

Abstract
Dihydropyridine-insensitive Ca channels are subject to direct receptor G-protein-mediated inhibition to differing extents. α1B channels are much more strongly modulated than α1E channels. To understand the structural basis for this difference, we have constructed and expressed various α1Band α1E chimeric Ca channels and examined their regulation by κ-opioid receptors. Replacement of the first membrane-spanning domain of α1E with the corresponding region of α1B resulted in a chimeric Ca channel that was modulated by κ-opioid receptors to a significantly greater extent than α1E. Transfer of the N terminus and I/II loop from α1B in addition to domain I resulted in a chimeric channel that was modulated to the same extent as α1B. Other regions of the molecule do not appear to contribute significantly to the degree of inhibition obtained, although the C terminus may contribute to facilitation.
Keywords: calcium channels, G-proteins, opioid receptors, modulation, structure–function, inverse agonism
Voltage-sensitive Ca channels are one of the most important classes of molecules used by excitable cells for the transduction of cellular excitability into the intracellular Ca signals that regulate events such as muscle contraction and the release of neurotransmitters. Inhibition of Ca influx through these channels by G-protein-linked receptors is an important means by which neurotransmitters and drugs can regulate the strength of synaptic transmission (Miller, 1990; Rhim et al., 1994). Elucidation of the primary structure of Ca channels has demonstrated that they consist of a family of related proteins that has two branches. Each branch consists of three different proteins that differ in their pharmacology and, in particular, in their sensitivity to dihydropyridine (DHP) drugs. It is thought that the subfamily of DHP-insensitive Ca channels can be regulated by receptors through the direct interaction of G-protein βγ subunits with the major pore-forming (α1) subunit of the channel (Herlitze et al., 1996; Ikeda, 1996; De Waard, 1997; Zamponi et al., 1997). Expression studies have indicated that all three types of DHP-insensitive Ca channels (α1A, α1B, and α1E) are regulated by G-proteins, although there are large differences in their susceptibility to modulation. Thus, α1B is the most sensitive to G-protein modulation, and α1E is the least sensitive (Toth et al., 1996; Yassin et al., 1996).
The structural basis for the G-protein regulation of Ca channels has received considerable attention, and recent studies have started to reveal the sites of interaction between G-protein βγ subunits and Ca channels. Initially, it was shown that G-protein βγ subunits could interact with two distinct sites in the intracellular loop connecting the first two domains of the α1 subunit (I/II loop) (De Waard et al., 1997; Qin et al., 1997; Zamponi et al., 1997). One of these sites contains the QXXER motif, which has been shown to be important in the binding of βγ subunits to other effectors (Pragnell et al., 1994; Chen et al., 1995). However, although there is little doubt that βγ subunits can interact with this region of the channel, controversy exists as to the significance of this interaction. Experiments designed to demonstrate the importance of this interaction for G-protein regulation of Ca channels have yielded mixed results. Further studies have indicated that βγ subunits can also bind to a region in the C-terminal tail of sensitive Ca channels (Qin et al., 1997), and there are data suggesting that this interaction may be important in G-protein modulation (Qin et al., 1997; Zhang et al., 1996). In either case, because binding to these sites is a common element to all three DHP-insensitive Ca channels, further structural features are likely to be required to determine differential sensitivity to G-proteins. To identify structural elements involved in determining the differential sensitivity of α1B compared with α1E channels, we have created a variety of chimeric Ca channels in which different parts of α1B-1 were transferred into an α1E-3 background. Exchange of domain I alone yields a chimeric channel with considerably increased sensitivity to modulation, but the further exchange of the I/II linker and the N terminus is required to obtain a chimeric channel with sensitivity to modulation equivalent to native α1B-1. Transfer of the C terminus of α1B-1 appears to enhance facilitation but not inhibition.
MATERIALS AND METHODS
Cell culture. tsA-201 cells (a gift from Dr. W. A. Horne, Stanford University) were cultured in MEM (Life Technologies, Gaithersburg, MD), 10% fetal bovine serum (Life Technologies), and 1% penicillin-streptomycin (Life Technologies) at 37°C in 5% CO2. When cells reached 40–70% confluency they were replated in 35 mm dishes and were transfected 4–10 hr later with a total of 5–7 μg of DNA (2–5 μg of α1, 1.7 μg of α2B, 0.8 μg of β1B, 1.5 μg of κ1R or δR, and 1 μg of CD8-α) by polyethyleneimine-mediated transfection (Boussif et al., 1996). Cells were washed with culture medium 1–2 hr after transfection, and fresh 10% serum-containing medium was added. Cells were detached from dishes with HBSS (Ca- and Mg-free; Life Technologies) 30–40 hr later and were replated on poly-l-lysine-coated coverslips. Cells were recorded from 36 to 80 hr after transfection. cDNA for CD8α was a gift from Dr. J. Bluestone (University of Chicago). The κ1R and δR constructs were gifts from Dr. G. I. Bell (Howard Hughes Medical Institute, University of Chicago) and have been described previously (Yasuda et al., 1993). Expression of α2B and β1B were verified by reverse transcription-PCR and Northern blotting (data not shown).
Electrophysiology. Currents were recorded in a solution containing 15 mm BaCl2 (5 mmBaCl2 in the case of the current–voltage data), 150 mm TEA-Cl, and 10 mm HEPES, pH 7.4, with TEA-OH or N-methyl-d-glucamine. Pipette solutions were made according to the method of Bean (1992). U69593 (Sigma, St. Louis, MO; or Research Biochemicals, Natick, MA) and norbinaltrophimine (norBNI; Research Biochemicals) were made as concentrated stock solutions in 100% ethanol. ICI-174,864 was made as a 10 mmstock solution in DMSO. [d-Pen2,5]-enkephalin (DPDPE) was made as a 10 mm stock solution in sterile water. U69593, DPDPE, and ICI-174864 were added to the bath solution at concentrations of 1 μm, and norBNI was added at 100 nm. Data were recorded under the control of pClamp6 software (Axon Instruments, Foster City, CA) with an Axon 200A amplifier at an acquisition rate of 10 kHz and were filtered with a four-pole Bessel filter at 2 kHz. Pipettes were typically 1–3 mΩ in resistance, and series resistance was compensated >80% in all cases. To assay G-protein modulation, currents were elicited by a dual-pulse protocol consisting of two 50 msec depolarizations to 0 mV from a holding potential of −90 mV, separated by 800 msec at the holding potential, with a 30 msec, 90 mV depolarization (prepulse) ending 5 msec before the second pulse. Cells expressing CD8α were identified by incubation with anti-CD8α-coated beads (Dynal, Great Neck, NY), and bead-decorated cells were chosen exclusively for recordings.
