

Abstract
The extracellular acidity that accompanies brain hypoxia–ischemia is known to reduce both NMDA and AMPA–kainate receptor-mediated currents and NMDA receptor-mediated neurotoxicity. Although a protective effect of acidic pH on AMPA–kainate receptor-mediated excitotoxicity has been assumed, such has not been demonstrated. Paradoxically, we found that lowering extracellular pH selectively increased AMPA–kainate receptor-mediated neurotoxicity in neocortical cell cultures, despite reducing peak elevations in intracellular free Ca2+. This injury potentiation may, at least in part, be related to a slowed recovery of intracellular Ca2+ homeostasis, observed after AMPA–kainate receptor activation, but not after NMDA receptor activation or exposure to high K+. The ability of acidic pH to selectively augment AMPA–kainate receptor-mediated excitotoxicity may contribute to the prominent role that these receptors play in selective neuronal death after transient global ischemia.
Keywords: AMPA, murine neuronal culture, pH, excitotoxicity, cyclothiazide, acidosis
Excitotoxic glutamate receptor overactivation likely contributes to the neuronal death induced by brain hypoxia–ischemia (Meldrum, 1985; Rothman and Olney, 1987; Choi, 1988). NMDA-type glutamate receptors appear to play a prominent role in mediating this neuronal death (Simon et al., 1984; Albers et al., 1992), likely reflecting their special ability to mediate rapid Ca2+ influx leading to cellular Ca2+ overload (MacDermott et al., 1986; Choi, 1992). However, pharmacological blockade of AMPA–kainate-type glutamate receptors also produces substantial protective effects in the ischemic brain. Neuroprotective efficacy has been demonstrated in both global ischemia (Sheardown et al., 1990, 1993; Balchen and Diemer, 1992;Le-Peillet et al., 1992; Pellegrini-Giampietro et al., 1992; for review, see Gill, 1994) and focal ischemia (Buchan et al., 1991a; Smith and Meldrum, 1992) models. The most striking observations are the ability of AMPA antagonists to reduce neuronal loss in the CA1 hippocampal subfield, cortex, striatum, and cerebellum after global ischemia, a setting where NMDA antagonists are relatively ineffective (Sheardown et al., 1990; Buchan et al., 1991b). However, the possibility has been raised that the dramatic protection provided by delayed treatment with AMPA–kainate receptor antagonists observed in early studies may have been attributable in part to drug-induced brain hypothermia (Nurse and Corbett, 1996).
The prominent role of AMPA–kainate receptor-mediated injury in animal models of ischemia contrasts with the low profile role of AMPA–kainate receptor-mediated injury found in several in vitro models of excitotoxicity (Choi, 1992). In mouse cortical cell cultures, AMPA–kainate antagonists do not increase neuronal survival after either glutamate exposure (Koh and Choi, 1991) or oxygen–glucose deprivation (Goldberg and Choi, 1993). The reason for this appears to be masking of the relatively slowly triggered AMPA–kainate receptor-mediated injury by more fulminant, rapidly triggered NMDA receptor-mediated injury (Choi, 1992). Although exposure to AMPA or kainate will kill most cultured cortical neurons, much longer exposure (hours) is needed to induce widespread lethal injury by this route compared with the 3–5 min required to induce lethal injury by NMDA receptor overactivation. If NMDA receptors are blocked and the duration of oxygen–glucose deprivation extended to overcome associated neuroprotection, then AMPA–kainate antagonists produce substantial additional neuroprotective effects (Kaku et al., 1991).
How to reconcile these in vitro observations with data obtained in animal models of brain ischemia? A key may be the acidosis associated with lactic acid accumulation and subsequent reduction of pH that occurs in vivo, but not in vitro. Reduction of extracellular pH to levels found in ischemia in vivo(6.4–6.8) (Nemoto and Frinak, 1981; Siemkowicz and Hansen, 1981;Siesjo, 1988) markedly reduces NMDA receptor-mediated current (Tang et al., 1990), as well as NMDA receptor-mediated injury in cultured neurons (Giffard et al., 1990a; Tombaugh and Salpolsky, 1990; Kaku et al., 1993), and thus could help unmask more slowly triggered AMPA–kainate receptor-mediated injury. Extrapolating from studies of AMPA–kainate receptor-mediated currents, it has been assumed that AMPA–kainate receptor-mediated injury is attenuated by extracellular acid shift, but to a lesser degree than NMDA receptor-mediated injury (Giffard et al., 1990a; Christensen and Hida, 1990).
The purpose of the present experiments was to test this assumption directly.
An abstract has been published previously (McDonald et al., 1996a).
MATERIALS AND METHODS
Cortical neuronal cultures. Mixed cortical cell cultures containing both neurons and astrocytes were prepared from fetal mice (15–16 d gestation) as previously described (Rose et al., 1992). Animals were handled in accordance with a protocol approved by our institutional animal care committee. Briefly, dissociated cortical cells were plated on a preexisting astrocyte monolayer (see below) at 2.5 hemispheres per 24-well plate (a plating density of ∼2.5 × 105 cells per well). Plating media consisted of Eagle’s minimal essential medium (MEM) (Earle’s salts, supplied glutamine-free) supplemented with 5% fetal bovine serum, 5% horse serum, 2 mm glutamine and glucose to 20 mmfinal concentration. Non-neuronal cell division was halted at 3–5 din vitro (DIV) by 3 d exposure to 10−5m cytosine arabinoside. Cultures were maintained in humidified 5% CO2 incubators at 37°C and were used for experiments at 13–16 DIV. Medium was exchanged twice weekly with a growth medium identical to the plating medium, except lacking fetal bovine serum. Pure neuronal cultures were prepared in a similar manner with some modification (Rose et al., 1992).
Astrocyte cultures. Neocortical astrocyte cell cultures were prepared from postnatal mice aged 1–3 d (Rose et al., 1992) and plated at 0.25–0.5 hemispheres per 24-well plate, in plating medium supplemented with 10 ng/ml epidermal growth factor, 10% fetal bovine serum, and 10% horse serum. After 2 weeks in vitro, astrocyte cultures were fed weekly with growth medium and used over the next 2 weeks as a substrate for subsequent neuronal plating.
Neuronal toxicity experiments. Mixed neuron–astrocyte cultures (13–16 DIV) were exposed to AMPA in a physiological salt solution (at 37°C) with the following composition (in mm): NaCl, 120; KCl, 5.4; MgSO4, 0.8; NaH2PO4, 1.0; CaCl2, 1.8; glucose, 5.5; PIPES, 20; and phenol red 10 mg/l. The NaHCO3 concentration was varied as appropriate for the chosen pH in a 5% CO2 atmosphere at 37°C (Dawson et al., 1986). Total [Na+] was maintained at 120 mm. Saturating concentrations of AMPA (300 μm) and/or cyclothiazide (100 μm) were used to avoid additive injury from secondary glutamate release. (+)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohept-5,10-imine hydrogen maleate (MK-801) (10 μm) was added with AMPA incubations to prevent NMDA receptor activation. Exposure was terminated by sequential washes (>1000-fold dilution) and transferred into Eagle’s MEM supplemented with glucose (25 mm). All final incubations in experimental conditions contained saturating concentrations of NMDA and AMPA receptor antagonists [10 μm MK-801 and 30 μm6,7-dinitroquinoxaline-2,3-dione (NBQX)] to limit delayed secondary glutamate-induced injury. Although prolonged blockade of calcium influx may be associated with delayed apoptotic neuronal death, none was observed over 24 hr; addition of cycloheximide (0.5 μg/ml) or IGF-1 (100 ng/ml) did not provide additive protection when combined with MK-801 and NBQX. Cell death was assessed qualitatively by examination under phase contrast microscopy, and quantitatively by measuring lactate dehydrogenase (LDH) efflux into the extracellular medium (Koh and Choi, 1987). Background levels of LDH present in the medium of sham-washed sister cultures (<10% total LDH) were subtracted from values measured in treatment conditions to yield the signal specifically associated with each condition. The LDH signal associated with near complete neuronal death was determined in sister cultures exposed to 300 μm NMDA for 24 hr, an exposure that reliably destroyed the neuronal population without concurrent glial death. All data are presented relative to LDH measurements associated with complete neuronal death in sister cultures (100). In additional experiments, cell viability was assessed by exclusion of 0.4% trypan blue dye.
