissected and embedded together (up to 30 specimens) in a cryomold containing OCT compound (Sakura Finetechnical, Tokyo, Japan). The cryomold was frozen in isopentane cooled with liquid nitrogen, and frontal serial sections (10 μm) were cut with a motor-driven cryostat (Micron HM 500 M, Walldorf, Germany).
Quantitative immunocytochemistry of CRH and AVP. Serial sections (50 μm interval) were incubated with rabbit antiserum to rat/human CRH (5Bo, 1:500) for 48 hr at 4°C. Adjacent serial sections were incubated with rabbit antiserum to AVP (Truus, 1:1000). Subsequently, sections were incubated with goat anti-rabbit antiserum conjugated to FITC (Tago, Burlingame, CA) for 2 hr at room temperature (21°C). Sections were enclosed in Vectashield anti-fading medium (Vector Laboratories, Burlingame, CA). All incubations were performed in 0.1 m Tris-buffered saline (TBS), pH 7.6, containing 0.5% Triton X-100, 0.2% BSA, 1% normal goat serum (Dakopatts, Glostrup, Denmark), and 0.01% sodium azide.
The immunofluorescence staining intensity was quantified by using a computer-controlled microfluorometer (Leitz-Combi, Leitz, Wetzlar, Germany) as described previously (De Goeij et al., 1991; Schmidt et al., 1995). Three sections of each median eminence (50 μm interval) were analyzed. Earlier studies in rats demonstrated a linear relationship between quantitative immunocytochemical data and those of radioimmunoassayable CRH in median eminence tissue extracts (r = 0.86–0.99) (Berkenbosch and Tilders, 1988). For specificity of the AVP antiserum, see De Goeij and colleagues (1991). Quantification of AVP stores in the internal zone of the median eminence (ZIME) served as control for the specificity of the AVP signal in the external zone of the median eminence (ZEME). The CRH antiserum 5Bo displays characteristics identical to those of 3B3 described earlier (Berkenbosch et al., 1986; De Goeij et al., 1991). Preincubation of 5Bo with 10 μm r/hCRH41 (Peninsula Laboratories, Belmont, CA) for 2 hr at 37°C prevented CRH immunostaining of murine hypothalami. Preincubation of 5Bo with up to 100 μm AVP (Sigma, St Louis, MO), oxytocin (Bachem Feinchemicalien, Torrance, CA), or αMSH (Ciba Geigy, Basel, Switzerland) did not inhibit CRH immunostaining.
CRH immunoreactive neurons in the PVN. To estimate the relative numbers of CRH-producing neurons in the PVN, we embedded hypothalami of WT and TG mice together and incubated serial 10 μm sections (50 μm apart) throughout the full rostrocaudal extension of the PVN with rabbit antiserum to CRH (5Bo, 1:500) for 2 d at 4°C, followed by incubation with goat anti-rabbit antiserum conjugated to CY3 (Jackson ImmunoResearch, West Grove, PA) for 2 hr at room temperature (21°C). Sections were enclosed as described above. The numbers of nucleated CRH immunoreactive neural profiles in the PVN of WT and TG mice were counted bilaterally by two independent investigators on photographs of coded preparations following a blinded protocol. Only those animals with a complete series of sections throughout the PVN were used. The data represent cumulative numbers of nucleated CRH profiles in three sections, 50 μm apart, in the medial part of the PVN.
In vitro CRH release from mediobasal hypothalami. After dissection, each MBH was incubated in 250 μl of Krebs’ Ringer’s bicarbonate (KRB) buffer supplemented with 20 μmbacitracin to inhibit peptidases. The tissues were incubated at 37°C in an atmosphere of 95% O2/5% CO2 in a metabolic shaker (50 cycles/sec) for 45 min. Next, the medium was discarded, and the tissues were incubated with 250 μl of KRB or KRB containing 56 mm K+ for 30 min to determine basal and depolarization-induced CRH release, respectively. Medium was collected and stored at −80°C until it was assayed for CRH.
Tissue processing for in situ hybridization histochemistry and 125I-CRH binding by autoradiography.After decapitation, brains were frozen in isopentane at −40°C and stored at −70°C. For in situ hybridization, serial sections (12 μm) were cut in a cryostat (Jung Frigocut, Leica, Germany) at −22°C. Selected regions were thaw-mounted on poly-l-lysine-coated slides and stored at −70°C until they were processed for CRH binding or in situhybridization. Serial sections (60 μm apart) of the PVN were collected on separate slides. Pituitary glands were mounted in OCT compound, and serial sections (12 μm) were cut by using a cryostat at −22°C. Five consecutive sections (12 μm) were collected per slide.
125I-CRH binding by autoradiography in brain and anterior pituitary. Brain and pituitary sections were placed in a dessicator for 8 hr at 4°C before the binding assay. Sections were preincubated for 15 min at room temperature in 50 mmTris-HCl, pH 7.4, containing (in mm) 2 EGTA, 5 MgCl2, 0.1 PMSF, and 140 NaCl plus 0.1% BSA and 100 KIU/ml aprotinin, were rinsed twice in 50 mm Tris-HCl, and were incubated with 3.0 × 105 cpm/8 ml125I-Tyr-oCRH (DuPont-New England Nuclear, Boston, MA) in 50 mm Tris-HCl, pH 7.4, containing (in mm) 2 EGTA, 5 MgCl2, 0.1 PMSF, and 1 DTT plus 0.1% BSA and 100 KIU/ml aprotinin at room temperature for 1 hr. For nonspecific binding, sections were incubated in the presence of 1 μmunlabeled CRH. Next, the sections were washed three times in ice-cold 50 mm Tris-HCl, rinsed in distilled water, and dried immediately. Sections were exposed to Hyperfilm β-Max (Amersham, Arlington Heights, IL).
Sections of control groups and experimental groups were processed and exposed to film simultaneously. Light transmittance of the autoradiographs was quantitated via a computerized image analysis system (Imaging Research, Ontario, Canada). Values obtained from three to four sections of each mouse were averaged before the nonspecific binding was subtracted. The data are presented as the mean transmittance of the number of mice indicated in Results or in the figures.
In situ hybridization histochemistry. Before hybridization, brain and pituitary sections were allowed to thaw at room temperature for 10 min and then fixed in 4% formaldehyde solution, acetylated, dehydrated, and air-dried.
CRH mRNA levels in the PVN were measured with [35S]deoxy-ATP-labeled 48-mer synthetic oligonucleotide probes directed to exonic sequences from bp 1254 to 1302 of murine CRH cDNA. CRH receptor mRNA levels in the anterior pituitary were measured by using a 35S-labeled cRNA probe prepared by transcription of a full cDNA clone of the murine type 1 CRH receptor (provided by Dr. A. Abou Samra, Massachusetts Institute of Technology, Boston, MA). POMC mRNA in the anterior pituitary was measured with [35S]deoxy-ATP-labeled 48-mer synthetic oligonucleotide probes directed to exonic sequences of rat POMC, as described previously (Harbuz and Lightman, 1989). This probe shows 98% homology to the equivalent fragment of murine POMC. POMC hnRNA in the pituitaries was measured with a 35S-labeled cRNA intronic probe corresponding to 1.2 kb of intron B of the murine POMC gene. The 1.2 kb DNA was generated by PstI digestion of the genomic clone JA3 (kindly provided by Dr. J. Drouin, Montreal, Canada) and subcloned into PGEM-4Z plasmid. For synthetic oligonucleotide probes, sections were covered with 90–100 μl of hybridization solution containing [35S]deoxy-ATP-labeled probe (3 × 106 cpm/ml), 50% formamide, 4× SSC, 500 μg/ml sheared single-stranded DNA, 250 μg/ml yeast tRNA, 1× Denhardt’s solution, 10% dextran sulfate, and 100 mm DTT. Slides were covered with coverslips and incubated for 18 hr at 42°C in a humidified chamber. After removal of coverslips and three rinses in 1× SSC, slides were washed consecutively four times for 15 min in 1× SSC at 55°C, twice for 30 min in 1× SSC at room temperature, rinsed in water followed by ethanol, and blow-dried. For cRNA probes, the sections were covered with 90–100 μl of hybridization solution containing [35S]-labeled cRNA probe (1.3 × 106 cpm/ml), 50% formamide, 50 mmTris-HCl buffer, 2.5 mm EDTA, 250 μg/ml yeast tRNA, 1× Denhardt’s solution, 10% dextran sulfate, 200 mm NaCl, and 100 mm DTT. Slides were covered with coverslips and incubated for 18 hr at 55°C in a humidified chamber. After the removal of the coverslips, the slides were washed three times in 4× SSC for 5 min at room temperature, dehydrated, washed in 50% formamide/250 mm NaCl at 60°C for 10 min, RNase-A-treated (20 μg/ml) at 37°C for 30 min, washed in 2× to 0.1× SSC, dehydrated, and blow-dried.