Data analysis. Data were analyzed off-line using Clampfit (Axon Instruments), Systat (SPSS Inc., Chicago, IL), MATLAB (MathWorks Inc.), and custom-written software. Current inhibition was estimated as follows. Peak current amplitudes were measured and were adjusted to remove any slow linear “runup” or “rundown” by subtraction of linear function fits to the baselines. A series of adjacent current amplitudes was averaged just before drug exposure, during U69593 exposure at the peak of the drug effect, and during the peak of the norBNI response. Currents in the presence of U69593 were taken as an estimate of current in the presence of maximally activated receptor I(R*). When norBNI caused amplitudes to increase beyond baseline values before drug exposure, currents were taken as an estimate of current in the presence of maximally inactive receptor I(R). Otherwise, baseline values were used to estimate I(R). “Total modulation” was then defined as [I(R) − I(R*)]/I(R).
Relief of inhibition by the depolarizing prepulse was quantified by calculating a “corrected prepulse ratio” to allow for comparisons between constructs while correcting for differences in inactivation caused by the prepulse. Current ratios in the presence of U69593,I(+pp)/I(−pp), were calculated and corrected for differences in inactivation between constructs by multiplying by the ratio I(−pp)/I(+pp) in the presence of norBNI. Facilitation was expressed as a simple current ratio,I(+pp)/I(−pp), for the purpose of describing the intrinsic receptor activity of the κ1R with respect to α1B-1 currents.
Unless otherwise noted, comparisons between the constructs in terms of current inhibition and relief of inhibition were conducted by means of a one-way ANOVA followed by post hoc analysis by the Tukey multiple-comparison procedure (Neter et al., 1985). Averages were expressed as mean ± SEM.
Construction of cDNAs.The chimeric contructs were created using overlap extension PCR (Ho et al., 1989; Horton et al., 1989,1990) and PCR-mediated site-directed mutagenesis. PCR products were cloned into pGEM-T (Promega, Madison, WI), pGEM-T-EZ (Promega), or pT7-Blue (Novagen, Madison, WI) and sequenced using semiautomated DNA sequencing (ABI 377; Perkin-Elmer, Oak Brook, IL). All α1subunit expression contructs were created by ligation of recombinant and native sequences into the pcDNA3.1 expression vector (Invitrogen, San Diego, CA). Final constructs were confirmed by a combination of restriction analysis and DNA sequencing. The native α1B-1construct bBbBbBbBb consisted of residues 1–2340 of GenBank accession number 284339 (a gift from Dr. R. Harpold, SIBIA Neurosciences). The native α1E-3 construct eEeEeEeEe consisted of residues 1–2271 of GenBank accession number 1082919 (a gift from Dr. R. Harpold, SIBIA Neurosciences). The construct bBbBbEeEb consisted of α1B-1 1–1145, α1E-3 1149–1724, and α1B-1 1710–2340. The construct bBbBbEeEe consisted of α1B-1 1–1145 and α1E-3 1149–2271. The construct bBbEeEeEe consisted of α1B-1 1–482 and α1E-3 477–2271. The construct eBbEeEeEe consisted of α1E-3 1–89, α1B-1 96–482, and α1E-3 477–2271. The construct eBeEeEeEe consisted of α1E-3 1–89, α1B-1 96–355, and α1E-3 351–2271. The construct eEbEeEeEe consisted of α1E-3 1–350, α1B-1 356–482, and α1E-3 477–2271. The construct eEbBbEeEe consisted of α1E-3 1–350, α1B-1 356–1145, and α1E-3 1149–2271. The construct eEeEbEeEe consisted of α1E-3 1–703, α1B-1 710–1145, and α1E-3 1149–2271. The construct eEbEbEeEe consisted of α1E-3 1–350, α1B-1 356–482, α1E-3 477–703, α1B-1 710–1145, and α1E-3 1149–2271. The construct e(E90–309B315–355)bEeEeEe consisted of α1E-3 1–309, α1B-1 315–482, and α1E-3 477–2271. The α2B/δ and β1B Ca channel subunit cDNAs were gifts from Dr. R. Harpold (Sibia Neurosciences) and were subcloned into the pCMV6c expression vector.
RESULTS
Intrinsic activity of the κ-opioid receptor and effects on α1B and α1E
After transfection of tsA201 cells (Margolskee et al., 1993) with the Ca channel subunits α1-B1, β1B, and α2B, the κ1-opioid receptor (κ1R), and CD8α, we observed that α1-B1 currents often showed a degree of prepulse facilitation, suggestive of G-protein-mediated inhibition, even without previous exposure to κ1R agonists (Fig.1A,B). Overall, 62% of α1B-1- and κ1R-transfected cells responsive to κ1R agonists showed some degree of facilitation before drug exposure. These effects were not seen in cells incubated overnight with 100 ng/ml pertussis toxin (n = 5) or in cells that were not transfected with the κ1R receptor (n = 6), suggesting a receptor-mediated, G-protein (Gi and/or Go)-dependent mechanism (data not shown). Similar G-protein receptor-dependent modulation of Ca channels in the absence of agonist has been described by other groups (Ikeda, 1992; Zhang et al., 1996). The effects seen in the present study did not diminish when cells remained in a rapidly flowing bath for longer than 20 min, suggesting that agonist-independent modulation was not caused by the secretion of opioid agonists by the cells or the presence of opioid agonists in the culture media (Doupnik et al., 1994). U69593 is a κ1R-selective agonist with an affinity for κ1 receptors in the range of 1–2 nm(Avidor-Reiss et al., 1995; Simonin et al., 1995) and an EC50 for adenylyl cyclase inhibition of ∼8 nm(Avidor-Reiss et al., 1995). When U69593 was added to the bath solution at a supramaximal concentration of 1 μm, α1-B1-expressing cells showed a marked reduction in current (Fig. 1A) (47 ± 6.4%;n = 13), similar to the results of a number of other groups (Kaneko et al., 1994; Tallent et al., 1994; Carpenter et al., 1996; Toth et al., 1996). When the κ1R-selective “antagonist” norBNI (Portoghese et al., 1987) was added directly after U69593 exposure at a concentration of 100 nm, the inhibition produced by the agonist was always relieved within 3–4 min (Fig. 1A,B), and currents often increased in magnitude beyond those seen before U69593 exposure (Fig.1A,B) (n = 13). When U69593 was added to cells without subsequent addition of norBNI, current inhibition and facilitation washed out in ∼10 min, and no enhancement of current amplitudes was observed (data not shown) (n = 5). When norBNI was added to κ1R expressing cells that showed facilitation without previous agonist exposure, this facilitation was always reversed, and currents were always observed to increase in amplitude (Fig. 1C) (n = 5). However, when the opioid receptor antagonist naloxone (20 μm) was added to such cells, no current increase was observed (n = 3) (data not shown), suggesting that naloxone, unlike norBNI, is a neutral antagonist. norBNI is known to bind at κ1 receptors with affinities in the range of 0.5–1.2 nm (Prather et al., 1995; Simonin et al., 1995) and completely blocks opioid agonist stimulation of adenylyl cyclase at 100 nm (Avidor-Reiss et al., 1995). Because norBNI was applied in the absence of agonist and reduced prepulse ratios to that seen in untransfected cells, we believe that maximal relief of agonist-independent modulation was obtained with 100 nm norBNI.
Fig. 1.