Oxygen–glucose deprivation experiments. Combined oxygen–glucose deprivation was initiated by exposing neuronal cultures to a deoxygenated, glucose-free solution at pH 6.4 or 7.4 [glucose-free Earle’s balanced salt solution (BSS)], in an air-tight chamber flooded with 95% N2 and 5% CO2(Goldberg and Choi, 1993). BSS contained (in mm): Na+, 143.6; K+, 5.4; Ca2+, 1.8; Mg2+, 0.8; Cl−, 125.3; HCO3−, 26.2; H2SO42−, 1.0; SO42−, 0.8; and phenol red, 10 mg/l. Exposure times of 50 min, pH 7.4, and 75 min, pH 6.4, were chosen to match total neuronal injury between pH exposures (70–80% death), because acid pH reduces oxygen–glucose deprivation injury (Giffard et al., 1990a). Deprivation was terminated by replacing the exposure medium with oxygenated BSS, pH 7.4, containing 5.5 mmglucose, and cultures were returned to normoxic incubator. Cell injury was assessed 24 hr later by LDH efflux and phase-contrast microscopy as described above.
45Ca2+ accumulation studies.Cultures were washed, then incubated in the physiological buffer described above containing (in μm): 300 AMPA, 100 cyclothiazide, 10 MK-801, and 45Ca2+(New England Nuclear; final activity, 0.5–1 mCi/ml) at pH 7.4 or 6.6, for 5, 15, or 30 min (Hartley et al., 1993). Exposure was terminated by thorough washout of extracellular45Ca2+. Cells were lysed with 0.2% SDS solution at 37°C to measure intracellular45Ca2+ accumulation.45Ca2+ accumulation in sham-washed sister cultures was subtracted from all values to yield the45Ca2+ accumulation specific to each condition tested. The mean value of45Ca2+ accumulation for each condition was scaled to that induced by 500 μm NMDA (100).
Intracellular free Ca2+ determination.Intracellular free Ca2+([Ca2+]i) was measured using fura-2 fluorescence video microscopy (Grynkiewicz et al., 1985). Neuronal cultures for [Ca2+]i imaging experiments were prepared as previously described, and experiments were performed between 12–17 DIV (Csernansky et al., 1994). Cells were loaded with 5 μm fura-2 AM plus 0.1% Pluronic F-127 for 30 min at room temperature (24°C), washed, and incubated for an additional 30 min in HBSS. All experiments were performed using the same bicarbonate-buffered salt solution as used for the toxicity studies. Experiments were performed at room temperature on the stage of a Nikon Diaphot inverted microscope equipped with a 75 W Xenon lamp and a Nikon 40×, 1.3 NA epifluorescence oil immersion objective under continuous perfusion (perfusion rate, 2 ml/min). Although temperature may alter many cellular functions, we have observed previously that temperature does not substantially alter NMDA or kainate-stimulated peak [Ca2+]i responses or recovery (Bruno et al., 1994). In addition to room temperature experiments, a set of additional fura-2 measurements was performed at 37°C to coincide with toxicity studies. Fura-2 (Ex λ = 340, 380 nm, Em λ = 510 nm) ratio images were acquired with an intensified CCD (Quantex) camera and digitized (256 × 512 pixels) using an Image-1 (Universal Imaging Corporation, West Chester, PA) system. Calibrated [Ca2+]i values were obtained using the ratio method of Grynkiewicz et al. (1985) by determining Fmin and Fmaxin situ using EGTA (10 mm) with 0 Ca2+ buffer and ionomycin (10 μm) forFmin and 10 mmCa2+ with ionomycin (10 μm) for Fmax. A Kd value of 225 nm Ca2+ was used for fura-2.
Materials. Cyclothiazide was obtained from Eli Lilly and Co. (Indianapolis, IN). Comparison toxicity studies were performed with cyclothiazide from another source (Research Biochemicals International, Natick, MA). Cyclothiazide was dissolved in DMSO as a 30 mmstock solution and stored at 4°C. Similar experimental observations were obtained with drug from both sources. All other chemicals were obtained from commercial sources.
RESULTS
Effects of extracellular pH on AMPA- and kainate-induced neuronal injury
We previously reported that multihour exposure to AMPA was required to induce widespread neuronal death in murine cortical cell cultures at pH 7.4 (Koh et al., 1990). At pH 7.4, 3–4 hr exposure of mixed (neuronal plus astrocyte) cultures (13–16 DIV) to 300 μm AMPA in the presence of 10 μm MK-801 (added to block secondary NMDA receptor-mediated injury) produced extensive acute neuronal cell swelling but little late neuronal death and LDH release by the next day (Figs. 1,2). At pH 6.6, the same exposure induced both acute neuronal cell swelling and substantial late neuronal death by the next day (still without astrocyte death) (Figs. 1, 2). Neuronal death was prevented by coapplication of the selective AMPA–kainate receptor antagonist NBQX (30 μm). Exposure to pH 6.6 alone for 3–4 hr produced little cell death. Continued exposure of cultures to acid pH, beyond the period of AMPA or kainate exposure, did not prevent neuronal death (data not shown). To examine if the potentiation of AMPA–kainate receptor toxicity at acidic pH was related to additive effects of two sublethal injuries, similar pH exposures were performed with another Ca2+-overload injury paradigm. Intermediate levels of neuronal death induced by a 3 hr exposure to the Ca2+ ionophore A23187 (100–500 μm) at pH 7.4 was not potentiated by coexposure to acidic pH (6.6) when examined 24 hr later (data not shown).
Fig. 1.
