

Abstract
We have reported recently a high density of transient A-type K+ channels located in the distal dendrites of CA1 hippocampal pyramidal neurons and shown that these channels shape EPSPs, limit the back-propagation of action potentials, and prevent dendritic action potential initiation (Hoffman et al., 1997). Because of the importance of these channels in dendritic signal propagation, their modulation by protein kinases would be of significant interest. We investigated the effects of activators of cAMP-dependent protein kinase (PKA) and the Ca2+-dependent phospholipid-sensitive protein kinase (PKC) on K+ channels in cell-attached patches from the distal dendrites of hippocampal CA1 pyramidal neurons. Inclusion of the membrane-permeant PKA activators 8-bromo-cAMP (8-br-cAMP) or forskolin in the dendritic patch pipette resulted in a depolarizing shift in the activation curve for the transient channels of ∼15 mV. Activation of PKC by either of two phorbol esters also resulted in a 15 mV depolarizing shift of the activation curve. Neither PKA nor PKC activation affected the sustained or slowly inactivating component of the total outward current. This downregulation of transient K+ channels in the distal dendrites may be responsible for some of the frequently reported increases in cell excitability found after PKA and PKC activation. In support of this hypothesis, we found that activation of either PKA or PKC significantly increased the amplitude of back-propagating action potentials in distal dendrites.
Keywords: potassium channels, IK(A), dendrite, protein kinase A, protein kinase C, hippocampus, phorbol esters, cAMP, action potential
Voltage-dependent K+ channels are the primary regulators of membrane excitability. As a group, they constitute the most diverse of the voltage-gated channels (Rudy, 1988). The large repertoire of K+ channel subtypes available may underlie the distinctive firing patterns expressed by different neurons and by the same cells under different conditions. We have recently found a transient A-type K+ channel to be expressed at high densities in the distal dendrites of CA1 pyramidal neurons (Hoffman et al., 1997). Modulation of these channels could provide the neuron with the means to dynamically and selectively control signal propagation through the dendrites.
The molecular identity of the transient K+ channels recorded in the CA1 hippocampal dendrites is not known for certain, although there is strong evidence in favor of the Shalchannel Kv4.2. Of the two transient K+ channels Kv4.2 and Kv1.4 found in the hippocampus by immunohistochemical techniques, Kv4.2 is primarily located in the soma and dendrites (with the highest degree of immunostaining occurring in dendrites), whereas Kv1.4 is found mainly in axons (Sheng et al., 1992; Maletic-Savatic et al., 1995; Serôdio et al., 1996). Kv4.2 has also been found clustered on the postsynaptic membrane directly apposed to the presynaptic terminal (Alonso and Widmer, 1997). Additionally, both Kv4.2 and the transient K+ channels recorded from CA1 somata are inhibited by arachidonic acid (Villaroel and Schwarz, 1996; Keros and McBain, 1997). Finally, Serôdio et al. (1994)demonstrate that hydrogen peroxide blocks the inactivation of Kv1.4 but not of Kv4.2 channels. Inactivation of the transient channels recorded in CA1 hippocampal neurons is unaffected by externally applied hydrogen peroxide (D. Hoffman, unpublished observations). In our previous report (Hoffman et al., 1997), transient K+ channels in distal dendrites were found to have an activation curve shifted 10–15 mV hyperpolarized compared with those recorded from the soma and proximal (up to 100 μm) dendrites. This result suggested that either a different type of channel is expressed in the two regions or that there is only one channel type, but one that is differentially modulated in the two regions. The immunohistochemical studies that found Kv4.2 located in both the soma and dendrites would support the later conclusion (Sheng et al., 1992; Maletic-Savatic et al., 1995;Serôdio et al., 1996).
Activators of protein kinase A (PKA) and protein kinase C (PKC) have been found to increase the amplitude of EPSPs and population spikes in hippocampal neurons (Malenka et al., 1986; Hu et al., 1987; Storm, 1987; Hvalby et al., 1988; Heginbotham and Dunwiddie, 1991; Slack and Pockett, 1991; Dunwiddie et al., 1992; Pockett et al., 1993). Because the amino acid sequence of Kv4.2 has been shown to contain a potential site for PKA phosphorylation and numerous potential PKC sites (Baldwin et al., 1991; Blair et al., 1991; Anderson et al., 1997), we hypothesized that phosphorylation of transient dendritic K+ channels could potentially account for some of these electrophysiological changes. To test this hypothesis, we made cell-attached patch-clamp recordings of K+ currents from CA1 hippocampal dendrites (180–340 μm from the soma) with and without the inclusion of membrane-permeant activators of both PKA and PKC in the recording pipette. The results suggest that activation of either PKA or PKC can downregulate these channels by shifting the activation curve to more positive potentials. As a test for the functional significance of this downregulation, dendritic action potentials were recorded before and after bath application of PKA and PKC activators. After activation of either kinase, dendritic action potential amplitude was found to increase over 50% in distal dendrites, consistent with a decrease in K+ channel activation.
MATERIALS AND METHODS
Preparation and solutions. Sprague Dawley rats, 5–8 weeks old, were anesthetized with a lethal dose of a combination of ketamine, xylazine, and acepromazine. After they were deeply anesthetized, they were perfused through the heart with cold-modified artificial CSF (ACSF) containing (in mm): 110 CholineCl, 2.5 KCl, 1.2 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7.0 MgCl2, 2.4 pyruvate, 1.3 ascorbic acid, and 20 dextrose. After removal of the brain, 400-μm-thick slices were cut using a vibratome, incubated submerged in a holding chamber for 10 min at 34°C, and stored and used for recordings at room temperature.
