

Abstract
We examined the expression, activation, and cellular localization of caspase-3 (CPP32) using immunohistochemistry, immunoblots, and cleavage of the fluorogenic substrateN-benzyloxycarbonyl-Asp-Glu-Val-Asp-7-amino-4-trifluoromethyl coumarin (zDEVD-afc) in adult mouse brain after temporary (2 hr) middle cerebral artery occlusion produced by filament insertion into the carotid artery. Immunoreactive caspase-3p32 but not its cleavage product caspase-3p20 was constitutively expressed in neurons throughout brain and was most prominent in neuronal perikarya within piriform cortex. Caspase-like enzyme activity was elevated in brain homogenate 0–3 hr after reperfusion and reached a peak within 30 to 60 min. Caspase-3p20 immunoreactivity became prominent in neuronal perikarya within the middle cerebral artery territory at the time of reperfusion and on immunoblots 1–12 hr later. DNA laddering (agarose gels) and terminal deoxynucleotidyl transferase-mediated dUTP—biotin nick-end labeling (TUNEL)-stained cells were detected 6–24 hr after reperfusion. At 12–24 hr, immunoreactive p20 was visualized in TUNEL-positive cells, a finding also observed in apoptotic mouse cerebellar granule cells on postnatal day 5. Together, these observations suggest the existence of a time-dependent evolution of ischemic injury characterized by the close correspondence between caspase-like enzyme activation and an associated increase in immunoreactive product (caspase-3p20) beginning at or before reperfusion and followed several hours later by morphological and biochemical features of apoptosis.
Keywords: ischemia, caspase, CED-3/ICE proteases, mouse brain, apoptosis, caspase-3 cleavage, caspase-like enzyme activity, TUNEL, CNS development
Apoptosis or programmed cell death (PCD) is a prominent feature of the developing nervous system (Oppenheim et al., 1990). Several lines of evidence suggest that apoptosis is also an important mechanism of cell death in adult brain in acute or chronic diseases, including stroke or Alzheimer’s disease (Loo et al., 1993; Thompson, 1995). In animal models of stroke, markers of apoptosis, such as cytoplasmic and nuclear condensation, and DNA fragmentation appear in neurons (MacManus et al., 1994; Li et al., 1995b).
The genetic control of PCD has been studied most completely in the nematode Caenorhabditis elegans and involves the death-promoting genes ced-3 and ced-4 (Ellis and Horvitz, 1986; Ellis et al., 1991; Yuan et al., 1993). Cysteine proteases called caspases are mammalian homologs of theced-3 gene product (Alnemri et al., 1996) and are essential for the execution step in apoptosis (Cohen, 1997; Nicholson and Thornberry, 1997). All caspases are synthesized as proenzymes activated by proteolytic cleavage at Asp-X sites and contain a conserved pentapeptide QACXG sequence. The first identified family member caspase-1 [interleukin-1β-converting enzyme (ICE)] cleaves pro-interleukin-1β (pro-IL-1β) to generate the mature cytokine (Cerretti et al., 1992; Thornberry et al., 1992).
Among the caspases, caspase-3 (CPP32) (Fernandes-Alnemri et al., 1994;Nicholson et al., 1995; Tewari et al., 1995) has the highest homology to CED-3 in terms of both amino acid sequence and substrate specificity (Xue et al., 1996). Caspase-3p32 is activated by cleavage into p20 and p12 fragments and cleaves several intracellular substrates (Nicholson and Thornberry, 1997). Caspase-3-deficient mice show grossly abnormal brain development and die within 3 weeks after birth (Kuida et al., 1996). Caspase-1 knockout mice by contrast show phenotypically normal brain development but are protected from endotoxin shock (Kuida et al., 1995; Li et al., 1995a). Transgenic mice that express a dominant negative mutant caspase-1 gene (Friedlander et al., 1997; Hara et al., 1997a) and caspase-1 knockout mice are resistant to ischemic cell damage (Schielke et al., 1998).
We recently reported that two irreversible caspase inhibitors,N-benzyloxycarbonyl-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-fluoromethyl ketone (zDEVD-fmk) andN-benzyloxycarbonyl-Val-Ala-Asp(OMe)-fluoromethyl ketone (zVAD-fmk) protected brain from ischemic injury and improved neurological deficits in mice and rats (Hara et al., 1997b). Because caspase-1 and its substrate pro-IL-1β exhibit both pro-inflammatory and pro-apoptotic actions (Rothwell and Relton, 1993), we sought more direct evidence for the linkage of the caspases to ischemic cell death by studying caspase-3 in an ischemia model in which zDEVD-fmk was neuroprotective. We chose to study reversible ischemia because injury tends to be milder and apoptosis more prominent, and because free radicals generated during reperfusion are associated with apoptosis (Bonfoco et al., 1995). We extend our preliminary findings (Namura et al., 1997), demonstrate that caspase-3p32 protein is constitutively expressed in mammalian adult brain, and provide biochemical and immunochemical evidence for caspase-3 activation in ischemic brain.
MATERIALS AND METHODS
Ischemia model. Adult male SV-129 mice (18–20 gm) (Taconic Farms, Germantown, NY) were anesthetized for induction with 1.5% halothane and maintained in 1.0% halothane in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH). Ischemia was induced with an 8.0 nylon monofilament coated with a silicone resin–hardener mixture (Xantopren and Elastomer Activator; Bayer Dental, Osaka, Japan) as described previously (Hara et al., 1996). The filament was introduced into the left common carotid artery, advanced into the anterior cerebral artery, and left in this position for 2 hr. For reperfusion, animals were reanesthetized briefly and the filament was withdrawn. Core temperature was maintained at ∼37°C with a thermostat (FHC, Brunswick, ME) and a heating lamp (Skytron; Daiichi Shomei, Tokyo, Japan) until 1 hr after reperfusion. Neurological deficits caused by ischemia were scored according to Bederson et al. (1986): 0, no observable neurological deficits (normal); 1, failure to extend the right forepaw (mild); 2, circling to the contralateral side (moderate); 3, loss of walking or righting reflex (severe). Animals were included only if their neurological deficit score was 2 or higher during reperfusion.
Protocols. Mice were killed 0–5 min after reperfusion or 1, 3, 6, 12, or 24 hr after reperfusion for characterization of enzyme activity, immunoblots, or immunohistochemistry as described below. One additional time point (30 min reperfusion) was studied only in the DEVDase activity experiments.
Caspase-like DEVDase activity assay. We initially established the caspase-like DEVDase activity assay in preliminary experiments using homogenates from HeLa cells exposed to staurosporine (Bertrand et al., 1994). After 3 hr exposure, caspase-like enzyme activity measured by cleavage of the fluorogenic substrateN-benzyloxycarbonyl-Asp-Glu-Val-Asp-7-amino-4-trifluoromethyl coumarin (zDEVD-afc) (Enzyme Systems, Dublin, CA) increased from 114 ± 32 pmol · mg−1protein · min−1 in control cells (n = 3) to 227 ± 18 pmol · mg−1protein · min−1, and this increase was blocked completely by 10 μm zDEVD-fmk (n = 2) (Enzyme Systems). We then measured activity in cerebellar homogenates from mouse pups at postnatal day 5, which is when granule cells undergo extensive apoptosis (Wood et al., 1993), and compared it with activity in brain homogenates from postnatal day 25 when developmental apoptosis is no longer present. Activity was increased approximately twofold at postnatal day 5, and this increase was inhibited 62% by adding 100 μm zDEVD-fmk. Mouse forebrain, excluding frontal pole (2 mm) and occipital pole (1 mm), was homogenized in 4 vol of 25 mm HEPES buffer, pH 7.5, containing 0.1% Triton X-100, 5 mm MgCl2, 2 mmdithiothreitol (DTT), 74 μm antipain, 0.15 μm aprotinin, 1.3 mm EDTA, 1 mmEGTA, 15 μm pepstatin, and 20 μm leupeptin, and centrifuged at 50,000 × g. One hundred microliters of the supernatant were added to 900 μl of 100 mm HEPES buffer, pH 7.4, containing 2 mm DTT. In preliminary experiments, enzyme activity was substantially reduced (>80%) by incubating at pH 6.8–7.0 instead of 7.4. After addition of the fluorogenic substrate zDEVD-afc (12.5 μm), fluorescence (400–505 nm; Hitachi F-4500 fluorometer) was measured at 5 min intervals. The increase in fluorescence was linear between 5 and 35 min after zDEVD-afc was added. Caspase-like activity was calculated from the slope as fluorescence units per milligram of protein per minute of reaction time and converted to picomoles of substrate cleaved per milligram of protein per minute based on a standard curve for amino-4-trifluoromethyl coumarin. Protein concentration in the supernatant was determined by the Bradford assay. Enzyme activity is expressed as mean ± SEM. Significant differences between zDEVD-fmk treatment and control were determined by Student’st test. Differences between caspase-like enzyme activity in ischemic versus sham brains were determined by one-way ANOVA followed by Dunnett’s post hoc tests. p < 0.05 was considered statistically significant.