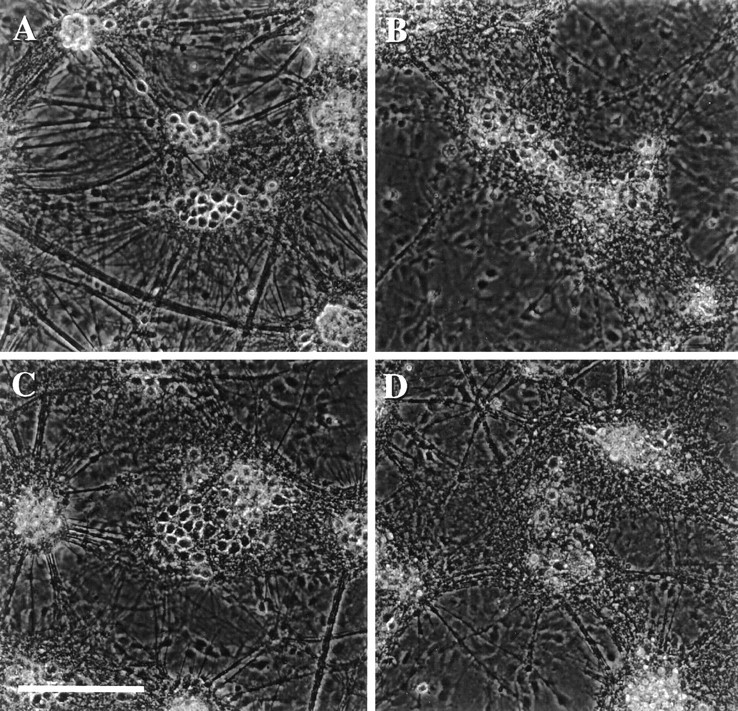
Acidic extracellular pH exacerbates slowly triggered AMPA toxicity. Phase-contrast micrographs of mixed cortical and glial cell cultures taken 18 hr after (A) 4 hr exposure to pH 6.6 alone (no change from sham wash controls); (B) 24 hr exposure to 300 μm NMDA at pH 7.4; (C) 4 hr exposure to 300 μm AMPA plus 10 μm MK-801 at pH 7.4 (this exposure, here and subsequently, was terminated by washing out AMPA and adding 10 μm MK-801 plus 30 μm NBQX); (D) 4 hr exposure to 300 μm AMPA plus 10 μm MK-801 at pH 6.6. Scale bar, 200 μm. All data are representative of at least three separate experiments.
Fig. 2.

Time course of AMPA-induced neuronal death at pH 7.4 and 6.6. Sister cultures were incubated in HBSS containing 300 μm AMPA and 10 μm MK-801 for the indicated periods. LDH release to the bathing medium was measured 24 hr later (mean ± SEM, n = 4) and scaled to the level (100) measured in sister cultures exposed to 300 μm NMDA for 24 hr (a condition that produced near complete neuronal death without astrocyte death). Background LDH release in sister cultures exposed to sham wash alone was subtracted from each condition to yield the signal specific to experimental injury. *Difference at p < 0.05, from corresponding time value at pH 7.4, by Student’s two-tailed independent t test.
To determine whether the same injury potentiation could be detected with shorter exposure duration, we exploited the ability of cyclothiazide to inhibit AMPA receptor desensitization and potentiate AMPA receptor-mediated neuronal death (Yamada and Rothman, 1992; May and Robinson, 1993; Patneau et al., 1993; Zorumski et al., 1993; Moudy et al., 1994). Exposure of mixed neuronal–astrocyte cultures for 1.5 hr to (in μm): 300 AMPA, 100 cyclothiazide, and 10 MK-801 resulted in increasing neuronal death without astrocyte death (trypan blue exclusion; data not shown) as bath pH was shifted from 7.4 to 6.6 (Fig. 3A). When bath was further shifted to pH 6.2, substantial astrocyte death also occurred (Fig. 3A; David et al., 1996). A 1.5 hr exposure to pH 6.6 alone produced little neuronal injury (4 ± 6% LDH release, n = 8).
Fig. 3.
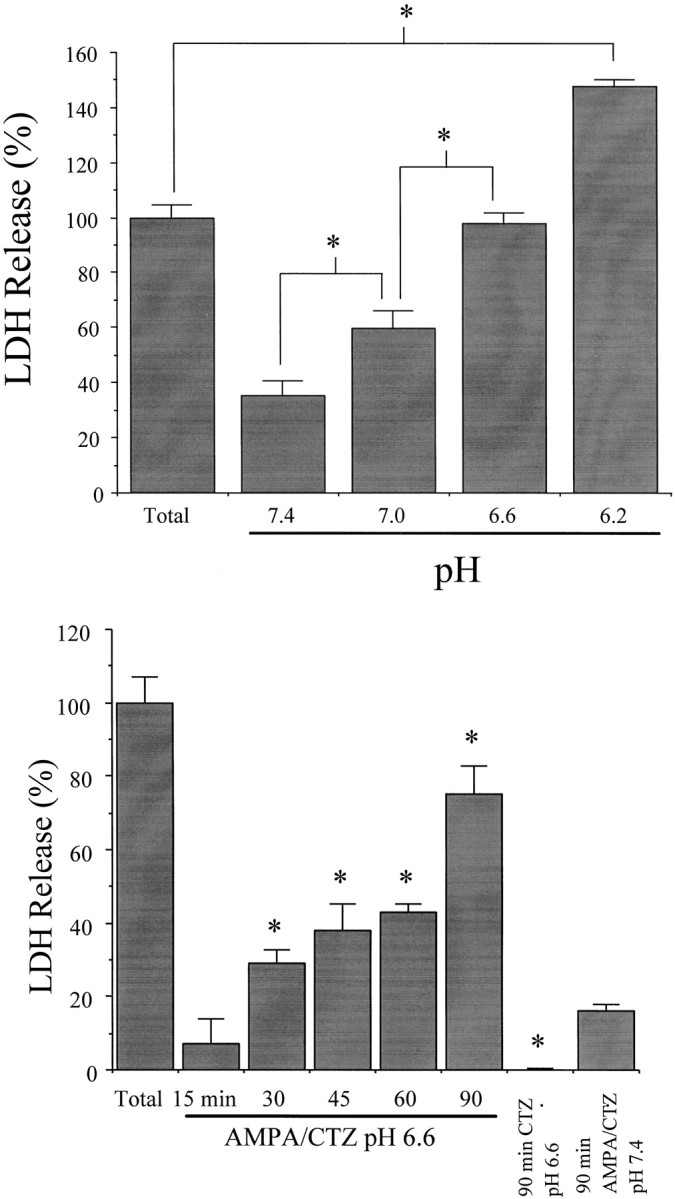
Acidic extracellular pH exacerbates rapidly triggered AMPA toxicity. A, Mixed cultures were exposed for 1.5 hr to 300 μm AMPA plus 100 μmcyclothiazide and 10 μm MK-801 at the indicated extracellular pH (mean ± SEM, n = 4). At pH 6.2 (but not at higher pH), some astrocyte death also occurred, accounting for the total LDH release exceeding 100. *Difference atp < 0.05, as indicated, by one-way ANOVA with Student Newman–Keuls test. B, Time course of rapidly triggered AMPA toxicity at pH 6.6. Neuronal cultures were exposed to 300 μm AMPA plus 100 μm cyclothiazide and 10 μm MK-801 at pH 6.6 for the indicated duration. *Difference at p < 0.05, one-way ANOVA with Student Newman–Keuls test, indicated time AMPA–cyclothiazide (CTZ) at pH 6.6 versus 90 min exposure AMPA–CTZ at pH 7.4 (far right bar). Exposure to cyclothiazide alone at pH 6.6 for 90 min produced no neuronal death (*difference atp < 0.05, 90 min CTZ at pH 6.6 vs 90 min AMPA–CTZ at pH 7.4, Student’s independent t test). LDH release to the bathing medium was measured 24 hr later (mean ± SEM,n = 4) and scaled to the level (100) measured in sister cultures exposed to 300 μm NMDA for 24 hr.
Lowering extracellular pH to 6.6 permitted relatively brief (30 min or more) duration exposure to 300 μm AMPA plus 100 μm cyclothiazide to become neurotoxic (Fig.3B). Nearly complete neuronal death was induced by a 1.5 hr exposure to AMPA and cyclothiazide at pH 6.6 (Fig. 3B). Application of cyclothiazide alone at pH 6.6 for 1.5 hr did not produce neuronal death.
Reduction of extracellular pH to 6.6 also potentiated (twofold) neuronal death resulting from exposure to 200 μmkainate (Table 1). Neuronal death was prevented by coexposure to 30 μm NBQX.