Opioid agonist and inverse agonist effects on Ba currents for α1B-1- and κ1R-expressing tsA-201 cells. A, Peak current amplitudes and representative currents before prepulse (green) and after a 30 msec depolarizing prepulse (black), illustrating the effects of the addition to the bath of 1 μm U69593 or 100 nm norBNI. Currents were taken from the points indicated. Currents were evoked by a 50 msec depolarization to 0 mV from a holding potential of −90 mV. Calibration: 250 pA, 10 msec. B, Representative currents illustrating relatively small current inhibition after U69593 exposure and relatively large current enhancement after norBNI exposure in cells with a large degree of modulation (as indicated by a relatively large increase in current amplitudes after a prepulse) before agonist exposure. C, Representative currents illustrating the effects of 100 nm norBNI added before 1 μmU69593. D, Currents evoked from a α1B-1- and δR-expressing cell in the presence of 100 nm norBNI, 1 μm ICI-174864, and 1 μm DPDPE.
Both the U69593 (inhibitory) and norBNI (enhancing) effects in these experiments could be completely blocked by pertussis toxin (n = 5), and cells responsive to one drug were always responsive to the other. When 50 μm GF109203, a specific PKC inhibitor, was included in the intracellular solution (n = 3), the response of α1B-1- and κ1R-expressing cells to U69593 and norBNI was unaltered relative to cells without GF109203 in the intracellular solution, suggesting that PKC-mediated phosphorylation plays no role in the observed effects on the currents (data not shown).
To further verify that norBNI does in fact act by relieving agonist-independent G-protein modulation, the effects of receptor expression, U69593, and norBNI on current facilitation as determined by current ratios were examined (Ikeda, 1991) and were defined as the ratio of the current amplitude after a prepulse, I(+pp) to that before a prepulse, I(−pp). Baseline current ratios (before drug exposure) for α1B-1- and κ1R-expressing cells (ratio, 1.05 ± 0.08;n = 13) were significantly greater than for cells not expressing κ1R (data pooled from κ1R-untransfected cells and U69593- and norBNI-unresponsive cells; ratio, 0.89 ± 0.05; n= 13; p < 0.05, one-tailed t test). These ratios were significantly correlated with the degree of current enhancement obtained after the addition of norBNI (baseline relative to peak of norBNI effect; r = 0.84; p < 0.001; n = 13) (Fig. 1, compare A,B), suggesting that cells most responsive to norBNI expressed the highest levels of agonist independent modulation. The effects of U69593 and norBNI on current amplitudes were mirrored in terms of the current ratios. At baseline, before drug exposure, a small degree of facilitation was seen (ratio, 1.05 ± 0.08), facilitation increased in the presence of U69593 (ratio, 1.70 ± 0.13), and currents inactivated to a small extent after a prepulse in the presence of norBNI (ratio, 0.78 ± 0.03; n = 13 in all cases) (Fig. 1). This current inactivation after a prepulse, as a consequence of a reduction in active G-protein, is similar to the results of Ikeda (1992), who found that internal perfusion of rat sympathetic neurons with GDPβS caused inactivation after a prepulse at most prepulse potentials. In summary, these results suggest that the currents were inhibited to an intermediate degree before drug exposure, that the agonist U69593 rapidly caused additional current inhibition, and that norBNI is actually an inverse agonist, relieving both the agonist-independent and agonist-dependent inhibition of the current.
Because this agonist-independent receptor activity is likely to complicate estimates of receptor effects on Ca channels, we used U69593 to observe currents in the presence of maximum receptor activity,I(R*), and norBNI to observe currents in the presence of inactive receptor, I(R). An index of total modulation was estimated from the peak of the U69593 response [an estimate ofI(R*)] and the peak of the norBNI response (in the case of cells showing agonist-independent modulation) or baseline currents (in the case of cells without agonist-independent modulation) to estimate I(R). Total modulation was then defined as [I(R) − I(R*)]/I(R). In the case of α1B-1- expressing cells, modulation was 68.9 ± 3.7% (n = 13) (see Fig. 5) when defined in this way and 47 ± 6.4% (n = 13) when defined as [I(baseline) − I(R*)]/I(baseline), suggesting a nearly 50% reduction in the SEm of the estimated effect size. Consistent with the involvement of heterotrimeric G-proteins (Marchetti et al., 1986; Ikeda, 1991; Pollo et al., 1992), this current reduction was greater with respect to currents evoked before a voltage prepulse than for currents evoked after a prepulse (total modulation, 38.6 ± 3.7% after prepulse;n = 13) (Fig. 1). Kinetic slowing was evident in the majority of cells (Fig. 1) but was not examined in detail for the purpose of this study.
Fig. 5.

Summary of inhibition and facilitation seen in native α1B-1- and α1E-3-expressing cells as well as for various chimeric Ca channels. A, Summary of κ1R inhibition obtained for Ba currents evoked before a depolarizing prepulse from native α1B-1- and α1E-3-expressing cells, as well as cells expressing various α1B-1 and α1E-3 chimeras. Values are plotted as mean ± SEM. Sample sizes for each construct were ≥6 as detailed in Results. Asterisks indicate modulation significantly different from native α1E-3(p < 0.05). B, Summary of corrected κ1R-mediated prepulse facilitation ratios from native α1B-1- and α1E-3-expressing cells, as well as cells expressing various α1B-1 and α1E-3 chimeras. Values are plotted as mean ± SEM. Ratios were calculated by multiplying prepulse ratios in the presence of U69593 [I(+pp)/I(−pp)] by an index of inactivation [I(−pp)/I(+pp)] calculated from currents in the presence of norBNI to adjust for differences in inactivation between the various constructs. Sample sizes for each construct were ≥6 as detailed in Results.Asterisks indicate facilitation significantly different from native α1E-3 (p < 0.05).
To further explore the specificity of agonist-independent opioid receptor effects on Ca currents, we transfected cells with α1-B1, β1B, α2B, CD8α, and the mouse δ-opioid receptor (δR). This receptor has well characterized agonist-independent activity; ICI174,864 is known to act as an inverse agonist, and DPDPE (enkephalin, [d-Pen2,5]) is known to act as an agonist (Chiu et al., 1996; Mullaney et al., 1996; Merkouris et al., 1997). These cells showed inhibition and prepulse facilitation qualitatively similar to κ1R-expressing cells before drug exposure (Fig. 1D). When norBNI was added to the bath, δR-expressing cells showed no alteration in their α1B-1 currents (Fig. 1D;n = 3). However, when ICI174,864 was added to the bath, an increase in current amplitudes was noted (28.0 ± 14.0%;n = 3), and facilitation decreased (current ratio, 1.4 ± 0.2 at baseline; current ratio, 1.0 ± 0.1 in the presence of ICI174,864). When DPDPE was subsequently added to the bath, current amplitudes rapidly decreased and facilitation increased (current ratio, 1.5 ± 0.4), and the DPDPE-mediated inhibition could be reversed by reapplication of ICI174,864 over a time frame of minutes (total modulation, 37.0 ± 13.0%; n = 3). The response of these cells to agonist and inverse agonist was therefore qualitatively similar to cells expressing κ1R but selective for δR-specific agents.