Cleavage activity did not differ between sham-operated brain and the contralateral hemisphere during reperfusion. To show that the addition of a caspase inhibitor reduced enzyme activity in ischemic brain, supernatant was incubated for 15 min with zDEVD-fmk (100 μm) before the addition of fluorogenic substrate zDEVD-afc. Enzyme activity was inhibited by 64% after 100 μm zDEVD-fmk was added. We used 100 μm in subsequent experiments because this concentration (and higher) was used in previous studies to characterize caspase-3-like activity in vitro (Enari et al., 1996; Armstrong et al., 1997; Eldadah et al., 1997). In more recent preliminary studies, the addition of 10 μm zDEVD-fmk inhibited the reaction to the same extent (∼55–60%), indicating that adding a lower concentration of inhibitor works as well and might provide greater specificity; 100 μm is tenfold higher than the Kmfor caspase-3 (Nicholson et al., 1995) and tenfold higher than the concentration required to completely inhibit enzyme activity in staurosporine-treated HeLa cells.
Immunoblots and immunohistochemistry. Two antisera were used to identify either caspase-3p32 or its cleavage product caspase-3p20. Caspase-3p32 antiserum was raised against a synthetic peptide C12SIKNFEVKT20 residing near the N terminus of the hamster caspase-3 prodomain (Wang et al., 1996) and was a gift from Dr. J. L. Goldstein (University of Texas Southwestern Medical Center). Caspase-3p20 antiserum was raised against163CRGTELDCGIETD175 spanning cysteine at the active site to the C terminus of the human and mouse p20 fragment and was affinity-purified against the peptide (Armstrong et al., 1997) (A. Srinivasan, unpublished observations). On immunoblots, this antibody recognizes processed caspase-3 preferentially to unprocessed zymogen. Antigen binding was examined in ischemic tissue by immunoblots as well as by immunohistochemistry.
To test antiserum specificity, caspase-3p32 antiserum was preadsorbed with a specific synthetic peptide (SIKNFEVKT) (Enzyme Systems). To demonstrate specificity of caspase-3p20 staining, preadsorption with the peptide immunogen (CRGTELDCGIETD) was performed.
Immunoblots. Extracts from mouse forebrain enriched in ischemic tissue (excluding 2 mm frontal and 1 mm occipital pole) were prepared by lysis in 10 mm HEPES buffer, pH 7.6, containing 42 mm KCl, 5 mm MgCl2, 1 mm phenylmethylsulfonyl fluoride (PMSF), 1 mmEDTA, 1 mm EGTA, 1 mm DTT, 1% lauryl sulfate sodium salt (SDS), 1.5 μm pepstatin, 2 μm leupeptin, and 0.7 μm aprotinin, and centrifugation at 21,000 × g. Ten micrograms of total protein of each sample were loaded on a 15% SDS-PAGE gel. After electrophoresis, protein was transferred to nitrocellulose membrane. Blots were blocked with 5% nonfat milk in TBST (10 mmTris, pH 7.5, 150 mm NaCl, 0.05% Tween 20) and probed with 1:1500 dilutions of either caspase-3p32 or caspase-3p20 antiserum in 5% nonfat milk/TBST. Immunoblots were then processed with horseradish–peroxidase-conjugated anti-rabbit immunoglobulin G (IgG) using the enhanced chemiluminescence (ECL) Western blotting detection system kit (Amersham, Arlington Heights, IL). The blots were exposed to Hyperfilm (ECL, Amersham) at room temperature.
Densitometry was performed on a total of four Western blots (Bio-Rad GS-700; Bio-Rad, Hercules, CA). Optical density (OD) values at each time point were obtained by comparing the absolute OD value of the 32 kDa band with that of α-tubulin. Differences between OD values for the 32 kDa band in ischemic hemisphere over time were determined by one-way ANOVA followed by Bonferroni’s post hoc test.p < 0.05 was considered statistically significant. OD values for the contralateral hemisphere were also analyzed in this way.
Immunohistochemistry. Mice were anesthetized deeply with an overdose of sodium pentobarbital (100 mg/kg, i.p.) and then transcardially perfused with 0.9% saline solution, followed by 4% paraformaldehyde in 0.1 m PBS, pH 7.4. The brains were removed quickly and stored in the same fresh buffer containing 20% sucrose. The brains were cut into coronal sections of 40 μm thickness on a freezing microtome. The sections were processed by the free-floating method. After they were washed three times in PBS, pH 7.4, the sections were successively incubated with 0.3% H2O2 in PBS, pH 7.4, for 30 min, 10% normal goat serum (NGS) in PBS, pH 7.4, for 30 min, and primary antisera (1:1000 for either caspase-3p32 or caspase-3p20 antisera) in 2% NGS, 0.3% Triton X-100, 0.05% NaN3 in PBS, pH 7.4, at 4°C overnight. After they were rinsed three times in PBS, the sections were incubated with biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA) in PBS, pH 7.4, containing 2% NGS, 0.3% Triton X-100 for 2 hr at room temperature. The sections were incubated with avidin–biotin–peroxidase solution (Vectastain ABC kit, Vector Laboratories) in PBS, pH 7.4, for 30 min at room temperature after they were washed three times in PBS, pH 7.4. After three additional washes in PBS, pH 7.4, the sections were incubated in 50 mmTris-HCl, pH 7.5, for 10 min and treated with 0.05% 3,3′-diaminobenzidine tetrahydrochloride (DAB) (Sigma, St. Louis, MO) with 0.003% H2O2 in 50 mmTris-HCl, pH 7.5. The sections were mounted on gelatin-coated glass slides, air-dried, dehydrated in ascending ethanol series, immersed in xylene, and coverslipped with Permount (Fisher Scientific, Pittsburgh, PA).