Table 1.
Kainate-induced cortical neuronal death is enhanced at acidic pH
| Treatment | % Neuronal death ± SEM |
|---|---|
| pH 7.4 | |
| Kainate | 19 ± 5 |
| Kainate + NBQX | 1 ± 0.3* |
| pH 6.6 | |
| Kainate | 50 ± 4** |
| Kainate + NBQX | 2 ± 0.4* |
Reducing extracellular pH from 7.4 to 6.6 enhanced cortical neuron death induced by 20 min exposure to 200 μmkainate. MK-801 (10 μm) was included with each exposure. Coapplication of NBQX (30 μm) prevented death induced by kainate under both conditions. LDH release to the bathing medium was measured 24 hr later (mean ± SEM, n = 8), scaled to the level (100) measured in sister cultures exposed to 300 μM NMDA for 24 hr (a condition that produced near complete neuronal death without astrocyte death). Background LDH release in sister cultures exposed to sham wash alone was subtracted from each condition to yield the signal specific to experimental injury.
*Difference at p < 0.05 from kainate exposure alone at same pH, by Student’s independent t test.
**Difference at p < 0.05 from kainate exposure at pH 7.4.
Effect of reduced extracellular pH on oxygen–glucose deprivation-induced neuronal injury
Oxygen–glucose deprivation-induced death in cortical neuronal cultures at pH 7.4 is predominantly attributable to NMDA receptor-mediated toxicity (Choi, 1992). We examined whether the contribution of AMPA receptors to this death would increase as a result of reducing extracellular pH. Although neuronal death resulting from 50 min exposure to oxygen–glucose deprivation at pH 7.4 was not altered by AMPA receptor blockade with 30 μmNBQX, death induced by 75 min exposure at pH 6.4 was sensitive to NBQX (Table 2). We have previously shown that acidic extracellular pH (6.4) attenuates oxygen–glucose deprivation-induced death of cortical neurons. Because reducing pH reduced overall neuronal death, in the latter condition the duration of oxygen–glucose deprivation was increased to 75 min to recover widespread neuronal death comparable to corresponding injury with 50 min oxygen–glucose deprivation at pH 7.4
Table 2.
Acidic extracellular pH increases the contribution of AMPA–kainate receptors to oxygen–glucose deprivation injury
| Treatment | % Neuronal death ± SEM |
|---|---|
| pH 7.4 | |
| OGD (50 min) | 65 ± 3 |
| OGD (50 min) + NBQX | 66 ± 5 |
| pH 6.4 | |
| OGD (75 min) | 84 ± 6 |
| OGD (75 min) + NBQX | 63 ± 6* |
Addition of 30 μm NBQX did not reduce neuronal death in cultures exposed to 50 min of oxygen-glucose deprivation at pH 7.4 but did partially reduce neuronal death in cultures exposed to oxygen–glucose deprivation at pH 6.4. Because reducing pH reduced NMDA receptor-mediated toxicity and overall neuronal death, in the latter condition the duration of oxygen–glucose deprivation was increased to 75 min to recover widespread neuronal death comparable to corresponding injury with 50 min oxygen–glucose deprivation at pH 7.4. Data are mean ± SEM (n = 8). Neuronal cell death was quantitated by measuring LDH released into the bathing medium 24 hr later.
*Difference at p < 0.05 from 75 min oxygen–glucose deprivation at pH 6.4, by Student’s two-tailed independentt test.
45Ca2+ accumulation
Because toxic Ca2+ influx appears to be a critical event in excitotoxic necrosis (Choi, 1987, 1992), we tested whether the potentiating effect of reduced extracellular pH on the toxicity of AMPA plus cyclothiazide was accompanied by increases in Ca2+ influx, as measured by a45Ca2+ uptake assay (Hartley et al., 1993). Paradoxically, the enhanced neuronal vulnerability to AMPA toxicity observed in mixed cultures at reduced extracellular pH was not associated with increased 45Ca2+accumulation but rather was associated with reduced45Ca2+ accumulation (Fig.4), consistent with previous electrophysiological data indicating reduced kainate-activated whole-cell current in cultured cortical neurons and cone horizontal cells at acidic pH (Christensen and Hida, 1990; Giffard et al., 1990a).
Fig. 4.

Acidic pH exacerbation of AMPA toxicity is associated with reduced 45Ca uptake. Mixed cultures were incubated with 300 μm AMPA plus 100 μmcyclothiazide and 10 μm MK-801 at pH 7.4 or 6.6, for the indicated times. Cellular 45Ca accumulation (mean ± SEM, n = 7) was scaled to the levels obtained in sister cultures exposed to 500 μm NMDA for 15 min (100). Basal 45Ca uptake in sham-washed sister cultures was subtracted from all measurements. *Difference atp < 0.05, from corresponding time point at pH 7.4, by one-way ANOVA and Student Newman–Keuls test.
Because reduced astrocyte 45Ca2+accumulation in response to acid pH shift could mask an increase in neuronal 45Ca2+ accumulation, we also examined 45Ca2+ accumulation in pure neuronal cultures. A similar decrease in45Ca2+ accumulation occurred at acid pH. Sister cultures of near-pure neurons (13 DIV) were exposed to (in μm): 300 AMPA, 100 cyclothiazide, 10 μmMK-801, and 45Ca2+, pH 7.4 or 6.6.45Ca2+ accumulation was examined 5 min later and values were normalized to45Ca2+ accumulation induced by 500 μm NMDA at pH 7.4. 45Ca2+accumulation was reduced by 44% at pH 6.6 compared with pH 7.4 (difference at p < 0.05 by Student’s two tailed independent t test; pH 7.4, 61 ± 2 vs pH 6.6, 34 ± 3; mean ± SEM, n = 24 per condition).
Intracellular free Ca2+ determination
To assess whether impaired [Ca2+]i buffering at reduced pH may render neurons more vulnerable to AMPA toxicity, we performed intracellular free [Ca2+]i imaging using fura-2. Neuronal cultures exposed for 5 sec to 10 μm AMPA (plus 100 μm cyclothiazide and 10 μm MK-801) at pH 6.6 (24°C), exhibited a marked delayed recovery of [Ca2+]i to baseline compared with sister cultures exposed to AMPA at pH 7.4, despite a predictable reduction of the peak [Ca2+]i response (Figs.5,6A; Table3). Similar observations were also made when experiments were performed at 37°C (data not shown). This delayed recovery was apparent across the whole neuronal population, and it was not dependent on stimulus sequence. A delayed recovery of [Ca2+]i was also seen in cultures exposed to kainate plus MK-801 (Fig. 6B). The impaired recovery was long lasting, and extended at least 1.5 hr after removal of kainate or AMPA. Delayed recovery persisted despite return of extracellular pH to 7.4 (data not shown).
Fig. 5.
