

Abstract
Treatment with proinflammatory prostaglandin E2(PGE2) produced a transient sensitization of whole-cell currents elicited by the vanilloid capsaicin. The intracellular signaling pathways that mediate the initiation of this PGE2-induced sensitization of the capsaicin-elicited current in rat sensory neurons are not well established. Treatment with either forskolin (100 nm to 10 μm) or membrane-permeant analogs of cAMP, 8-bromo-cAMP (8-Br-cAMP) and chlorphenylthio-cAMP (10 μm to 1 mm), transiently sensitized neuronal responses elicited by capsaicin in a manner analogous to that produced by PGE2. The duration of sensitization was lengthened with increasing concentrations of forskolin; however, higher concentrations of 8-Br-cAMP or chlorphenylthio-cAMP led to a shortening of sensitization. The inactive analog of forskolin, dideoxy-forskolin, had no effect on capsaicin responses. Inclusion of the inhibitor of protein kinase A in the recording pipette completely suppressed the sensitization produced by PGE2 or forskolin. In recordings from membrane patches in the cell-attached configuration, the bath application of capsaicin evoked single-channel currents in which the level of channel activity was concentration-dependent and had an EC50 of 1.4 μm. These single-channel currents evoked by capsaicin exhibited an apparent reversal potential of +4 mV and were blocked by the capsaicin antagonist capsazepine. Exposure of the sensory neuron to either PGE2 or forskolin produced a large and transient increase in the mean channel activity (NPo) elicited by capsaicin, although the unitary conductance remained unaltered. Taken together, these observations suggest that modulation of the capsaicin-gated channel by the cAMP–protein kinase A signaling pathway enhanced the gating of these channels and consequently resulted in the sensitization of the whole-cell currents.
Keywords: prostaglandin E2, cAMP, protein kinase A, capsaicin, sensitization, neuronal excitability
A specific population of mammalian sensory neurons can be distinguished by their excitatory response to the vanilloid capsaicin. These capsaicin-sensitive A-δ and C fibers are believed to be involved in the perception and signaling of nociceptive information (for review, see Holzer, 1991; Szallasi and Blumberg, 1996). Indeed, the intradermal injection of capsaicin in human subjects provokes intense pain (LaMotte et al., 1991, 1992). In responsive sensory neurons, capsaicin generates an inward current that results in the depolarization of the neuron (Heyman and Rang, 1985;Marsh et al., 1987; Bevan and Forbes, 1988; Bevan and Szolcsanyi, 1990). This inward current results from the opening of a nonselective cationic channel that is largely permeable to Na+and Ca2+ (Marsh et al., 1987; Wood et al., 1988;Bevan and Szolcsanyi, 1990; Oh et al., 1996a). Recently, the capsaicin receptor has been cloned, and the expressed protein has electrophysiological properties that are quite similar to the currents conducted by the native receptor (Caterina et al., 1997). Although there is considerable information concerning the excitatory properties of capsaicin, the exact role of this receptor in nociception remains to be elucidated. Similarly, little is known about the capacities of intracellular signaling pathways to modulate either the capsaicin-elicited whole-cell current or the activity of the capsaicin-gated ion channel.
The sensitivity or excitability of mammalian sensory neurons in response to various forms of noxious stimulation, such as mechanical pressure or chemical agents, can be facilitated by exposure to proinflammatory prostaglandins (Higgs et al., 1984; Foreman, 1987;Salmon and Higgs, 1987). In behavioral studies, sensitization is manifested as a heightened perception of pain (for review, seeHandwerker and Kobal 1993; Kress and Reeh, 1996). Furthermore, in electrophysiological recordings obtained from isolated sensory neurons grown in culture, treatment with prostaglandin E2(PGE2) enhances the number of action potentials generated by exposure to either elevated levels of potassium (Baccaglini and Hogan, 1983) or focally applied bradykinin (Nicol and Cui, 1994). The increased excitability results, in part, from the modulation of specific membrane currents. Treatment with PGE2 results in the facilitation of a tetrodotoxin-resistant sodium current (Gold et al., 1996; England et al., 1996) and the suppression of a potassium current (Nicol et al., 1997).
Recent studies indicate that this PGE2-induced sensitization results from activation of the cAMP transduction cascade. Experimental findings that support this notion are threefold. First, PGE2 increases intracellular levels of cAMP in sensory neurons (Hingtgen et al., 1995). Second, the exogenous application of membrane-permeant analogs of cAMP mimics the sensitizing effects of PGE2 in studies examining either the excitatory response elicited by bradykinin (Cui and Nicol, 1995) or the release of neuropeptides from sensory neurons (Hingtgen et al., 1995). Last, inhibition of the cAMP-dependent protein kinase A (PKA) blocks sensitization by PGE2 (Cui and Nicol, 1995). These studies indicate that the cAMP transduction cascade plays a critical role in the PGE2-induced sensitization; however, the role of the cAMP transduction pathway in mediating the onset of sensitization and its capacity to modulate the excitatory response to capsaicin remain to be defined. Therefore, in this study we examined the capacity of both the proinflammatory prostaglandin PGE2 and activators of the cAMP transduction cascade to sensitize the response to capsaicin recorded from rat sensory neurons grown in culture. Our findings demonstrate that PGE2, through activation of the cAMP–PKA signaling pathway, increases the whole-cell currents elicited by capsaicin and that this enhancement results from the increased activity of the capsaicin-gated ion channel.
Portions of this work have been published previously (Lopshire and Nicol, 1997a).
MATERIALS AND METHODS
Isolation and culture of embryonic rat sensory neurons. The procedures used for the isolation and maintenance of embryonic rat sensory neurons have been described previously (Vasko et al., 1994). Briefly, pregnant Harlan Sprague Dawley (Indianapolis, IN) rats were rendered unconscious by exposure to CO2 and killed by cervical dislocation. The dorsal root ganglia (DRG) were removed from the embryos (days 15–17 of gestation) and placed in a dish containing sterile calcium-free, magnesium-free HBSS at 4°C. The DRG were incubated in HBSS containing 0.025% trypsin for 30 min at 37°C. The digestion was terminated with the addition of 0.25% trypsin inhibitor. The ganglia were washed once with HBSS, centrifuged, and resuspended in growth medium (DMEM; Life Technologies, Grand Island, NY) supplemented with 2 mm glutamine, 50 μg/ml penicillin and streptomycin, 10% (v/v) heat-inactivated fetal bovine serum, 50 μm 5-fluoro-2′-deoxyuridine, 150 μm uridine, and 250 ng/ml 7S nerve growth factor (Harlan Bioproducts, Indianapolis, IN). Individual cells were obtained by dissociation with a fire-polished pipette. Approximately 300,000 cells were plated on a collagen-coated culture dish (35 mm) containing small plastic coverslips. Cells were maintained at 37°C in a 5% CO2 atmosphere, and the media were changed every 2 d. All procedures were approved by the Animal Care and Use Committee at Indiana University School of Medicine.
Electrophysiology. The procedures for whole-cell patch-clamp recording from rat sensory neurons have been described previously (Lopshire and Nicol, 1997b). Briefly, a coverslip of sensory neurons (typically after 4–5 d in culture) was placed in a recording chamber where the neurons were superfused with normal Ringer’s solution of the following composition (in mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.4, with NaOH. Small-diameter sensory neurons were selected for these recordings. Membrane currents or voltages were recorded with a List EP-7 patch-clamp amplifier (List Electronic, Darmstadt, Germany) using the whole-cell patch-clamp technique (Hamill et al., 1981). Recording pipettes were pulled from borosilicate glass and typically had resistances of 2–4 MΩ when filled with the following solution (in mm): 140 KCl, 5 MgCl2, 4 Na2 ATP, 0.3 Na3GTP, 2.5 CaCl2, 5 EGTA (calculated free calcium concentration of ∼150 nm), and 10 HEPES, pH 7.2, with KOH. After establishing the whole-cell configuration and allowing a 5–10 min equilibration period, the cell capacitance was compensated by the nulling circuitry of the amplifier. The series resistance was compensated (average value, 38.2 ± 1.5%), which yielded uncompensated series resistances that ranged from 2.2 to 7.9 MΩ (average value, 3.7 ± 0.4 MΩ). The membrane was voltage-clamped at −60 mV, which is close to the normal resting potential of this neuronal population (Nicol and Cui, 1994). The whole-cell currents evoked by capsaicin were filtered at 3 kHz and sampled at 500 Hz using the Fetchex program of pClamp 6.0.3 (Axon Instruments, Foster City, CA). In those experiments examining inhibition of PKA, the recording pipettes were backfilled with the normal internal solution containing 20 μm PKI14–24. Before any recording was initiated, a period of 10 min was used to permit the equilibration of PKI between the pipette and the cytoplasm of the neuron.