Double labeling with in situ terminal deoxynucleotidyl transferase-mediated DNA nick-end labeling (TUNEL) and caspase-3p20 immunohistochemistry. TUNEL was based on the method of Gavrieli et al. (1992) with modifications (Wood et al., 1993). After they were immunostained with caspase-3p20 primary antibody and Bodipy fluorescein (Bodipy FL) -conjugated goat anti-rabbit IgG secondary antibody (Molecular Probes, Eugene, OR), the sections were passed through ethanol (70%, 95%, 100%, and 100% for 3 min each) and then immersed in chloroform for 2 min. The sections were rehydrated by passage through decreasing ethanol series (100%, 100%, 95%, 70% for 3 min each) followed by three washes in PBS, pH 7.4, at 5 min per wash. The sections were immersed in terminal deoxynucleotidyl transferase (TdT) buffer (30 mm Trizma base, 140 mm sodium cacodylate, 1 mm cobalt chloride) for 5 min at room temperature, and then incubated with TdT buffer containing 12.5 μm biotinylated dUTP (Boehringer Mannheim, Indianapolis, IN) and 0.15 U/l TdT (Boehringer Mannheim) at 37°C for 70 min. The reaction was stopped by transferring the sections to termination buffer (300 mm NaCl, 30 mm sodium citrate) for 15 min at room temperature. After three washes in PBS, pH 7.4, the sections were blocked with PBS containing normal goat serum (10%) and incubated with streptavidin-conjugated Cy3 (Jackson ImmunoResearch, West Grove, PA) in PBS, pH 7.4, for 25 min at room temperature. After three washes in PBS, pH 7.4, the sections were dehydrated in ascending ethanol series. After immersion in xylene, the sections were coverslipped in Permount. For control studies, the sections were treated the same way except that either TdT or biotinylated dUTP was omitted or the sections were treated with DNase before the delipidization.
To validate our method for double staining, we examined cerebellar granule cells from C57BL/6 mouse pups at postnatal day 5 when extensive apoptosis normally develops (Wood et al., 1993). By so doing, we could also determine whether double staining was a distinct event in ischemic apoptotic cell death or whether it occurred during normal development. Parasagittal sections of 40 μm thickness were incubated with caspase-3p20 antiserum followed by TUNEL. Fluorescent-stained sections were analyzed on a Leica DMRB/Bio-Rad MRC 1024 confocal microscope with krypton–argon laser. For Bodipy FL, the excitation filter was 488 nm and the emission was 522 nm. For Cy3, excitation and emission filters were 568 and 605 nm, respectively.
Double labeling with Hoechst 33342 and caspase-3p20 immunohistochemistry. After fluorescence immunostaining as described above, sections were incubated with 10 mg/ml Hoechst 33342 (Sigma) in PBS for 5 min, washed three times in PBS, coverslipped in Aqua-Mount (Lerner Laboratories, Pittsburgh, PA), and examined under an Olympus BH-2 fluorescence microscope with ultraviolet and fluorescein isothiocyanate (FITC) filters for Hoechst 33342 and Bodipy FL, respectively.
DNA analysis. The brain was cut into 2 mm slices with a brain matrix (RBM-2000C, Activational Systems), and ischemic tissue was obtained 4–6 mm from the frontal pole 6, 12, and 24 hr after reperfusion. DNA laddering was visualized with a radioactive end-labeling method by terminal transferase according to Tilly and Hsueh (1993), with minor modifications according to Hara et al. (1997a). Three micrograms of DNA per animal were labeled with [32P]-ddATP, electrophoresed on a 2.0% agarose gel (agarose 3:1) (Amresco, Solon, OH), and autoradiographed.
RESULTS
Detection of caspase-like activity in ischemic brain lysate
We used a fluorogenic substrate zDEVD-afc, which may be preferentially cleaved by caspase-like proteases (Sarin et al., 1996;Armstrong et al., 1997; Keane et al., 1997) to measure enzyme activity in control as well as in ischemic brain lysate. Cleavage of zDEVD-afc was much higher in homogenates of apoptotic HeLa cells as compared with homogenates from ischemic brain (Fig.1A).
Fig. 1.
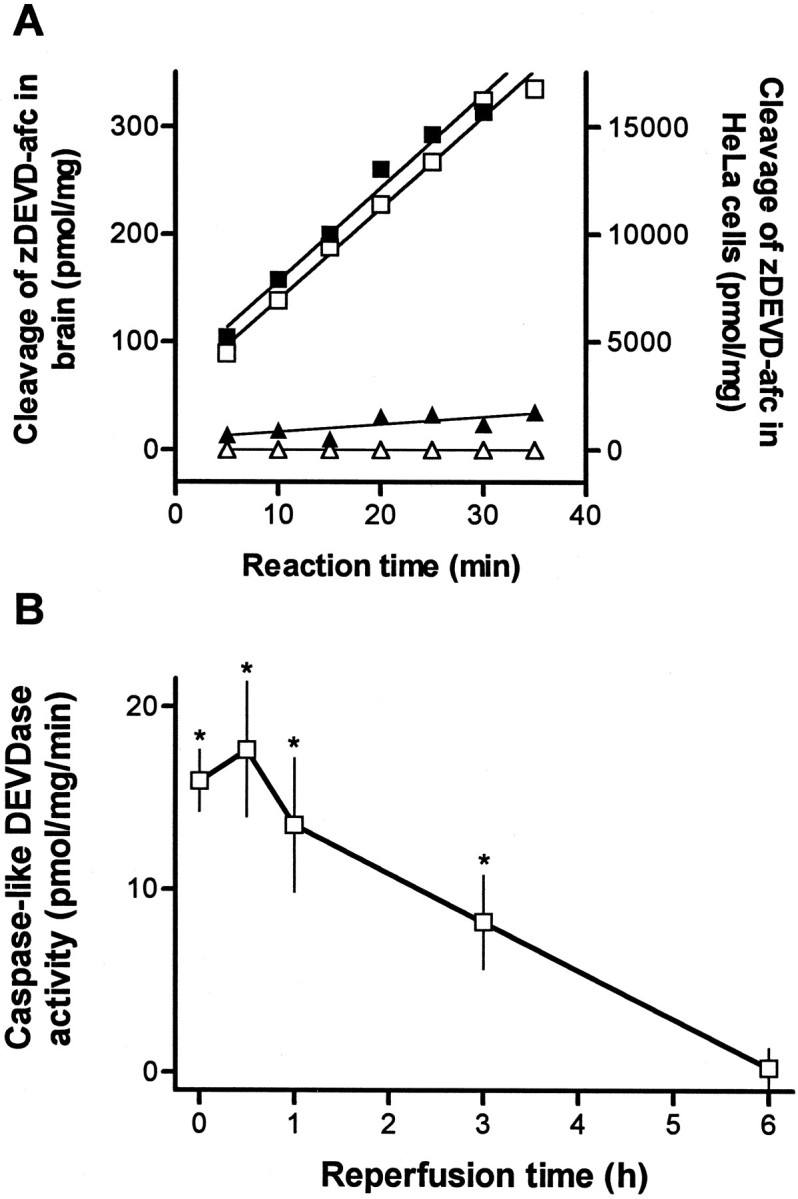
A, Cleavage of the caspase fluorogenic substrate zDEVD-afc (12.5 μm) in homogenates of HeLa cells and brain tissue homogenates. Reaction was monitored every 5 min for 35 min by spectrofluorometry in apoptotic HeLa cell lysate, which was isolated after treatment with 1 μmstaurosporine for 3 hr (□) or in control cell lysate (▵). Caspase-like DEVDase activity was also measured in brain tissue homogenates from the ischemic (▪) or contralateral (▴) hemisphere 1 hr after reperfusion after 2 hr of middle cerebral artery occlusion. The data are from a single set of representative experiments that were highly reproducible. B, Caspase-like enzyme activity increases in brain homogenates after reperfusion after 2 hr occlusion of left middle cerebral artery. Enzyme activity measured as described above is expressed as picomole of substrate cleaved per milligram of protein per minute. Data show mean ± SEM of four to eight experiments assayed in duplicate. *p < 0.05 indicates a significant difference compared with sham-operated animals.