When κ1R- and α1E-3-expressing cells were exposed to U69593 and norBNI, much less current inhibition (25.3 ± 1.9%; n = 11) (Fig.2; see Fig. 5) was observed than for α1B-1-expressing cells (p < 0.001), consistent with previous findings (Toth et al., 1996; Yassin et al., 1996). α1E-3-expressing cells also showed partial voltage-dependent relief of inhibition such that inhibition after a prepulse was less than before a prepulse (total modulation, 21.9 ± 1.6% after prepulse), but cells fell far short of the ∼50% relief seen in α1B-1-expressing cells. Prepulse ratios in the presence of U69593 corrected for inactivation (see Materials and Methods) were 1.052 ± 0.02 for α1E-3-expressing cells but 2.184 ± 0.02 for α1B-1-expressing cells (p < 0.001; see Fig. 5).
Fig. 2.
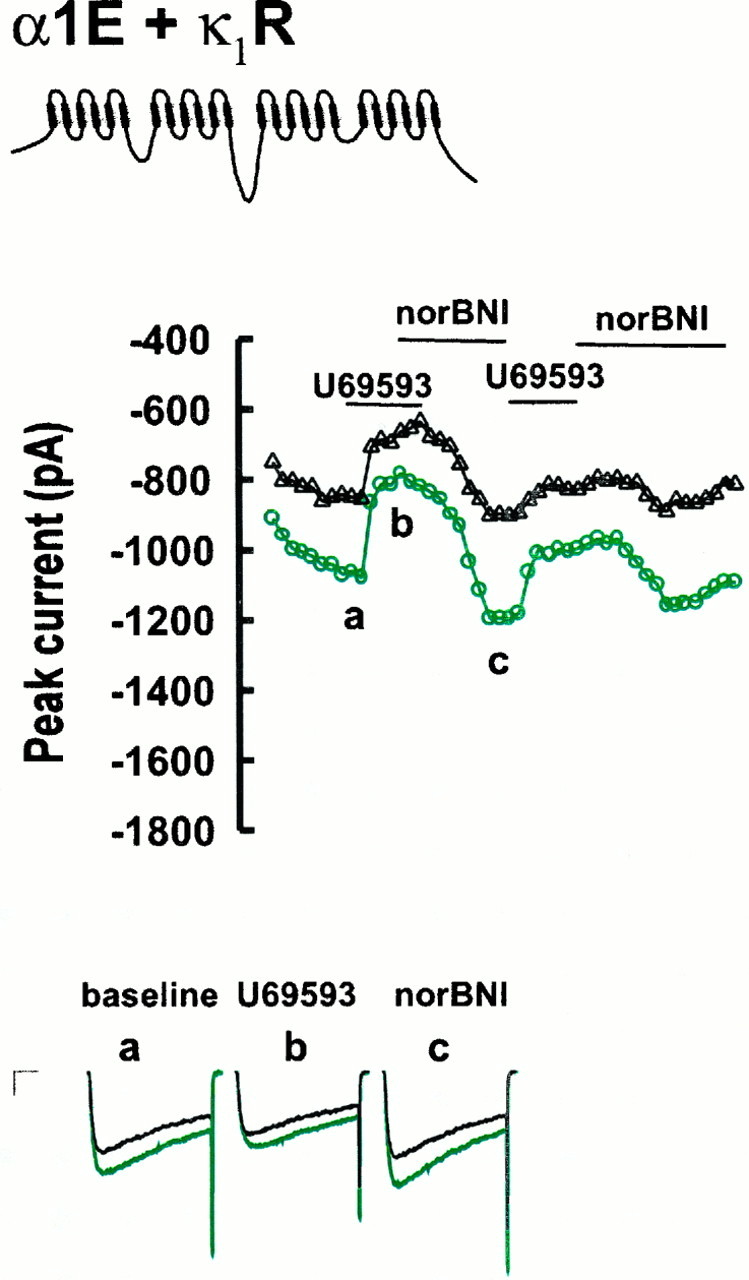
Opioid agonist and inverse agonist effects on Ba currents for α1E-3- and κ1R-expressing tsA-201 cells. Currents were taken from the points indicated. Calibration: 250 pA, 10 msec. See Figure 1 for details.
Structural determinants of G-protein regulation
To elucidate the structural basis for differences in the degree of G-protein regulation of α1B-1 and α1E-3 Ca channels, we constructed Ca channels in which portions of α1B-1 were inserted into an α1E-3background. When the entire N-terminal half of α1B-1 was transferred into the α1E-3 background to create the bBbBbEeEe chimera (where lowercase letters indicate intracellular sequence, and uppercase letters indicate transmembrane domains, written in the sequence N terminus, domain I, I/II loop, etc.), currents were markedly reduced by U69593 and markedly enhanced by norBNI (Fig.3; see Fig. 5) (n = 7). Overall modulation was 69.4 ± 4.3% and not significantly different from modulation seen with α1B-1. Inhibition was partially relieved by a voltage prepulse (to 46 ± 7.9%), and somewhat less relief was seen than for α1B-1 (corrected prepulse ratio, 1.78 ± 0.20; p > 0.05; see Fig.5).
Fig. 3.

Representative currents from various α1B-1 and α1E-3 chimeric Ca channels with sensitivity to modulation approaching that of native α1B-1 channels (Fig. 1) before prepulse (green) and after a 30 msec depolarizing prepulse (black). Maximal effects of U69593 and norBNI are illustrated. Calibration: 250 pA, 10 msec.
To determine the minimal sequence responsible for these observations, we transferred two of the intracellular loops contained in the bBbBbEeEe chimera into the α1E-3 background, individually and in combination. The I/II intracellular loop has been shown by GST fusion studies to bind βγ G-protein subunits (De Waard et al., 1997) and to posses a consensus sequence QXXER that it shares with adenyl cyclase and a number of other G-protein effectors (Pragnell et al., 1994). Somewhat consistent with the results of Page et al. (1997), the eEbEeEeEe chimera (Figs.4-6) showed current inhibition that was slightly greater but not significantly different from α1E-3 (33.8 ± 2.3%;n = 9) and much less than that of α1B-1(p < 0.001) or the bBbBeEeE chimera (p < 0.001). When the II/III intracellular loop of α1B-1 was transferred into the α1E-3background (Figs. 4, 5, eEeEbEeEe chimera), inhibition similar to that of α1E-3 was seen (23.1 ± 4.8%;n = 7). When the II/III loop was transferred into the α1E-3 background, in combination with the I/II loop (Figs. 4, 5, eEbEbEeEe chimera), the resulting construct also showed inhibition similar to that of α1E-3(24.6 ± 1.1%; n = 8). All three constructs showed prepulse facilitation similar to α1E-3(p > 0.05 in each case) and significantly less facilitation than α1B-1 (p < 0.001 for each).
Fig. 4.