Sections of control groups and experimental groups were processed and exposed to film simultaneously. For determination of mRNA levels, the sections were exposed to Hyperfilm β-Max (Amersham). Light transmittance of the autoradiographs was quantitated via a computerized image analysis system (Imaging Research). Values represent the mean of the number of mice indicated in Results or in the figures.
Hormone assays. Plasma corticosterone concentrations were measured by radioimmunoassay (RIA; ICN Biomedicals, Costa Mesa, CA). The detection limit was 1.5 ng/ml plasma. Intra- and interassay variations were 4% and 7%, respectively.
The CRH concentrations in the in vitro incubation medium were determined by RIA (Phoenix Pharmaceuticals, Mountain View, CA). The detection limit was 1 pg/tube (10 pg/ml). The intra- and interassay variations were 4% and 8%, respectively.
Statistical analysis. Statistical significance of differences between groups was determined by ANOVA (one- or two-way ANOVA where appropriate), followed by post hoc multiple comparisons with Bonferroni correction for treatment effects or independent samples t test for group comparisons. Statistical evaluation was performed with the SPSS for Windows statistical program (SPSS, Chicago, IL). Significance was defined at the p < 0.05 level.
RESULTS
CRH and AVP stores in the median eminence (ZEME) and numbers of CRH neurons in the PVN
Parvocellular CRH-producing neurons in the PVN project their secretory terminals to the ZEME (Fig.1A). CRH neurons are known to produce AVP as well, which is stored together with CRH in the ZEME (Whitnall et al., 1985; De Goeij et al., 1991; Bertini and Kiss, 1991). AVP in the ZIME is located in fibers en passant that originate from magnocellular AVP-producing neurons in the PVN and supraoptic nucleus (SON) and project to the posterior lobe of the pituitary gland.
Fig. 1.
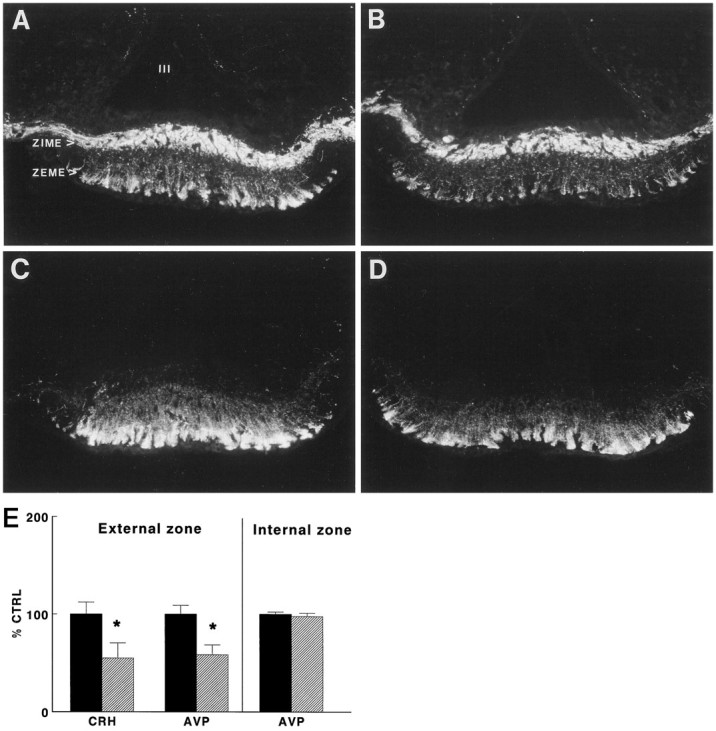
A–D, Photomicrographs showing immunocytochemical staining of AVP (A,B) and CRH (C, D) in representative frontal sections of the external zone of the median eminence (ZEME) of wild-type (A,C) and GR antisense transgenic (B,D) mice. ZIME, Internal zone of the median eminence; III, third ventricle. E, CRH and AVP stores in the external zone of the median eminence (ZEME) in TG (hatched bars) and WT mice (black bars). CRH and AVP immunoreactivity in the ZEME and ZIME was evaluated by quantitative immunocytochemistry. CRH and AVP are expressed as a percentage of the mean values found in WT mice (% CTRL). Data are expressed as mean and SEM (n = 6); *p < 0.05, TG versus WT.
As shown in Figure 1B, CRH and AVP stores in the ZEME of untreated TG mice were decreased (t test,p < 0.05) by 45% and 42%, respectively. Quantification of AVP in the ZIME revealed no differences between WT and TG animals, indicating a selective reduction of peptide stores in parvocellular CRH neurons in TG mice.
Parvocellular CRH neurons and magnocellular AVP neurons in TG and WT mice do not show a clear differential distribution within the PVN, as is the case in rats. Rostrocaudal extensions of these CRH and AVP neurons are similar in WT and TG mice (Fig.2A). As illustrated in Figure 2B, the numbers of nucleated CRH immunoreactive neural profiles in the PVN of TG mice were decreased by 54% (p < 0.001). The drop in the numbers of CRH neurons was found throughout the PVN.
Fig. 2.

Distribution of CRH and AVP neurons (A) and relative numbers of CRH immunoreactive neurons (B) in the PVN of transgenic (TG) and wild-type (WT) mice. A, Area containing vasopressin neurons (hatched fields) in frontal sections (50 μm apart) throughout the PVN of a TG (left panels) and a WT mouse (right panels). Note the absence of a clear separation of parvocellular CRH neurons and magnocellular AVP neurons.Dots indicate the positions of nucleated CRH immunoreactive neural profiles. B, Cumulative numbers of nucleated CRH immunoreactive neurons in the PVN of WT and TG mice. Data represent mean and SEM (n = 4–5); *p < 0.05, TG versus WT.
Stimulated CRH release from mediobasal hypothalamiin vitro
To study the capacity of hypothalamic CRH neurons to release CRH, we selected an in vitro approach. Unstimulated CRH release from MBH in vitro of both WT and TG animals was below the detection limit. Figure 3 shows that the K+-stimulated CRH release from MBH of TG micein vitro was 44% lower (p < 0.05) than that of WT animals.
Fig. 3.

Stimulated release of CRH from mediobasal hypothalami (MBH) in vitro of transgenic mice (TG) and wild-type mice (WT). After preincubation for 45 min, depolarization-induced CRH release was evoked by incubation of the MBHs in the presence of KRB containing 56 mmK+ for 30 min. Data are expressed as mean and SEM (n = 10–11); *p < 0.05 versus WT.
Unstimulated levels of plasma cort, CRH mRNA, and125I-CRH binding in the brain and POMC mRNA, POMC hnRNA,125I-CRH binding, and CRH-R1 mRNA in the anterior pituitary gland
As illustrated in Figure 4, baseline plasma cort concentrations were not significantly different in TG as compared with those of WT mice. Under basal conditions (UNT) the mean CRH mRNA levels in the PVN of TG animals were reduced somewhat but were not statistically different from those in WT mice (Fig.5).
Fig. 4.

Effect of interleukin-1β on plasma concentrations of corticosterone (Cort) in wild-type mice (black bars) and GR antisense transgenic mice (hatched bars). Groups of mice were injected intraperitoneally with hrIL-1β (1 μg/mouse; IL-1β) or vehicle (SAL) or were left untreated (UNT) and then were decapitated 4 hr later. Data are expressed as mean and SEM (n = 9–10); *p < 0.05 versus SAL and UNT.
Fig. 5.