In the ischemic hemisphere, enzyme activity was detected in brain homogenates from 0 to 3 hr after reperfusion (Fig.1B) (12 or 24 hr data not shown). Highest increases were observed after 30 min, and preincubation with zDEVD-fmk (100 μm) reduced activity by 64% (p < 0.03; n = 5). Addition of zVAD-fmk (100 μm) (Enzyme Systems) to the zDEVD-fmk-containing reaction mixture did not further decrease enzyme activity. Cleavage of zDEVD-afc in sham-operated brain was not inhibited by adding 100 μmzDEVD-fmk and was subtracted as nonspecific activity.
Constitutive expression of caspase-3 immunoreactivity in normal brain and activation of caspase-3 in ischemic brain
Caspase-3 is synthesized as a 32 kDa pro-form that is cleaved during activation into a large subunit of 20 or 18 kDa, depending on the apoptotic signal (Erhardt and Cooper, 1996), and a small subunit of 12 kDa (Nicholson et al., 1995). To determine whether caspase-3 is activated in ischemic brain, we examined for the presence of caspase-3p32 and its cleavage products using two different antisera.
The specificity and sensitivity of these two antisera were compared in mouse cerebellum at postnatal day 5, which is when extensive developmental apoptosis occurs (Wood et al., 1993). Caspase-3p32 antiserum detected only one band at 32 kDa that disappeared after preadsorption with synthetic peptide (Fig.2A), whereas caspase-3p20 antiserum identified a single band at 20 kDa but did not recognize a band corresponding to caspase-3p32 (Fig.2B). The 20 kDa band detected with caspase-3p20 antiserum was markedly diminished by preadsorption with synthetic peptide.
Fig. 2.

The specificity of antisera detecting either caspase-3p32 (A) or caspase-3p20 (B) is shown by immunoblots of lysates from mouse cerebellum on postnatal day 5 (P5). Ten micrograms of protein were loaded per lane, and the membrane was probed with 1:1500 dilution.A, A band corresponding to caspase-3p32 was detected (lane 1) that disappeared after preadsorption with peptide immunogen (lane 2). B, Immunoblots detected caspase-3p20 in lysates from mouse cerebellum (P5) (lane 1) but did not detect caspase-3p32. The p20 band disappeared after preadsorption with peptide immunogen (lane 2).
On the basis of the above results, we investigated the expression and activation of caspase-3 and its cleavage products in the mouse MCA occlusion and reperfusion model. Full length caspase-3 (p32) was detected by caspase-3p32 antiserum in ischemic and contralateral brain tissue homogenates at every time point from 0 to 24 hr after reperfusion as well as in normal brain (Fig.3A). Significant time-dependent changes in caspase-3p32 expression were not detected by densitometric analysis of immunoblots over 24 hr (n = 4 experiments; data not shown). However, the expression of a 20 kDa fragment as detected by caspase-3p20 antiserum was increased at 1–12 hr after reperfusion compared with the contralateral hemisphere (Fig.3B). Thus, the processing of caspase-3 to an active form was detected in ischemic mouse brain early during reperfusion.
Fig. 3.

Time-dependent changes in caspase-3 during reperfusion after 2 hr middle cerebral artery occlusion. Brain lysates (10 μg/lane) were subjected to SDS-PAGE and immunoblot analysis using either caspase-3p32 (A) or caspase-3p20 (B) antiserum as described in Figure 2. Caspase-3p32 band was present in normal brain and did not change over time when measured in four different experiments (see Materials and Methods). Cleavage product (p20) increased 1–6 hr after reperfusion in the left (L, ischemic) compared with right (R, contralateral) and normal brain (N). A faint p20 band is also present at 12 hr. α-tubulin was used as an internal control (bottom). The experiment was repeated four times; a representative experiment is shown.
To provide more detailed information about cellular localization of caspase-3 in brain, we examined caspase-3p32 and caspase-3p20 immunoreactivity in normal and ischemic brain sections. Throughout normal brain, we observed caspase-3p32 immunoreactivity within neurons and axonal fibers (Fig.4A,B). Neurons in the piriform cortex (Fig. 4A) and neocortex (Fig.4B), as well as axons passing through the striatum (data not shown), showed intense immunoreactivity. A few large cells in the striatum showed caspase-3p32 immunoreactivity; otherwise, constitutively expressed protein appeared lower in striatum than in cortex (data not shown). Preadsorption with peptide immunogen markedly reduced immunostaining (Fig. 4C). By contrast, caspase-3p20 immunoreactivity was barely detectable above background when tissue sections from normal brain were incubated with caspase-3p20 antiserum (Fig. 4D).
Fig. 4.
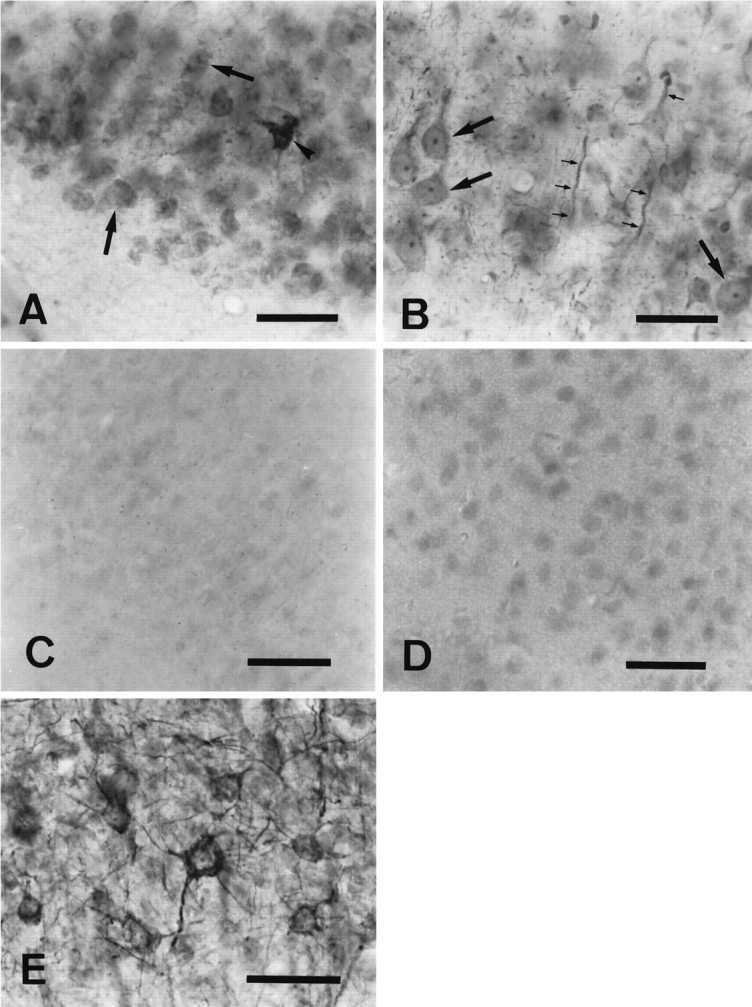
Immunohistochemical staining of normal (A–D) and ischemic (E) brains using caspase-3p32 or caspase-3p20 antiserum. Caspase-3p32 immunoreactivity (A, B) was detected in neuronal perikarya (large arrows) and dendrites (small arrows), in 40 μm sections from piriform (A) or in parietal cortex (B) counterstained with thionine hydrochloride. Frequently, cells in the piriform cortex contained intense caspase-3p32 immunoreactivity (A, arrowhead). Preadsorption with peptide immunogen (see Materials and Methods) markedly blocked the staining in piriform cortex (C). By contrast, normal brain stained poorly with anti-caspase-3p20 (piriform cortex) (D). Within the penumbra of ischemic cortex, caspase-3p32 staining was prominent in neuronal cytoplasm 24 hr after reperfusion (E). Scale bar, 15 μm.