Hippocampal slices were visualized with a Zeiss Axioskop using infrared video microscopy and differential interference contrast optics. For all recordings, the bath solution contained (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2.0 CaCl2, 1.0 MgCl2, and 25 dextrose. In whole-cell experiments, the following were included in the external solution to block synaptic transmission (in μm): 50 d,l-APV (Research Biochemicals, Natick, MA), 20 MK-801 (Research Biochemicals), 10 CNQX (Research Biochemicals), 10 bicuculline (Sigma, St. Louis, MO), and 10 picrotoxin (Sigma). The external solution was bubbled with 95% O2/5% CO2 at ∼22°C, pH ∼7.4. For cell-attached recordings, the normal pipette solution consisted of 125 mm NaCl, 10 mm HEPES, 2.0 mmCaCl2, 1.0 mm MgCl2, 2.5 mm KCl, and 1.0 μm tetrodotoxin (TTX), pH 7.4 with NaOH, to which 100 μm 8-bromo-cAMP (8-br-cAMP; Sigma) was included in certain experiments. For forskolin (Sigma) and phorbol ester (Sigma) recordings, drugs were dissolved in DMSO to 10 mm, kept frozen until use, and then diluted to the appropriate concentration. Whole-cell recording pipettes (10–15 MΩ) were filled with (in mm): 120 KGluconate, 20 KCl, 10 HEPES, 2 MgCl2, 4 Mg-ATP, 0.3 Mg-GTP, and 14 phosphocreatine, pH 7.25 with KOH; pipettes were coated with Sylgard. 1-(5-isoquinolinylsulfonyl)2methyl-piperazine dichloride (300 μm; Sigma) and 8-br-cAMP were dissolved directly into the external bath solution in whole-cell experiments.
Recording techniques and analysis. All neurons exhibited a resting membrane potential between −55 and −73 mV (mean, −66 mV). Cell-attached pipettes (10–20 MΩ) were pulled from borosilicate glass and coated with Sylgard. The tips were visually inspected before use and had uniform tip diameters of ∼1 μm. Channel recordings, using an Axopatch 1D amplifier (Axon Instruments), were analog filtered at 2 kHz and digitally filtered at 1 kHz off-line. Leakage and capacitive currents were digitally subtracted by averaging null traces or scaling traces of similar amplitude. Whole-cell patch-clamp recordings were made using an Axoclamp 2A (Axon Instruments) amplifier in “bridge” mode, low-pass filtered at 3 kHz, and digitized at 10 kHz. Series resistance was 15–40 MΩ. Antidromic action potentials were stimulated by constant current pulses (Neurolog; Digitimer Ltd.) through tungsten electrodes (AM Systems) placed in the alveus.
For activation plots, the chord conductance, as calculated from the peak ensemble current amplitude from a holding potential of approximately −85 mV, was normalized to the maximum value and plotted as a function of the test potential. In the case in which the maximal voltage step did not result in a saturated conductance, conductances were fit to a single Boltzmann, and the peak conductance was used for normalization. For inactivation, the peak ensemble current amplitude for a step to approximately +55 mV was normalized to maximum and plotted as a function of holding potential. Data were binned into either 10 or 20 mV compartments. Curves are nonlinear least-square fits of the data to Boltzmann functions. Inactivation time constants were fit using the Fourier expansion method DISCRETE (Provencher, 1976) and were well fit by a single exponential in most cases. Significance (p < 0.05) was determined by two-samplet tests in all cases except for the PDA effect on action potential amplitude in which a paired t test was used. Error bars represent SEM, and voltages were not corrected for the junction potential (approximately −7 mV).
RESULTS
Isolation of the transient current
Cell-attached patch-clamp recordings from CA1 hippocampal dendrites, in the presence of TTX to block voltage-gated Na+ channels, revealed a large outward current composed of a rapidly inactivating component along with a sustained or slowly inactivating component. Although the transient currentIK(A) was from three to five times the amplitude of the sustained current in the distal dendrites, the transient current was routinely isolated from the sustained current by the voltage protocol shown in Figure 1. In a typical experiment, the patch was held 20 mV hyperpolarized to rest and subsequently depolarized by 20–140 mV in 20 mV increments. Ensemble averages were constructed of 10–30 sweeps per step potential. At the conclusion of the experiment, the membrane was ruptured, and the resting membrane potential was measured, allowing for conversion of the voltage steps relative to rest into absolute voltages. The total outward current recorded for a voltage step from −89 to +51 mV is shown in Figure 1A. A 50 msec prepulse to −29 mV allowed the transient channels to inactivate, leaving only the sustained current (Fig. 1B). The sustained current was subsequently digitally subtracted from the total outward current, resulting in the isolated transient current (Fig. 1C). Occasionally, when a full prepulse ensemble was not obtained, the transient component was taken to be the peak of the total current measured 2 msec into the pulse (before substantial sustained component activation). Activation and inactivation curves constructed using this method were similar to those using prepulses. All channel recordings were made 180–340 μm from the soma.
Fig. 1.
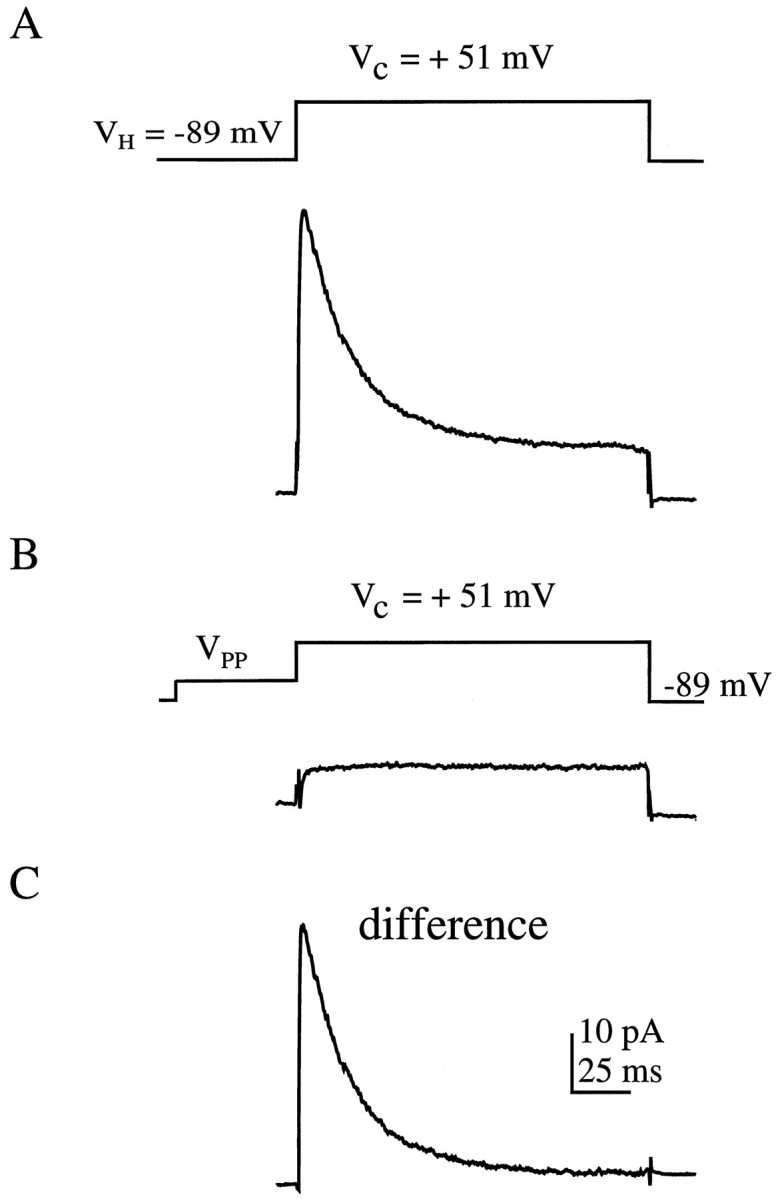
Procedure for isolating the transient current in cell-attached patches. A, The total outward current from a dendritic patch located 320 μm from the soma for a 150 msec voltage step from −89 to +51 mV is shown. Voltage protocol is shownabove the trace. Here and in subsequent figures, traces are ensemble averages of 10–30 sweeps.B, A 50 msec prepulse (VPP) to −29 mV results in transient channel inactivation, leaving only the sustained component of the total outward current. C, Digital subtraction of the current remaining in B from that in A results in the isolated transient component shown.