Effect of interleukin-1β on CRH mRNA levels in PVN of wild-type mice (black bars) and GR antisense transgenic mice (hatched bars). Groups of mice were injected intraperitoneally with hrIL-1β (1 μg/mouse;IL-1β) or vehicle (SAL) or were left untreated (UNT) and then were decapitated 4 hr later. CRH mRNA levels are given as transmittance, and data are expressed as mean and SEM (n = 5); *p < 0.05 versus SAL and UNT.
Autoradiographic analysis of 125I-Tyr-oCRH binding revealed a similar pattern in the brains of WT and TG mice (Table1). Two-way ANOVA revealed no strain differences in 125I-CRH binding in any of the brain regions studied, with the exception of the inner layers of the olfactory bulb (OB/layer 2–5).
Table 1.
125I-CRH binding in different brain areas of wild-type (WT) and glucocorticoid receptor antisense transgenic (TG) mice
| WT | TG | |||||
|---|---|---|---|---|---|---|
| UNT | SAL | IL-1β | UNT | SAL | IL-1β | |
| Olfactory bulb/layer 1 | 93 ± 2 | 88 ± 4 | 82 ± 6 | 85 ± 4 | 79 ± 5 | 79 ± 4 |
| Olfactory bulb/layer 2–5 | 120 ± 2 | 124 ± 1 | 110 ± 5 | 113 ± 51-159 | 111 ± 51-159 | 107 ± 61-159 |
| Frontal cortex/layer 1–3 | 31 ± 2 | 27 ± 2 | 25 ± 1 | 28 ± 2 | 27 ± 2 | 26 ± 3 |
| Frontal cortex/layer 4 | 55 ± 3 | 54 ± 2 | 47 ± 2 | 49 ± 4 | 53 ± 3 | 49 ± 5 |
| Cingulate cortex | 56 ± 3 | 51 ± 7 | 48 ± 3 | 61 ± 4 | 54 ± 3 | 51 ± 4 |
| Ventrolateral striatum | 32 ± 1 | 43 ± 4* | 31 ± 3 | 32 ± 3 | 38 ± 4* | 31 ± 4 |
| Dorsomedial striatum | 22 ± 1 | 30 ± 3* | 21 ± 2 | 23 ± 2 | 28 ± 3* | 21 ± 3 |
| Piriform cortex | 43 ± 2 | 47 ± 2 | 50 ± 6 | 49 ± 3 | 56 ± 4 | 45 ± 5 |
| Septum | 25 ± 7 | 31 ± 3 | 32 ± 4 | 31 ± 4 | 35 ± 3 | 31 ± 5 |
| Lateral amygdaloid nucleus | 58 ± 4 | 56 ± 3 | 43 ± 5 | 66 ± 4 | 48 ± 11 | 55 ± 5 |
| Anterior amygdaloid nucleus | 45 ± 2 | 50 ± 3 | 39 ± 3 | 46 ± 1 | 34 ± 7 | 38 ± 3 |
| Cortex | 43 ± 5 | 42 ± 6 | 33 ± 7 | 57 ± 3 | 35 ± 8 | 44 ± 4 |
| Hippocampus/Ammon horn | 13 ± 2 | 9 ± 1** | 6 ± 2** | 14 ± 3 | 7 ± 2** | 8 ± 3** |
| Subiculum | 15 ± 3 | 15 ± 2 | 11 ± 2 | 18 ± 4 | 9 ± 3 | 13 ± 3 |
| Colliculus superior | 109 ± 8 | 118 ± 6 | 110 ± 8 | 110 ± 7 | 108 ± 7 | 106 ± 9 |
| Cerebellum/granular layer | 123 ± 6 | 126 ± 6 | 125 ± 6 | 127 ± 7 | 119 ± 9 | 129 ± 9 |
| Cerebellum/olfactory layer | 58 ± 4 | 61 ± 4 | 58 ± 1 | 60 ± 2 | 61 ± 5 | 58 ± 5 |
WT and TG mice were left untreated (UNT) or given saline (SAL) or interleukin-1β (IL-1β) 4 hr before decapitation.
125I-CRH-binding given as transmittance. Data are expressed as mean ± SEM of four to five animals per group.
*p < 0.05 versus UNT and IL-1β (one-way ANOVA on pooled data of WT and TG animals).
**p < 0.05 versus UNT (one-way ANOVA on pooled data of WT and TG animals).
F1-159: p < 0.05 versus WT (two-way ANOVA).
As illustrated in Figures 6 and7, also anterior pituitary125I-CRH binding and anterior pituitary CRH-R1mRNA, POMC mRNA, and POMC hnRNA levels were similar in untreated WT and TG mice.
Fig. 6.

Effect of interleukin-1β on 125I-CRH binding (top panel) and CRH receptor mRNA levels (bottom panel) in anterior pituitary glands of wild-type mice (black bars) and GR antisense transgenic mice (hatched bars). Groups of mice were injected intraperitoneally with hrIL-1β (1 μg/mouse; IL-1β) or vehicle (SAL) or were left untreated (UNT) and then were decapitated 4 hr later.125I-CRH binding and CRH-R1 mRNA are given as transmittance, and data are expressed as mean and SEM (n = 9–10). *p < 0.05 versus UNT; **p < 0.05 versus SAL and UNT;+p < 0.05 versus wild type.
Fig. 7.

Effect of interleukin-1β on POMC mRNA levels (top panel) and POMC heteronuclear RNA (POMC hnRNA) levels (bottom panel) in anterior pituitary glands of wild-type mice (black bars) and GR antisense transgenic mice (hatched bars). Groups of mice were injected intraperitoneally with hrIL-1β (1 μg/mouse; IL-1β) or vehicle (SAL) or were left untreated (UNT) and then were decapitated 4 hr later. POMC mRNA and POMC hnRNA levels are given as transmittance, and data are expressed as mean and SEM (n = 5 and n = 9–10 forPOMC mRNA and POMC hnRNA, respectively); #p < 0.05 versus UNT (one-way ANOVA on pooled data of wild-type and transgenic mice).
Interleukin-1β-induced responses
As shown in Figure 4, plasma cort concentrations were not statistically different in TG animals as compared with those of WT animals 4 hr after intraperitoneal administration of saline. At this time, intraperitoneal administration of IL-1β had increased (p < 0.05) plasma cort concentrations to the same extent in WT and TG mice.
Like in the untreated animal groups, the mean CRH mRNA levels in the PVN of saline-treated TG animals were somewhat lower but were not statistically different from those in saline-treated WT mice (see Fig.5). Administration of IL-1β tended to increase CRH mRNA levels in the PVN but reached statistical significance only in TG mice (p < 0.05).
Two-way ANOVA of 125I-Tyr-oCRH binding to brain sections (see Table 1) showed an overall effect of treatment with saline or IL-1β on CRH receptor binding in the striatum and hippocampus (p < 0.05) 4 hr postinjection without any strain differences. Further analyses on pooled data of WT and TG animals revealed that saline injection increased125I-CRH binding in the ventrolateral and dorsomedial regions of the striatum (p < 0.05), as compared with binding in untreated and IL-1β-injected mice, whereas injection of either saline or IL-1β decreased 125I-CRH binding in the Ammon’s horn of the hippocampus (p < 0.05).
Autoradiographic analysis of 125I-Tyr-oCRH binding in the anterior pituitary revealed an effect of treatment (two-way ANOVA;p < 0.05), but no strain differences (see Fig. 6,top panel). Post hoc analysis revealed no group differences.
Saline and IL-1β treatment had differential effects on CRH-R1 mRNA levels (see Fig. 6, bottom panel) in WT and TG animals (two-way ANOVA; treatment effect, p < 0.001; strain effect, p < 0.001; interaction treatment and strain, p < 0.001). The injection of saline markedly reduced CRH-R1 mRNA levels in pituitaries of WT mice (p < 0.05), but not of TG mice. Thus, after saline treatment CRH-R1 mRNA expression was lower in WT than in TG mice (t test,p < 0.05). After IL-1β, both WT and TG mice showed reduced CRH-R1 mRNA levels (p < 0.05), as compared with those of untreated controls. Accordingly, the CRH-R1 mRNA attained similar levels in WT and TG animals after treatment with IL-1β.