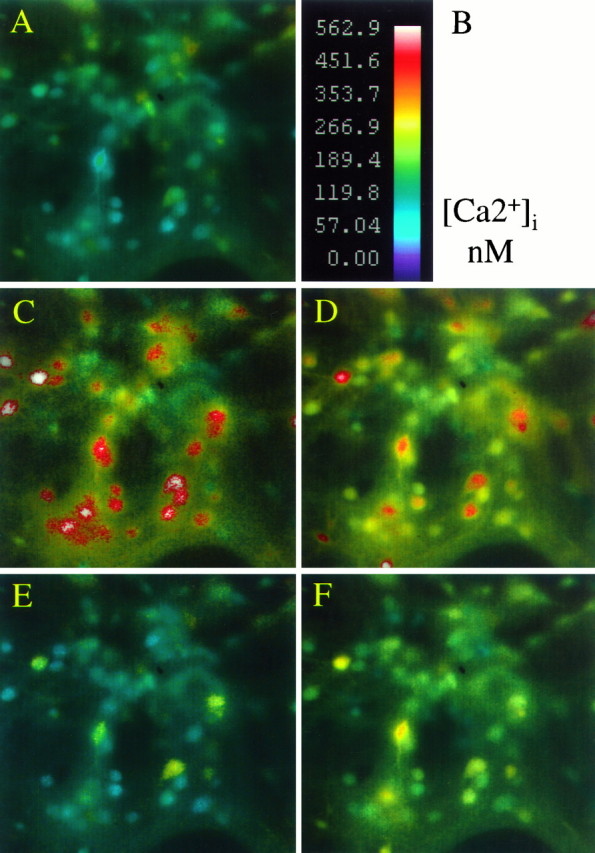
Reducing extracellular pH attenuates the peak [Ca2+]i response to AMPA but delays subsequent normalization. Pseudocolor images, shown as intensity modulated display, of intracellular free calcium in the same field of neurons exposed to 10 μm AMPA at pH 7.4 or 6.6 (24°C).A, Basal [Ca2+]imeasurements in cortical neurons at rest in pH 7.4 buffer.B, Calibration scale.C–D, Peak responses during a 5 sec application of 10 μm AMPA plus 10 μmcyclothiazide and 10 μm MK-801 at pH 7.4 (C) or at pH 6.6 (D).E–F, Corresponding fields asC–D, but measured 15 min after drug washout. Peak [Ca2+]i measurements are reduced by acidic pH, but response recovery is impaired.
Fig. 6.

Changes in [Ca2+]i after exposure to AMPA, kainate, NMDA, or high KCl at normal and acidic pH. Refer to Table 3for corresponding calcium integral measurements. All measurements were performed at 24°C. A, AMPA response; sequential averaged changes in [Ca2+]i in 35 neurons during and after a 5 sec exposure to 10 μm AMPA plus 100 μm cyclothiazide and 10 μm MK-801 at pH 7.4 or 6.6. Exposure to acidic pH attenuates peak AMPA responses but impairs recovery. B, Kainate-induced [Ca2+]i responses at pH 7.4 and 6.6 were similar to that observed for AMPA. Cultures were exposed to 100 μm kainate, as outlined in A. Peak responses are attenuated at pH 6.6, but again, normalization was markedly impaired. C, Reducing extracellular pH attenuates the peak [Ca2+]i response to NMDA with minimal change in subsequent normalization. The trace represents average (n = 27 neurons) changes in [Ca2+]i during and after 5 sec exposures to 50 μm NMDA at pH 7.4 or 6.6. In the low pH exposure experiments, neurons were washed for 5 min at pH 6.6 before the application of NMDA, exposed to NMDA at pH 7.4 for 5 sec, and immediately returned at pH 6.6 for 15 min. Under these conditions the intracellular pH remains below 6.6, and the brief extracellular exposure to NMDA at pH 7.4 allows activation of NMDA receptors (i.e., the pH-sensitive site on NMDA receptors is extracellular). D, Reducing extracellular pH attenuates the peak [Ca2+]i response to high KCl with minimal change in subsequent normalization. Thetrace represents average (n = 30 neurons) changes in [Ca2+]i during and after 5 sec exposures to 60 mm KCl plus 10 μmMK-801 at pH 7.4 or 6.6. In the low pH exposure experiments, neurons were washed for 5 min at pH 6.6 before the application of 60 mm KCl, exposed to high KCl at pH 7.4, and immediately returned to pH 6.6 for 15 min. Under these conditions, the intracellular pH remains below 6.6, and the brief extracellular exposure to 60 mm KCl at pH 7.4 allows activation of voltage-sensitive calcium channels. All the experiments are representative of at least three different experiments in three different cultures.
Table 3.
Comparison of area measurements of free [Ca2+]i responses to AMPA, kainate, NMDA, and KCl stimulation at pH 7.4 and 6.6
| Trigger | No. of cells tested | Area under curve (mean ± SEM, arbitrary units) | Value ratio 6.6/7.4 | Difference at p < 0.05 | |
|---|---|---|---|---|---|
| pH 7.4 | pH 6.6 | ||||
| AMPA | 167 | 11.0 ± 0.2 | 39.1 ± 0.4 | 3.5 | Yes |
| Kainate | 122 | 8.0 ± 0.1 | 15.1 ± 0.2 | 1.9 | Yes |
| NMDA | 212 | 8.2 ± 0.1 | 6.6 ± 0.1 | 0.8 | Yes |
| KCl | 187 | 7.0 ± 0.1 | 6.3 ± 0.1 | 0.9 | Yes |
See Figure 6 legend, Results and Materials and Methods for experimental details. Abbreviated stimulus conditions listed above were as follows: AMPA = 10 μm AMPA + 100 μm cyclothiazide, kainate = 100 μmkainate, NMDA = 20 μm NMDA, KCl = 60 mm KCl. Experiments were carried out at 24°C. All experiments, except the NMDA experiment, were performed in the presence of MK-801 (10 μm) to block secondary activation of NMDA receptors. Measurement of the calcium integral was over 15 min for all pH 7.4 values and NMDA and KCl values at pH 6.6 (all reached baseline measures by this time point) and was over 90 min for AMPA and kainate at pH 6.6. These values correspond to the second and third calcium responses (pH 7.4 vs pH 6.6) for each stimulus shown in Figure 6. Statistical comparisons were performed by Student’s two-tailed independent t test.
To determine whether this effect was selective for AMPA–kainate responses, we examined the effect of pH on NMDA- and KCl-induced [Ca2+]i. Cells were preincubated at pH 6.6 for 10 min, and then 50 μm NMDA was applied for 5 sec (at pH 7.4, because application at pH 6.6 produces little response). In control experiments, we demonstrated that this paradigm produces well maintained intracellular acidification despite the brief application of pH 7.4 buffer (data not shown). Application of NMDA induced a rapid and large increase in neuronal [Ca2+]i, but recovery was not delayed at pH 6.6 (Fig. 6C, Table 3). A >10-fold increase in exposure time to NMDA (60 sec), resulting in peak responses comparable with AMPA–kainate exposure, did not delay [Ca2+]i recovery. Despite even higher peak [Ca2+]i responses produced by 60 mm KCl at pH 6.6, [Ca2+]irecovery was not impaired (Fig. 6D, Table 3), suggesting a specific effect of reduced extracellular pH on AMPA receptor-mediated [Ca2+]iresponses.
DISCUSSION
The major finding of the present study is the novel and unexpected observation that reduction of extracellular pH to levels of acidity associated with ischemia in vivo (Nemoto and Frinak, 1981;Siemkowicz and Hansen, 1981; Tombaugh and Sapolsky, 1993; Siesjo, 1988) paradoxically potentiated AMPA–kainate receptor-mediated cortical neuronal death. This potentiation extended to the AMPA–kainate receptor-mediated component of oxygen–glucose deprivation-induced neuronal death. Acid pH potentiation of AMPA–kainate receptor-mediated toxicity occurred despite a reduction in both net influx of45Ca2+ and peak increase in [Ca2+]i, most likely because subsequent normalization of [Ca2+]iwas selectively impaired. Elevated [Ca2+]i persisted for >90 min after termination of AMPA or kainate exposure, even after return to normal extracellular pH. Similarly delayed normalization of [Ca2+]i was not seen after comparable peak elevations induced by exposure to NMDA or high K+. The brief, multisecond exposures to AMPA, NMDA, and high K+ used in the fura-2 studies were intended to probe calcium handling and were not sufficient to produce detectable injury or neuronal death. It is therefore unlikely that the delayed normalization of [Ca2+]i after AMPA receptor activation at acidic pH can be explained by lethal injury. The observation that acidic pH did not enhance intermediate levels of neuronal death induced by exposure to the calcium ionophore A23187, suggests that the potentiation of AMPA–kainate receptor toxicity at acidic pH is not a simple reflection of additive sublethal injury.