Modulation of transient K+ channels by activators of PKA
To test for modulation by PKA, we included 100 μm8-br-cAMP, a membrane soluble analog of cAMP, in the patch pipette in 12 dendritic cell-attached recordings. Representative traces (scaled to represent their fraction of maximal conductance) from both control and 8-br-cAMP recordings are shown in Figure2A. In control conditions, a voltage step to −5 mV (near the peak voltage reached during an action potential) activates nearly 50% of the available channels. The inclusion of 8-br-cAMP in the patch pipette reduced this fraction to ∼25%. In the same cell, subsequent bath application of 300 μm H7, a protein kinase inhibitor, increased the fraction back toward control levels. Steady-state activation and inactivation curves were generated from these experiments and are illustrated in Figure 2B. Adding 8-br-cAMP to the patch pipette produced a 13 mV rightward shift in the activation curve, presumably via the activation of PKA (V1/2, −2 and +11 mV for control and 8-br-cAMP, respectively; Fig.2B; Table 1). Similar effects were seen when 50 μm forskolin, an adenylyl cyclase activator, was included in the dendritic patch pipette (V1/2 = +7 mV; n = 5; Table 1). A rightward shift in the activation curve means that at any given potential there will be a decrease in the probability of channel opening. We also found the inactivation curve to be shifted slightly but significantly toward the right (V1/2, −56 vs −50 mV for control and 8-br-cAMP, respectively; Fig. 2B; Table 1). In three of the 12 8-br-cAMP experiments, the kinase inhibitor H7 was bath applied after obtaining a full activation curve. Under these conditions, the activation curve was found to shift progressively back toward control levels in a time-dependent manner (V1/2 = +3.5 mV after 30 min in H7; Fig. 2B; Table 1). Two of the three patches were lost before complete reversal of the 8-br-cAMP effect. On one occasion, however, the patch was held for nearly 1 hr after H7 application. In this patch the curve shifted completely back to control levels (V1/2 = −2 mV; Fig.2B, inset). These results indicate that the shift in the activation curve is attributable to kinase activation and not just a direct effect of 8-br-cAMP on the channel. No significant difference in resting membrane potential was found among groups (Table 1).
Fig. 2.

Downregulation of transient channel activation by PKA activation. A, Representative example of isolated transient currents for the control condition and for that with 100 μm 8-br-cAMP included in the patch pipette, scaled to demonstrate their fraction of maximal conductance, for a step from −85 to −5 mV. Under control conditions, nearly 50% of the available channels are activated by this voltage step. With PKA activation, only ∼25% are activated. In the same patch, subsequent H7 application returned the fraction of channels activated back toward control levels.B, Steady-state activation and inactivation curves for control and 8-br-cAMP conditions and an activation curve for the condition with 8-br-cAMP in the pipette after bath application of 300 μm H7. Half-activation with 8-br-cAMP (V1/2 = +11 mV; n = 12) was shifted 13 mV to theright of control levels (V1/2 = −2 mV;n = 12). There was also a small (6 mV) shift in the inactivation curve with 8-br-cAMP (V1/2 = −50 mV and n = 8 vs V1/2 = −56 mV andn = 5 for controls). Bath application of H7 shifted the 8-br-cAMP curve back toward control levels (V1/2= +3.5 mV after 30 min in H7; n = 3).Inset, Time course of reversal of PKA effect by H7 application (top horizontal bar). The activation and inactivation curves under control conditions are nearly identical to those reported previously for transient K+ channels in distal dendrites (Hoffman et al., 1997). Curves (here and see Figs.3, 5, 6) are least-square fits of the data to Boltzmann functions (see Materials and Methods).
Table 1.
Summary of transient channel properties
| Vm(mV) | Activation | Inactivation | |||
|---|---|---|---|---|---|
| V½(mV) | k | V½ (mV) | k | ||
| Control (12) | −65.8 ± 1.7 | −2 | 15 | −56 | −8 |
| PDA (7) | −67.7 ± 1.2 | 13* | 15 | −50* | −8 |
| PDBu (4) | −65.0 ± 3.4 | 12* | 15 | — | — |
| 4-alpha phorbol (8) | −64.4 ± 1.6 | −2 | 17 | — | — |
| 8-br-cAMP (12) | −65.9 ± 1.2 | 11* | 16 | −50* | −8 |
| Forskolin (5) | −67.2 ± 0.2 | 7* | 15 | — | — |
| 8-br-cAMP + H7 (3) | −65.7 ± 1.9 | 3.5 | 13 | — | — |
| 8-br-cAMP + PDA (6) | −67.2 ± 1.7 | 8* | 17 | −51 | −7 |
For each condition, membrane potential upon breakin (Vm), V½ for activation and inactivation, and fitted slope factors (k) are given. Membrane potential upon breakin at the conclusion of the experiment was similar for each condition. Half-activation was significantly different than controls for PDA, PDBu, 8-br-cAMP, forskolin, and 8-br-cAMP plus PDA experiments, although the slope factor was similar for each group. The voltage for half-inactivation was also significantly different than that of controls under 8-br-cAMP and PDA conditions, but the slope factor was again the same for each group. Asterisks denote significantly different than control. Numbers inparentheses are the n values for each group.Dashes indicate not determined.