Caspase-3p32 immunoreactivity in the ischemic area appeared diminished early after reperfusion (data not shown). This decrease, however, was not observed on immunoblot (a more quantitative method but less sensitive to spatial changes). At 6–24 hr after reperfusion, immunostaining increased within perikarya and dendrites of cortical neurons in the penumbral territory (Fig.4E).
Many caspase-3p20-immunoreactive neurons appeared in the ipsilateral piriform and parietal cortices and striatum after ischemia and reperfusion (Fig. 5) (and data not shown). Caspase-3p20 staining, which appeared as dense coarse granular immunodeposits in cortex, was less granular but nevertheless quite dense in striatum (Figs. 5E,F) (and data not shown). This immunostaining was dramatically reduced by preadsorption with peptide immunogen (data not shown). In 3 of 18 examined brains, p20 immunostaining appeared slightly increased over background levels in the contralateral cortex during reperfusion (data not shown), and this faint staining was observed on immunoblots as well. In the other 15 brains, however, caspase-3p20 immunoreactivity remained at background levels in the contralateral hemisphere during reperfusion (Fig.5A,C).
Fig. 5.
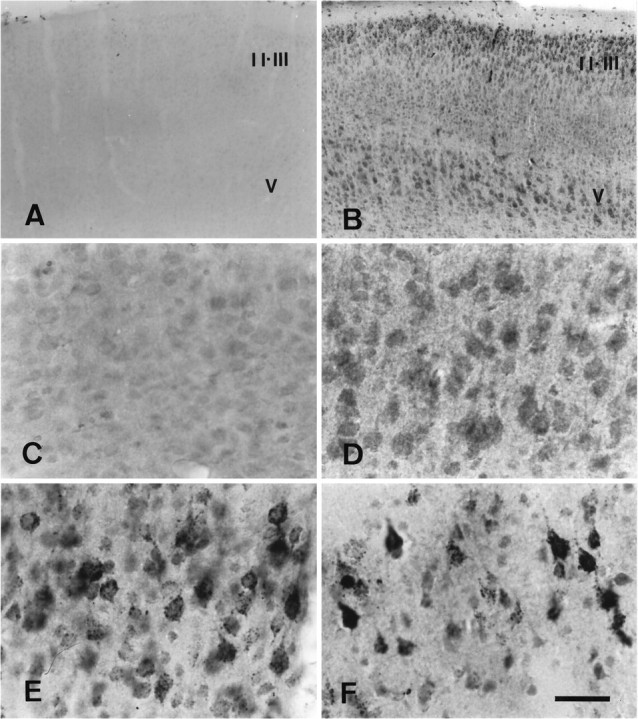
Caspase-3p20 immunohistochemical staining of tissue sections (40 μm) from ischemic mouse parietal cortex after 2 hr middle cerebral artery occlusion and reperfusion. High-power photomicrographs (C–F) were taken from lamina V. Caspase-3p20 immunoreactivity was detected as early as 5 min after reperfusion in ischemic cortex (B, D) compared with the contralateral side (A, C) and was found within cytoplasm of cortical neurons after 5 min (B, D), 1 hr (E), and 24 hr (F) of reperfusion. Caspase-3p20 staining did not change in the contralateral hemisphere at these times (not shown). At 1 and 24 hr, coarse granular immunoproducts were densely concentrated in neurons (E, F). II, III, and V refer to cortical laminae (n = 3 animals per time point). Scale bar (shown in F): A, B, 120 μm; C–F, 15 μm.
Double-fluorescence immunohistochemistry was used to detect the coexistence of caspase-3p20 and glial fibrillary acidic protein (GFAP) within single cells. Colocalization was not observed when tissue sections were examined from 5 min to 24 hr after reperfusion (data not shown).
Double staining with caspase-3 immunohistochemistry and TUNEL
To determine whether caspase-3p20-positive neurons also contain fragmented DNA, we stained tissue sections for caspase-3p20 immunoreactivity followed by TUNEL. Caspase-3p20 immunoreactivity was found within the same cells containing double-strand DNA breaks 12 to 24 hr after reperfusion (Fig.6D–I). Many caspase-3p20-positive cells contained TUNEL (∼80 and 60% in striatum and cortex, respectively, among 400 cells counted at 12 and 24 hr). A smaller subset of TUNEL-positive cells was caspase-3p20 positive (40 and 44% in striatum and cortex, respectively, at 12 hr among 500 counted cells). Some striatal nuclei appeared to contain both caspase-3p20 staining and Hoechst 33342 (Fig.6J,K). Within ischemic cortical cells, caspase-3p20 staining was found predominantly in the cytoplasm at the examined time point (Fig. 6L,M).
Fig. 6.
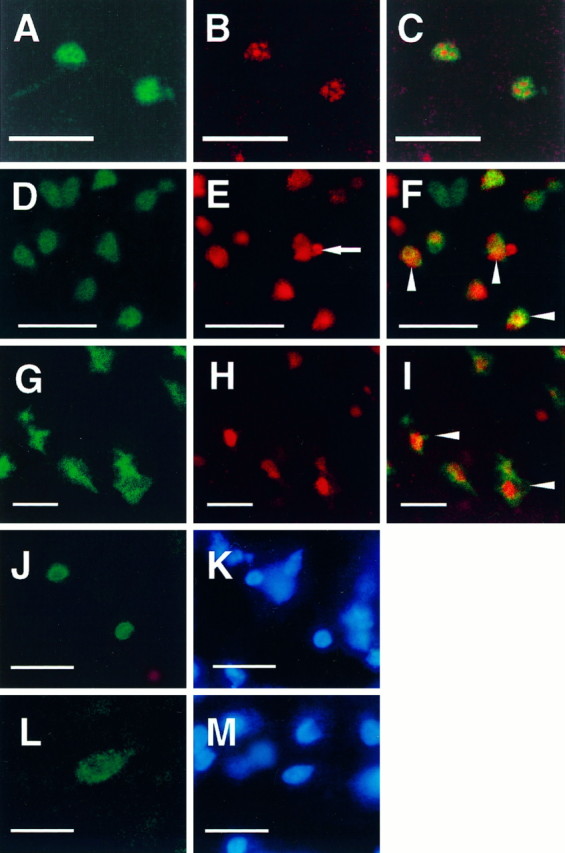
Confocal microscopic images document the localization of caspase-3p20 immunoreactivity and TUNEL in single tissue sections (40 μm) from normal cerebellar hemisphere (postnatal day 5, P5) (A–C) and ischemic striatum (D–F) and parietal cortex (G–I) after 2 hr middle cerebral artery occlusion and 12 hr (D–F) or 24 hr (G–I) reperfusion. Immunoreactivity and TUNEL were visualized with Bodipy fluorescein (A, D, G:green) and Cy3 (B, E, H:red), respectively. Almost every caspase-3p20 immunoreactive cerebellar granule cell at P5 was TUNEL positive. Two representative cells are shown here (C). Many ischemic cells contained both immunoreactive staining (p20) and TUNEL (F, I, arrowheads). Note a typical apoptotic body in a TUNEL-positive striatal cell (E, arrow). After 12 hr of reperfusion caspase-3p20 staining looked as if it was localized within the nucleus of some striatal cells [immunofluorescence staining (J) combined with Hoechst 33342 staining (K) using conventional fluorescence microscopy]. Within cortical cells, however, the pattern of p20 differed at 24 hr (L, p20 immunostaining; M, Hoechst 33342 staining), which may reflect the more rapid rate of ischemic evolution in striatum. Scale bar, 10 μm.
To determine whether caspase-3p20 immunoreactivity and TUNEL are present in nonischemic apoptotic cells, we stained single tissue sections from mouse pups (postnatal day 5) for TUNEL and caspase-3p20 immunoreactivity. Some cerebellar granule cells showed intense caspase-3p20 immunoreactivity and most of these were TUNEL positive (Fig. 6A–C). By contrast, cells stained negative for both when apoptosis was no longer present (postnatal day 25; data not shown).