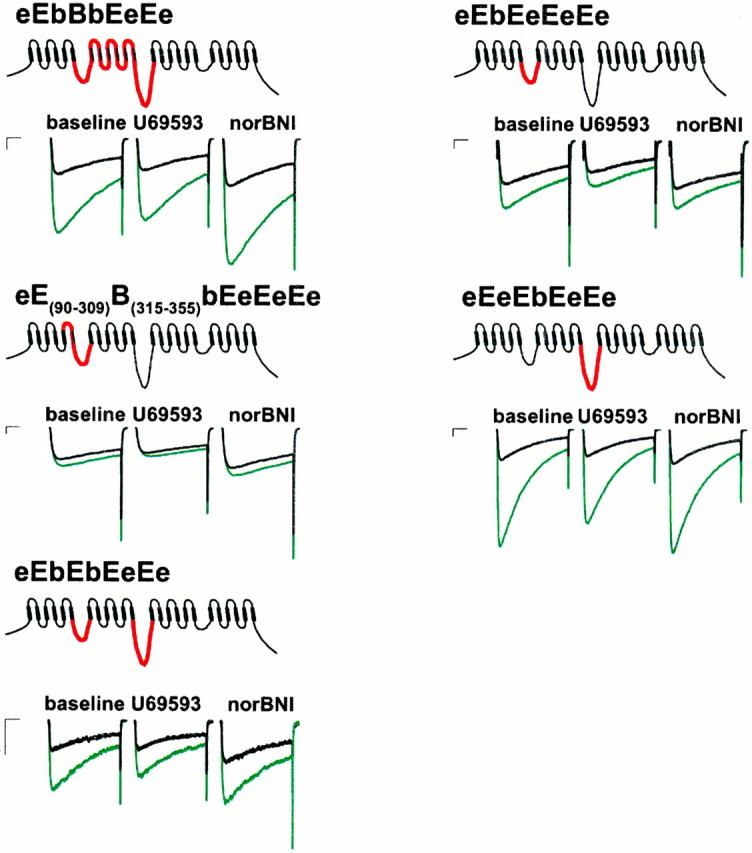
Representative currents from various α1B-1 and α1E-3 chimeric Ca channels with sensitivity to modulation approaching that of native α1E-3 channels (Fig. 2) before prepulse (green) and after a 30 msec depolarizing prepulse (black). Maximal effects of U69593 and norBNI are illustrated. Calibration: 250 pA, 10 msec.
Fig. 6.

Current–voltage relations for selected Ca channel constructs in the presence of U69593 (circles) and norBNI (triangles) before (green) and after (black) a depolarizing prepulse (pp). Sample sizes were ≥5 as detailed in Results.
Because the I/II loop alone failed to significantly enhance modulation but may play some role in mediating the binding of βγ subunits to Ca channels (De Waard et al., 1997), a series of additional constructs was created to assess the role of sequences near the I/II loop in determining effector sensitivity. When domain II, the I/II loop, and the II/III loop from α1B-1 were transferred into the α1E-3 background (eEbBbEeEe chimera), modulation and facilitation similar to that of α1E-3 was obtained (total modulation, 24.6 ± 1.1%; p > 0.05; corrected ratio, 1.03 ± 0.02; p > 0.05; n= 8). The addition of α1B-1 sequence from the N terminus to the C-terminal end of the I/II loop into the α1E-3background (Figs. 3, 5, 6, bBbEeEeEe chimera) resulted in a channel showing inhibition not significantly different from α1B-1 (66.6 ± 4.1%; n = 8). However, as in the case of the bBbBbEeEe chimera, this inhibition was less completely relieved by a prepulse (to 46.0 ± 7.9%; corrected prepulse ratio, 1.55 ± 0.11; p < 0.001) than in the case of α1B-1, although facilitation was greater than that seen in the case of α1E-3 (p < 0.05). When domain I and the I/II loop from α1B-1 were transferred alone into the α1E-3 background (Figs. 3, 5, eBbEeEeEechimera) modulation that was not significantly different from α1B-1 was also observed (56.8 ± 4.4%;n = 7; p > 0.05). These channels showed less facilitation (corrected prepulse ratio, 1.27 ± 0.05) than α1B-1-expressing cells (p < 0.001) and not significantly more facilitation than α1E-3-expressing cells (p > 0.05). We also attempted to examine the contribution of the N terminus in isolation by the chimera bEeEeEeEe but were unable to obtain adequate current expression from this chimera. When domain I of α1B-1 was transferred alone into the α1E-3background (Figs. 3, 5, eBeEeEeEe chimera), modulation significantly greater than α1E-3 was observed (44.6 ± 0.03%; n = 10; p < 0.001), but this was significantly less than that observed for α1B-1(p < 0.05). Facilitation (corrected ratio, 1.3 ± 0.02) was not significantly different from α1E-3 (p > 0.05).
Domain I of Ca channels is thought to be partly responsible for determining the voltage dependence of Ca channel inactivation (Zhang et al., 1994), although the C terminus (Klockner et al., 1995; Soldatov et al., 1997) and I/II loop (Herlitze et al., 1997) are also thought to play a role. The regions in domain I responsible for determining inactivation are thought to be a C-terminal portion of the Is5 sec6 linker as well as Is6 (Zhang et al., 1994). When this sequence alone was added along with the I/II loop (Figs. 4,5, eE(90–309)B(315–355)bEeEeEe chimera), the resulting construct showed modulation (30.0 ± 3.9%; n = 7) and facilitation (corrected ratio, 1.04 ± 0.01) similar to α1E-3 and the eEbEeEeEe chimera (p > 0.05 in both cases), although inactivation was reduced compared with the eEbEeEeEe chimera (p < 0.05).
Recently Qin et al. (1997) have provided biochemical and functional evidence that the C terminus of non-DHP-sensitive Ca channels is involved in βγ binding and current inhibition. To assess the role of the C terminus in our system, we exchanged the C terminus of α1B-1 into the bBbBbEeEe chimera to produce the bBbBbEeEb chimera (Figs. 3, 5). These channels showed inhibition (65.6 ± 4.2%; n = 7) not significantly different from α1B-1 or the bBbBbEeEe chimera (p > 0.05 in both cases). However, facilitation more closely approximated that of α1B-1 than was seen in the case of the bBbBbEeEe chimera (corrected ratio, 2.04 ± 0.12).
Because Ca channel modulation by G-proteins is voltage-dependent, we collected current–voltage data in the presence of norBNI and U69593 for a number of the constructs (Fig. 6). The bBbBbEeEb chimera (n = 7) closely resembled wild-type α1B-1(n = 5) both in terms of inhibition and facilitation (Fig. 6). As we observed using a single test potential of 0 mV, the bBbBbEeEe construct (n = 5) showed less facilitation than bBbBbEeEb across a range of activating potentials (Fig. 6). These results suggest that the C terminus may be partly responsible for the differences in facilitation between α1B-1 and α1E-3, but it does not appear to mediate the differences in inhibition between the two channels. The bBbEeEeEe chimera (n = 5) showed strong inhibition, similar in magnitude to α1B-1, but showed little facilitation at any activating potential. The eEbEeEeEe chimera (Fig. 6;n = 8) showed relatively little facilitation or inhibition at any activating potential. For example, at a potential of −10 mV, the current ratio in the presence of U69593 for α1B-1 (2.28 ± 0.34) was not significantly different from the bBbBbEeEb chimera (2.00 ± 0.20; p > 0.05) but was significantly greater than the ratios for the bBbEeEeEe (1.22 ± 0.11), eEbEeEeEe (0.92 ± 0.04), and bBbBbEeEe (1.40 ± 0.11) chimeras (all p < 0.05). These data further reinforce the conclusion that facilitation and inhibition are mediated by different structural elements.