Modulation of transient K+ channels by phorbol esters
We investigated the effects of PKC activation on these channels by including one of two different phorbol esters in the patch pipette. Both phorbol diacetate (PDA, 10 μm) and phorbol dibutyrate (PDBu, 10 μm) were found to produce a 15 mV depolarizing shift in the activation curve for transient K+ channels located in the distal dendrites, very similar to that found for PKA activation. Representative traces, scaled to represent their fraction of maximal conductance, from both control and PDA recordings are shown in Figure3A. The inclusion of PDA in the patch pipette led to a 15 mV rightward shift in the activation curve, presumably via the activation of PKC (V1/2, −2 and +13 mV for control and PDA, respectively; Fig. 3B; Table 1). Again, this shift would have the effect of decreasing the probability of channel opening at a given potential. As was the case for PKA activation, there was also a small but significant (6 mV) depolarizing shift in the inactivation curve in the presence of PDA (V1/2, −56 vs −50 mV for control and PDA, respectively; Fig. 3B; Table 1). The steady-state activation curve produced with PDBu in the pipette also was found to be shifted by ∼14 mV (V1/2 = +12 mV; Fig. 3B; Table 1). There was, however, no shift in the activation curve when the inactive phorbol ester 4-α-phorbol was included in the pipette (V1/2 = −2 mV; Fig. 3B; Table 1). These results indicate that the shift in the activation curve is attributable to kinase activation rather than to nonspecific effects of phorbol esters on the channels. Again, no significant difference in resting membrane potential was found among the groups (Table 1).
Fig. 3.

Downregulation of transient channel activation by PKC activation. A, Representative example of isolated transient currents for the control condition and for that with 10 μm PDA included in the patch pipette, scaled to demonstrate their fraction of maximal conductance, for a step from −85 to −5 mV. B, Steady-state activation and inactivation curves for control and two different phorbol ester conditions. Half-activation with PDA (V1/2 = +13 mV;n = 7) or PDBu (V1/2 = +12 mV;n = 4) was shifted ∼15 mV to theright of controls (V1/2 = −2 mV;n = 12). There was also a small (6 mV) shift in the inactivation curve with PDA (V1/2 = −50 mV andn = 6 vs V1/2 = −56 mV andn = 5 for controls). There was, however, no activation curve shift when the inactive phorbol ester 4-α-phorbol was included in the pipette (V1/2 = −2 mV;n = 8).
The voltage dependency of inactivation is altered by 8-br-cAMP and PDA
We have reported previously that the time constant of inactivation of the transient current increases with membrane potential (Hoffman et al., 1997). The same voltage dependency was found in the present study in which the time constant of inactivation for the transient component increased linearly with the amount of depolarization from 11.9 ± 0.9 msec for a step to −5 mV to 27.9 ± 2.3 msec for a step to +55 mV under control conditions (slope = 2.5 msec/10 mV; Fig.4). Inclusion of either 8-br-cAMP or PDA in the patch pipette resulted in a more shallow slope (1.9 msec/10 mV). The time constant at −5 mV was similar to the control value (11.3 ± 0.6 and 10.2 ± 0.9 msec for 8-br-cAMP and PDA, respectively) but was significantly faster than the control value at +55 mV (22.6 ± 2.1 and 22.1 ± 1.6 msec; Fig. 4). Traces from the PDBu group of experiments had time constants of inactivation similar to that of the PDA and 8-br-cAMP groups (slope = 2.0 msec/10 mV), and the inactivation rates in the 4-α-phorbol traces were similar to that of controls (2.5 msec/10 mV; data not shown).
Fig. 4.

Both 8-br-cAMP and PDA alter the voltage dependency of inactivation for the transient channels. Time constants for inactivation are plotted versus membrane potential for control, 8-br-cAMP, and PDA. Time constants for control and drug conditions were similar for the smallest depolarization measured (−5 mV) but were significantly different at higher potentials, resulting in a shallower slope of the lines fit to the data. Slope equals 2.5 msec/10 mV for control and 1.9 msec/10 mV for both 8-br-cAMP and PDA conditions. Inset, Two traces, scaled to the same amplitude, for a step to +55 mV under control (bottom trace) and 8-br-cAMP (top trace) conditions fit by a single exponential (τ, 30.7 and 20.6 msec for control and 8-br-cAMP, respectively).
The peak current level from which the time constant was measured is a result of both activation and inactivation kinetics. It seemed possible that this change in slope was the result of incomplete activation under the active kinase conditions. This possibility was ruled out, however, by also measuring the time constant 20 msec into the trace (when activation is presumably complete). The time constants measured in this manner were similar to those measured from the peak current level.
The effects of 8-br-cAMP and PDA on transient channel activation are not additive
In five patches, both 8-br-cAMP and PDA were included the patch pipette. The results, shown in Figure 5, indicate that the effects of the two agents are not additive but rather resemble the shifts seen with either 8-br-cAMP or PDA alone. Representative traces, scaled to represent their fraction of maximal conductance, from both control and PDA with 8-br-cAMP recordings are shown in Figure 5A. The activation curve for 8-br-cAMP plus PDA was shifted to the right by 10 mV compared with that for controls (Fig. 5B; Table 1).
Fig. 5.

The effects of PKA and PKC activation are not additive. A, Representative example of isolated transient currents for the control condition and for that with 100 μm 8-br-cAMP plus 10 μm PDA included in the patch pipette, scaled to demonstrate their fraction of maximal conductance, for a step from −85 to −5 mV. B, Steady-state activation and inactivation curves for control and 8-br-cAMP plus PDA. Half-activation with 8-br-cAMP plus PDA (V1/2 = +8 mV; n = 5) was shifted 10 mV to the right of control levels (V1/2 = −2 mV; n = 7). There was also a small (5 mV) shift in the inactivation curve with 8-br-cAMP plus PDA (V1/2 = −51 mV and n = 3 vs V1/2 = −56 mV andn = 5 for controls).