For the experiments examining single-channel activities, recording pipettes were coated with Sylgard (Dow Corning, Midland, MI) and heat-polished with a custom-built microforge. The pipette resistance was typically 3–5 MΩ after filling with a low-calcium external solution of the following composition (in mm): 140 NaCl, 5 KCl, 10 HEPES, 10 glucose, 2 MgCl2, 2 CaCl2, and 3.5 EGTA (calculated free calcium concentration of ∼150 nm), pH 7.4, with NaOH. The low-calcium external solution was used to superfuse the neurons. Single-channel currents were recorded using the cell-attached patch-clamp technique (Hamill et al., 1981). Currents evoked by capsaicin were filtered at 1 kHz using an eight-pole Bessel filter (Frequency Devices, Haverhill, MA), digitized at 5 kHz using the Fetchex program of pClamp, and stored in a personal computer. The traces of single-channel activity shown in the figures were filtered digitally at 500 Hz.
Capsaicin was administered to the neuron by either bath application or focal application via a large-bore pipette (5–10 μm tip diameter and positioned within 20–40 μm of the cell body) containing the desired external solution, the appropriate concentration of capsaicin, and 1 mm trypan blue dye. During continuous superfusion of the bath, positive pressure (1–2 sec duration for the whole-cell experiments and continuous for the single-channel experiments) was applied to the focal pipette, resulting in the delivery of capsaicin to the cell body. The trypan blue allowed for visual confirmation of drug delivery. The focal application of trypan blue alone had no effect on these neurons (Nicol and Cui, 1994;Lopshire and Nicol, 1997b). Two control responses to capsaicin, separated by ∼2 min, were obtained before the application of sensitizing agents. The response to capsaicin then was tested at the subsequent times indicated in each figure. To avoid desensitization, responses to capsaicin were obtained from only a single neuron on each coverslip.
Unless otherwise noted, all voltages are expressed as the membrane voltage, i.e., inside the cell relative to the outside for the whole-cell recordings or the negative of the pipette potential for the single-channel recordings. To determine the contribution of the resting membrane potential to the total membrane voltage of the cell-attached patch, the effects of a continuous exposure to capsaicin on the membrane potential were examined. Using the whole-cell configuration in current-clamp mode, the bath application of 1 μmcapsaicin in a low-calcium Ringer’s solution depolarized the membrane from −54 ± 1 to −5 ± 1 mV (n = 5) with an average onset latency of 19 ± 2 sec (expressed as the time elapsed from initiation of capsaicin exposure to steady-state depolarization; see Fig. 7B, inset, for a representative trace). This depolarization was maintained; after 4 and 20 min of continuous exposure to capsaicin the potentials were −4 ± 1 and −5 ± 1 mV, respectively. Because the voltage recorded under these conditions remained near a value of 0 mV, the small contribution of the resting membrane potential was neglected. The voltages indicated in the single-channel measurements are reported as the negative of the applied pipette voltage.
Fig. 7.
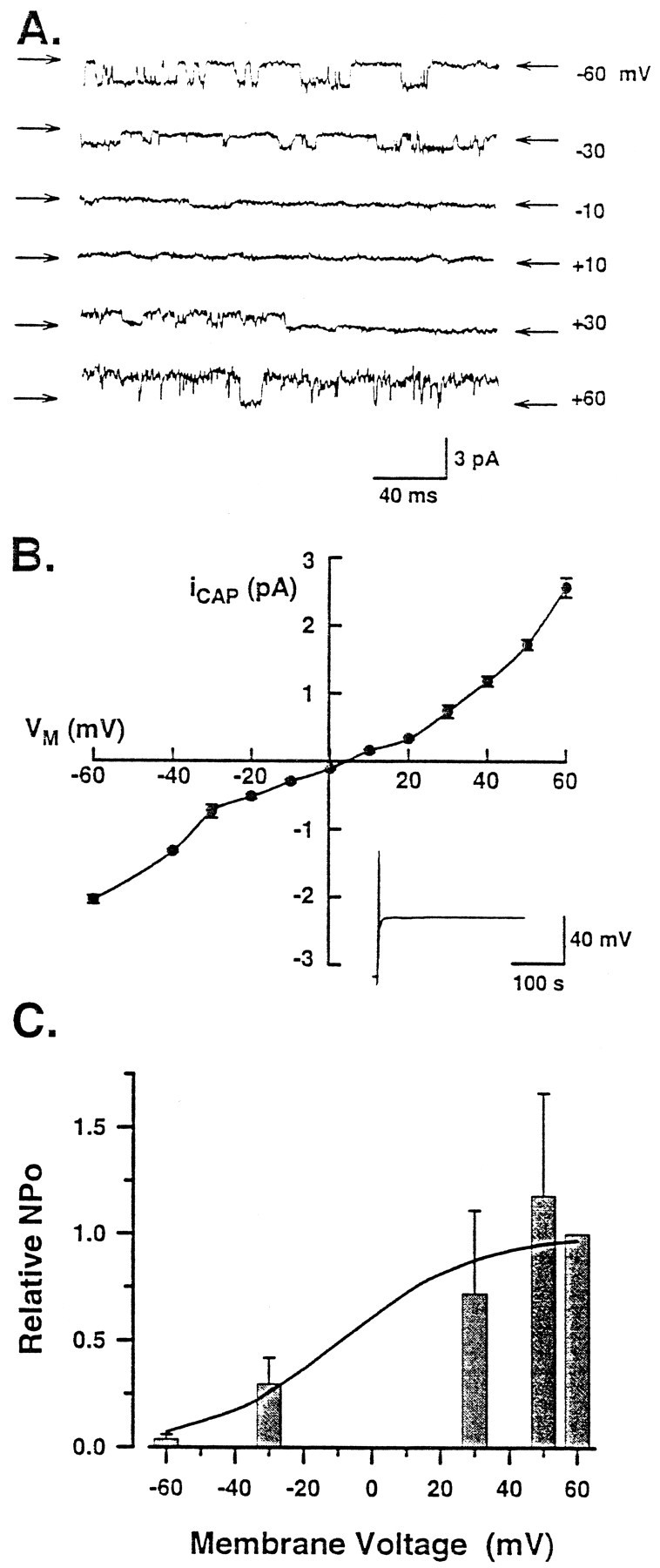
The capsaicin-gated channel exhibits outward rectification and increased NPo at positive membrane potentials. A, Effect of membrane potential on the single-channel activity elicited by bath exposure to 1 μmcapsaicin in a representative patch. The arrows indicate the closed level for each trace. The membrane potential for each trace is provided at the right. B, Current–voltage relation obtained from recordings in which the number of sensory neurons ranged between three and nine. Theinset shows a representative current-clamp recording for the depolarization (to −3 mV) elicited by treatment with 1 μm capsaicin. C, Dependence of the relative NPo elicited by 1 μm capsaicin (normalized to that obtained at +60 mV) on membrane potential (n = 3). The line through thebars represents the fit described by the Boltzmann relation.
For the whole-cell experiments, all neurons were required to maintain a zero-current potential more hyperpolarized than −45 mV for 4–5 min after establishing the whole-cell configuration, and the peak amplitudes of the two control currents elicited by capsaicin had to be within 10% of their mean value. If a neuron failed to satisfy these criteria, the recording was terminated. Depolarizing currents are shown as downward. All experiments were performed at room temperature (∼21°C).
Analysis. All values are reported as the mean ± SEM. In the whole-cell experiments, the time course of sensitization for each neuron was characterized by two parameters, the time to peak sensitization and the time for sensitization to return to half-maximal values (t½MAX). The values from individual neurons were then averaged to obtain the mean ± SEM for each experimental treatment. The time to peak sensitization is defined as the time elapsed from the delivery of sensitizing agent to the maximal sensitization, and thet½MAX is defined as the time elapsed from the maximally sensitized response to the point at which sensitization has returned to halfway between the peak and control values.
For the analysis of single-channel recordings, all-points amplitude histograms and events lists were constructed using the Fetchan program of pClamp. The all-points amplitude histograms were fit by Gaussian functions using the pStat program of pClamp. Channel openings were detected using the half-amplitude detection algorithm. A minimum estimate of the number of channels was obtained from the maximum number of overlapping events observed in each patch. NPo was calculated as follows:
| Equation 1 |
where i is the level of the open channel,N is the maximum number of overlapping events observed in that recording (assumed to be the maximum number of open channels), andPoi is the probability of i channels being open as reported by Fetchan. The total time of the recording was obtained from the events list and was typically ∼40 sec.
To determine the dependence of the relative NPo of the capsaicin-gated channel on membrane potential, these results were fitted by the Boltzmann relation:
| Equation 2 |
where Vm is the membrane potential, V0.5is the voltage at half-maximal activation, and k is a steepness factor.
The concentration–response relation was fitted with a ligand-binding isotherm of the following form:
| Equation 3 |
where CAPRESP is NPo at the test concentration, CAPMAX is the maximum NPoelicited by capsaicin, CAP0.5 is the concentration of capsaicin that produces a half-maximal response, CAP is the test concentration of capsaicin, and n is a steepness factor. Statistical differences were determined using a one-way ANOVA with a Student Newman–Keuls post hoc test. Values ofp < 0.05 were judged to be statistically significant.