The ability of acid pH to enhance AMPA–kainate receptor-mediated injury is especially striking when placed in the context of reduced net Ca2+ influx, as measured by45Ca2+ accumulation, and reduced somatic peak elevation in [Ca2+]i, as measured by fura-2 video microscopy. These reductions are predictable given electrophysiological data indicating that acid pH moderately reduces current through AMPA–kainate receptor-gated channels (Christensen and Hida, 1990; Giffard et al., 1990a; Tang et al., 1990). As a hypothesis suitable for future testing, we think it likely that acid pH-induced enhancement of toxicity reflects the marked impairment of [Ca2+]i normalization observed here after exposure to AMPA or kainate at acid pH. This acid pH-impaired recovery of [Ca2+]i homeostasis could reflect increased release of Ca2+ from intracellular stores, reduced intracellular buffering, or reduced extrusion of Ca2+. For example, cells challenged with an acid load can be expected to try to restore pH homeostasis by activating the Na+–H+ antiporter (Moody, 1981), thus increasing intracellular Na+ and impairing Ca2+ extrusion via the Na+–Ca2+ antiporter.
The selectivity of the pH-dependent delayed normalization of [Ca2+]i, seen after AMPA–kainate receptor stimulation but not after comparable NMDA- or high K+-induced elevations in somatic [Ca2+]i, is yet another reminder that all pools of intracellular free Ca2+are not equivalent (Lafon-Cazal et al., 1993; Tymianski et al., 1993). Specifically, a difference in the behavior of the [Ca2+]i elevation induced by AMPA–kainate receptor stimulation, compared with that induced by NMDA receptor stimulation, might be linked to spatial differences in Ca2+ entry points. However, it is more difficult to explain the observed difference in pH sensitivity of [Ca2+]i recovery after AMPA–kainate receptor stimulation versus exposure to high K+, because most Ca2+ entry in both cases presumably occurs via the same voltage-gated Ca2+ channels (Fig. 5, delayed recovery noted in the majority of cells, even those with modest [Ca2+]i responses). One possible explanation is that AMPA–kainate receptor activation might induce greater cellular Na+ loading compared with that induced by high K+ so that, as a result, the propensity of acid pH itself to induce cellular Na+-loading would have special impact on the Na+–Ca2+ antiporter (see above). It is unlikely that a pH paradox accounts for the observed pH enhancement of AMPA–kainate receptor-mediated neuronal death. Continued exposure to acid pH, beyond the period of AMPA exposure, did not protect against the injury.
Cerebral tissue acidosis is a well established feature of ischemic brain tissue, which has long been considered an important factor in the pathogenesis of the resultant brain damage, especially glia death (Plum, 1983; Kraig et al., 1987; Siesjo, 1988; Giffard et al., 1990b;Nedergaard et al., 1991; Tombaugh and Sapolsky, 1993). However, moderate tissue acidosis may also provide some protective effects against neuronal injury by reducing NMDA receptor-mediated excitotoxicity. Both NMDA and AMPA–kainate receptor-mediated whole-cell currents in rat hippocampal neurons are attenuated by moderate extracellular acidity in the range of 6.2–7.2, with the most dramatic effects produced on NMDA receptor-mediated currents (Tang et al., 1990). Similar inhibition of NMDA receptor-mediated currents has been observed on cultured cortical and cerebellar neurons (Giffard et al., 1990a; Traynelis and Cull-Candy, 1990). Reduction of extracellular pH below 6.5 reduces both glutamate neurotoxicity and oxygen–glucose deprivation-induced neuronal death in vitro (Giffard et al., 1990a; Tombaugh and Sapolsky, 1990; Kaku et al., 1993).
Present data thus raise the important possibility that the extracellular acidity that accompanies brain ischemia increases the prominence of AMPA–kainate receptor-mediated injury relative to NMDA receptor-mediated injury, not only by blocking the latter, but also by enhancing the former. Both the inhibition of NMDA receptor activation and exacerbation of AMPA–kainate-mediated neuronal injury occur in the same clinically relevant acid pH range (6.2–7.4). We hypothesize that the toxic contribution of AMPA–kainate receptors might be most prominent during early stages of ischemia associated with acidic extracellular pH, and that subsequently, the balance might shift back toward NMDA receptor-mediated injury as reperfusion occurs and extracellular pH returns to normal, and the acid pH inhibition of NMDA receptors is relieved. The acid pH shifts that accompany reversible focal and global ischemia generally normalize within 0.5–2 hr, with intracellular pH lagging behind extracellular pH. However, normalization can be much slower in models of permanent focal ischemia (Siemkowicz and Hanson, 1981; Mabe et al., 1983; Smith et al., 1986; Von Hanweher et al., 1986; Silver and Erecinska, 1992; Tomlinson et al., 1993; Maruki et al., 1993; Dempsey et al., 1996). Our data suggest that brief periods of acid pH exposure, compatible with the pH changes observed in ischemia models, may importantly potentiate AMPA–kainate receptor-mediated neuronal death. This idea might have implications for the timing and selection of drug treatment in settings associated with prolonged tissue ischemia and acidosis such as stroke, status epilepticus, trauma, and subarachnoid hemorrhage (Becker, 1985;Siesjo and Wieloch, 1986; Siesjo, 1988; Brooke et al., 1994). The foregoing consideration is independent of the additional possibility that certain neuronal subpopulations expressing Ca2+-permeable AMPA or kainate receptors, e.g., Purkinje cells (Brorson et al., 1994) or NADPH-diaphorase–nitric oxide synthase-containing neurons (Koh and Choi, 1988; Weiss et al., 1994), may exhibit heightened baseline vulnerability to AMPA–kainate receptor-mediated injury. In addition, the possibility has been raised that sublethal ischemic insults may selectively depress the expression of the Ca2+-gatekeeper AMPA receptor subunit, gluR-B/gluR-2 (Hollman et al., 1991; Verdoorn et al., 1991) relative to other AMPA receptor subunits, perhaps enhancing AMPA–kainate receptor-mediated Ca2+ influx and death (Pellegrini-Giampietro et al., 1992; Gorter et al., 1997; Ying et al., 1996). Finally, we have recently found that oligodendrocytes maintained for >3 weeks in vitro develop prominent vulnerability to AMPA–kainate receptor-mediated excitotoxic death, comparable to that of neurons (McDonald et al., 1996b, 1998). The ability of AMPA–kainate antagonists to reduce brain damage in animal models of brain ischemia may reflect contributions from some or all of these factors.
Footnotes
This work was supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke grants NS01931 (J.W.M.), NS32636 (D.W.C.) and the American Paralysis Association (J.W.M., D.W.C.).
Correspondence should be sent to Dr. Dennis Choi, Department of Neurology, Box 8111, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110.