Genomic DNA extraction and analysis
To confirm that TUNEL reflected nucleosomal DNA fragmentation, we analyzed DNA from ischemic brain homogenates by gel electrophoresis. DNA laddering was first observed in ischemic tissue 6 hr after reperfusion. DNA laddering became more apparent 12 hr after reperfusion, and this sustained for at least 24 hr (Fig.7).
Fig. 7.
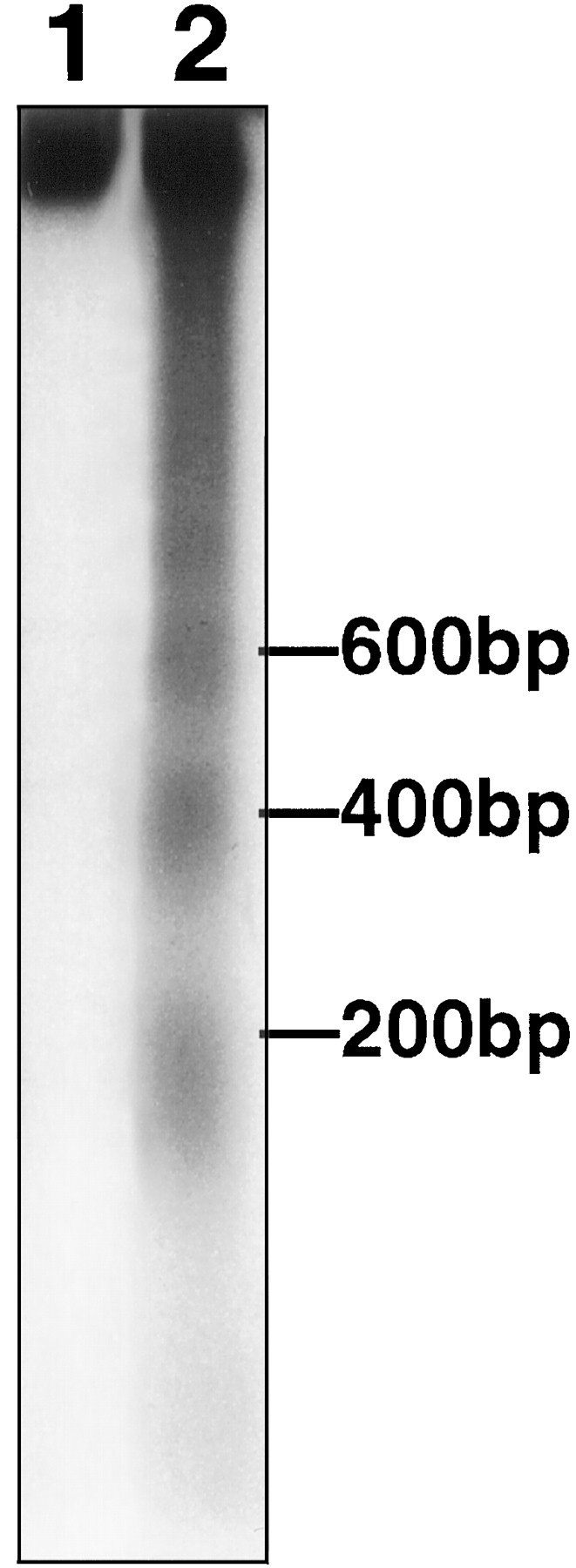
DNA laddering was detected by agarose gel electrophoresis in ipsilateral (lane 2) but not contralateral striatum (lane 1) 12 hr after reperfusion after 2 hr middle cerebral artery occlusion. Laddering appeared as early as 6 hr and was sustained at 24 hr as well (see Results). DNA was end-labeled with [32P]ddATP, electrophoresed on a 2% agarose gel, and autoradiographed. A ladder corresponding to 200, 400, and 600 bp is shown.
DISCUSSION
The present study provides evidence for constitutive expression of caspase-3 in normal adult mammalian brain. We found caspase-3p32 immunoreactivity diffusely distributed within neuronal perikarya, dendrites, and axons in cerebral cortex and striatum, more notably in cortex than striatum but particularly marked in piriform cortex. Constitutive expression was not particularly prominent in brain regions selectively vulnerable to ischemia, such as hippocampal CA1 pyramidal cells, although immunoreactivity was localized predominantly in neurons that are known to be more susceptible to ischemic insult than glial or endothelial cells.
We also documented caspase-3 cleavage (appearance of caspase-3p20) and DEVDase enzymatic activity in brain homogenate during the reperfusion phase of ischemia. Both enzymatic assay and immunohistochemistry showed enhanced signal within the MCA territory during reperfusion (0–5 min) and were temporally consistent early during reperfusion. In fact, caspase-3 appears to have been activated during the occlusion period because the p20 antigen and enzyme activity were already evident upon reperfusion. (In a preliminary experiment, DEVDase activity was elevated 1 hr after MCA occlusion and before reperfusion.) When examined 5 min after reperfusion, p20 cleavage product appeared within the cytoplasm of GFAP-negative cells exhibiting the morphology of neurons and remained in those cells for at least 24 hr. At 1 hr, the immunoproducts appeared as cytoplasmic granules reminiscent of the immunostaining pattern observed for the lysosomal protease cathepsin B in ischemic hippocampus (Nitatori et al., 1995).
Caspase-3p20 was found within cells showing features of apoptosis, and more than half of the caspase-3p20 labeled cells were TUNEL positive. The relatively large fraction of double-labeled cells suggests the importance of caspase-3 activation to ischemia-induced apoptosis in mammalian brain and is consistent with previously published data indicating that blocking caspase activation alters the outcome of experimental cerebral ischemia (Friedlander et al., 1997; Hara et al., 1997a,b) and markers of apoptosis (Endres et al., 1998). A similar staining pattern was observed for Reaper, an apoptosis inducer, and TUNEL-positive cells in Drosophila embryos: there are cells positive for Reaper only, for TUNEL only, and for Reaper and TUNEL (White et al., 1994). Caspase-3 activation may not be required for apoptosis in all cells because we observed ∼50% of TUNEL-positive cells that did not stain for the caspase-3 cleavage product.
Several regional differences were observed between striatum and parietal cortex that may reflect more severe striatal injury or region-specific differences in constitutive caspase-3p32 expression. Constitutive caspase-3p32 immunoreactivity was low in striatal cells, but by 6 hr p20 immunoreactivity was marked in numerous striatal cells and colocalized with TUNEL after reperfusion. By contrast, constitutive caspase-3p32 staining was prominent within parietal cortex, as was p20 immunoreactivity early after reperfusion. The onset of TUNEL, however, was more delayed and less dramatic than in striatum. In every instance though, activation of caspase-3 preceded the appearance of TUNEL. The findings suggest that the apoptotic cell death pathway shows regional differences conditioned by cell type and local factors.
Methodological issues
Enzyme activity
We adapted an enzyme activity assay for caspase that has been applied successfully in cell culture supernatants (Sarin et al., 1996;Armstrong et al., 1997; Eldadah et al., 1997; Keane et al., 1997). We determined that cleavage of the fluorogenic substrate was temperature and protein dependent, could be partially inhibited by zDEVD-fmk, and was abolished by previous boiling of brain homogenate. Recently, it was reported that caspase-7 as well as caspase-1, -3, and -4 cleave substrates after DEVD sequence (Talanian et al., 1997; Thornberry et al., 1997). Hence, we believe that caspase-3 is not the only enzyme cleaving zDEVD-afc in ischemic brain homogenate, and we use the term caspase-like enzyme activity in this report.