Chemicals. Prostaglandins were obtained from Cayman Chemical Co. (Ann Arbor, MI). Protein kinase A inhibitor 14–24 amide (PKI14–24) was obtained from Peninsula Laboratories, Inc. (Belmont, CA). All other chemicals were obtained from Sigma (St. Louis, MO). Prostaglandins, capsaicin, forskolin, and dideoxyforskolin were first dissolved in methyl-2-pyrolidinone (HPLC grade; Aldrich, Milwaukee, WI) and subsequently diluted in the appropriate solution to the desired concentration. The concentrations of free Ca2+ were calculated from the binding constants described by Caldwell (1970).
RESULTS
Prostaglandin E2 and forskolin transiently sensitize sensory neurons to capsaicin excitation
The focal application of capsaicin evoked an inward current when applied to small-diameter sensory neurons voltage-clamped at −60 mV in the whole-cell configuration. The peak amplitude of the capsaicin-elicited current (ICAP) was increased transiently after treatment with PGE2. Control and experimental responses after PGE2 treatment obtained from a representative neuron are shown in Figure1A. The left trace illustrates the current response to a 2 sec application of 100 nm capsaicin. This concentration of capsaicin was used in all whole-cell experiments because it is slightly less than the EC50 (156 nm) established for the whole-cell current evoked by capsaicin in these cultured sensory neurons (Lopshire and Nicol, 1997b). In this neuron, the peak amplitude of the control response was 240 pA. After a 10 min exposure to 1 μm PGE2, the amplitude ofICAP was increased to 822 pA, a 3.4-fold increase. After a 25 min exposure to PGE2, the transient nature of the PGE2-induced sensitization was evident. In the continued presence of PGE2,ICAP was reduced to 246 pA and was similar to the control response. Another series of experiments examined the notion that the PGE2-induced increase in the amplitude ofICAP might result from an acceleration of the response kinetics to these brief applications of capsaicin. Capsaicin (100 nm) was applied focally to the neuron to obtain a response that reached a stable plateau; the application was then continued for an additional 2 sec. In six neurons, the average times to peak for the two control responses were 6.6 ± 0.9 and 6.2 ± 0.4 sec (peak amplitudes of 109 ± 31 and 112 ± 29 pA, respectively) in which the average times of capsaicin exposure were 8.7 ± 0.9 and 8.8 ± 0.5 sec, respectively. Using this stimulus protocol, treatment with 1 μm PGE2significantly enhanced ICAP by 2.2 ± 0.3-fold (range, 1.5 to 3.1-fold; data not shown). The time course of sensitization was similar to that shown in Figure 1Afor the shorter exposures to capsaicin in which the maximal sensitization occurred after a 10 min treatment with PGE2. These results corroborate our previous observations in which treatment of sensory neurons with PGE2 produced a transient increase in the amplitude of ICAP. AlthoughICAP was enhanced, PGE2 had no significant effect on the holding current. In those experiments using both short and long capsaicin exposures, the average holding current under control conditions was −55 ± 11 pA (n = 15) compared with −71 ± 17 pA (n = 15) after a 10 min exposure to 1 μm PGE2 (the time of peak sensitization). In the absence of PGE2, the repeated application of 100 nm capsaicin at 2 min intervals did not alter the response, indicating that these brief exposures to a low concentration of capsaicin produced neither sensitization nor desensitization of ICAP (Lopshire and Nicol, 1997b).
Fig. 1.

PGE2 and forskolin treatment enhance the amplitude of the whole-cell current elicited by capsaicin.A, Voltage-clamp recording (holding potential −60 mV) obtained from a representative neuron in which PGE2treatment increased the amplitude of the current elicited by a 2 sec focal application of capsaicin. B, Sensitizing effects of 100 nm and 1 μm forskolin treatment (top and bottom traces, respectively) on capsaicin responses in two other neurons. The times indicate when these recordings were obtained after the onset of prostaglandin or forskolin treatment. The extent and time courses of sensitization induced by different concentrations of forskolin are summarized inC. The amplitude of the capsaicin response after forskolin treatment was normalized to the average of the two control responses and represented as the fold increase relative to the control value. The bar beginning at 3 min represents the change to forskolin-containing Ringer’s solution. D, Effects of dideoxyforskolin on the capsaicin response. Thehatched and open bars indicate the applications of dideoxyforskolin and PGE2, respectively. Asterisks indicate significant differences at p < 0.05 compared with control.
The response of sensory neurons to bradykinin undergoes a PGE2-induced sensitization in which this enhancement was mediated by the cAMP transduction cascade (Cui and Nicol, 1995). To investigate whether sensitization of capsaicin responses by PGE2 involved similar cellular mechanisms, we determined the ability of forskolin, a diterpene that directly activates adenylyl cyclase (Seamon et al., 1981), to produce an analogous sensitization of capsaicin responses. The forskolin concentrations (100 nmto 10 μm) used in these experiments elevated the intracellular cAMP content by 2- and 10-fold, respectively, in cultured sensory neurons (Fig. 1, Hingtgen et al., 1995). Figure1B (top panel) shows that 100 nm forskolin sensitized capsaicin responses. The control response in this representative neuron was 510 pA. After a 10 min exposure to 100 nm forskolin, the amplitude ofICAP was increased to 1356 pA, a 2.7-fold enhancement. Analogous to the PGE2-induced increase inICAP, the sensitization produced by forskolin was transient; after a 35 min exposure to forskolin,ICAP (530 pA) returned to control levels.
Higher concentrations of forskolin produced a similar level of sensitization; however, the duration of this sensitization was increased. The bottom panel in Figure 1B illustrates the effect of 1 μm forskolin onICAP in another neuron. After a 10 min exposure to forskolin, ICAP was 836 pA, a threefold increase over the control response (280 pA). AlthoughICAP was enhanced, the holding current was unaltered by forskolin; after a 10 min application, the holding current was −79 ± 45 pA compared with the control value of −68 ± 23 pA (n = 7). This higher concentration of forskolin produced a sensitization of greater duration. After exposure to forskolin for 50 min, ICAP remained almost twofold larger than the control response. This concentration-dependent effect of forskolin on the duration of the enhanced capsaicin response was obtained in all neurons and is summarized in Figure 1C. The responses obtained after forskolin treatment were normalized to the mean of the two control responses. Exposure to 100 nmforskolin caused a significant and transient 2.7 ± 0.2-fold increase in the amplitude of ICAP with a time to peak of 11.3 ± 1.0 min and at½MAX of 5.3 ± 0.6 min (n = 3). In neurons exposed to either 1 or 10 μm forskolin, ICAP exhibited a 3.7 ± 0.4-fold (n = 7) or 3.1 ± 0.4-fold (n = 6) increase, respectively. The times to peak of 14.7 ± 3.3 min and 20.5 ± 3.3 min were similar for either 1 or 10 μm forskolin, respectively. However, compared with the results obtained with 100 nm forskolin, the sensitization remained significantly elevated for longer times. For 1 and 10 μm forskolin,t½MAX was increased to 19.2 ± 4.0 and 19.8 ± 5.0 min, respectively. Therefore, 100 nmforskolin appears to elicit maximal sensitization ofICAP, whereas higher concentrations prolong the period of sensitization.
To determine whether the sensitizing effects of forskolin were attributable to a specific action on adenylyl cyclase or reflected some other nonspecific action, we tested the effects of dideoxyforskolin, a forskolin analog that does not stimulate adenylyl cyclase (Seamon et al., 1983). As shown in Figure 1D, treatment with 1 μm dideoxyforskolin had no effect onICAP (n = 3). To determine whether dideoxyforskolin somehow influenced the capacity of neurons to be sensitized, another series of experiments was conducted in which neurons were first exposed to dideoxyforskolin and then to PGE2. As before, dideoxyforskolin had no effect onICAP; however, in the continued presence of dideoxyforskolin, treatment with 1 μm PGE2produced a significant and transient 2.0 ± 0.1-fold increase inICAP (n = 4). In all four neurons examined, the time to peak was at 6 min with at½MAX of 3.9 ± 0.2 min. Taken together, these experiments indicate that forskolin treatment, in a manner analogous to the sensitization evoked by PGE2, produced a transient increase in the magnitude of ICAP. This sensitization resulted from stimulation of adenylyl cyclase because of the lack of effect of dideoxyforskolin. Additionally, the duration of sensitization produced by forskolin was concentration-dependent, suggesting that increased concentrations of intracellular cAMP lead to greater durations of sensitization of ICAP.
Analogs of cAMP also sensitize the capsaicin-elicited current
To explore the notion that the sensitization produced by PGE2 and forskolin was mediated by the cAMP pathway, the capacity of a membrane-permeable analog of cAMP, 8-bromo-cAMP (8-Br-cAMP), to enhance ICAP was examined. The concentrations of 8-Br-cAMP used in these experiments facilitated the release of neuropeptides evoked by excitatory chemical agents in cultured rat sensory neurons (Hingtgen et al., 1995). In a representative neuron, the control response to capsaicin was 220 pA (Fig. 2A, top panel). After a 10 min exposure to 10 μm 8-Br-cAMP, ICAP was increased by twofold (434 pA). Unlike the results obtained with 100 nm forskolin, the duration of sensitization was prolonged in this neuron and remained elevated by 1.4-fold (314 pA) compared with the control responses, even after a 35 min exposure to 8-Br-cAMP. To determine whether the duration of the 8-Br-cAMP-induced sensitization was also concentration-dependent, 1 mm 8-Br-cAMP was used in an attempt to produce longer periods of sensitization. The results from a representative neuron are shown in the bottom panel of Figure2A. After a 6 min exposure to 1 mm8-Br-cAMP, the amplitude of ICAP was increased to 449 pA, a 2.3-fold increase from control response of 199 pA. Surprisingly, the duration of the sensitization produced by this concentration of 8-Br-cAMP was shortened, wherein the response had returned to control levels after only 20 min (response amplitude of 229 pA).