The sustained component is not affected by either 8-br-cAMP or PDA
We also examined the effect of including 8-br-cAMP and PDA in the patch pipette on the sustained component of the total outward current. Ensemble averages of the current remaining at the end of the pulse were used to construct an activation curve for the sustained component. The half-activation values of +2, +7, and +5 mV for control (n = 10), 8-br-cAMP (n = 6), and PDA (n = 8) conditions, respectively, were not significantly different (Fig. 6).
Fig. 6.

The sustained current was unaffected by either 8-br-cAMP or PDA. Steady-state activation curves for the sustained component are plotted for control, 8-br-cAMP, and PDA. Half-activation with 8-br-cAMP (V1/2 = +7 mV; k = 10.5;n = 6) and PDA (V1/2 = +5 mV;k = 8.5; n = 8) was similar to that with controls (V1/2 = +2 mV;k = 8.5; n = 10). Note that the difference in V1/2 between control and 8-br-cAMP conditions was primarily caused by the small difference in fitted slope, although this slope change was not significant.
PKA or PKC activation increases the back-propagating action potential amplitude
Dendritic, whole-cell patch recordings of antidromically initiated action potentials were made to assess the functional impact of decreasing the transient K+ channel activity by protein kinase activation. Action potential amplitude, known to decrease with distance from the soma because of an increasing density of A-channels (Turner et al., 1991; Andreasen and Lambert, 1995;Spruston et al., 1995; Hoffman et al., 1997; Magee and Johnston, 1997), was measured before and after bath application of PKA and PKC activators. In distal dendrites, where the amplitude is significantly attenuated by A-channels, both PKA and PKC activation led to a substantial increase in action potential amplitude (Fig.7). Action potential amplitude increased by an average of 78 ± 15% after 10 μm PDA application in distal recordings (Fig.7A,C). In Figure 7A, PKC activation increased action potential amplitude by 62% in a recording 240 μm from the soma. In these experiments, the cell was hyperpolarized to −80 mV to remove residual Na+channel inactivation, which has been shown to be affected by PKC activation (Colbert et al., 1997; Colbert and Johnston, 1998). It has been reported previously that PKC activation does not lead to a significant increase in action potential amplitude in more proximal recordings (Colbert and Johnston, 1998). Similarly, in proximal dendrites, 100 μm 8-br-cAMP had little effect on action potential amplitude in contrast to distal recordings in which the action potential was increased by an average of 51 ± 14% (Fig.7B,C). In Figure 7B, PKA activation increased action potential amplitude by 42% in a recording 280 μm from the soma but had no effect on the action potential recorded 150 μm from the soma. Recordings made at locations in between those shown in Figure 7B resulted in an intermediate but significant increase in action potential amplitude (Fig.7C). Inclusion of H7 in the external solution to oppose kinase activation prevented the effect of 8-br-cAMP on action potential amplitude (Fig. 7C).
Fig. 7.

PKA and PKC activation increases the back-propagating action potential amplitude in the distal dendrites.A, Antidromically initiated action potentials before (pre) and after (+PDA) bath application of 10 μm PDA. In this recording 240 μm from the soma, application of PDA lead to a 62% increase in action potential amplitude (from 44 to 71 mV). B, Antidromically initiated action potentials before (pre) and after (post and +8-br-cAMP) bath application of 100 μm8-br-cAMP. Left, In a more proximal recording (150 μm from the soma), in which A-channel density is smaller, action potential amplitude was large to begin with, and 8-br-cAMP did not lead to an increase in amplitude. Right, In this distal recording (280 μm from the soma), in which action potential amplitude is attenuated because of high A-channel density, PKA activation increased the amplitude over 40% (from 33 to 47 mV). C, Summary data. Percent change in action potential amplitude and maximal rate of rise are plotted for five conditions: distal recordings after PDA application, proximal (150 μm) recordings after 8-br-cAMP application, mid (180–200 μm) recordings after 8-br-cAMP application, distal (250–320 μm) recordings after 8-br-cAMP application, and distal and mid recordings after 8-br-cAMP application with 300 μm H7 included in the external solution. Thenumber of cells for each group is inparentheses. Asterisks denote a significant percent increase over the controls (see Materials and Methods).
The increase in action potential amplitude occurred without a significant increase in the rate of rise. This along with the lack of PKA effect on proximally recorded action potentials indicates that the kinase-dependent increase in amplitude is not caused by an increase in Na+ conductance (Hodgkin and Katz, 1949). This action potential augmentation without an effect on rate of rise was also found after blockade of A-channels by 4-aminopyridine (Hoffman et al., 1997). These data suggest that depolarizing the A-channel activation curve via phosphorylation by PKA or PKC allows dendritic action potentials to propagate farther out into the dendrites than would occur under control conditions.
DISCUSSION
In the present study, we report the downregulation of a voltage-gated K+ channel by activators of two protein kinases, PKA and PKC. Both 8-br-cAMP and forskolin, which activate PKA, shifted the activation curve for dendritic, transient K+ channels by ∼15 mV toward depolarizing potentials. A decrease in transient K+ channel activation in the presence of cAMP activators was also suggested byDeadwyler et al. (1995), although the degree to which activation was decreased was not reported. We also report here that activation of PKC results in a 15 mV shift in the same direction. A recent report has found a PKC effect on Kv4.2 channels expressed in Xenopusoocytes (Nakamura et al., 1997). However, they do not find a shift in the activation curve but, rather, a simple suppression of currents. Although these two findings both result in a decrease in A-current, they are quite different. A shift in the activation curve is a change in the voltage dependence of the probability of channel opening, which indicates an effect on the activation gate of the channel. The decrease in Kv4.2 currents in oocytes, however, was without an effect on the steady-state inactivation curve or the time course of recovery from inactivation, indicating a direct inhibition of the channels by PKC (Nakamura et al., 1997). This difference may mean that transient currents are somehow differentially regulated in the two preparations (perhaps by auxiliary subunits) or that the currents recorded in CA1 dendrites are not the result of Kv4.2 expression. It seems unlikely, however, that Kv4.2 is not at least partially responsible for the dendritic A-currents given the numerous immunohistochemical studies that find Kv4.2 expressed in dendrites, with much less staining of other transient channels (Sheng et al., 1992; Maletic-Savatic et al., 1995; Serôdio et al., 1996). Especially relevant is a recent report showing that both PKA and PKC phosphorylate Kv4.2 (Anderson et al., 1997). Both the C and N terminals of Kv4.2 were shown to be substrates for PKA phosphorylation, and the C terminal is a substrate for PKC phosphorylation.