We defined agonist-independent modulation as the difference in the current amplitude in the presence of norBNI compared with that at baseline. The degree of such modulation would be expected to correlate with the index of total modulation for a particular construct if both indices reflect sensitivity to inhibition by G-proteins. We computed this correlation across all of our observations and found that the correlation was large and highly significant (r = 0.74;p < 0.00001). In addition, the magnitude of the agonist-independent modulation seen for each construct was well correlated with the magnitude of the total modulation seen for the construct (0.03 ± 0.01%, α1E-3; 0.02 ± 0.01%, eEbEeEeEe; 0.22 ± 0.03%, eBeEeEeEe; 0.38 ± 0.06%, α1B-1) This provides additional support for the notion that the index of total modulation is indeed a valid index of sensitivity to current inhibition by G-proteins.
We were concerned that the observed differences in facilitation between the constructs were attributable to differences in the kinetics of G-protein association with the channels. We measured the G-protein reassociation rates for a number of our constructs using a variable interval between the prepulse and the second test pulse. Facilitation decayed as a single exponential with time constants between the limits of 15.9 ± 2.3 msec (eEbEeEeEe; n = 4) (data not shown) and 41.6 ± 2.4 msec (α1B-1;n = 6) (data not shown). Facilitation will have decayed to 73–89% of its maximum after a 5 msec interval for our constructs. Therefore, the large differences in facilitation observed (Figs. 5, 6) cannot be accounted for by differences in reassociation kinetics.
As noted above, current ratios (Fig. 5) were corrected for inactivation to facilitate comparisons between constructs that differed markedly in terms of inactivation. An adequate kinetic model that can account for modulation, activation, and inactivation of Ca channels has not yet been elaborated, and the relationship between inactivation and modulation therefore remains unclear. However, with respect to the most important constructs used in the present study, it is clear that modulation and inactivation are relatively independent. For example, the constructs bBbBbEeEb, bBbBbEeEe, bBbEeEeEe, eBbEeEeEe, eBeEeEeEe, and e(E90–309B315–355)bEeEeEe did not differ in terms of inactivation compared with bBbBbBbBb (all p> 0.9). However, these constructs differed markedly from one another in terms of facilitation. The chimeras bBbBbEeEb and bBbBbEeEe did not differ significantly from α1B-1 in terms of facilitation (p > 0.1), but bBbEeEeEe, eBbEeEeEe, eBeEeEeEe, and e(EB)bEeEeEe did differ significantly (all p < 0.0001). Because these constructs differ greatly in terms of facilitation but not in terms of inactivation, it seems clear that the two are not highly correlated.
DISCUSSION
We have identified a domain in the family of DHP-insensitive Ca channels that appears to largely dictate the efficacy of G-protein-induced channel inhibition. The results of the present study also suggest that the κ1R, like the δR (Chiu et al., 1996; Mullaney et al., 1996; Merkouris et al., 1997), has agonist-independent activity that can be suppressed by drugs with inverse-agonist properties. Therefore, it appears that these pharmacological properties may be general characteristics of the opioid receptor family. Agonist-independent activity of the κ1R has not been previously reported. It is likely that we observed such activity in our experiments because of the high transient expression levels obtainable in tsA-201 cells or because of some other cell-dependent variables. The observation of this agonist-independent activity allowed us to identify norBNI as an inverse agonist at the κ1R. The use of inverse agonists in combination with agonists appears to have important utility in that receptor states near maximal and minimal activity can be readily generated, allowing for more precise estimates of receptor effects on G-protein regulated effectors. This consideration would theoretically be of greatest importance for systems with the highest levels of receptor expression, that would be expected to exhibit significant agonist-independent receptor activity. We achieved substantial improvements in the precision of our estimates of G-protein modulation by using this approach.
With regard to the structural basis for the differences in G-protein sensitivity between α1B-1 and α1E-3 Ca channels, the results of the present study suggest that domain I of α1-B1 is the single most important structural feature required for determining the higher sensitivity to opioid receptor modulation of α1B-1 compared with α1E-3. Whereas κ1R-mediated modulation of α1E-3was only 37% of that obtained for native α1B-1 channels, exchange of domain I alone yielded a chimeric channel with 65% of the modulation of α1B-1. The further addition of the I/II linker increased modulation to 83% of native α1B-1channels, and the further addition of the N terminus increased modulation to 100% of α1B-1 (Fig. 5). These same regions appear largely responsible for facilitation, although the determinants of facilitation appear to be complex and may also include the C terminus (Figs. 5, 6). The lack of significant differences in modulation between the eEbEeEeEe chimera and native α1E-3(Fig. 5) suggests that the I/II loop is not sufficient to determine the differential sensitivity to modulation between α1B-1 and α1E-3. Likewise, the lack of significant differences in inhibition between the bBbBbEeEe and bBbBbEeEb chimeras (Fig. 5) suggests that the C terminus is not involved in determining differences in sensitivity to modulation. Therefore, the two structural elements shown to be involved in G-protein βγ binding to Ca channels (De Waard et al., 1997; Qin et al., 1997; Zamponi et al., 1997) do not appear to account for differences in sensitivity to inhibition between α1B-1 and α1E-3.
The role of the I/II loop in G-protein modulation of Ca channels is controversial. A number of other studies have also suggested that the I/II loop is not sufficient to account for high sensitivity to G-protein modulation. Page et al. (1997) showed that transfer of the I/II loop from α1B to α1E or from α1B to α1A conferred kinetic slowing but not inhibition on the recipient channel. Qin et al. (1997)showed that transfer of the I/II loop from α1C to α1E yielded a chimeric channel that was still modulated like α1E. Zhang et al. (1996) showed that transfer of the I/II loop from α1A or α1C into α1B yielded a channel that was still modulated like α1B. In contrast, Zamponi et al. (1997) showed that transfer of the I/II loop from α1B into α1Ayielded a chimeric channel with significantly more modulation than native α1A. Herlitze et al. (1997) and De Waard et al. (1997) showed that mutations in the sequence QXXER of α1Acan disrupt modulation, and peptides made according to sequences in the I/II loop can block modulation of both channels (Zamponi et al., 1997). In addition, a number of groups have demonstrated that the I/II loop can indeed bind βγ subunits (De Waard et al., 1997; Qin et al., 1997; Zamponi et al., 1997), and this may be important in mediating the effects of βγ subunits in some general way. The results of Qin et al. (1997) and Zhang et al. (1996) also suggest that the C terminus plays a critical role in mediating modulation, and the latter study suggests a role of the N terminal portion of the Ca channel that is N-terminal to the I/II loop. Overall, the results of the present study are most consistent with those of Zhang et al. (1996) in terms of the involvement of the N terminal portion of the channel, although we find that the C terminus plays little if any role in determining inhibition.