POMC mRNA levels were not affected by saline injection or IL-1β injection in WT or in TG mice (see Fig. 7, top panel). However, two-way ANOVA revealed a treatment effect (p < 0.05) on primary POMC transcript RNA (POMC hnRNA) levels in the anterior pituitary (see Fig. 7,bottom panel) that was similar in WT and TG mice. Further analysis on pooled data of WT and TG animals revealed that IL-1β increased POMC hnRNA levels, as compared with those of untreated mice (p < 0.05).
DISCUSSION
In the present study we have demonstrated that the numbers of CRH immunoreactive neurons in the PVN of TG mice are reduced substantially, as compared with those of their WT control. Moreover, CRH and AVP stores in the ZEME are decreased in adult TG mice, as is the stimulated CRH release from their MBH in vitro. The reduction in CRH neurons in TG mice is not reflected in a decrease in basal morning plasma ACTH and cort levels (Barden et al., 1997; Karanth et al., 1997). Earlier reports of elevated hormone levels in TG mice may have been attributable to nonspecific stress effects at the time of death (Pepin et al., 1992a,b; Beaulieu et al., 1994; Morale et al., 1995). As reported previously (Beaulieu et al., 1994; Barden et al., 1997;Karanth et al., 1997), we found similar plasma cort concentrations in WT and TG animals in the early afternoon (this study). Evening plasma ACTH and cort levels in TG animals have been reported to be either normal (Barden et al., 1997) or decreased (Beaulieu et al., 1994), which is surprising in view of the presumed role of GR in the regulation of diurnal peak HPA activity (De Kloet et al., 1993;Bradbury et al., 1994). Because at least in rats HPA axis activity in the evening, but not in the morning, is driven by CRH (Van Oers and Tilders, 1991; Schmidt et al., 1997), the “normal” or decreased evening levels of cort in TG animals may be attributable to the decreased activity of hypothalamic CRH neurons rather than to the impaired GR function per se.
Despite GR impairment and decreased numbers of CRH-expressing neurons in the PVN of TG mice, HPA hormone levels are not altered severely either under baseline conditions or after stress, although poststress ACTH responses overshoot in these animals. These observations indicate that secondary changes may have occurred at the level of the adrenal and/or pituitary glands. However, TG animals display no evident adrenal hyperplasia and, rather, show reduced adrenocortical output to ACTH stimulation (Barden et al., 1997). In rats, an intra-adrenal CRH system has been demonstrated that, in conjunction with the sympathetic input, plays an important role in the regulation of adrenal sensitivity to ACTH (Van Oers et al., 1992; Jasper and Engeland, 1994). Therefore, hyposensitivity of the adrenal glands to ACTH in TG mice may relate to a generalized hypoactivity of CRH systems in these animals and/or reduced sympathetic activity. Indeed, sympathetic function was found to be reduced in TG mice (see below) (Richard et al., 1993; A. C. E. Linthorst, unpublished data). Thus, it is clear that the GR impairment in TG mice is not compensated for by increased adrenal cort secretion, which may result in a functional glucocorticoid insufficiency predominantly at the CNS and pituitary level (Karanth et al., 1997). In contrast to these TG animals, mice with a complete GR knock-out exhibit adrenocortical hypertrophy and hypersecretion of HPA hormones (Cole et al., 1995), suggesting that a complete ablation of GR function entails distinct (counter-) regulatory mechanisms from those in TG mice.
In the pituitary gland of TG mice, ACTH responses to various stimuliin vivo and in vitro are enhanced (Montkowski et al., 1995; Barden et al., 1997; Karanth et al., 1997). Accordingly, CRH-stimulated ACTH release from pituitary glands in vitroand in vivo was found to be larger in TG than in WT animals, which is in line with an overall reduced drive of endogenous CRH. On the other hand, pituitary glands of WT and TG animals contain comparable amounts of ACTH (Barden et al., 1997; Karanth et al., 1997), which is consistent with similar levels of both POMC mRNA and POMC hnRNA found in the present study. Because 125I-CRH binding and CRH-R1 mRNA expression levels are not different in TG and WT mice, we considered the possibility that postreceptor mechanisms may be enhanced, which may relate to reduced pituitary GR function. Because of the finding that K+-induced ACTH secretion from the pituitary gland in vitro also is enhanced in TG animals, as compared with that of WT animals, we believe that reduced GR function possibly interfere with mechanisms involved in ACTH secretion rather than with receptor activation-dependent mechanisms.
We observed markedly different changes in anterior pituitary CRH-R1 mRNA levels between WT and TG mice after stressful stimuli. An intraperitoneal saline injection, which may be considered as a weak stimulus, substantially reduced CRH-R1 mRNA levels in WT mice, but not in TG mice. Peripheral administration of IL-1β, which is a strong stimulus for CRH neurons in the PVN, substantially reduced CRH-R1 mRNA levels in the anterior pituitary of both WT and TG untreated mice. These data indicate that only strong stimuli were able to decrease anterior pituitary CRH-R1 mRNA levels in TG mice, suggesting that stimulus-induced reduction of these mRNA levels appears less sensitive in TG mice than in WT mice. It has been shown that CRH induces acute downregulation of CRH-R1 mRNA in the pituitary gland (Pozzoli et al., 1996; Rabadan-Diehl et al., 1996; Sakai et al., 1996). CRH appears to play an important role in the IL-1β-induced decrease in CRH-R1 mRNA levels in the rat pituitary, because this response can be prevented by immunoneutralization of CRH (F. Tilders, G. Aguilera, E. Schmidt, A. Kiss, S. Lolait, unpublished data). Thus, the relative resistance of TG mice to stimulus-induced reduction in CRH-R1 mRNA levels in the anterior pituitary may be attributable to the decreased CRH secretion in these animals. In addition to CRH, GRs have been shown to mediate the glucocorticoid-induced decline in pituitary CRH-R1 mRNA levels (Luo et al., 1995; Makino et al., 1995; Pozzoli et al., 1996). Thus, both impaired GR function and reduced hypothalamic CRH expression are likely to contribute to the mechanisms underlying the relative resistance of CRH-R1 mRNA to stressor-induced downregulation in TG mice.
In keeping with observations during acute stress (Rabadan-Diehl et al., 1996), the decreases in pituitary CRH-R1 mRNA after IL-1 injection were not associated with downregulation of CRH binding. This is probably attributable to rapid increases in receptor synthesis or recruitment of cryptic receptors to the plasma membrane in response to acute stimulation. In contrast to the effects of chronic glucocorticoid deficiency induced by adrenalectomy (Wynn et al., 1985; Holmes et al., 1987; Anderson et al., 1993), there was no downregulation of resting CRH binding levels in the pituitary gland of TG mice. Because pituitary CRH receptor downregulation after adrenalectomy is attributable mainly to increased CRH and AVP exposure (Holmes et al., 1987; Wynn et al., 1988; Hauger and Aguilera, 1993), normal levels of CRH binding in TG mice are consistent with the hypoactivity of hypothalamic CRH neurons, as shown in the present study.
Several studies have indicated that alterations in central CRH systems may lead to reciprocal changes in CRH receptors in the brain. For example, the density of CRH binding sites in the frontal cortex of suicide victims was found to be decreased, possibly as a consequence of CRH hyperactivity (Nemeroff et al., 1988), whereas in Alzheimer’s disease CRH receptor densities were increased in frontal, temporal, occipital, and cingulate cortex, possibly because of decreased central CRH activity (De Souza et al., 1993). In rats, prolonged central administration of CRH decreased CRH receptor levels in the amygdala (Hauger et al., 1993). In view of these observations, we were interested to see whether reduced hypothalamic CRH activity in TG mice would affect CRH receptors in the brain. Our findings show that, with the exception of the olfactory bulb, no differences in125I-CRH binding exist between WT and TG animals in any of the brain regions that were studied. Although we cannot exclude the possibility that compensatory changes in the expression of CRH-BP in these brain areas may have occurred during development, this may indicate that in TG animals extrahypothalamic CRH neuronal systems are less affected than that in the PVN.