The source of zDEVD-afc cleavage activity unblocked by the addition of zDEVD-fmk or zVAD-fmk remains unknown but was present in homogenates from adult ischemic forebrain and developing cerebellum as well as HeLa cells after staurosporine treatment. This probably reflects activation of an unidentified enzyme or enzymes.
Antibody specificity
Caspase-3 was detected using two polyclonal antisera recognizing different epitopes in mature and cleaved proteins. Both constitutively expressed immunoreactivity and binding after ischemia were markedly diminished by preadsorption with synthetic immunogens. Caspase-3p20 antiserum detected p20 in ischemic mouse brain and p18 in cultured mouse cerebellar cells after K+–serum withdrawal (Armstrong et al., 1997) (A. Srinivasan, unpublished observations). Hence, p18 formed by cleavage at 28DS29may not be formed or may be unstable in mouse brain. This antiserum detected caspase-3p20 antigen during apoptosis induced by ischemia and in mouse pup cerebellar granule cells together with TUNEL at a developmental stage when apoptosis is prominent.
Changes in enzyme activity and immunochemistry during reperfusion
Caspase-like enzyme activity increased upon reperfusion and returned to baseline between 3 and 6 hr later. The onset of cleavage activity was similar to the appearance of p20 immunoreactivity, although there were certain differences in time course, which may reflect methods of detection or activity of other DEVD-cleaving enzymes (Talanian et al., 1997; Thornberry et al., 1997). For example, p20 was first detected during reperfusion by immunohistochemistry, compared with 1 hr after reperfusion, using a less sensitive but more specific method: immunoblotting. Methodological differences may partly explain why the p20 cleavage product was detected by immunohistochemical method but not reflected by DEVDase activity 6–24 hr after reperfusion. A decrease in DEVDase activity coinciding with the appearance of DNA fragmentation (both at 3–6 hr) may reflect cell death as a reduction of caspase-like activity. Moreover, caspase activity is highly concentration dependent, especially in cell–tissue extracts (Thornberry et al., 1992) (J. Yuan, unpublished observations); a partial reduction of immunoreactivity could translate into a loss of assayable DEVD cleaving activity.
We recently observed that zDEVD-fmk administration 1 hr, but not later, after reperfusion following 2 hr ischemia decreased infarction volume (Ma et al., 1998). The therapeutic time window is quite consistent with the peak of caspase-like enzyme activity (30 min to 1 hr), which lends support to the notion that putative caspase inhibitors in fact may decrease infarction volume and improve neurological deficits by irreversible enzyme inactivation.
Caspase-3 activation in related models
As noted above, Armstrong et al. (1997) reported that K+–serum withdrawal activates caspase-3 and induces apoptosis in cultured mouse cerebellar granule neurons by zVAD-fmk-inhibitable mechanisms. Keane et al. (1997) observed similar findings after staurosporine treatment of cultured mouse cortical neurons. Ni et al. (1997) recently found upregulation in a cloned caspase-3-related rat protease gene in cultured cerebellar granule neurons after K+–serum withdrawal. The mRNA expression of this gene was robust in adult piriform cortex, which agrees with our data showing strong constitutive expression of caspase-3p32 in this brain area. Asahi and colleagues (1997) found upregulation of caspase-3 mRNA after permanent MCA occlusion in rat brain at 16–24 hr, although protein expression, activation, and localization were not determined. In the present study, we demonstrate that ischemic insult upregulates caspase-3p32 in the penumbra, as evidenced by the increase in caspase-3p32 immunoreactivity in this region after 24 hr of reperfusion. Recently, activation of caspase-3 enzyme activity was identified after traumatic brain injury in rats; improvement in neurological deficits after zDEVD-fmk treatment was reported in that model as well (Yakovlev et al., 1997), consistent with our previous report in brain ischemia (Hara et al., 1997b).
Colocalization of TUNEL and caspase-3p20 immunoreactivity
We observed colocalization of p20 immunoreactivity and TUNEL in individual cells within the ischemic striatum 12–24 hr after reperfusion and in postnatal day 5 cerebellum in normal mouse brain. In some cells, these two stains appear to localize within individual nuclei (e.g., TUNEL and caspase-3p20), although additional studies are required to clarify this point. Our data provide evidence for caspase-3 activation in neurons undergoing ischemic cell death, although we have not directly shown that caspase-3 activation causes cell death. Linkage is suggested by the protective effects of caspase inhibition in ischemic cell death (Friedlander et al., 1997; Hara et al., 1997a,b;Schielke et al., 1998) and by the observation that apoptotic neurons contain both p20 and TUNEL and that caspases activate hours before the appearance of TUNEL-positive cells. The recent discovery of a caspase-activated deoxyribonuclease (CAD) establishes an important linkage between cysteine protease activity and DNA laddering (Enari et al., 1998; Sakahira et al., 1998), which could be highly relevant to the appearance of p20 immunoreactivity within TUNEL-positive cells. By characterizing events both upstream and downstream from caspase-3-like activation (including activation of other caspase family members in addition to caspase-1), new therapeutic opportunities might emerge beyond those suggested previously for zVAD-fmk and zDEVD-fmk (Hara et al., 1997b). Taken together, our findings support an important role for caspase-3 in apoptotic signal transduction in injured brain.
Footnotes
Studies were supported by Massachusetts General Hospital Interdepartmental Stroke Project Grants (NS10828) and an unrestricted award in Neuroscience from Bristol Myers Squibb (M.A.M.). S.N. was supported by Uehara Memorial Foundation. K.F. and M.E. were supported by the Deutsche Forschungsgemeinschaft (Fi600/2-1 and En343/1-1, respectively). J.Y. was supported by the National Institute of Neurological Disorders and Stroke and an American Heart Association Established Investigatorship Award. We thank Drs. Turgay Dalkara, Jianya Ma, Zhihong Huang, and Christian Waeber for assistance, and Dr. Joseph L. Goldstein, University of Texas Southwestern Medical Center, for the kind gift of caspase-3 antiserum.
Correspondence should be addressed to Michael A. Moskowitz, Massachusetts General Hospital, Harvard Medical School, 149 13th Street, Room 6403, Charlestown, MA 02129.