Fig. 2.
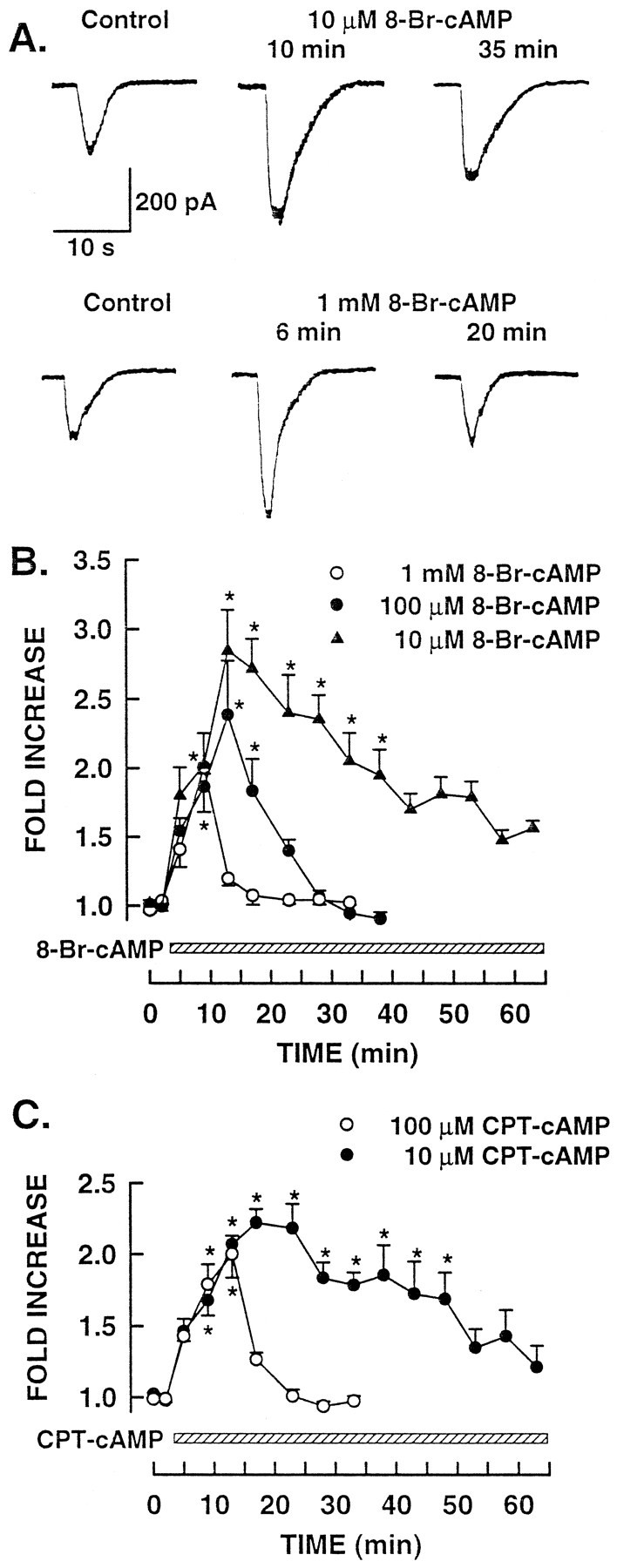
Treatment with membrane-permeant analogs of cAMP enhance the whole-cell responses to capsaicin. A, Sensitizing effect of 10 μm and 1 mm8-Br-cAMP treatment (top and bottom traces, respectively) on capsaicin responses in two different neurons. B, Extent and time course of the sensitization produced by these concentrations of 8-Br-cAMP. C, Sensitization induced by 10 and 100 μmchlorphenylthio-cAMP. In B and C, thebar beginning at 3 min represents the change to Ringer’s solution containing the indicated concentrations of cAMP analogs. Asterisks indicate significant differences atp < 0.05 compared with control.
The apparent inverse concentration effect (lower concentrations of 8-Br-cAMP producing a longer duration of sensitization) was obtained in all neurons exposed to 8-Br-cAMP. These experimental results are summarized in Figure 2B. Exposure to 10 μm 8-Br-cAMP produced a significant 2.8 ± 0.3-fold increase in the amplitude of ICAP with a time to peak of 11.6 ± 0.1 min and at½MAX of 22.3 ± 2.1 min (n = 5). In neurons exposed to 100 μm8-Br-cAMP, ICAP was increased significantly by 2.4 ± 0.4-fold above the control responses with a time to peak of 9.3 ± 1.5 min (n = 6). However, the duration of sensitization was decreased significantly wheret½MAX was reduced to 3.8 ± 0.4 min. Increasing the concentration of 8-Br-cAMP to 1 mmsignificantly enhanced ICAP by 2.0 ± 0.1-fold and had a time to peak of 6.0 ± 0.1 min (n = 3). This concentration of 8-Br-cAMP produced a further decrease in t½MAX to 2.3 ± 0.1 min. Although the exact mechanism(s) for the inverse-concentration effect obtained with 8-Br-cAMP remains unclear (but see below), treatment with 8-Br-cAMP sensitized the capsaicin response in a manner analogous to that produced by PGE2 and forskolin. These results then suggest that increased levels of intracellular cAMP likely mediate the sensitization produced by PGE2.
The shortened duration of sensitization obtained with higher concentrations of the cAMP analog 8-Br-cAMP suggests that this agent may have other cellular actions. Therefore, to explore this possibility, the capacity of another membrane-permeant cAMP analog, chlorphenylthio-cAMP (cpt-cAMP) to sensitize sensory neurons was determined. As presented in Figure 2C, the results obtained with cpt-cAMP were similar to those obtained with 8-Br-cAMP. Exposure to 10 μm cpt-cAMP produced a 2.2 ± 0.1-fold increase in the amplitude of ICAP with a time to peak of 16.0 ± 2.1 min (n = 4). The sensitization produced by 10 μm cpt-cAMP remained significantly elevated for a sustained period, as evidenced by thet½MAX of 26.7 ± 5.0 min. Similarly, ICAP was enhanced significantly by 2.0 ± 0.2-fold after treatment with 100 μmcpt-cAMP; the sensitization had a time to peak of 10.0 ± 0.1 min (n = 4). However, with this higher concentration of cpt-cAMP, t½MAX was reduced to 3.1 ± 0.3 min. This series of experiments suggests that, like forskolin, elevations in intracellular cAMP enhanced the sensitivity of the neuron to capsaicin. However, unlike forskolin, higher concentrations of the cAMP analogs somehow led to a decreased duration of sensitization.
The above studies raise the possibility that the more transient sensitization observed with higher concentrations of the cAMP analogs may reflect the activity of these agents at an effector that normally is not activated by physiological increases in the concentration of cAMP. Previous studies observed that cAMP analogs and high concentrations of cAMP were capable of activating cyclic GMP-dependent protein kinase (PKG); this pattern of stimulation was termed cross-activation (Jiang et al., 1992). In support of this notion, our previous results demonstrated that recovery of PGE2-induced sensitization of ICAP was mediated by activation of PKG (Lopshire and Nicol, 1997b). To investigate the possibility that these cAMP analogs reverse the sensitization through the cross-activation of PKG, we examined the action of 1 mm8-Br-cAMP on ICAP in neurons that were sensitized fully by consecutive treatments with 1 and 10 μm forskolin. These results are shown in Figure3. After exposure to 1 μmforskolin, ICAP was enhanced by 2.1-fold compared with the control responses and had a time to peak of 7.3 ± 1.1 min (n = 3). The enhanced response to capsaicin stabilized at this level. If the reversal or inactivation of sensitization was produced by an additional large increase in cAMP, then treatment of these neurons with 10 μm forskolin should cause the sensitization to recover to control levels. However, 10 μm forskolin produced no further change in the level of sensitization. After ∼21 min in 10 μm forskolin,ICAP was maintained at a twofold increase over the control levels, suggesting that maximal sensitization had been attained. At this time, exposure to 1 mm 8-Br-cAMP caused the enhanced ICAP to return rapidly to control levels with a t½MAX of 2.7 ± 0.3 min (relative to the time of the last capsaicin response before 8-Br-cAMP addition). These results suggest that higher concentrations of cAMP analogs may act at site(s) distinct from those usually stimulated by normal physiological changes in the levels of cAMP.
Fig. 3.