Brain CRH neurons are involved not only in the control of HPA responses to stress, but they also play a major role in a wide variety of other stress-induced neuroendocrine, metabolic, autonomic, and behavioral responses (Owens and Nemeroff, 1991). Central administration of CRH induces behavioral activation in a familiar environment (Sutton et al., 1982; Dunn and Berridge, 1990; Linthorst et al., 1997), induces anxiety-related behavior in unfamiliar conditions (Menzaghi et al., 1994; Heinrichs et al., 1995), increases sympathetic activity (Brown et al., 1982, 1985; Brown and Fisher, 1983; Diamant et al., 1992b), increases metabolic activity (Rothwell, 1990), induces hyperthermia (Rothwell, 1990; Diamant et al., 1992a; Linthorst et al., 1997), and leads to immune suppression (Labeur et al., 1995). Some of these observations correspond with changes seen in transgenic mice constitutively overexpressing CRH (Stenzel-Poore et al., 1992, 1994,1996). In contrast to CRH-overexpressing mice and CRH-treated rats, GR-impaired TG mice show a number of physiological and behavioral characteristics that accord with a generally reduced activity of hypothalamic CRH systems rather than with CRH hyperactivity. Accordingly, TG animals show an overall decreased locomotor activity in their home cage (A. C. E. Linthorst, unpublished data), decreased oxygen consumption (Richard et al., 1993), cognitive deficits (Montkowski et al., 1995), and decreased indices of sympathetic activity in peripheral fat tissue and heart (Richard et al., 1993). Accordingly, the greatly increased fat deposition in TG animals (after the age of 4 months), especially accumulated in adipose tissue (Pepin et al., 1992a), may be attributable to the decreased sympathetic nervous activity rather than to GR dysfunction. Because sympathetic activity enhances ACTH-dependent cort secretion from the adrenal gland (Edwards and Jones, 1987; Engeland and Gann, 1989; Bornstein et al., 1990; Jasper and Engeland, 1994; Dijkstra et al., 1996), the decreased sensitivity of the adrenal gland for ACTH (Barden et al., 1997) may be a consequence of the reduced sympathetic outflow in TG mice. Thus, it is important to note that the phenotype of the GR defunct mice appears for a large part to be determined by the reduced hypothalamic CRH activity.
Originally, the TG mice had been developed to serve as a model for the HPA axis aberrations seen in major depression. However, as discussed previously in detail (Barden et al., 1997; Karanth et al., 1997), these TG mice show similarities as well as major differences with the neuroendocrine features during this psychiatric illness (and familial glucocorticoid resistance, as well). The primary goal of the present study was to gain insight in the cause-and-effect relationship between CRH hypersecretion and GR dysfunction. It is clear from our data that long-term GR dysfunction does not necessarily generate CRH hyperexpression, thus challenging the concept of CRH hypersecretion in major depression as the consequence of impaired GR function. Rather, it may be postulated that chronic CRH hypersecretion caused by chronic stress in conjunction with genetically determined vulnerabilities is the primary determinant producing GR dysfunction in addition to the observed neurochemical, physiological, and behavioral disturbances (Linthorst et al., 1997).
The hypoactivity of hypothalamic CRH neurons in TG mice clearly differs from the enhancement observed in rats with complete deletion of GR function that is caused by adrenalectomy (Swanson et al., 1983;Whitnall et al., 1985; Whitnall, 1988). The difference could be the result of the timing of the glucocorticoid deficiency (from early fetal life vs adult). However, this is unlikely because complete GR knock-out mice show increases in CRH mRNA in the PVN (Reichardt and Schutz, 1996). Because the control of CRH expression involves interaction of glucocorticoids with other regulators, it is possible that the degree of glucocorticoid dysfunction (total in adrenalectomy and GR knock-out animals vs partial in this TG model) may determine the outcome. Furthermore, reduced GR function, in addition to direct effects in hypothalamic CRH neurons, is likely to affect stimulatory as well as inhibitory brain circuits, neurotransmitters, and receptors that affect CRH neurons (De Kloet et al., 1997; Herman and Cullinan, 1997). Therefore, the suppression of hypothalamic CRH may result from a GR dysfunction-induced shift in the balance between excitatory and inhibitory input.
In addition, changes in mineralocorticoid receptors (MR) may have to be taken into consideration. Although MRs are virtually absent in the PVN (Reul and De Kloet, 1985), they are abundantly present in the hippocampus and mediate transsynaptic tonic inhibition on PVN CRH neurons (Reul and De Kloet, 1985; De Kloet and Reul, 1987). However, preliminary observations indicate that hippocampal MR levels are decreased in TG mice (J. Reul, unpublished observations), which is in line with the role of GR in the regulation of MR expression (Reul et al., 1987, 1989). Accordingly, reduced MR levels in the hippocampus would result in enhanced PVN CRH expression, which is clearly not the case in TG animals. Changes in MR do not, therefore, provide an explanation for the reduced CRH expression in these animals.
In conclusion, we demonstrate that transgenic mice expressing antisense RNA directed against GR develop reduced activity of hypothalamic CRH neurons, indicating that defunct GR, at least if present since an early embryonic stage, is not necessarily associated with CRH hyperactivity. Counter-regulatory mechanisms at the level of CRH-R1 mRNA regulation may be involved in maintaining the responsiveness of pituitary corticotrophs during stress. Finally, it seems that a number of phenotypic changes in the physiology and behavior of the TG animals are a consequence of hypothalamic CRH hypoactivity rather than of GR dysfunction per se.
Footnotes
This work was supported by the Dutch Foundation for Scientific Research (Nederlands Wetenschappelijk Onderzoek, Grant 900-564-034) and the Volkswagen Foundation (Germany; Grant I/70 543). We thank Drs. R. M. Buijs and F. W. van Leeuwen (Amsterdam, The Netherlands) for providing the anti-AVP antiserum “Truus,” Dr. E. B. De Souza and the DuPont-Merck Pharmaceutical Company for providing hrIL-1β, Drs. J. Drouin and A. Abou Samra for providing the murine POMC genomic clone and the murine CRH receptor clone, respectively, and Mr. A. W. J. W. Janszen for his expert technical assistance.
Correspondence should be addressed to Dr. F. J. H. Tilders, Research Institute Neurosciences Vrije Universiteit, Faculty of Medicine, Department of Pharmacology, van der Boechorststraat 7, 1081 BT Amsterdam, The Netherlands.
Dr. Kiss’s present address: Institute of Experimental Endocrinology, Slovak Academy of Sciences, Bratislava, Slovak Republic.