After PKA or PKC activation, we also report a small but significant depolarizing shift in the inactivation curves. Comparing the inactivation curves for control and 8-br-cAMP conditions in Figure2B, it seems that this shift will have little effect on the channels when the cell is at rest (approximately −65 to −70 mV), with ∼90% of the channels being available for activation. With sustained depolarization, however, there would be a greater percent of channels available for activation with the kinase activated. For example, at −55 mV, only 50% of the channels are available in control conditions compared with 70% with PKA activated. This shift may act to partially compensate for the increased excitability produced by PKA and PKC activation.
It is now well established that back-propagating action potentials in dendrites of CA1 neurons become progressively smaller in amplitude the farther they travel from the soma and may even fail to propagate beyond distal branch points. This decrement in action potential amplitude is found in vivo and in vitro in both the hippocampus and neocortex (Turner et al., 1991; Andreasen and Lambert, 1995; Spruston et al., 1995; Buzsáki et al., 1996; Magee and Johnston, 1997; Svoboda et al., 1997). Although the function of this decrementing action potential is unclear, the high density of transient K+ channels is thought to be primarily responsible for this phenomenon in the hippocampus, because blocking the channels with 4-aminopyridine results in back-propagating action potentials that decrement much less with distance (Hoffman et al., 1997). A decrease in action potential amplitude could also be accomplished by a decreasing density of Na+ channels. By increasing an outward conductance rather than decreasing an inward conductance, however, the cell is able to selectively propagate full amplitude action potentials under certain conditions. Here, we demonstrate one such condition, in which the downregulation of the A-channels by PKA or PKC activation resulted in larger back-propagating action potentials. In a model of a CA1 pyramidal neuron, even a 5 mV shift in the A-channel activation curve was found to significantly increase action potential amplitude in the distal dendrites (M. Migliore, D. A. Hoffman, J. C. Magee, D. Johnston, unpublished observations). If A-channel modulation were to occur selectively in specific regions of the dendritic tree, action potential amplitude may be increased in those regions only, possibly to the extent of preferentially invading one branch over another. Neuromodulatory inputs into the distal dendrites (activating PKA via dopamine, norepinephrine, or serotonin or PKC via muscarinic receptors) may then be able to direct action potentials to specific regions of the dendrite.
It has been reported that the pairing of back-propagating action potentials with EPSPs increases dendritic action potential amplitude and associated Ca2+ influx supralinearly. This pairing was found to facilitate the induction of long-term potentiation (LTP) at CA1 synapses (Magee and Johnston, 1997). We have suggested previously that the mechanism for this EPSP-spike boosting could involve A-channel inactivation (Hoffman et al., 1997). The rapid rate of inactivation of A-channels occurring for small depolarizations such as EPSPs can lead to the amplification of back-propagating action potentials near the site of synaptic input. The present results suggest that downregulation of the A-channels by protein kinase activation, yielding larger dendritic action potentials, might also increase the probability of LTP induction. With high concentrations of 4-AP, which block ∼80% of the total A-channel population, large EPSPs can induce dendritic action potential initiation (Hoffman et al., 1997). It seems unlikely, however, that the 15 mV shift in the activation curve reported here would in itself reduce the A-channel population to the degree necessary for dendritic action potential initiation under normal conditions. If the cell were to undergo a sustained depolarization, such as occurs during bursting or during the tetanus used to elicit LTP, the decreased population of A-channels resulting from kinase activation might increase the likelihood of dendritic action potential initiation.
Given the possible implications of A-channel modulation listed above, persistent downregulation of dendritic, transient K+channels might also contribute to the long-lasting increase in neuronal excitability observed after PKA and PKC activation (Malenka et al., 1986; Hu et al., 1987; Storm, 1987; Hvalby et al., 1988; Heginbotham and Dunwiddie, 1991; Slack and Pockett, 1991; Dunwiddie et al., 1992;Pockett et al., 1993). Many of these effects, including enhancement of EPSPs and increased probability of action potential firing, are similar to those seen in LTP. These observations, along with occlusion experiments and the fact that both PKC and PKA levels increase as a result of the high-frequency stimulation used to induce LTP, strongly suggest that both kinases play important roles in LTP (Malenka et al., 1986; Malinow et al., 1989; Matthies et al., 1991; Wang and Feng, 1992;Klann et al., 1993; Roberson and Sweatt, 1996). Recently, it has been reported that genetic downregulation of PKA results in a decrease in LTP, along with long-term memory deficits (Abel et al., 1997). Also, dopamine acting via the D1/D5 receptor has been shown to increase the magnitude of LTP and to inhibit depotentiation via a cAMP-dependent mechanism (Otmakhova and Lisman, 1996, 1998). The transient K+ channels, as substrates for PKA and PKC, may then play a role in the induction and/or the expression of LTP. An additional association between A-channels and LTP comes from studies involving the mitogen-activated protein kinase (MAPK). Transient K+ channels recorded in CA1 dendrites were found to have their activation curve shifted to the left after blockade of MAPK activation, and Kv4.2 was shown to be phosphorylated by MAPK (Adams et al., 1997) (J. P. Adams, A. E. Anderson, D. A. Hoffman, J. D. English, R. G. Cook, D. Johnston, P. Pfaffinger, J. D. Sweatt, unpublished observations). Other studies show that blocking MAPK activation severely reduces LTP induction in the hippocampus and facilitation inAplysia (English and Sweatt, 1996, 1997; Bailey et al., 1997; Martin et al., 1997).
The downregulation of transient K+ channels in CA1 hippocampal dendrites by activators of PKA and PKC reported here is one of several demonstrations of A-channel modulation. A-type K+ channels are modulated or blocked by neurotransmitters, including serotonin, glutamate, and GABA, by cation concentrations, by auxiliary subunits, by β-amyloid peptide, by arachidonic acid, and by oxidative states (Rudy, 1988; Saint et al., 1990; Chen and Wong, 1991; Wang et al., 1992; Covarrubias et al., 1994;Rettig et al., 1994; Serôdio et al., 1994; Talukder and Harrison, 1995; Good et al., 1996; Keros and McBain, 1997). The vast number and diversity of agents that modulate these channels suggest that they are highly regulated and thus able to finely, dynamically, and selectively control signals as they propagate through the dendrites.