Treatment with 8-Br-cAMP reverses the forskolin-induced sensitization. The consecutive application of 1 and 10 μm forskolin is indicated by thehatched and solid bars, respectively. In the presence of 10 μm forskolin, the addition of 1 mm 8-Br-cAMP (open bar) rapidly reversed the forskolin-induced sensitization. The inset tracesillustrate the capsaicin responses from a representative neuron at the indicated times. Asterisks indicate significant differences at p < 0.05 compared with control.
Inhibition of protein kinase A prevents PGE2- and forskolin-induced sensitization
Both forskolin and cAMP analogs gave rise to a sensitization ofICAP that was similar to that produced by PGE2. To establish whether this increased sensitivity to capsaicin was attributable to a direct action of cAMP or was dependent on cAMP activation of PKA, a series of experiments was conducted in which sensory neurons were perfused intracellularly with the peptide inhibitor of PKA, PKI14–24 (Cheng et al., 1986). In the presence of 20 μm PKI14–24, the peak amplitudes of the control responses to 100 nm capsaicin were not different significantly from control responses obtained in earlier experiments (PKI + PGE2 series: 325 ± 111 pA;n = 8; PKI + forskolin series: 284 ± 44 pA;n = 4; and no PKI series: 255 ± 18 pA;n = 50). As shown in Figure4, intracellular perfusion of PKI14–24 completely blocked the capacity of either 1 μm PGE2 or 1 μm forskolin (Fig.4A,B, respectively) to enhanceICAP. The insets illustrate representative responses obtained at the indicated times during both experiments. These findings demonstrate that activation of PKA was necessary and sufficient for the sensitization of ICAPproduced by either PGE2 or forskolin.
Fig. 4.
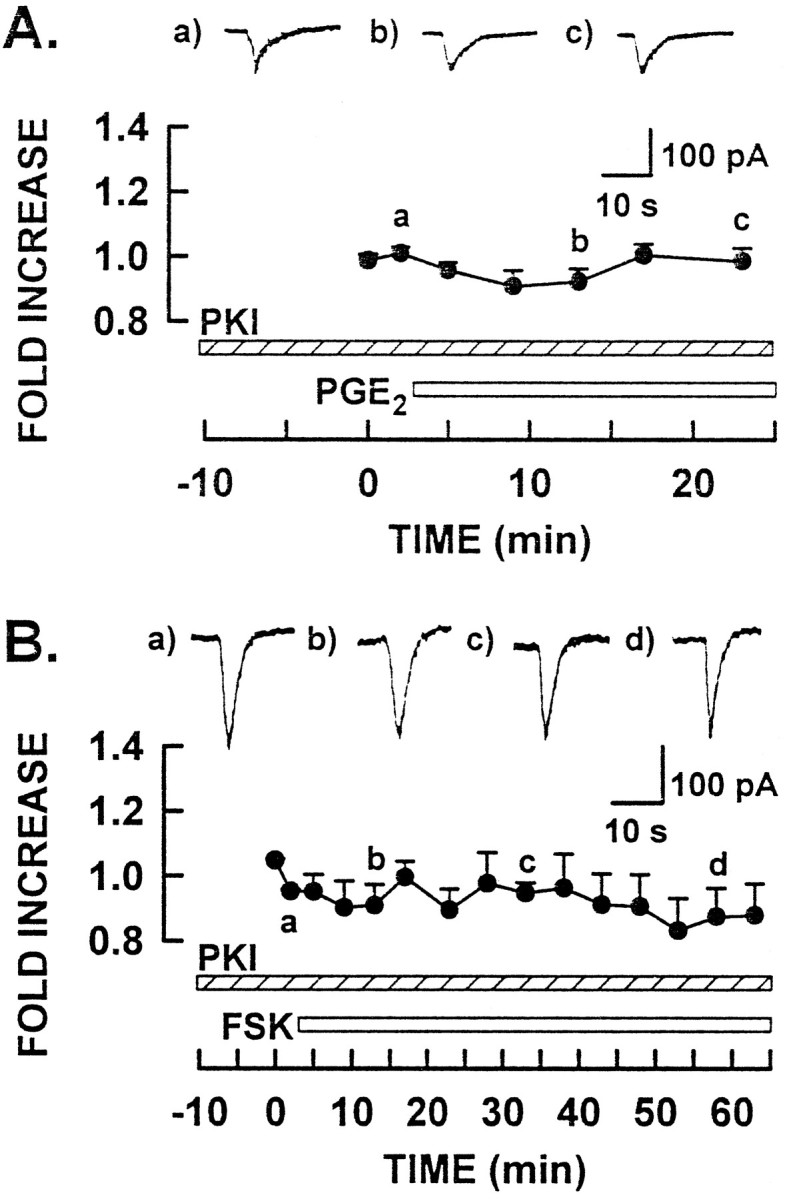
PKI14–24 treatment blocks both the PGE2- and forskolin-induced sensitization.A, Results from experiments with PKI14–24and PGE2 treatment. The hatched andopen bars indicate the addition of PKI14–24and PGE2, respectively. B, Results from experiments with PKI14–24 and forskolin treatments. The hatched and open bars indicate the presence of PKI14–24 and forskolin, respectively. Theinset traces illustrate representative responses to capsaicin from two different neurons for each experimental condition at the times indicated. Asterisks indicate significant differences atp < 0.05 compared with control.
Capsaicin elicits single-channel activity in cell-attached membrane patches that is antagonized by capsazepine
The cAMP-mediated sensitization of ICAPlikely is manifested by a change in one or more of the single-channel properties of the capsaicin-gated channel. Oh et al. (1996a,b) reported recently that, in cell-attached recordings, extracellularly applied capsaicin elicited single-channel currents in the membrane patch. Thus, these investigators proposed that capsaicin crosses the plasma membrane and binds to the channel at the intracellular surface of the membrane. Based on these observations, the cell-attached configuration was used to explore the modulation of the capsaicin-gated ion channel by PGE2 and the cAMP pathway. An additional advantage of this approach is that normal intracellular signaling pathways of the neuron remain intact.
Initially, the properties of the capsaicin-gated channel in these cultured sensory neurons were characterized. In a representative recording in which the membrane potential was maintained at −60 mV (Fig.5A,B), changing the superfusate to low-calcium external solution containing 1 μm capsaicin evoked the appearance of inward currents, which was indicative of the activation of capsaicin-gated ion channels. Figure 5B illustrates selected portions of the compressed traces shown in Figure 5A on an expanded time scale. Each trace is taken from that particular segment of the recording indicated by its respective number. The unitary nature of the multiple channel openings is evident in traces 2 and 4. After washout of capsaicin, the inward currents were no longer present. Inclusion of 10 μm capsazepine, the competitive antagonist of capsaicin (Bevan et al., 1992), with the 1 μm capsaicin low-calcium external solution, produced a nearly complete suppression of the currents elicited by capsaicin. With the removal of capsazepine, the introduction of capsaicin again elicited the channel currents. The decreased channel activity observed during this second capsaicin application may reflect the presence of residual capsazepine or desensitization of the capsaicin response. Desensitization seems less likely, because this process should be diminished at the reduced calcium concentrations (∼150 nm) used in these experiments. This notion was based on previous observations in which desensitization of the capsaicin response was attenuated greatly as the external calcium was lowered (Cholewinski et al., 1993; Liu and Simon, 1996; Koplas et al., 1997).
Fig. 5.

Capsaicin elicits single-channel activity that is blocked by capsazepine. A, Representative recording from a cell-attached patch held at −60 mV. Changing the superfusate from the low-calcium external solution (trace 1) to one containing 1 μm capsaicin (CAP) promoted single-channel activity that was blocked reversibly by 10 μm capsazepine (CAP + CZP).B, Selected portions of the compressed traces shown inA at greater time resolution. The numbered traces in B represent the channel activity observed for those specific portions indicated by their respective numbers under the traces of A. The arrowsindicate the closed level for each trace. C, All-points histogram generated from a 40 sec recording during the first application of capsaicin to this neuron. D, Effect of capsaicin and capsaicin-capsazepine treatment on the mean NPo in three cell-attached patches.Asterisks indicate significant differences atp < 0.05 compared with control.
An all-points amplitude histogram was constructed from the initial capsaicin exposure in this neuron and is shown in Figure 5C. The peaks in this histogram were fitted with a Gaussian distribution in which the values corresponded to one closed level and four open levels. This suggested that there were at least four active capsaicin channels in this patch. The amplitude of the unitary current obtained for the open levels was −2.1 ± 0.1 pA and yielded a single-channel conductance of 35.1 ± 1.5 pS. The mean values for NPoobtained under these different experimental conditions in three different neurons are summarized in Figure 5D. These results demonstrate that the addition of 1 μm capsaicin produced a large increase in NPo that was blocked by the addition of capsazepine. Taken together, our observations support the previous results of Oh et al. (1996,a,b) that capsaicin is capable of diffusing through the plasma membrane to elicit single-channel activity in these cell-attached patches.
Capsaicin-elicited channel activity is concentration-dependent
To ascertain the appropriate concentration of capsaicin with which to probe the activity of single channels in cell-attached patches, we determined the concentration–response relationship for capsaicin applied extracellularly. The patch was held at −60 mV and exposed to increasing bath concentrations of capsaicin that ranged from 30 nm to 10 μm. Between capsaicin applications the cell was washed extensively with low-calcium external solution. NPo at each concentration of capsaicin was determined, and the results from nine neurons were pooled to obtain the mean NPo for a given concentration (Fig.6). The results were fitted by a ligand-binding isotherm (described in Materials and Methods) that yielded an EC50 of 1.4 μm and a steepness factor of 2.5.