There are at least three models that could account for the findings of the present study. First, differences in the kinetics of the two channels, probably determined by multiple and complex structural features, could account for at least some of the differences in modulation. For example, we numerically integrated the kinetic model of G-protein modulation proposed by Patil et al. (1996) while systematically varying the rate constants of the model a small amount near their fitted values and have observed alterations in predicted inhibition for each of the rate constants in the model (data not shown). Apart from rate constants directly introduced to model modulation (e.g., “willing” to “reluctant” transitions), predicted inhibition is particularly sensitive to the values of the rate constants for exit from the inactivated state and for transitions between “willing” closed states.
A second model that could account for these data is an allosteric model in which the I/II loop and/or C terminus binds G-protein βγ subunits and changes conformation but then must induce conformational changes in the gating apparatus of the channel. The C terminus and I/II loop may interact directly with the gating apparatus or may be coupled to these domains through other structures. According to this view, it is possible that domain I of α1B-1, which may itself contain sequence involved in voltage-dependent channel gating, is more susceptible to structural alteration by βγ binding to the I/II loop and/or C terminus than is domain I of α1E-3, and that the N terminus somehow facilitates such interactions. Alternatively, domain I may play some role in linking binding to the C terminus and/or I/II linker to regions of the channel involved in gating to generate modulation.
Finally, it is possible that Ca channel regions other than the I/II loop and C terminus play a role in βγ binding. Biochemical binding studies performed to date (Qin et al., 1997; De Waard et al., 1997) have demonstrated that the I/II loop and C terminus have affinities that are by themselves sufficient for tight binding but have not shown that other sequences do not play an important role in βγ binding to the intact channel. Although domain I (and the N terminus) may not be competent to bind βγ subunits in isolation, it is possible that it does contribute to the affinity of βγ subunit binding to the intact channel and that domain I of α1B-1 is differentially capable of stabilizing βγ binding in comparison to domain I of α1E-3.
The notion that different structural elements account for facilitation and inhibition is similar to the results of Zhang et al. (1996), who showed that both the first domain and the C terminus of α1B were required within an α1A background to confer facilitation and inhibition, but N- or C-terminal halves of α1B in an α1A background were sufficient for inhibition alone. A physical interaction of the C terminus of α1B-1 with Gβγ subunits may play some role in the functional uncoupling of Gβγ and Ca channels after a depolarizing pulse.
Whatever the precise mechanism by which domain I and the N terminus contribute to Ca channel modulation, the findings of the present study suggest that Ca channel sensitivity to G-protein modulation not only is determined by sequences with high-affinity interactions with βγ subunits but also requires another sequence that is perhaps not directly involved in high-affinity βγ binding. Domain I and the N terminus of the channel appear largely responsible for these differences. The N-terminal tails of the two channels are fairly dissimilar (60% identity), especially in their N terminal halves. The only significant regions of dissimilarity between α1B-1and α1E-3 in domain I fall within the extracellular loops (s1–s2, s3–s4, and s5–s6). One or both of these regions of dissimilarity may therefore account for the observed effects of exchanging domain I between α1B-1 and α1E-3.
Footnotes
This work was supported by Public Service Grants DA02121, DA02575, MH40165, NS33502, DK42086, and DK44840. A.A.S. was supported by Public Service Grants HD07009 and DA02575. We are grateful to Dr. R. Harpold (Sibia Neurosciences) for the Ca channel subunits used in these studies and to Dr. G. I. Bell (Howard Hughes Medical Institute, University of Chicago) for helpful discussions regarding the molecular techniques used in these studies, for the use of his laboratory facilities, and for the opioid receptor clones used in this work.
Correspondence should be addressed to Dr. Richard J. Miller, Department of Pharmacological and Physiological Sciences, University of Chicago, 947 East 58th Street, Chicago IL 60637.
REFERENCES
- 1.Avidor-Reiss T, Zippel R, Levy R, Saya D, Ezra V, Barg J, Matus-Leibovitch N, Vogel Z. kappa-Opioid receptor-transfected cell lines: modulation of adenylyl cyclase activity following acute and chronic opioid treatments. FEBS Lett. 1995;361:70–74. doi: 10.1016/0014-5793(95)00154-2. [DOI] [PubMed] [Google Scholar]
- 2.Bean BP. Whole-cell recording of Ca channel currents. Methods Enzymol. 1992;207:181–193. doi: 10.1016/0076-6879(92)07013-e. [DOI] [PubMed] [Google Scholar]
- 3.Boussif O, Zanta MA, Behr JP. Optimized galenics improve in vitro gene transfer with cationic molecules up to 1000-fold. Gene Ther. 1996;3:1074–1080. [PubMed] [Google Scholar]
- 4.Carpenter E, Gent JP, Peers C. Opioid receptor independent inhibition of Ca2+ and K+ currents in NG108–15 cells by the kappa opioid receptor agonist U50488H. NeuroReport. 1996;7:1809–1812. doi: 10.1097/00001756-199607290-00024. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, DeVivo M, Dingus J, Harry A, Li J, Sui J, Carty DJ, Blank JL, Exton JH, Stoffel RH, Inglese J, Lefkowitz RJ, Logothetis DE, Hildebrandt JD, Iyengar RA. A region of adenylyl cyclase 2 critical for regulation by G-protein beta gamma subunits. Science. 1995;268:1166–1169. doi: 10.1126/science.7761832. [DOI] [PubMed] [Google Scholar]
- 6.Chiu TT, Yung LY, Wong YH. Inverse agonist effect of ICI-174,864 on the cloned delta-opioid receptor: role of G-protein and adenylyl cyclase activation. Mol Pharmacol. 1996;50:1651–1657. [PubMed] [Google Scholar]
- 7.De Waard M, Liu H, Walker D, Scott VE, Gurnett CA, Campbell KP. Direct binding of G-protein beta-gamma complex to voltage-dependent Ca channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- 8.Doupnik CA, Pun RYK. G-protein activation mediates prepulse facilitation of Ca2+ channel currents in bovine chromaffin cells. J Membr Biol. 1994;140:47–56. doi: 10.1007/BF00234485. [DOI] [PubMed] [Google Scholar]
- 9.Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;21:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- 10.Herlitze S, Hockerman GH, Scheuer T, Catterall WA. Molecular determinants of inactivation and G-protein modulation in the intracellular loop connecting domains I and II of the Ca channel alpha1A subunit. Proc Natl Acad Sci USA. 1997;94:1512–1516. doi: 10.1073/pnas.94.4.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 12.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 13.Horton RM, Cai ZL, Ho SN, Pease LR. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques. 1990;8:528–535. [PubMed] [Google Scholar]