Footnotes
This work was supported by National Institutes of Health Grants NS11535, MH44754, and MH48432, and by Human Frontiers Science Program. We thank Jeff Magee and Rick Gray for comments on this manuscript.
Correspondence should be addressed to Dr. Dan Johnston, Division of Neuroscience, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030.
REFERENCES
- 1.Abel T, Nguyen PV, Barad M, Deuel TAS, Kandel ER. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997;88:615–626. doi: 10.1016/s0092-8674(00)81904-2. [DOI] [PubMed] [Google Scholar]
- 2.Adams JP, Anderson AE, Johnston D, Pfaffinger PJ, Sweatt JD. Kv4.2: a novel substrate for MAP kinase phosphorylation. Soc Neurosci Abstr. 1997;23:1176. [Google Scholar]
- 3.Alonso G, Widmer H. Clustering of Kv4.2 potassium channels in postsynaptic membrane of rat supraoptic neurons: an ultrastructural study. Neuroscience. 1997;77:617–621. doi: 10.1016/s0306-4522(96)00561-1. [DOI] [PubMed] [Google Scholar]
- 4.Anderson AE, Adams JP, Swann JW, Johnston D, Pfaffinger PJ, Sweatt JD. Kv4.2, a fast transient A-type potassium channel is a substrate for PKA and PKC. Soc Neurosci Abstr. 1997;23:1394. [Google Scholar]
- 5.Andreasen M, Lambert JDC. Regenerative properties of pyramidal cell dendrites in area CA1 of the rat hippocampus. J Physiol (Lond) 1995;483:421–441. doi: 10.1113/jphysiol.1995.sp020595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey CH, Kaang B-K, Chen M, Martin KC, Lim C-S, Casadio A, Kandel ER. Mutation in the phosphorylation sites of MAP kinase blocks learning-related internalization of apCAM in Aplysia sensory neurons. Neuron. 1997;18:913–924. doi: 10.1016/s0896-6273(00)80331-1. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin TJ, Tsaur M-L, Lopez GA, Jan YN, Jan LY. Characterization of a mammalian cDNA for an inactivating voltage-sensitive K+ channel. Neuron. 1991;7:471–483. doi: 10.1016/0896-6273(91)90299-f. [DOI] [PubMed] [Google Scholar]
- 8.Blair TA, Roberds SL, Tamkun MM, Hartshorne RP. Functional characterization of RK5, a voltage-gated K+ channel cloned from the rat cardiovascular system. FEBS Lett. 1991;295:211–213. doi: 10.1016/0014-5793(91)81420-d. [DOI] [PubMed] [Google Scholar]
- 9.Buzsáki G, Penttonen M, Nádasdy Z, Bragin A. Pattern and inhibition-dependent invasion of pyramidal cell dendrites by fast spikes in the hippocampus in vivo. Proc Natl Acad Sci USA. 1996;93:9921–9925. doi: 10.1073/pnas.93.18.9921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen QX, Wong RKS. Intracellular Ca2+ suppressed a transient potassium current in hippocampal neurons. J Neurosci. 1991;11:337–343. doi: 10.1523/JNEUROSCI.11-02-00337.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colbert CM, Johnston D. Protein kinase C activation decreases activity-dependent attenuation of dendritic Na+ current in hippocampal CA1 pyramidal neurons. J Neurophysiol. 1998;79:491–495. doi: 10.1152/jn.1998.79.1.491. [DOI] [PubMed] [Google Scholar]
- 12.Colbert CM, Magee JC, Hoffman DA, Johnston D. Slow recovery from inactivation of Na+ channels underlies the activity-dependent attenuation of dendritic action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 1997;17:6512–6521. doi: 10.1523/JNEUROSCI.17-17-06512.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Covarrubias M, Wei A, Salkoff L, Vyas TB. Elimination of rapid potassium channel inactivation by phosphorylation of the inactivation gate. Neuron. 1994;13:1403–1412. doi: 10.1016/0896-6273(94)90425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deadwyler SA, Hampson RE, Mu J, Whyte A, Childers S. Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP-dependent process. J Pharmacol Exp Ther. 1995;273:734–743. [PubMed] [Google Scholar]
- 15.Dunwiddie TV, Taylor M, Heginbotham LR, Proctor WR. Long-term increases in excitability in the CA1 region of rat hippocampus induced by β-adrenergic stimulation: possible mediation by cAMP. J Neurosci. 1992;12:506–517. doi: 10.1523/JNEUROSCI.12-02-00506.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- 17.English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 18.Good TA, Smith DO, Murphy RM. β Amyloid peptide blocks the fast-inactivating K+ current in rat hippocampal neurons. Biophys J. 1996;70:296–304. doi: 10.1016/S0006-3495(96)79570-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heginbotham LR, Dunwiddie TV. Long-term increases in the evoked population spike in the CA1 region of rat hippocampus induced by β-adrenergic receptor. J Neurosci. 1991;11:2519–2527. doi: 10.1523/JNEUROSCI.11-08-02519.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hodgkin AL, Katz B. The effect of sodium ions on the electrical activity of the giant axon of the squid. J Physiol (Lond) 1949;108:37–77. doi: 10.1113/jphysiol.1949.sp004310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- 22.Hu G-Y, Hvalby O, Walaas SI, Albert KA, Skjeflo P, Andersen P, Greengard P. Protein kinase C injection into hippocampal pyramidal cells elicits features of long term potentiation. Nature. 1987;328:426–429. doi: 10.1038/328426a0. [DOI] [PubMed] [Google Scholar]
- 23.Hvalby Ø, Reymann K, Andersen P. Intracellular analysis of potentiation of CA1 hippocampal synaptic transmission by phorbol ester application. Exp Brain Res. 1988;71:588–596. doi: 10.1007/BF00248751. [DOI] [PubMed] [Google Scholar]
- 24.Keros S, McBain CJ. Arachidonic acid inhibits transient potassium currents and broadens action potentials during electrographic seizures in hippocampal pyramidal and inhibitory interneurons. J Neurosci. 1997;17:3476–3487. doi: 10.1523/JNEUROSCI.17-10-03476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klann E, Chen SJ, Sweatt JD. Mechanism of protein kinase-c activation during the induction and maintenance of long-term potentiation probed using a selective peptide substrate. Proc Natl Acad Sci USA. 1993;90:8337–8341. doi: 10.1073/pnas.90.18.8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magee JC, Johnston D. A synaptically controlled, associative signal for hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- 27.Malenka RC, Madison DV, Nicoll RA. Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature. 1986;321:175–177. doi: 10.1038/321175a0. [DOI] [PubMed] [Google Scholar]