REFERENCES
- 1.Anderson SM, Kant GJ, De Souza EB. Effects of chronic stress on anterior pituitary and brain corticotropin-releasing factor receptors. Pharmacol Biochem Behav. 1993;44:755–761. doi: 10.1016/0091-3057(93)90002-b. [DOI] [PubMed] [Google Scholar]
- 2.Banki CM, Karmacsi L, Bissette G, Nemeroff CB. Cerebrospinal fluid neuropeptides in mood disorder and dementia. J Affect Disord. 1992;25:39–46. doi: 10.1016/0165-0327(92)90091-j. [DOI] [PubMed] [Google Scholar]
- 3.Barden N, Reul JMHM, Holsboer F. Do antidepressants stabilize mood through actions on the hypothalamic-pituitary-adrenocortical system? Trends Neurosci. 1995;18:6–11. doi: 10.1016/0166-2236(95)93942-q. [DOI] [PubMed] [Google Scholar]
- 4.Barden N, Stec ISM, Holsboer F, Reul JMHM. Endocrine profile and neuroendocrine challenge tests in transgenic mice expressing antisense RNA against the glucocorticoid receptor. Neuroendocrinology. 1997;66:212–220. doi: 10.1159/000127240. [DOI] [PubMed] [Google Scholar]
- 5.Beaulieu S, Rousse I, Gratton A, Barden N, Rochford J. Behavioral and endocrine impact of impaired type II glucocorticoid receptor function in a transgenic mouse model. Ann NY Acad Sci. 1994;746:388–391. doi: 10.1111/j.1749-6632.1994.tb39263.x. [DOI] [PubMed] [Google Scholar]
- 6.Berkenbosch F, Tilders FJ. Effect of axonal transport blockade on corticotropin-releasing factor immunoreactivity in the median eminence of intact and adrenalectomized rats: relationship between depletion rate and secretory activity. Brain Res. 1988;442:312–320. doi: 10.1016/0006-8993(88)91517-x. [DOI] [PubMed] [Google Scholar]
- 7.Berkenbosch F, Linton EA, Tilders FJ. Colocalization of peptide histidine isoleucine amine and corticotropin-releasing factor immunoreactivity in neurons of the rat hypothalamus: a surprising artefact. Neuroendocrinology. 1986;44:338–346. doi: 10.1159/000124666. [DOI] [PubMed] [Google Scholar]
- 8.Berkenbosch F, van Oers J, del Rey A, Tilders F, Besedovsky H. Corticotropin-releasing factor-producing neurons in the rat activated by interleukin-1. Science. 1987;238:524–526. doi: 10.1126/science.2443979. [DOI] [PubMed] [Google Scholar]
- 9.Bertini LT, Kiss JZ. Hypophysiotrophic neurons are capable of altering the ratio of co-packaged neurohormones. Neuroscience. 1991;42:237–244. doi: 10.1016/0306-4522(91)90161-g. [DOI] [PubMed] [Google Scholar]
- 10.Bornstein SR, Ehrhart-Bornstein M, Scherbaum WA, Pfeiffer EF, Holst JJ. Effects of splanchnic nerve stimulation on the adrenal cortex may be mediated by chromaffin cells in a paracrine manner. Endocrinology. 1990;127:900–906. doi: 10.1210/endo-127-2-900. [DOI] [PubMed] [Google Scholar]
- 11.Bradbury MJ, Akana SF, Dallman MF. Roles of type I and II corticosteroid receptors in regulation of basal activity in the hypothalamo-pituitary-adrenal axis during the diurnal trough and the peak: evidence for a nonadditive effect of combined receptor occupation. Endocrinology. 1994;134:1286–1296. doi: 10.1210/endo.134.3.8119168. [DOI] [PubMed] [Google Scholar]
- 12.Brown MR, Fisher LA. Central nervous system effects of corticotropin-releasing factor in the dog. Brain Res. 1983;280:75–79. doi: 10.1016/0006-8993(83)91175-7. [DOI] [PubMed] [Google Scholar]
- 13.Brown MR, Fisher LA, Spiess J, Rivier C, Rivier J, Vale W. Corticotropin-releasing factor: actions on the sympathetic nervous system and metabolism. Endocrinology. 1982;111:928–931. doi: 10.1210/endo-111-3-928. [DOI] [PubMed] [Google Scholar]
- 14.Brown MR, Fisher LA, Webb V, Vale WW, Rivier JE. Corticotropin-releasing factor: a physiologic regulator of adrenal epinephrine secretion. Brain Res. 1985;328:355–357. doi: 10.1016/0006-8993(85)91048-0. [DOI] [PubMed] [Google Scholar]
- 15.Carroll BJ, Martin FI, Davies B. Resistance to suppression by dexamethasone of plasma 11-O.H.C.S. levels in severe depressive illness. Br Med J. 1968;3:285–287. doi: 10.1136/bmj.3.5613.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chrousos GP, Gold PW. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA. 1992;267:1244–1252. [PubMed] [Google Scholar]
- 17.Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, Fantuzzi G, Hummler E, Unsicker K, Schutz G. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995;9:1608–1621. doi: 10.1101/gad.9.13.1608. [DOI] [PubMed] [Google Scholar]
- 18.De Goeij DC, Kvetnansky R, Whitnall MH, Jezova D, Berkenbosch F, Tilders FJ. Repeated stress-induced activation of corticotropin-releasing factor neurons enhances vasopressin stores and colocalization with corticotropin-releasing factor in the median eminence of rats. Neuroendocrinology. 1991;53:150–159. doi: 10.1159/000125712. [DOI] [PubMed] [Google Scholar]
- 19.De Kloet ER, Reul JMHM. Feedback action and tonic influence of corticosteroids on brain function: a concept arising from heterogeneity of brain receptor systems. Psychoneuroendocrinology. 1987;12:83–105. doi: 10.1016/0306-4530(87)90040-0. [DOI] [PubMed] [Google Scholar]
- 20.De Kloet ER, Oitzl MS, Joels M. Functional implications of brain corticosteroid receptor diversity. Cell Mol Neurobiol. 1993;13:433–455. doi: 10.1007/BF00711582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. Glucocorticoid feedback resistance. Trends Endocrinol Metab. 1997;8:26–33. doi: 10.1016/s1043-2760(96)00205-6. [DOI] [PubMed] [Google Scholar]
- 22.De Souza EB, Bissette G, Whitehouse PJ, Powers RE, Price DL, Vale WW, Nemeroff CB. Abnormalities in corticotropin-releasing factor (CRF) in neurodegenerative diseases. In: De Souza EB, Nemeroff CB, editors. Corticotropin-releasing factor: basic and clinical studies of a neuropeptide. CRC; Boca Raton, FL: 1993. pp. 335–349. [Google Scholar]
- 23.Deuschle M, Schweiger U, Weber B, Gotthardt U, Korner A, Schmider J, Standhardt H, Lammers CH, Heuser I. Diurnal activity and pulsatility of the hypothalamus-pituitary-adrenal system in male depressed patients and healthy controls. J Clin Endocrinol Metab. 1997;82:234–238. doi: 10.1210/jcem.82.1.3689. [DOI] [PubMed] [Google Scholar]
- 24.Diamant M, Croiset G, de Wied D. The effect of corticotropin-releasing factor (CRF) on autonomic and behavioral responses during shock-prod burying test in rats. Peptides. 1992a;13:1149–1158. doi: 10.1016/0196-9781(92)90022-u. [DOI] [PubMed] [Google Scholar]
- 25.Diamant M, Kashtanov SI, Fodor M, de Wied D. Corticotropin-releasing factor induces differential behavioral and cardiovascular effects after intracerebroventricular and lateral hypothalamic/perifornical injections in rats. Neuroendocrinology. 1992b;56:750–760. doi: 10.1159/000126303. [DOI] [PubMed] [Google Scholar]
- 26.Dijkstra I, Binnekade R, Tilders FJH. Diurnal variation in resting levels of corticosterone is not mediated by variation in adrenal responsiveness to adrenocorticotropin but involves splanchnic nerve integrity. Endocrinology. 1996;137:540–547. doi: 10.1210/endo.137.2.8593800. [DOI] [PubMed] [Google Scholar]
- 27.Dunn AJ, Berridge CW. Physiological and behavioral responses to corticotropin-releasing factor administration: is CRF a mediator of anxiety or stress responses? Brain Res Rev. 1990;15:71–100. doi: 10.1016/0165-0173(90)90012-d. [DOI] [PubMed] [Google Scholar]