Fig. 6.

NPo depends on the concentration of capsaicin. These results summarize the effect of increasing concentrations of bath-applied capsaicin on mean NPo in eight cell-attached patches. The line through the data points represents the fit obtained for the ligand-binding isotherm as described in Materials and Methods.
The capsaicin-gated channel exhibits outward rectification and increased activity at positive potentials
Figure 7A illustrates representative single-channel currents elicited by 1 μmcapsaicin at several membrane voltages. At −60 mV, the currents were inward; the currents remained inward at −30 and −10 mV but were reduced in amplitude. At positive membrane potentials, the currents became outward and increased in amplitude with depolarizing potentials. NPo increased from 0.11 to 1.27 as the potential changed from −60 to +60 mV values, respectively. The current–voltage relation obtained for the single-channel currents is summarized in Figure7B. The current–voltage relationship appeared linear between −60 and +20 mV with an average conductance of 33 ± 1 pS at −60 mV. At more positive potentials, the current began to rectify with the single-channel conductance increasing to 46 ± 3 pS at +60 mV. The apparent reversal potential (uncorrected for membrane potential) obtained from this relation was +4 ± 1 mV, which was similar to values reported previously for capsaicin-elicited currents (Oh et al., 1996a; Caterina et al., 1997).
In cell-attached patches from embryonic sensory neurons, NPo of the capsaicin-gated channel increased as the membrane potential was depolarized. Because NPo was maximal at +60 mV, values for NPo obtained at other membrane voltages were normalized to this maximum for each neuron. Figure6C shows that the relative NPo increased from 0.04 ± 0.02 to 1 (n = 3) as the membrane potential was changed from −60 to +60 mV. The dependence of the relative NPo on membrane potential was described by the Boltzmann relation where V0.5 was −9 and k was 20 mV. Previous results demonstrated similar findings for the capsaicin-gated channel in sensory neurons of the neonatal rat (Oh et al., 1996a).
PGE2 and forskolin treatment transiently increase capsaicin-gated channel activity
To investigate the capacity of PGE2 and forskolin to sensitize the capsaicin-gated channel, we used a concentration of 300 nm capsaicin to evoke recordings of only single-level openings of the channel. This choice was based on two observations. First, 300 nm was well below the EC50 for the concentration–response relation (Fig. 6). Second, in preliminary studies, recordings obtained with 100 nm capsaicin failed to exhibit reliable levels of activity under control conditions (data not shown). The results obtained from a cell-attached patch exposed to 300 nm capsaicin are shown in Figure8A. However, in this trace, two overlapping open events were detected, indicating that at least two active channels were present. The mean unitary current was −1.9 pA, which corresponds to a single-channel conductance of 31 pS; NPo in this trace was 0.13. After exposure to 1 μm PGE2 for 12 min, there were clearly three overlapping open levels observed, and NPo was increased to 0.53. There was no change in the unitary current after PGE2treatment; it remained at −2.0 pA (Fig. 8B). In this neuron, the PGE2-induced increase in NPo was transient when, after a 20 min exposure to PGE2, NPo was reduced to 0.03. The unitary current remained unchanged at −2.0 pA (Fig. 8C). Interestingly, the application of 10 μm capsaicin (Fig.8D) after a 25 min exposure to PGE2produced a large increase in NPo to 0.85 with three detectable open levels. The unitary current was still unchanged at −2.0 pA.
Fig. 8.

PGE2 treatment enhances the channel activity elicited by capsaicin. A, Representative recording of the single-channel activity elicited by 300 nmcapsaicin under control conditions. In B, the single-channel activity elicited by capsaicin is increased after treatment with 1 μm PGE2 in the same neuron. The inset trace shows, at greater time resolution, the capsaicin response during the period indicated by thebar. The transient nature of the PGE2-induced increase in channel activity is shown inC. The trace illustrates the recording obtained after 20 min of continuous exposure to PGE2.D, Effect of 10 μm capsaicin treatment in this same neuron after a 25 min exposure to PGE2. Theinset illustrates the response to capsaicin obtained during the time indicated by the bar. In all traces, thearrows indicate the closed level. The all-points histogram generated for each trace is provided at theright.
As summarized in Figure 9, PGE2 significantly enhanced NPo in all seven neurons examined. After a 12 min exposure, PGE2 increased NPo to 0.79 ± 0.1 from the control level of 0.07 ± 0.03. Furthermore, this increase was transient wherein NPo returned to control levels after only 20 min in the continuous exposure to PGE2. The unitary conductance of the capsaicin-gated channel remained unaltered throughout the exposure to PGE2, having values of 33 ± 0.7, 33 ± 0.4, and 33 ± 0.6 pS for the control condition and for 12 and 20 min exposures to PGE2, respectively. In all of these recordings, multiple channel openings were noted after PGE2exposure, thereby complicating a kinetic analysis of channel activity. Similarly, treatment with 1 μm forskolin resulted in a significant and transient increase in NPo evoked by 300 nm capsaicin (Fig. 9), whereas the unitary conductance was unaltered (33 ± 0.8 and 34 ± 0.4 pS for the control and after a 12 min exposure to forskolin, respectively). In the absence of PGE2 or forskolin, neither the unitary conductance nor NPo elicited by 300 nm capsaicin was changed over the 23 min time course (Fig. 9). Thus, these findings suggest that the PGE2- or forskolin-induced enhancement of NPo without a concomitant change in unitary conductance results from an increase in the number of active channels and/or an increased open probability of the channel.
Fig. 9.

PGE2 or forskolin treatment transiently increases NPo of the capsaicin-gated channel. The hatched bar indicates the time and duration for the application of each agent. Asterisks indicate significant differences at p < 0.05 compared with control.
DISCUSSION
Capsaicin-elicited currents in sensory neurons
In this report we demonstrated that the application of the vanilloid capsaicin to embryonic sensory neurons elicits whole-cell currents that are similar to those observed in previous studies (Marsh et al., 1987; Bevan and Szolcsanyi, 1990; Koplas et al., 1997; Lopshire and Nicol, 1997b). Likewise, the properties of the capsaicin-gated ion channel described in embryonic sensory neurons are comparable to characterizations of this channel in sensory neurons of the neonatal rat (Oh et al., 1996a) as well as the cloned vanilloid receptor expressed in mammalian HEK293 cells (Caterina et al., 1997). For example, the EC50 (1.4 μm) and the steepness factor n (2.5) determined for the single-channel recordings in Figure 6are similar to those values (1.1 μm and 1.8) reported byOh et al. (1996a) for neonatal rat sensory neurons. Our observation that n > 2 indicates that two or more molecules of capsaicin bind to the channel to promote opening. In addition, our estimation of the single-channel conductance at −60 mV is similar to previously described values that ranged from 30 to 45 pS (Bevan and Szolcsanyi, 1990; Oh et al., 1996a; Caterina et al., 1997). The conductance exhibits rectification wherein the value increased by nearly twofold (ranging from 70 to 80 pS) at +60 mV (Bevan and Szolcsanyi, 1990; Oh et al., 1996a; Caterina et al., 1997). However, the increase in conductance at +60 mV determined in our studies was smaller than that described in these previous studies. This may result from the higher concentration of extracellular calcium used in our study (∼150 nm) compared with other studies in which no added calcium was used (Oh et al., 1996a; Caterina et al., 1997). Indeed, both Forbes and Bevan (1988) and Oh et al. (1996a) reported that the conductance of the capsaicin-gated channel can be reduced by increased levels of extracellular calcium.
The relative NPo increased with depolarizing potentials (Fig. 7C); these results were described by the Boltzmann relationship in which the values of V0.5 and kwere −9 and 20 mV, respectively. These values corroborate a previous report by Oh et al. (1996a) in which V0.5 was −9 andk was 29 mV. Thus, the channel properties determined in embryonic rat sensory neurons are similar to those published by other investigators using different preparations. This then suggests that the electrophysiological properties of the capsaicin receptor are not regulated or modified differentially during development.
Sensitization of the response to capsaicin
Treatment with the proinflammatory prostaglandin PGE2enhanced by twofold to threefold the amplitude of whole-cell currents elicited by capsaicin (Lopshire and Nicol, 1997b). This sensitization was transient wherein the capsaicin response returned to control levels after ∼20–25 min in the continued presence of PGE2 (Fig.1A,D). This reversal of sensitization does not involve desensitization of the capsaicin response because the recovery was blocked by inhibitors of nitric oxide synthase (Lopshire and Nicol, 1997b), whereas desensitization was insensitive to inhibitors of nitric oxide synthase but was suppressed by calcineurin inhibitors (Docherty et al., 1996). In addition, this eicosanoid significantly increased the value of NPo for the capsaicin-gated channel; however, the conductance was unaltered. Thus, the enhanced whole-cell currents appear to be a direct result of the PGE2-induced modification of the capsaicin receptor that leads to increased levels of activity without modulation of the conductance.