- 28.Maletic-Savatic M, Lenn NJ, Trimmer JS. Differential spatiotemporal expression of K+ channel polypeptides in rat hippocampal neurons developing in situ and in vitro. J Neurosci. 1995;15:3840–3851. doi: 10.1523/JNEUROSCI.15-05-03840.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–866. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 30.Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- 31.Matthies H, Jr, Behnisch T, Kase H, Matthies H, Reymann KG. Differential effects of protein kinase inhibitors on pre-established long-term potentiation in rat hippocampal neurons in vitro. Neurosci Lett. 1991;121:259–262. doi: 10.1016/0304-3940(91)90699-t. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura TY, Coetzee WA, Miera EV-SD, Artman M, Rudy B. Modulation of KV4 channels, key components of rat ventricular transient outward K+ current, by PKC. Am J Physiol. 1997;42:H1775–H1786. doi: 10.1152/ajpheart.1997.273.4.H1775. [DOI] [PubMed] [Google Scholar]
- 33.Otmakhova NA, Lisman JE. D1/D5 dopamine receptor activation increases the magnitude of early long-term potentiation at CA1 hippocampal synapses. J Neurosci. 1996;16:7478–7486. doi: 10.1523/JNEUROSCI.16-23-07478.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otmakhova NA, Lisman JE. D1/D5 dopamine receptors inhibit depotentiation at CA1 synapses via cAMP-dependent mechanism. J Neurosci. 1998;18:1270–1279. doi: 10.1523/JNEUROSCI.18-04-01270.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pockett S, Slack JR, Peacock S. Cyclic amp and long-term potentiation in the CA1 region of rat hippocampus. Neuroscience. 1993;52:229–236. doi: 10.1016/0306-4522(93)90151-5. [DOI] [PubMed] [Google Scholar]
- 36.Provencher SW. A Fourier method for the analysis of exponential decay curves. Biophys J. 1976;16:27–41. doi: 10.1016/S0006-3495(76)85660-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O. Inactivation properties of voltage-gated K+ channels altered by presence of β-subunit. Nature. 1994;369:289–294. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- 38.Roberson ED, Sweatt JD. Transient activation of cyclic AMP-dependent protein kinase during hippocampal long-term potentiation. J Biol Chem. 1996;271:30436–30441. doi: 10.1074/jbc.271.48.30436. [DOI] [PubMed] [Google Scholar]
- 39.Rudy B. Diversity and ubiquity of K channels. Neuroscience. 1988;25:729–749. doi: 10.1016/0306-4522(88)90033-4. [DOI] [PubMed] [Google Scholar]
- 40.Saint DA, Thomas T, Gage PW. GABAβ agonists modulate a transient potassium current in cultured mammalian hippocampal neurons. Neurosci Lett. 1990;118:9–13. doi: 10.1016/0304-3940(90)90236-3. [DOI] [PubMed] [Google Scholar]
- 41.Serôdio P, Kentros C, Rudy B. Identification of molecular components of A-type channels activation at subthreshold potentials. J Neurophysiol. 1994;72:1516–1529. doi: 10.1152/jn.1994.72.4.1516. [DOI] [PubMed] [Google Scholar]
- 42.Serôdio P, Miera EV-S de, Rudy B. Cloning of a novel component of A-type K+ channels operating at subthreshold potentials with unique expression in heart and brain. J Neurophysiol. 1996;75:2174–2179. doi: 10.1152/jn.1996.75.5.2174. [DOI] [PubMed] [Google Scholar]
- 43.Sheng M, Tsaur M, Jan YN, Jan LY. Subcellular segregation of two A-type K+ channel proteins in rat central neurons. Neuron. 1992;9:271–284. doi: 10.1016/0896-6273(92)90166-b. [DOI] [PubMed] [Google Scholar]
- 44.Slack JR, Pockett S. Cyclic AMP induces long-term increase in synaptic efficacy in CA1 region of rat hippocampus. Neurosci Lett. 1991;130:69–72. doi: 10.1016/0304-3940(91)90229-m. [DOI] [PubMed] [Google Scholar]
- 45.Spruston N, Schiller Y, Stuart G, Sakmann B. Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science. 1995;268:297–300. doi: 10.1126/science.7716524. [DOI] [PubMed] [Google Scholar]
- 46.Storm JF. Phorbol esters broaden the action potential in CA1 hippocampal pyramidal cells. Neurosci Lett. 1987;75:71–74. doi: 10.1016/0304-3940(87)90077-2. [DOI] [PubMed] [Google Scholar]
- 47.Svoboda K, Denk W, Kleinfeld D, Tank DW. In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature. 1997;385:161–165. doi: 10.1038/385161a0. [DOI] [PubMed] [Google Scholar]
- 48.Talukder G, Harrison NL. On the mechanism of modulation of transient outward current in cultured rat hippocampal neurons by di- and trivalent cations. J Neurophysiol. 1995;73:73–79. doi: 10.1152/jn.1995.73.1.73. [DOI] [PubMed] [Google Scholar]
- 49.Turner RW, Meyers DER, Richardson TL, Barker JL. The site for initiation of action potential discharge over the somatodendritic axis of rat hippocampal CA1 pyramidal neurons. J Neurosci. 1991;11:2270–2280. doi: 10.1523/JNEUROSCI.11-07-02270.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Villaroel A, Schwarz TL. Inhibition of the Kv4 (Shal) family of transient K+ currents by arachidonic acid. J Neurosci. 1996;16:2522–2531. doi: 10.1523/JNEUROSCI.16-08-02522.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang JH, Feng DP. Postsynaptic protein kinase-C essential to induction and maintenance of long-term potentiation in the hippocampal CA1 region. Proc Natl Acad Sci USA. 1992;89:2576–2580. doi: 10.1073/pnas.89.7.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y, Strahlendorf JC, Strahlendorf HK. Serotonin reduces a voltage-dependent transient outward potassium current and enhances excitability of cerebellar Purkinje cells. Brain Res. 1992;571:345–349. doi: 10.1016/0006-8993(92)90675-y. [DOI] [PubMed] [Google Scholar]