REFERENCES
- 1.Albers GW, Goldberg MP, Choi DW. Do NMDA antagonists prevent neuronal injury? Yes. Arch Neurol. 1992;49:418–420. doi: 10.1001/archneur.1992.00530280112031. [DOI] [PubMed] [Google Scholar]
- 2.Balchen T, Diemer NH. The AMPA antagonist, NBQX, protects against ischemia-induced loss of cerebellar Purkinje cells. NeuroReport. 1992;3:785–788. doi: 10.1097/00001756-199209000-00016. [DOI] [PubMed] [Google Scholar]
- 3.Becker DP. Brain acidosis in head injury: a clinical trial. In: Becker DP, Povlishock JT, editors. Central nervous system trauma status report 1985. Byrd; Richmond: 1985. pp. 229–242. [Google Scholar]
- 4.Brooke NSR, Ouwerkerk R, Adams CBT, Radda GK, Ledingham JGG, Rajagopalan B. Phosphorus-31 magnetic resonance spectra reveal prolonged intracellular acidosis in the brain following subarachnoid hemorrhage. Proc Natl Acad Sci. 1994;91:1903–1907. doi: 10.1073/pnas.91.5.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brorson JR, Manzolillo PA, Miller RJ. Ca2+ entry via AMPA/KA receptors and excitotoxicity in cultured cerebellar Purkinje cells. J Neurosci. 1994;14:187–197. doi: 10.1523/JNEUROSCI.14-01-00187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruno VMG, Goldberg MP, Dugan LL, Giffard RG, Choi DW. Neuroprotective effect of hypothermia in cortical cultures exposed to oxygen-glucose deprivation or excitatory amino acids. J Neurochem. 1994;63:1398–1406. doi: 10.1046/j.1471-4159.1994.63041398.x. [DOI] [PubMed] [Google Scholar]
- 7.Buchan AM, Xue D, Haung ZG, Smith KH, Lesiuk H. Delayed AMPA receptor blockade reduces cerebral infarction induced by focal ischemia. NeuroReport. 1991a;2:473–476. doi: 10.1097/00001756-199108000-00016. [DOI] [PubMed] [Google Scholar]
- 8.Buchan AM, Li H, Pulsinelli WA. The N-methyl-d-aspartate antagonist, MK-801, fails to protect against neuronal damage caused by transient, severe forebrain ischemia in adult rats. J Neurosci. 1991b;11:1049–1056. doi: 10.1523/JNEUROSCI.11-04-01049.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi DW. Ionic dependence of glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 11.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 12.Christensen BN, Hida E. Protonation of histidine groups inhibits gating of the quisqualate/kainate channel protein in isolated catfish cone horizontal cells. Neuron. 1990;5:471–478. doi: 10.1016/0896-6273(90)90086-u. [DOI] [PubMed] [Google Scholar]
- 13.Csernansky CA, Canzoniero LMT, Sensi SL, Yu SP, Choi DW. Delayed application of aurintricarboxylic acid reduces glutamate-induced cortical neuronal injury. J Neurosci. 1994;38:101–108. doi: 10.1002/jnr.490380113. [DOI] [PubMed] [Google Scholar]
- 14.David JC, Yamada KA, Bagwe MR, Goldberg MP. AMPA receptor activation is rapidly toxic to cortical astrocytes when desensitization is blocked. J Neurosci. 1996;16:200–209. doi: 10.1523/JNEUROSCI.16-01-00200.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dawson RMC, Elliot DC, Elliot WH, Jones KM. Data of biochemical research, 3rd ed, p 433. Oxford Science Publications; Oxford: 1986. [Google Scholar]
- 16.Dempsey RJ, Baskaya MK, Combs DJ, Donaldson D, Rao AM, Prasad MR. Delayed hyperglycemia and intracellular acidosis during focal cerebral ischemia in cats. Acta Neurochir. 1996;138:745–751. doi: 10.1007/BF01411482. [DOI] [PubMed] [Google Scholar]
- 17.Giffard RG, Monyer H, Christine CW, Choi DW. Acidosis reduces NMDA receptor activation, glutamate neurotoxicity, and oxygen-glucose deprivation neuronal injury in cortical cultures. Brain Res. 1990a;506:339–342. doi: 10.1016/0006-8993(90)91276-m. [DOI] [PubMed] [Google Scholar]
- 18.Giffard RG, Monyer H, Choi DW. Selective vulnerability of cultured cortical glia to injury by extracellular acidosis. Brain Res. 1990b;530:138–141. doi: 10.1016/0006-8993(90)90670-7. [DOI] [PubMed] [Google Scholar]
- 19.Gill R. The pharmacology of 181-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA)/kainate antagonists and their role in cerebral ischemia. Cerebrovasc Brain Metab Rev. 1994;6:225–256. [PubMed] [Google Scholar]
- 20.Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gorter JA, Petrozzino JJ, Aronica EM, Rosenbaum DM, Opitz T, Bennett MVL, Connor JA, Zukin RS. Global ischemia induces down regulation of Glur2 mRNA and increases AMPA receptor-mediated Ca2+ influx in hippocampal CA1 neurons of gerbil. J Neurosci. 1997;17:6179–6188. doi: 10.1523/JNEUROSCI.17-16-06179.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grynkiewicz G, Poenie M, Tsien R. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 23.Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hollman M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 25.Kaku DA, Goldberg MP, Choi DW. Antagonism of non-NMDA receptors augments the neuroprotective effect of NMDA receptor blockade in cortical cultures subjected to prolonged deprivation of oxygen and glucose. Brain Res. 1991;554:344–347. doi: 10.1016/0006-8993(91)90214-g. [DOI] [PubMed] [Google Scholar]
- 26.Kaku DA, Giffard RG, Choi DW. Neuroprotective effects of glutamate antagonists and extracellular acidity. Science. 1993;260:1516–1518. doi: 10.1126/science.8389056. [DOI] [PubMed] [Google Scholar]
- 27.Koh J, Choi DW. Quantitative determination of glutamate-mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 28.Koh JY, Choi DW. Vulnerability of cultural cortical neurons to damage by excitotoxins: differential susceptibility of neurons containing NADPH-diaphorase. J Neurosci. 1988;8:2153–2163. doi: 10.1523/JNEUROSCI.08-06-02153.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koh JY, Choi DW. Selective blockade of non-NMDA receptors does not block rapidly triggered glutamate-induced neuronal death. Brain Res. 1991;548:318–321. doi: 10.1016/0006-8993(91)91140-v. [DOI] [PubMed] [Google Scholar]
- 30.Koh JY, Goldberg MP, Hartley DM, Choi DW. Non-NMDA receptor-mediated neurotoxicity in cortical culture. J Neurosci. 1990;10:693–705. doi: 10.1523/JNEUROSCI.10-02-00693.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kraig RP, Petito CK, Plum F, Pulsinelli WA. Hydrogen ions kill brain at concentrations reached in ischemia. J Cereb Blood Flow Metab. 1987;7:379–386. doi: 10.1038/jcbfm.1987.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 33.Le-Peillet E, Arvin B, Moncada C, Meldrum BS. The non-NMDA antagonists, NBQX and GYKI 52466, protect against cortical and striatal cell loss following transient global ischemia in the rat. Brain Res. 1992;571:115–120. doi: 10.1016/0006-8993(92)90516-c. [DOI] [PubMed] [Google Scholar]