Previously, we demonstrated that the duration of PGE2-induced sensitization of the capsaicin response depended on the concentration of extracellular calcium wherein lowered levels lengthened the period of sensitization (Lopshire and Nicol, 1997b). We observed that the duration of the transient sensitization (Fig. 9) of single-channel activity (performed at ∼150 nmextracellular calcium) was similar to the transient sensitization of whole-cell currents (performed at 2 mm external calcium). This correspondence may result from the different procedures used to deliver capsaicin. In the whole-cell studies, capsaicin was applied focally in puffs of 1–2 sec, whereas in the single-channel studies, capsaicin was administered continuously during the recording. Despite the lowered calcium concentration, this persistent depolarization occurring with the continuous delivery (Fig. 7B,inset) might permit a sufficient calcium influx to activate pathways that reverse sensitization.
Signaling pathways that mediate sensitization
Previous studies demonstrated that activation of the cAMP transduction pathway played a necessary and sufficient role in the PGE2-induced sensitization of the bradykinin response in sensory neurons (Cui and Nicol, 1995; Hingtgen et al., 1995). In our studies described above, forskolin, an activator of adenylyl cyclase, or membrane-permeant analogs of cAMP enhanced the amplitude of the whole-cell current elicited by capsaicin in a manner comparable to PGE2. In support of our findings, Pitchford and Levine (1991) reported that exposure to PGE2 or analogs of cAMP increased the amplitude of the capsaicin-evoked current in adult rat sensory neurons. However, given the high concentrations of PGE2 (1 mm) or 8-Br-cAMP (20 mm) used in that study, it is difficult to assess directly the physiological significance of such findings. We also observed that treatment with either PGE2 or forskolin increased NPo of the channel in response to activation by capsaicin. After treatment with either PGE2 or forskolin, the amplitude histograms revealed openings to additional levels (Fig.8B) that were not observed in the control recording (Fig. 8A). These results raise the possibility that PGE2 or forskolin increased the sensitivity of the channel to activation by capsaicin. In support of this notion is the observation that after treatment with PGE2, the histogram obtained for 300 nm capsaicin (well below the EC50) was very similar to that obtained with a maximal concentration of 10 μm capsaicin (Fig. 8, compareB,D).
Sensitization of the capsaicin response results from activation of adenylyl cyclase and the consequent activation of PKA. This idea is corroborated by two additional observations. First, dideoxyforskolin, which retains all of the properties of forskolin except the capacity to activate adenylyl cyclase, did not sensitize capsaicin-elicited currents, and it failed to alter the PGE2-induced sensitization of capsaicin responses (Fig. 1D). Second, intracellular perfusion of the neuron with the inhibitor of PKA, PKI14–24, blocked both the PGE2- and forskolin-induced sensitization, suggesting that a cAMP-dependent phosphorylation played a key role in the sensitization of the capsaicin response. The notion that the activity of the capsaicin-gated ion channel can be modulated by a phosphorylation reaction mediated by PKA is supported by the amino acid sequence determined for the cloned vanilloid receptor. The receptor cDNA predicted three putative sites for phosphorylation by PKA on the cytoplasmic side of the protein (Caterina et al., 1997). The role of PKA in modulating the sensitivity of sensory neurons to noxious stimulation was defined further in studies examining mice with a targeted mutation of the type I regulatory subunit (RIβ) of PKA. The heightened thermal sensitivity after injection of PGE2 into the hindpaw of the targeted mice was reduced in comparison to the wild-type mice (Malmberg et al., 1997). Taken together, these findings suggest that elevated levels of intracellular cAMP lead to the activation of PKA. It is this activated PKA that subsequently modifies the capsaicin receptor–ion channel complex resulting in an altered sensitivity to capsaicin and/or increasing the open probability of the channel.
It is interesting that increasing concentrations of forskolin lengthened the duration of sensitization, whereas higher concentrations of either 8-Br-cAMP or cpt-cAMP decreased the duration (compare Figs.1C, 2B,C). In a previous study (Lopshire and Nicol, 1997b), we demonstrated that the PGE2-induced sensitization of the capsaicin-elicited current was inactivated or reversed by stimulation of PKG. Based on these findings, we speculate that the rapid inactivation of sensitization observed with higher concentrations of 8-Br-cAMP or cpt-cAMP results from the cross-activation of PKG. As suggested by Figures 1-3, we assume that the onset and amplitude of sensitization are correlated directly with the intracellular levels of cAMP. Both 1 and 10 μm forskolin produced maximal levels of sensitization (Fig. 1C), and, in fact, there was no further change in the level of sensitization when treatment with 1 μm forskolin was followed subsequently by 10 μm forskolin (Fig. 3). This implies that the physiological effects of the elevated cAMP were maximized and that any additional increases in cAMP did not produce greater levels of sensitization. Also shown in Figure 3, addition of 1 mm8-Br-cAMP rapidly reversed the sensitization, suggesting that this recovery was mediated by a pathway independent of the activation of PKA by cAMP. The rapid reversal of sensitization was similar to that previously observed after treatment with either nitric oxide donors or 8-Br-cGMP (Lopshire and Nicol, 1997b). In that study, inhibition of PKG prevented the cGMP-induced inactivation of sensitization. Taken together, these findings suggest that the reversal of sensitization by high concentrations of cAMP analogs may result from their cross-activation of PKG.
Biochemical studies examining the activity of purified PKG isolated from bovine lung (PKG type Iα) demonstrated that cAMP was capable of activating PKG, although 50–100 times greater concentrations of cAMP were required to produce activation levels equivalent to cGMP (Lincoln et al., 1977). In a later study, Francis et al. (1988) found that 8-Br-cGMP activated purified PKG with a Ka of 0.025 μm and had an EC50 of 47 μm for the relaxation of coronary artery. However, both cpt-cAMP and 8-Br-cAMP activated PKG and produced relaxation with cpt-cAMP being more effective (Ka, 0.2 μm; EC50, 30 μm) than 8-Br-cAMP (Ka, 5.8 μm; EC50, 1670 μm). Activation of PKG by cAMP itself was much less effective compared with cpt-cAMP in which theKa was 16 μm (Wolfe et al., 1989).
These values for the effectiveness of cAMP, cpt-cAMP, and 8-Br-cAMP to activate PKG could account for our observations regarding the recovery times shown in Figures 1C and 2, B andC. Increasing forskolin concentrations, which elevate intracellular cAMP (a poor activator of PKG), produced a sensitization of longer durations. Increasing concentrations of cAMP analogs, which are better activators of PKG, cause the duration of sensitization to be shortened. Furthermore, 100 μm cpt-cAMP inactivated sensitization on a time course that was similar to 1 mm8-Br-cAMP; this may result from the greater ability of cpt-cAMP relative to 8-Br-cAMP to activate PKG.
In conclusion, PGE2 sensitizes the capsaicin response through activation of the cAMP transduction cascade. Increased levels of intracellular cAMP lead to the activation of PKA. This kinase enhances the activity of the capsaicin-gated ion channel by increasing the sensitivity of the receptor to capsaicin and/or the open probability. A direct consequence of the facilitation is the increased whole-cell current that may give rise to a larger and/or longer depolarization, thereby enhancing the excitability of the sensory neuron. This cAMP-mediated sensitization ultimately augments the capacity of the sensory neuron to signal the reception of nociceptive stimulation to the spinal cord.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS-30527 to G.D.N. J.C.L. was supported by grants from the Indiana University–Purdue University, Indianapolis Research Investment Fund and the American Heart Association, Indiana Affiliate. We are grateful to Dr. Jim Kenyon for his advice regarding the analysis of single-channel activity.
Correspondence should be addressed to Dr. Grant Nicol, Department of Pharmacology and Toxicology, Indiana University School of Medicine, Indianapolis, IN 46202.