REFERENCES
- 1.Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong RC, Aja TJ, Hoang KD, Gaur S, Bai X, Alnemri ES, Litwack G, Karanewsky DS, Fritz LC, Tomaselli KJ. Activation of the CED3/ICE-related protease CPP32 in cerebellar granule neurons undergoing apoptosis but not necrosis. J Neurosci. 1997;17:553–562. doi: 10.1523/JNEUROSCI.17-02-00553.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Asahi M, Hoshimaru M, Uemura Y, Tokime T, Kojima M, Ohtsuka T, Matsuura N, Aoki T, Shibahara K, Kikuchi H. Expression of interleukin-1β converting enzyme gene family and bcl-2 gene family in the rat brain following permanent occlusion of the middle cerebral artery. J Cereb Blood Flow Metab. 1997;17:11–18. doi: 10.1097/00004647-199701000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- 5.Bertrand R, Solary E, O’Connor P, Kohn KW, Pommier Y. Induction of common pathway of apoptosis by staurosporine. Exp Cell Res. 1994;211:314–321. doi: 10.1006/excr.1994.1093. [DOI] [PubMed] [Google Scholar]
- 6.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cerretti DP, Kozlosky CJ, Mosley B, Nelson N, Van Ness K, Greenstreet TA, March CJ, Kronheim SR, Druck T, Cannizzaro LA, Huebner K, Black RA. Molecular cloning of the interleukin-1β converting enzyme. Science. 1992;256:97–100. doi: 10.1126/science.1373520. [DOI] [PubMed] [Google Scholar]
- 8.Cohen GM. Caspases: the executioners of apoptosis. Biochem J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eldadah BA, Yakovlev AG, Faden AI. The role of CED-3-related cysteine proteases in apoptosis of cerebellar granule cells. J Neurosci. 1997;17:6105–6113. doi: 10.1523/JNEUROSCI.17-16-06105.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- 11.Ellis RE, Yuan J, Horvitz HR. Mechanisms and functions of cell death. Annu Rev Cell Biol. 1991;7:663–698. doi: 10.1146/annurev.cb.07.110191.003311. [DOI] [PubMed] [Google Scholar]
- 12.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 13.Enari M, Talanian RV, Wong WW, Nagata S. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature. 1996;380:723–726. doi: 10.1038/380723a0. [DOI] [PubMed] [Google Scholar]
- 14.Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gómez-Isla T, Hyman BT, Moskowitz MA. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Erhardt P, Cooper GM. Activation of the CPP32 apoptotic protease by distinct signaling pathways with differential sensitivity to Bcl-XL. J Biol Chem. 1996;271:17601–17604. doi: 10.1074/jbc.271.30.17601. [DOI] [PubMed] [Google Scholar]
- 16.Fernandes-Alnemri T, Litwack G, Alnemri ES. CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1β-converting enzyme. J Biol Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- 17.Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, MacDonald G, Fishman MC, Greenberg AH, Moskowitz MA, Yuan J. Expression of a dominant negative mutant of interleukin-1β converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J Exp Med. 1997;185:933–940. doi: 10.1084/jem.185.5.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hara H, Huang PL, Panahian N, Fishman MC, Moskowitz MA. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J Cereb Blood Flow Metab. 1996;16:605–611. doi: 10.1097/00004647-199607000-00010. [DOI] [PubMed] [Google Scholar]
- 20.Hara H, Fink K, Endres M, Friedlander RM, Gagliardini V, Yuan J, Moskowitz MA. Attenuation of transient focal cerebral ischemic injury in transgenic mice expressing a mutant ICE inhibitory protein. J Cereb Blood Flow Metab. 1997a;17:370–375. doi: 10.1097/00004647-199704000-00002. [DOI] [PubMed] [Google Scholar]
- 21.Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J, Moskowitz MA. Inhibition of interleukin 1β converting enzyme family proteases reduces ischemic and excitotoxic damage. Proc Natl Acad Sci USA. 1997b;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keane RW, Srinivasan A, Foster LM, Testa M-P, Örd T, Nonner D, Wang H-G, Reed JC, Bredesen DE, Kayalar C. Activation of CPP32 during apoptosis of neurons and astrocytes. J Neurosci Res. 1997;48:168–180. doi: 10.1002/(sici)1097-4547(19970415)48:2<168::aid-jnr9>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 23.Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1β converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 24.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 25.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, Towne E, Tracey D, Wardwell S, Wei F-Y, Wong W, Kamen R, Seshadri T. Mice deficient in IL-1β-converting enzyme are defective in production of mature IL-1β and resistant to endotoxic shock. Cell. 1995a;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Chopp M, Jiang N, Yao F, Zaloga C. Temporal profile of in situ DNA fragmentation after transient middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1995b;15:389–397. doi: 10.1038/jcbfm.1995.49. [DOI] [PubMed] [Google Scholar]
- 27.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by β-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma J, Endres M, Moskowitz MA (1998) Synergistic effects of caspase inhibitors and MK-801 in brain injury after transient focal cerebral ischemia in mice. Br J Pharmacol, in press. [DOI] [PMC free article] [PubMed]
- 29.MacManus JP, Hill IE, Huang Z-G, Rasquinha I, Xue D, Buchan AM. DNA damage consistent with apoptosis in transient focal ischaemic neocortex. NeuroReport. 1994;5:493–496. doi: 10.1097/00001756-199401120-00031. [DOI] [PubMed] [Google Scholar]
- 30.Namura S, Zhu J, Fink K, Endres M, Tomaselli KJ, Srinivasan A, Yuan J, Moskowitz MA. CPP32 activation in neuronal apoptosis after transient focal cerebral ischemia in mice. Soc Neurosci Abstr. 1997;23:850. [Google Scholar]
- 31.Ni B, Wu X, Du Y, Su Y, Hamilton-Byrd E, Rockey PK, Rosteck P, Jr, Poirier GG, Paul SM. Cloning and expression of a rat brain interleukin-1β-converting enzyme (ICE)-related protease (IRP) and its possible role in apoptosis of cultured cerebellar granule neurons. J Neurosci. 1997;17:1561–1569. doi: 10.1523/JNEUROSCI.17-05-01561.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 33.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, Yamin T-T, Yu VL, Miller DK. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 34.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J Neurosci. 1995;15:1001–1011. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oppenheim RW, Prevette D, Tytell M, Homma S. Naturally occurring and induced neuronal death in the chick embryo in vivo requires protein and RNA synthesis: evidence for the role of cell death genes. Dev Biol. 1990;138:104–113. doi: 10.1016/0012-1606(90)90180-q. [DOI] [PubMed] [Google Scholar]
- 36.Rothwell NJ, Relton JK. Involvement of interleukin-1 and lipocortin-1 in ischaemic brain damage. Cerebrovasc Brain Metab Rev. 1993;5:178–198. [PubMed] [Google Scholar]
- 37.Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- 38.Sarin A, Wu ML, Henkart PA. Different interleukin-1β converting enzyme (ICE) family protease requirements for the apoptotic death of T lymphocytes triggered by diverse stimuli. J Exp Med. 1996;184:2445–2450. doi: 10.1084/jem.184.6.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schielke GP, Yang G-Y, Shivers BD, Betz AL. Reduced ischemic brain injury in interleukin-1β converting enzyme-deficient mice. J Cereb Blood Flow Metab. 1998;18:180–185. doi: 10.1097/00004647-199802000-00009. [DOI] [PubMed] [Google Scholar]
- 40.Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificities of caspase family proteases. J Biol Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- 41.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32β, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 42.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 43.Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, Elliston KO, Ayala JM, Casono FJ, Chin J, Ding GJ-F, Egger LA, Graffney EP, Limjuco G, Palyha O, Raju SM, Rolando AM, Salley JP, Yamin T-T, Lee TD, Shively JE, MacCross M, Mumford RA, Schmidt JA, Tocci MJ. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 44.Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW. A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 45.Tilly JL, Hsueh AJW. Microscale autoradiographic method for the qualitative and quantitative analysis of apoptotic DNA fragmentation. J Cell Physiol. 1993;154:519–526. doi: 10.1002/jcp.1041540310. [DOI] [PubMed] [Google Scholar]
- 46.Wang X, Zelenski NG, Yang J, Sakai J, Brown MS, Goldstein JL. Cleavage of sterol regulatory element binding proteins (SREBPs) by CPP32 during apoptosis. EMBO J. 1996;15:1012–1020. [PMC free article] [PubMed] [Google Scholar]
- 47.White K, Grether ME, Abrams JM, Young L, Farrell K, Steller H. Genetic control of programmed cell death in Drosophila. Science. 1994;264:677–683. doi: 10.1126/science.8171319. [DOI] [PubMed] [Google Scholar]
- 48.Wood KA, Dipasquale B, Youle RJ. In situ labeling of granule cells for apoptosis-associated DNA fragmentation reveals different mechanisms of cell loss in developing cerebellum. Neuron. 1993;11:621–632. doi: 10.1016/0896-6273(93)90074-2. [DOI] [PubMed] [Google Scholar]
- 49.Xue D, Shaham S, Horvitz HR. The Caenorhabditis elegans cell-death protein CED-3 is a cysteine protease with substrate specificities similar to those of the human CPP32 protease. Genes Dev. 1996;10:1073–1083. doi: 10.1101/gad.10.9.1073. [DOI] [PubMed] [Google Scholar]
- 50.Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1β-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]