- 34.MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurons. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 35.May PC, Robinson PM. Cyclothiazide treatment unmasks AMPA excitotoxicity in rat primary hippocampal cultures. J Neurochem. 1993;60:1171–1174. doi: 10.1111/j.1471-4159.1993.tb03272.x. [DOI] [PubMed] [Google Scholar]
- 36.McDonald JW, Bhattacharyya T, Sensi SL, Lobner D, Ying HS, Choi DW. Extracellular acidic pH exacerbates AMPA receptor-mediated neuronal injury. Ann Neurol. 1996a;46:A468. doi: 10.1523/JNEUROSCI.18-16-06290.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDonald JW, Althomsons SP, Hyrc KL, Goldberg MP, Choi DW. Oligodendrocytes are highly susceptible to AMPA/kainate receptor- and hypoxia-induced death. J Neurotrauma. 1996b;13:599. [Google Scholar]
- 38.McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nature Med. 1998;4:291–297. doi: 10.1038/nm0398-291. [DOI] [PubMed] [Google Scholar]
- 39.Meldrum B. Possible therapeutic applications of antagonists of excitatory amino acid neurotransmitters. Clin Sci. 1985;68:113–122. doi: 10.1042/cs0680113. [DOI] [PubMed] [Google Scholar]
- 40.Moody WJ., Jr The ionic mechanism of intracellular pH regulation in crayfish neurones. J Physiol (Lond) 1981;316:293–308. doi: 10.1113/jphysiol.1981.sp013788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moudy AM, Yamada KA, Rothman SM. Rapid desensitization determines the pharmacology of glutamate neurotoxicity. Neuropharmacology. 1994;33:953–962. doi: 10.1016/0028-3908(94)90153-8. [DOI] [PubMed] [Google Scholar]
- 42.Nedergaard M, Goldman SA, Desai S, Pulsinelli WA. Acid-induced death in neurons and glia. J Neurosci. 1991;11:2489–2497. doi: 10.1523/JNEUROSCI.11-08-02489.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nemoto EM, Frinak S. Brain tissue pH after global brain ischemia and barbituate loading in rats. Stroke. 1981;12:77–82. doi: 10.1161/01.str.12.1.77. [DOI] [PubMed] [Google Scholar]
- 44.Nurse S, Corbett D. Neuroprotection after several days of mild, drug-induced hypothermia. J Cereb Blood Flow Metab. 1996;16:474–480. doi: 10.1097/00004647-199605000-00014. [DOI] [PubMed] [Google Scholar]
- 45.Patneau DK, Vyklicky L, Jr, Mayer ML. Hippocampal neurons exhibit cyclothiazide-sensitive rapidly desensitizing responses to kainate. J Neurosci. 1993;13:3496–3509. doi: 10.1523/JNEUROSCI.13-08-03496.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pellegrini-Giampietro DE, Zukin RS, Bennett MV, Cho S, Pulsinelli WA. Switch in glutamate receptor subunit gene expression in CA1 subfield of hippocampus following global ischemia in rats. Proc Natl Acad Sci USA. 1992;89:10499–10503. doi: 10.1073/pnas.89.21.10499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Plum F. What causes infarction in ischemia brain? Neurology. 1983;33:222–233. doi: 10.1212/wnl.33.2.222. [DOI] [PubMed] [Google Scholar]
- 48.Rose K, Goldberg MP, Choi DW. Cytotoxicity in murine cortical cell culture. In: Tyson CA, Frazier JM, editors. In vitro biological methods, methods in toxicology. Academic; San Diego: 1992. pp. 46–60. [Google Scholar]
- 49.Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor. Trends Neurosci. 1987;10:299–302. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- 50.Sheardown MJ, Nielsen EO, Hansen AJ, Jacobsen P, Honore T. 2,3-Dihydroxy-6-nitro-7-sulfamoylbenzo(F)quinoxaline: a neuroprotectant for cerebral ischemia. Science. 1990;247:571–574. doi: 10.1126/science.2154034. [DOI] [PubMed] [Google Scholar]
- 51.Sheardown MJ, Suzdak PD, Nordholm L. AMPA, but not NMDA, receptor antagonism is neuroprotective in gerbil global ischemia, even when delayed 24 hr. Eur J Pharmacol. 1993;236:347–353. doi: 10.1016/0014-2999(93)90470-3. [DOI] [PubMed] [Google Scholar]
- 52.Siemkowicz E, Hansen AJ. Brain extracellular ion composition and EEG activity following 10 min ischemia in normo- and hyperglycemic rats. Stroke. 1981;12:236–240. doi: 10.1161/01.str.12.2.236. [DOI] [PubMed] [Google Scholar]
- 53.Siesjo BK, Wieloch T. Epileptic brain damage: pathophysiology and neurochemical pathology. Adv Neurol. 1986;44:813–847. [PubMed] [Google Scholar]
- 54.Siesjo BK. Acidosis and ischemic brain damage. Neurochem Pathol. 1988;9:31–88. doi: 10.1007/BF03160355. [DOI] [PubMed] [Google Scholar]
- 55.Simon RP, Swan JH, Griffiths T, Meldrum BS. Blockade of N-methyl-d-aspartate receptors may protect against ischemic damage in brain. Science. 1984;226:850–852. doi: 10.1126/science.6093256. [DOI] [PubMed] [Google Scholar]
- 56.Smith SE, Meldrum BS. Cerebroprotective effect of a non-N-methyl-d-aspartate antagonist, GYKI 52466, after focal ischemia in the rat. Stroke. 1992;23:861–864. doi: 10.1161/01.str.23.6.861. [DOI] [PubMed] [Google Scholar]
- 57.Tang CM, Dichter M, Morad M. Modulation of the N-methyl-d-aspartate channel by extracellular H+. Proc Natl Acad Sci USA. 1990;87:6445–6449. doi: 10.1073/pnas.87.16.6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tombaugh GC, Sapolsky RM. Mild acidosis protects hippocampal neurons from injury induced by oxygen and glucose deprivation. Brain Res. 1990;506:343–345. doi: 10.1016/0006-8993(90)91277-n. [DOI] [PubMed] [Google Scholar]
- 59.Tombaugh GC, Sapolsky RM. Evolving concepts about the role of acidosis in ischemic neuropathology. J Neurochem. 1993;61:793–803. doi: 10.1111/j.1471-4159.1993.tb03589.x. [DOI] [PubMed] [Google Scholar]
- 60.Traynelis SF, Cull-Candy SG. Proton inhibition of N-methyl-d-aspartate receptors in cerebellar neurons. Nature. 1990;345:347–350. doi: 10.1038/345347a0. [DOI] [PubMed] [Google Scholar]
- 61.Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 1991;252:1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- 63.Weiss JH, Turetsky DM, Wilke G, Choi DW. AMPA/kainate receptor-mediated damage to NADPH-diaphorase containing neurons is Ca2+ dependent. Neurosci Lett. 1994;167:93–96. doi: 10.1016/0304-3940(94)91035-9. [DOI] [PubMed] [Google Scholar]
- 64.Yamada KA, Rothman SM. Diazoxide blocks glutamate desensitization and prolongs excitatory postsynaptic currents in rat hippocampal neurons. J Physiol (Lond) 1992;458:409–423. doi: 10.1113/jphysiol.1992.sp019424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ying HS, Weishaupt J, Grabb M, Canzoniero SMT, Sensi SL, Monyer H, Choi DW. AMPA receptor expression in cultured rat hippocampal neurons following sublethal oxygen-glucose deprivation. Soc Neurosci Abstr. 1996;22:597. doi: 10.1523/JNEUROSCI.17-24-09536.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zorumski C, Yamada KA, Price MT, Olney JW. A benzodiazepine recognition site associated with the non-NMDA glutamate receptor. Neuron. 1993;10:61–67. doi: 10.1016/0896-6273(93)90242-j. [DOI] [PubMed] [Google Scholar]