REFERENCES
- 1.Baccaglini PI, Hogan PG. Some rat sensory neurons in culture express characteristics of differentiated pain sensory cells. Proc Natl Acad Sci USA. 1983;80:594–598. doi: 10.1073/pnas.80.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bevan S, Forbes CA. Membrane effects of capsaicin on rat dorsal root ganglion neurones in cell culture. J Physiol (Lond) 1988;398:28P. [Google Scholar]
- 3.Bevan S, Szolcsanyi J. Sensory neuron-specific actions of capsaicin: mechanisms and applications. Trends Pharmacol Sci. 1990;11:330–333. doi: 10.1016/0165-6147(90)90237-3. [DOI] [PubMed] [Google Scholar]
- 4.Bevan S, Hothi S, Hughes G, James IF, Rang HP, Shah K, Walpole CSJ, Yeats JC. Capsazepine: a competitive antagonist of the sensory neurone excitant capsaicin. Br J Pharmacol. 1992;107:544–552. doi: 10.1111/j.1476-5381.1992.tb12781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caldwell PC. Calcium chelation and buffers. In: Cuthbert AW, editor. Calcium and cell function. St. Martin’s; New York: 1970. pp. 11–16. [Google Scholar]
- 6.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 7.Cheng H-C, Kemp BE, Pearson RB, Smith AJ, Misconi L, Van Patten SM, Walsh DA. A potent synthetic peptide inhibitor of the cAMP-dependent protein kinase. J Biol Chem. 1986;261:989–992. [PubMed] [Google Scholar]
- 8.Cholewinski A, Burgess GM, Bevan S. The role of calcium in capsaicin-induced desensitization in rat cultured dorsal root ganglion neurons. Neuroscience. 1993;55:1015–1023. doi: 10.1016/0306-4522(93)90315-7. [DOI] [PubMed] [Google Scholar]
- 9.Cui M, Nicol GD. Cyclic AMP mediates the prostaglandin E2-induced potentiation of bradykinin excitation in rat sensory neurons. Neuroscience. 1995;66:459–466. doi: 10.1016/0306-4522(94)00567-o. [DOI] [PubMed] [Google Scholar]
- 10.Docherty RJ, Yeats JC, Bevan S, Boddeke HWGM. Inhibition of calcineurin inhibits the desensitization of capsaicin-evoked currents in cultured dorsal root ganglion neurones from adult rats. Pflügers Arch. 1996;431:828–837. doi: 10.1007/s004240050074. [DOI] [PubMed] [Google Scholar]
- 11.England S, Bevan S, Docherty RJ. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J Physiol (Lond) 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forbes CA, Bevan S. Properties of single capsaicin-activated channels. Soc Neurosci Abstr. 1988;14:642. [Google Scholar]
- 13.Foreman JC. Peptides and neurogenic inflammation. Br Med Bull. 1987;43:386–400. doi: 10.1093/oxfordjournals.bmb.a072189. [DOI] [PubMed] [Google Scholar]
- 14.Francis SH, Noblett BD, Todd BW, Wells JN, Corbin JD. Relaxation of vascular and tracheal smooth muscle by cyclic nucleotide analogs that preferentially activate purified cGMP-dependent protein kinase. Mol Pharmacol. 1988;34:506–517. [PubMed] [Google Scholar]
- 15.Gold MS, Reichling DB, Schuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci USA. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 17.Handwerker HO, Kobal G. Psychophysiology of experimentally induced pain. Physiol Rev. 1993;73:639–671. doi: 10.1152/physrev.1993.73.3.639. [DOI] [PubMed] [Google Scholar]
- 18.Heyman I, Rang HP. Depolarizing responses to capsaicin in a subpopulation of rat dorsal root ganglion cells. Neurosci Lett. 1985;56:69–75. doi: 10.1016/0304-3940(85)90442-2. [DOI] [PubMed] [Google Scholar]
- 19.Higgs GA, Moncada S, Vane JR. Eicosanoids in inflammation. Ann Clin Res. 1984;16:287–299. [PubMed] [Google Scholar]
- 20.Hingtgen CM, Waite KJ, Vasko MR. Prostaglandins facilitate peptide release from rat sensory neurons by activating the adenosine 3′,5′-cyclic monophosphate transduction cascade. J Neurosci. 1995;15:5411–5419. doi: 10.1523/JNEUROSCI.15-07-05411.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holzer P. Capsaicin: cellular targets, mechanisms of action, and selectivity for thin sensory neurons. Pharmacol Rev. 1991;43:143–201. [PubMed] [Google Scholar]
- 22.Jiang H, Colbran JL, Francis SH, Corbin JD. Direct evidence for cross-activation of cGMP-dependent protein kinase by cAMP in pig coronary arteries. J Biol Chem. 1992;267:1015–1019. [PubMed] [Google Scholar]
- 23.Koplas PA, Rosenberg RL, Oxford GS. The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J Neurosci. 1997;17:3525–3537. doi: 10.1523/JNEUROSCI.17-10-03525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kress M, Reeh PW. Chemical excitation and sensitization in nociceptors. In: Belmonte C, Cervero F, editors. Neurobiology of nociceptors. Oxford UP; Oxford: 1996. pp. 258–297. [Google Scholar]
- 25.LaMotte RH, Shain CN, Simone DA, Tsai EF. Neurogenic hyperalgesia: psychophysical studies of underlying mechanisms. J Neurophysiol. 1991;66:190–211. doi: 10.1152/jn.1991.66.1.190. [DOI] [PubMed] [Google Scholar]
- 26.LaMotte RH, Lunfberg LE, Torebjork HE. Pain, hyperalgesia and activity in nociceptive C units in humans after intradermal injection of capsaicin. J Physiol (Lond) 1992;448:749–764. doi: 10.1113/jphysiol.1992.sp019068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lincoln TM, Dills WL, Corbin JD. Purification and subunit composition of guanosine 3′:5′-monophosphate-dependent protein kinase from bovine lung. J Biol Chem. 1977;252:4269–4275. [PubMed] [Google Scholar]
- 28.Liu L, Simon SA. Capsaicin-induced currents with distinct desensitization and Ca2+ dependence in rat trigeminal ganglion cells. J Neurophysiol. 1996;75:1503–1514. doi: 10.1152/jn.1996.75.4.1503. [DOI] [PubMed] [Google Scholar]
- 29.Lopshire JC, Nicol GD. The cAMP transduction cascade enhances capsaicin-elicited currents in rat sensory neurons. Soc Neurosci Abstr. 1997a;23:374. doi: 10.1523/JNEUROSCI.18-16-06081.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopshire JC, Nicol GD. Activation and recovery of the PGE2-mediated sensitization of the capsaicin response in rat sensory neurons. J Neurophysiol. 1997b;78:3154–3164. doi: 10.1152/jn.1997.78.6.3154. [DOI] [PubMed] [Google Scholar]
- 31.Malmberg AB, Brandon EP, Idzerda RL, Liu H, McKnight GS, Basbaum AI. Diminished inflammation and nociceptive pain with preservation of neuropathic pain in mice with a targeted mutation of the type I regulatory subunit of cAMP-dependent protein kinase. J Neurosci. 1997;17:7462–7470. doi: 10.1523/JNEUROSCI.17-19-07462.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marsh SJ, Stansfeld CE, Brown DA, Davey R, McCarthy D. The mechanism of action of capsaicin on sensory C-type neurons and their axons in vitro. Neuroscience. 1987;23:275–289. doi: 10.1016/0306-4522(87)90289-2. [DOI] [PubMed] [Google Scholar]
- 33.Nicol GD, Cui M. Prostaglandin E2 enhances bradykinin activation of embryonic rat sensory neurones. J Physiol (Lond) 1994;480:485–492. doi: 10.1113/jphysiol.1994.sp020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicol GD, Vasko MR, Evans AR. Prostaglandins suppress an outward potassium current in embryonic rat sensory neurons. J Neurophysiol. 1997;77:167–176. doi: 10.1152/jn.1997.77.1.167. [DOI] [PubMed] [Google Scholar]
- 35.Oh U, Hwang SW, Kim D. Capsaicin activates a nonselective cation channel in cultured neonatal rat dorsal root ganglion neurons. J Neurosci. 1996a;16:1659–1667. doi: 10.1523/JNEUROSCI.16-05-01659.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh U, Hwang SW, Kwak JY, Yang J. Capsaicin (CAP) binds to the intracellular surface of capsaicin-activated ion channel. Soc Neurosci Abstr. 1996b;22:876. doi: 10.1523/JNEUROSCI.19-02-00529.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pitchford S, Levine JD. Prostaglandins sensitize nociceptors in cell culture. Neurosci Lett. 1991;132:105–108. doi: 10.1016/0304-3940(91)90444-x. [DOI] [PubMed] [Google Scholar]
- 38.Salmon JA, Higgs GA. Prostaglandins and leukotrienes as inflammatory mediators. Br Med Bull. 1987;43:285–296. doi: 10.1093/oxfordjournals.bmb.a072183. [DOI] [PubMed] [Google Scholar]
- 39.Seamon KB, Padgett W, Daly JW. Forskolin: unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci USA. 1981;78:3363–3367. doi: 10.1073/pnas.78.6.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seamon KB, Daly JW, Metzger H, de Souza NJ, Reden J (1983) Structure-activity relationships for activation of adenylate cyclase by the diterpene forskolin and its derivatives. J Med Chem 436–439. [DOI] [PubMed]
- 41.Szallasi A, Blumberg PM. Vanilloid receptors: new insights enhance potential as a therapeutic target. Pain. 1996;68:195–208. doi: 10.1016/s0304-3959(96)03202-2. [DOI] [PubMed] [Google Scholar]
- 42.Vasko MR, Campbell WB, Waite KJ. Prostaglandin E2 enhances bradykinin-stimulated release of neuropeptides from rat sensory neurons in culture. J Neurosci. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wolfe L, Corbin JD, Francis SH. Characterization of a novel isozyme of cGMP-dependent protein kinase from bovine aorta. J Biol Chem. 1989;264:7734–7741. [PubMed] [Google Scholar]
- 44.Wood JN, Winter J, James IF, Rang HP, Yeats J, Bevan S. Capsaicin-induced ion fluxes in dorsal root ganglion cells in culture. J Neurosci. 1988;8:3208. doi: 10.1523/JNEUROSCI.08-09-03208.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]