- 28.Edwards AV, Jones CT. The effect of splanchnic nerve section on the sensitivity of the adrenal cortex to adrenocorticotrophin in the calf. J Physiol (Lond) 1987;390:23–31. doi: 10.1113/jphysiol.1987.sp016683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engeland WC, Gann DS. Splanchnic nerve stimulation modulates steroid secretion in hypophysectomized dogs. Neuroendocrinology. 1989;50:124–131. doi: 10.1159/000125211. [DOI] [PubMed] [Google Scholar]
- 30.Gold PW, Loriaux DL, Roy A, Kling MA, Calabrese JR, Kellner CH, Nieman LK, Post RM, Pickar D, Gallucci W. Responses to corticotropin-releasing hormone in the hypercortisolism of depression and Cushing’s disease. Pathophysiologic and diagnostic implications. N Engl J Med. 1986;314:1329–1335. doi: 10.1056/NEJM198605223142101. [DOI] [PubMed] [Google Scholar]
- 31.Harbuz MS, Lightman SL. Responses of hypothalamic and pituitary mRNA to physical and psychological stress in the rat. J Endocrinol. 1989;122:705–711. doi: 10.1677/joe.0.1220705. [DOI] [PubMed] [Google Scholar]
- 32.Hauger RL, Aguilera G. Regulation of pituitary corticotropin-releasing hormone (CRH) receptors by CRH: interaction with vasopressin. Endocrinology. 1993;133:1708–1714. doi: 10.1210/endo.133.4.8404613. [DOI] [PubMed] [Google Scholar]
- 33.Hauger RL, Irwin MR, Lorang M, Aguilera G, Brown MR. High intracerebral levels of CRH result in CRH receptor downregulation in the amygdala and neuroimmune desensitization. Brain Res. 1993;616:283–292. doi: 10.1016/0006-8993(93)90219-d. [DOI] [PubMed] [Google Scholar]
- 34.Heinrichs SC, Menzaghi F, Pich EM, Britton KT, Koob GF. The role of CRF in behavioral aspects of stress. Ann NY Acad Sci. 1995;771:92–104. doi: 10.1111/j.1749-6632.1995.tb44673.x. [DOI] [PubMed] [Google Scholar]
- 35.Herman JP, Cullinan WE. Neurocircuitry of stress: central control of the hypothalamo-pituitary-adrenal axis. Trends Neurosci. 1997;20:78–84. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- 36.Heuser I, Yassouridis A, Holsboer F. The combined dexamethasone/CRH test: a refined laboratory test for psychiatric disorders. J Psychiatr Res. 1994;28:341–356. doi: 10.1016/0022-3956(94)90017-5. [DOI] [PubMed] [Google Scholar]
- 37.Holmes MC, Catt KJ, Aguilera G. Involvement of vasopressin in the down-regulation of pituitary corticotropin-releasing factor receptors after adrenalectomy. Endocrinology. 1987;121:2093–2098. doi: 10.1210/endo-121-6-2093. [DOI] [PubMed] [Google Scholar]
- 38.Holsboer F, Von Bardeleben U, Gerken A, Stalla GK, Muller OA. Blunted corticotropin and normal cortisol response to human corticotropin-releasing factor in depression. N Engl J Med. 1984;311:1127. doi: 10.1056/NEJM198410253111718. [DOI] [PubMed] [Google Scholar]
- 39.Holsboer F, Grasser A, Friess E, Wiedemann K. Steroid effects on central neurons and implications for psychiatric and neurological disorders. Ann NY Acad Sci. 1994;746:345–359. doi: 10.1111/j.1749-6632.1994.tb39255.x. [DOI] [PubMed] [Google Scholar]
- 40.Jasper MS, Engeland WC. Splanchnic neural activity modulates ultradian and circadian rhythms in adrenocortical secretion in awake rats. Neuroendocrinology. 1994;59:97–109. doi: 10.1159/000126645. [DOI] [PubMed] [Google Scholar]
- 41.Karanth S, Linthorst ACE, Stalla GK, Barden N, Holsboer F, Reul JMHM. Hypothalamic-pituitary-adrenocortical axis changes in a transgenic mouse with impaired glucocorticoid receptor function. Endocrinology. 1997;138:3476–3485. doi: 10.1210/endo.138.8.5331. [DOI] [PubMed] [Google Scholar]
- 42.Labeur MS, Arzt E, Wiegers GJ, Holsboer F, Reul JMHM. Long-term intracerebroventricular corticotropin-releasing hormone administration induces distinct changes in rat splenocyte activation and cytokine expression. Endocrinology. 1995;136:2678–2688. doi: 10.1210/endo.136.6.7750492. [DOI] [PubMed] [Google Scholar]
- 43.Lamberts SWJ, Koper JW, Biemond P, den Holder FH, de Jong FH. Cortisol receptor resistance: the variability of its clinical presentation and response to treatment. J Clin Endocrinol Metab. 1992;74:313–321. doi: 10.1210/jcem.74.2.1309833. [DOI] [PubMed] [Google Scholar]
- 44.Linkowski P, Mendlewicz J, Leclerq R, Brasseur M, Mubain P, Golstein J, Copinschi G, Cauter EV. The 24 hr profile of adrenocorticotropin and cortisol in major depressive illness. J Clin Endocrinol Metab. 1985;61:429–438. doi: 10.1210/jcem-61-3-429. [DOI] [PubMed] [Google Scholar]
- 45.Linthorst ACE, Flachskamm C, Hoadley M, Hopkins S, Labeur MS, Holsboer F, Reul JMHM. Long-term intracerebroventricular infusion of corticotropin-releasing hormone alters neuroendocrine, neurochemical, autonomic, behavioral, and cytokine responses to a systemic inflammatory challenge. J Neurosci. 1997;17:4448–4460. doi: 10.1523/JNEUROSCI.17-11-04448.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo X, Kiss A, Rabadan-Diehl C, Aguilera G. Regulation of hypothalamic and pituitary corticotropin-releasing hormone receptor messenger ribonucleic acid by adrenalectomy and glucocorticoids. Endocrinology. 1995;136:3877–3883. doi: 10.1210/endo.136.9.7649095. [DOI] [PubMed] [Google Scholar]
- 47.Makino S, Schulkin J, Smith MA, Pacak K, Palkovits M, Gold PW. Regulation of corticotropin-releasing hormone receptor messenger ribonucleic acid in the rat brain and pituitary by glucocorticoids and stress. Endocrinology. 1995;136:4517–4525. doi: 10.1210/endo.136.10.7664672. [DOI] [PubMed] [Google Scholar]
- 48.Marchetti B, Peiffer A, Morale MC, Batticane N, Gallo F, Barden N. Transgenic animals with impaired type II glucocorticoid receptor gene expression: a model to study aging of the neuroendocrine-immune system. Ann NY Acad Sci. 1994;746:308–327. doi: 10.1111/j.1749-6632.1994.tb56839.x. [DOI] [PubMed] [Google Scholar]
- 49.Menzaghi F, Heinrichs SC, Merlo-Pich E, Tilders FJ, Koob GF. Involvement of hypothalamic corticotropin-releasing factor neurons in behavioral responses to novelty in rats. Neurosci Lett. 1994;168:139–142. doi: 10.1016/0304-3940(94)90435-9. [DOI] [PubMed] [Google Scholar]
- 50.Montkowski A, Barden N, Wotjak C, Stec I, Ganster J, Meaney M, Engelmann M, Reul JMHM, Landgraf R, Holsboer F. Long-term antidepressant treatment reduces behavioural deficits in transgenic mice with impaired glucocorticoid receptor function. J Neuroendocrinol. 1995;7:841–845. doi: 10.1111/j.1365-2826.1995.tb00724.x. [DOI] [PubMed] [Google Scholar]
- 51.Morale MC, Batticane N, Gallo F, Barden N, Marchetti B. Disruption of hypothalamic-pituitary-adrenocortical system in transgenic mice expressing type II glucocorticoid receptor antisense ribonucleic acid permanently impairs T cell function: effects on T cell trafficking and T cell responsiveness during postnatal development. Endocrinology. 1995;136:3949–3960. doi: 10.1210/endo.136.9.7649104. [DOI] [PubMed] [Google Scholar]
- 52.Nemeroff CB, Widerlov E, Bissette G, Walleus H, Karlsson I, Eklund K, Kilts CD, Loosen PT, Vale W. Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science. 1984;226:1342–1344. doi: 10.1126/science.6334362. [DOI] [PubMed] [Google Scholar]
- 53.Nemeroff CB, Owens MJ, Bissette G, Andorn AC, Stanley M. Reduced corticotropin-releasing factor receptor binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry. 1988;45:577–579. doi: 10.1001/archpsyc.1988.01800300075009. [DOI] [PubMed] [Google Scholar]
- 54.Owens MJ, Nemeroff CB. Physiology and pharmacology of corticotropin-releasing factor. Pharmacol Rev. 1991;43:425–473. [PubMed] [Google Scholar]