- 14.Ikeda SR. Double-pulse calcium channel current facilitation in adult rat sympathetic neurons. J Physiol (Lond) 1991;439:181–214. doi: 10.1113/jphysiol.1991.sp018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikeda SR. Prostaglandin modulation of Ca2+ channels in rat sympathetic neurones is mediated by guanine nucleotide binding proteins. J Physiol (Lond) 1992;458:339–359. doi: 10.1113/jphysiol.1992.sp019421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- 17.Kaneko S, Fukuda K, Yada N, Akaike A, Mori Y, Satoh M. Ca2+ channel inhibition by kappa opioid receptors expressed in Xenopus oocytes. NeuroReport. 1994;5:2506–2508. doi: 10.1097/00001756-199412000-00025. [DOI] [PubMed] [Google Scholar]
- 18.Klockner U, Mikala G, Varadi M, Varadi G, Schwartz A. Involvement of the carboxyl-terminal region of the alpha 1 subunit in voltage-dependent inactivation of cardiac Ca channels. J Biol Chem. 1995;270:17306–17310. doi: 10.1074/jbc.270.29.17306. [DOI] [PubMed] [Google Scholar]
- 19.Labrecque J, Fargin A, Bouvier M, Chidiac P, Dennis M. Serotinergic antagonists differentially inhibit spontaneous activity and decrease ligand binding capacity of the rat 5-hydroxytryptamine type 2C receptor in Sf9 cells. Mol Pharmacol. 1995;48:150–159. [PubMed] [Google Scholar]
- 20.Marchetti C, Carbone E, Lux HD. Effects of dopamine and noradrenaline on Ca channels of cultured sensory and sympathetic neurons of chick. Pflügers Arch. 1986;406:104–111. doi: 10.1007/BF00586670. [DOI] [PubMed] [Google Scholar]
- 21.Margolskee RF, McHendry-Rinde B, Horn R. Panning transfected cells for electrophysiological studies. Biotechniques. 1993;15:906–911. [PubMed] [Google Scholar]
- 22.Merkouris M, Mullaney I, Georgoussi Z, Milligan G. Regulation of spontaneous activity of the δ-opioid receptor: studies of inverse agonism in intact cells. J Neurochem. 1997;69:2115–2122. doi: 10.1046/j.1471-4159.1997.69052115.x. [DOI] [PubMed] [Google Scholar]
- 23.Miller RJ. Receptor-mediated regulation of calcium channels and neurotransmitter release. FASEB J. 1990;4:3291–3299. [PubMed] [Google Scholar]
- 24.Mullaney I, Carr IC, Milligan G. Analysis of inverse agonism at the delta opioid receptor after expression in Rat 1 fibroblasts. Biochem J. 1996;315:227–234. doi: 10.1042/bj3150227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neter J, Wasserman W, Kutner M. Applied linear statistical models, Ed 2. Irwin; Homewood, IL: 1985. [Google Scholar]
- 26.Page KM, Stephens GJ, Berrow NS, Dolphin AC. The intracellular loop between domains I and II of the B-type Ca channel confers aspects of G-protein sensitivity to the E-type Ca channel. J Neurosci. 1997;17:1330–1338. doi: 10.1523/JNEUROSCI.17-04-01330.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patil PG, de Leon M, Reed RR, Dubel S, Snutch TP, Yue DT. Elementary events underlying voltage-dependent G-protein inhibition of N-type calcium channels. Biophys J. 1996;71:2509–2521. doi: 10.1016/S0006-3495(96)79444-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pollo A, Lovallo M, Sher E, Carbone E. Voltage-dependent noradrenergic modulation of omega-conotoxin-sensitive Ca2+ channels in human neuroblastoma IMR32 cells. Pflügers Archiv. 1992;422:75–83. doi: 10.1007/BF00381516. [DOI] [PubMed] [Google Scholar]
- 29.Portoghese PS, Lipkowski AW, Takemori AE. Binaltorphimine and nor-binaltorphimine, potent and selective kappa-opioid receptor antagonists. Life Sci. 1987;40:1287–1292. doi: 10.1016/0024-3205(87)90585-6. [DOI] [PubMed] [Google Scholar]
- 30.Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Ca channel beta-subunit binds to a conserved motif in the I-II cytoplasmic linker of the alpha 1-subunit. Nature. 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- 31.Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Direct interaction of Gβγ with a c-terminal Gβγ-binding domain of the Ca channel α1 subunit is responsible for channel inhibition by G-protein-coupled receptors. Proc Natl Acad Sci USA. 1997;94:8866–8871. doi: 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rhim H, Miller RJ. Opioid receptors modulate diverse types of calcium channels in the nucleus tractus solitarius of the rat. J Neurosci. 1994;14:7608–7615. doi: 10.1523/JNEUROSCI.14-12-07608.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simonin F, Gaveriaux-Ruff C, Befort K, Matthes H, Lannes B, Micheletti G, Mattei MG, Charron G, Bloch B, Kieffer B. kappa-Opioid receptor in humans: cDNA and genomic cloning, chromosomal assignment, functional expression, pharmacology, and expression pattern in the central nervous system. Proc Natl Acad Sci USA. 1995;92:7006–7010. doi: 10.1073/pnas.92.15.7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soldatov NM, Zuhlke RD, Bouron A, Reuter H. Molecular structures involved in L-type Ca channel inactivation. Role of the carboxyl-terminal region encoded by exons 40–42 in alpha1C subunit in the kinetics and Ca dependence of inactivation. J Biol Chem. 1997;272:3560–3566. doi: 10.1074/jbc.272.6.3560. [DOI] [PubMed] [Google Scholar]
- 35.Tallent M, Dichter MA, Bell GI, Reisine T. The cloned kappa opioid receptor couples to an N-type calcium current in undifferentiated PC-12 cells. Neuroscience. 1994;63:1033–1040. doi: 10.1016/0306-4522(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 36.Toth PT, Shekter LR, Ma GH, Philipson LH, Miller RJ. Selective G-protein regulation of neuronal calcium channels. J Neurosci. 1996;16:4617–4624. doi: 10.1523/JNEUROSCI.16-15-04617.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yassin M, Zong S, Tanabe T. G-protein modulation of neuronal class E (alpha 1E) Ca channel expressed in GH3 cells. Biochem Biophys Res Commun. 1996;220:453–458. doi: 10.1006/bbrc.1996.0426. [DOI] [PubMed] [Google Scholar]
- 38.Yasuda K, Raynor K, Kong H, Breder CD, Takeda J, Reisine T, Bell GI. Cloning and functional comparison of kappa and delta opioid receptors from mouse brain. Proc Natl Acad Sci USA. 1993;90:6736–6740. doi: 10.1073/pnas.90.14.6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G-proteins and protein kinase C mediated by the calcium channel alpha1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- 40.Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Molecular determinants of voltage-dependent inactivation in Ca channels. Nature. 1994;372:97–100. doi: 10.1038/372097a0. [DOI] [PubMed] [Google Scholar]
- 41.Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Multiple structural elements in voltage-dependent Ca2+ channels support their inhibition by G-proteins. Neuron. 1996;17:991–1003. doi: 10.1016/s0896-6273(00)80229-9. [DOI] [PubMed] [Google Scholar]