- 55.Pepin M-C, Pothier F, Barden N. Impaired type II glucocorticoid receptor function in mice bearing antisense RNA transgene. Nature. 1992a;355:725–728. doi: 10.1038/355725a0. [DOI] [PubMed] [Google Scholar]
- 56.Pepin M-C, Pothier F, Barden N. Antidepressant drug action in a transgenic mouse model of the endocrine changes seen in depression. Mol Pharmacol. 1992b;42:991–995. [PubMed] [Google Scholar]
- 57.Pozzoli G, Bilezikjian LM, Perrin MH, Blount AL, Vale WW. Corticotropin-releasing factor (CRF) and glucocorticoids modulate the expression of type 1 CRF receptor messenger ribonucleic acid in rat anterior pituitary cell cultures. Endocrinology. 1996;137:65–71. doi: 10.1210/endo.137.1.8536643. [DOI] [PubMed] [Google Scholar]
- 58.Raadsheer FC, Hoogendijk WJG, Stam FC, Tilders FJH, Swaab DF. Increased numbers of corticotropin-releasing hormone-expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology. 1994;60:436–444. doi: 10.1159/000126778. [DOI] [PubMed] [Google Scholar]
- 59.Raadsheer FC, Van Heerikhuize JJ, Lucassen PJ, Hoogendijk WJ, Tilders FJH, Swaab DF. Corticotropin-releasing hormone (CRH) mRNA levels in the paraventricular nucleus of patients with Alzheimer’s disease and depression. Am J Psychiatry. 1995;152:1372–1376. doi: 10.1176/ajp.152.9.1372. [DOI] [PubMed] [Google Scholar]
- 60.Rabadan-Diehl C, Kiss A, Camacho C, Aguilera G. Regulation of messenger ribonucleic acid for corticotropin-releasing hormone receptor in the pituitary during stress. Endocrinology. 1996;137:3808–3814. doi: 10.1210/endo.137.9.8756551. [DOI] [PubMed] [Google Scholar]
- 61.Reichardt HM, Schutz G. Feedback control of glucocorticoid production is established during fetal development. Mol Med. 1996;2:735–744. [PMC free article] [PubMed] [Google Scholar]
- 62.Reul JMHM, De Kloet ER. Two receptor systems for corticosterone in rat brain: microdistribution and differential occupation. Endocrinology. 1985;117:2505–2511. doi: 10.1210/endo-117-6-2505. [DOI] [PubMed] [Google Scholar]
- 63.Reul JMHM, Van den Bosch FR, De Kloet ER. Differential response of type I and type II corticosteroid receptor to changes in plasma steroid level and circadian rhythmicity. Neuroendocrinology. 1987;45:407–412. doi: 10.1159/000124766. [DOI] [PubMed] [Google Scholar]
- 64.Reul JMHM, Pearce PT, Funder JW, Krozowski ZS. Type I and type II corticosteroid receptor gene expression in the rat: effect of adrenalectomy and dexamethasone administration. Mol Endocrinol. 1989;3:1674–1680. doi: 10.1210/mend-3-10-1674. [DOI] [PubMed] [Google Scholar]
- 65.Richard D, Chapdelaine S, Deshaies Y, Pepin M-C, Barden N. Energy balance and lipid metabolism in transgenic mice bearing an antisense GCR gene construct. Am J Physiol. 1993;265:R146–R150. doi: 10.1152/ajpregu.1993.265.1.R146. [DOI] [PubMed] [Google Scholar]
- 66.Rothwell NJ. Central effects of CRF on metabolism and energy balance. Neurosci Biobehav Rev. 1990;14:263–271. doi: 10.1016/s0149-7634(05)80037-5. [DOI] [PubMed] [Google Scholar]
- 67.Rubin RT, Poland RE, Lesser IM, Winston RA, Blodgett ALN. Neuroendocrine aspects of primary endogenous depression. I. Cortisol secretory dynamics in patients and matched controls. Arch Gen Psychiatry. 1987;44:328–336. doi: 10.1001/archpsyc.1987.01800160032006. [DOI] [PubMed] [Google Scholar]
- 68.Sakai K, Horiba N, Sakai Y, Tozawa F, Demura H, Suda T. Regulation of corticotropin-releasing factor receptor messenger ribonucleic acid in rat anterior pituitary. Endocrinology. 1996;137:1758–1763. doi: 10.1210/endo.137.5.8612512. [DOI] [PubMed] [Google Scholar]
- 69.Sapolsky R, Rivier C, Yamamoto G, Plotsky P, Vale W. Interleukin-1 stimulates the secretion of hypothalamic corticotropin-releasing factor. Science. 1987;238:522–524. doi: 10.1126/science.2821621. [DOI] [PubMed] [Google Scholar]
- 70.Schmidt ED, Janszen AW, Wouterlood FG, Tilders FJH. Interleukin-1-induced long-lasting changes in hypothalamic corticotropin-releasing hormone (CRH) neurons and hyperresponsiveness of the hypothalamus–pituitary–adrenal axis. J Neurosci. 1995;15:7417–7426. doi: 10.1523/JNEUROSCI.15-11-07417.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmidt ED, Janszen AWJW, Binnekade R, Tilders FJH. Transient suppression of resting corticosterone levels induces sustained increase of AVP stores in hypothalamic CRH neurons of rats. J Neuroendocrinol. 1997;9:69–77. doi: 10.1046/j.1365-2826.1997.00618.x. [DOI] [PubMed] [Google Scholar]
- 72.Stec I, Barden N, Reul JMHM, Holsboer F. Dexamethasone nonsuppression in transgenic mice expressing antisense RNA to the glucocorticoid receptor. J Psychiatr Res. 1994;28:1–5. doi: 10.1016/0022-3956(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 73.Stenzel-Poore MP, Cameron VA, Vaughan J, Sawchenko PE, Vale W. Development of Cushing’s syndrome in corticotropin-releasing factor transgenic mice. Endocrinology. 1992;130:3378–3386. doi: 10.1210/endo.130.6.1597149. [DOI] [PubMed] [Google Scholar]
- 74.Stenzel-Poore MP, Heinrichs SC, Rivest S, Koob GF, Vale WW. Overproduction of corticotropin-releasing factor in transgenic mice: a genetic model of anxiogenic behavior. J Neurosci. 1994;14:2579–2584. doi: 10.1523/JNEUROSCI.14-05-02579.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stenzel-Poore MP, Duncan JE, Rittenberg MB, Bakke AC, Heinrichs SC. CRH overproduction in transgenic mice: behavioral and immune system modulation. Ann NY Acad Sci. 1996;780:36–48. doi: 10.1111/j.1749-6632.1996.tb15110.x. [DOI] [PubMed] [Google Scholar]
- 76.Sutton RE, Koob GF, Le Moal M, Rivier J, Vale W. Corticotropin-releasing factor produces behavioural activation in rats. Nature. 1982;297:331–333. doi: 10.1038/297331a0. [DOI] [PubMed] [Google Scholar]
- 77.Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- 78.Van Oers JWAM, Tilders FJH. Non-adrenocorticotropin-mediated effects of endogenous corticotropin-releasing factor on the adrenocortical activity in the rat. J Neuroendocrinol. 1991;3:119–121. doi: 10.1111/j.1365-2826.1991.tb00251.x. [DOI] [PubMed] [Google Scholar]
- 79.Van Oers JWAM, Hinson JP, Binnekade R, Tilders FJH. Physiological role of corticotropin-releasing factor in the control of adrenocorticotropin-mediated corticosterone release from the rat adrenal gland. Endocrinology. 1992;130:282–288. doi: 10.1210/endo.130.1.1309333. [DOI] [PubMed] [Google Scholar]
- 80.Whitnall MH. Distribution of pro-vasopressin-expressing and pro-vasopressin deficient CRH neurons in the paraventricular hypothalamic nucleus of colchicine-treated normal and adrenalectomized rats. J Comp Neurol. 1988;275:13–28. doi: 10.1002/cne.902750103. [DOI] [PubMed] [Google Scholar]
- 81.Whitnall MH, Mezey E, Gainer H. Co-localization of corticotropin-releasing factor and vasopressin in median eminence neurosecretory vesicles. Nature. 1985;317:248–250. doi: 10.1038/317248a0. [DOI] [PubMed] [Google Scholar]
- 82.Wynn PC, Harwood JP, Catt KJ, Aguilera G. Regulation of corticotropin-releasing factor (CRF) receptors in the rat pituitary gland: effects of adrenalectomy on CRF receptors and corticotroph responses. Endocrinology. 1985;116:1653–1659. doi: 10.1210/endo-116-4-1653. [DOI] [PubMed] [Google Scholar]
- 83.Wynn PC, Harwood JP, Catt KJ, Aguilera G. Corticotropin-releasing factor (CRF) induces desensitization of the rat pituitary CRF receptor–adenylate cyclase complex. Endocrinology. 1988;122:351–358. doi: 10.1210/endo-122-1-351. [DOI] [PubMed] [Google Scholar]