

Abstract
The cerebral cortex of mice with a targeted disruption in the gene for cyclin-dependent kinase 5 (cdk5) is abnormal in its structure. Bromodeoxyuridine labeling reveals that the normal inside-out neurogenic gradient is inverted in the mutants; earlier born neurons are most often found superficial to those born later. Despite this, the early preplate layer separates correctly and neurons with a normal, pyramidal morphology can be found between true marginal zone and subplate. Consistent with their identity as layer VI corticothalamic neurons, they can be labeled by DiI injections into thalamus. The DiI injections also reveal that the trajectories of thecdk5−/− thalamocortical axons are oblique and cut across the entire cortical plate, instead of being oriented tangentially in the subcortical white matter. We propose a model in which the cdk5−/− defect blocks cortical development at a heretofore undescribed intermediate stage, after the splitting of the preplate, but before the migration of the full complement of cortical neurons.
Keywords: neuronal migration, cdk5, reeler, cerebral cortical development, neuronal morphology, BrdU
The power of genetic mutations to reveal the central aspects of a variety of developmental processes has often been demonstrated. In recent years, an excellent example of this power has been provided by a rapid succession of studies describing mutations that influence the development of the cerebral cortex. These mutations have occurred in both experimental animals (reelerand scrambler) and in humans [X-linked subcortical laminar heterotopia (des Portes et al., 1998; Gleeson et al., 1998) and lissencephaly (Reiner et al., 1993; Hattori et al., 1994)]. As a whole, the resulting analyses have emphasized the importance of genetic contributions to the mechanics of corticogenesis, in particular the process of cell migration.
The cerebral cortex begins as a layer of cells in the early telencephalon known as the preplate (Boulder-Committee, 1970). As cells of the definitive cortex complete neurogenesis, they migrate into the preplate splitting it into two layers: a superficial marginal zone, containing Cajal-Retzius cells, and a deeper subplate layer (Marin-Padilla, 1978). Each additional cohort of cortical neurons migrates past subplate as well as earlier generated cortical plate neurons and assumes a position under the marginal zone. The result is an adult cerebral cortex that is built from inside-out according to neuronal birthdate (Angevine and Sidman, 1961). Afferent thalamocortical fibers usually run within subplate, superficial to efferents, such as the corticothalamics, which tend to run below subplate (Sheppard et al., 1991). Cells of the subplate may guide thalamocortical and corticothalamic axons (for review, see Allendoerfer and Shatz, 1994; Molnar and Blakemore, 1995), and provide trophic support for thalamic neurons (Ghosh and Shatz, 1993).
A well characterized mouse mutant with disruptions in the migration of cerebral cortical neurons is known as reeler (for review, see Caviness and Rakic, 1978; Goffinet, 1984; Gilmore and Herrup, 1997). All reeler alleles lead to a deficiency of reelin, a large extracellular protein produced by the Cajal-Retzius cells (D’Arcangelo et al., 1995; Ogawa et al., 1995) and subsequent inversion of neuronal layering with respect to birthdate: the earliest born neurons of the cortical plate are near the pial surface; later born neurons are found in deeper locations. In addition to this inversion of the normal inside-out neurogenic gradient, the preplate is never split into marginal zone and subplate. In simple terms, the mutation can be viewed as blocking cortical development at the preplate stage, because cells of the cortical plate never achieve their proper position between marginal zone and subplate. The reeler axon tracts are also disturbed. Whereas cortical efferents are found in their normal deep location, the thalamocortical afferent axons travel in abnormal bundles that run at oblique angles toward the pial surface through the cerebral cortex to connect with the displaced subplate, known as the superplate. The reeler mutation is mimicked by three newly described mutations that disrupt the gene for mDab-1, an src-family tyrosine kinase associated protein (Howell et al., 1997;Sheldon et al., 1997; Ware et al., 1997).
Normal corticogenesis is also blocked by engineered mutations in genes for cyclin dependent kinase 5 (Cdk5) and its activator, p35 (Ohshima et al., 1996; Chae et al., 1997). Cdk5 is a serine/ threonine kinase originally cloned via its homology to other cyclin dependent kinases (Hellmich et al., 1992; Lew et al., 1992; Meyerson et al., 1992). Although Cdk5 is expressed in most tissue types, enzymatically active kinase is only found within the brain (Tsai et al., 1993). Kinase activity requires the binding of neuronal specific activators, including p35 (Lew et al., 1994; Tsai et al., 1994) or its isoform p39 (Tang et al., 1995). Mutant cdk5−/−mice die in the perinatal period with severe defects in cerebral and cerebellar cortices and hippocampus, reminiscent of thereeler phenotype (Ohshima et al., 1996). It has been reported that p35−/− mice have an inverted layering (outside-in) neurogenic gradient and abnormal axonal patterns, but these mice are viable (Chae et al., 1997). The current report is a detailed description of the pattern of cortical development in cdk5−/− mice.
MATERIALS AND METHODS
Animals. Genotype was determined by Southern blot analysis (Ohshima et al., 1996). Because no phenotypic difference has been identified between Cdk5+/+ andCdk5+/− mice, it would appear that the wild-type allele is fully penetrant hence both +/+ and +/− mice are considered wild type for this study. The mice for this study were of a mixed 129/SV and C57BL/6 background. Little difference was observed among mutants from a number of different litters. Therefore, background is not thought to have a major effect on the phenotypes described in this manuscript. For all observations, at least three animals were used to confirm results. Embryos were immersion-fixed in 4% paraformaldehyde at room temperature, equilibrated in 20% sucrose/0.8% paraformaldehyde in PBS at 4°C, embedded in OCT (Tissue Tek), frozen in dry ice, and sectioned at 10 μm on a cryostat. For bromodeoxyuridine (BrdU) experiments, pregnant dams were injected with BrdU (100 μg/g, i.p.), and perinatal embryos were removed by cesarean section at embryonic day 18.5 (E18.5). The animals used in the preparation of this manuscript were housed and cared for in compliance with all relevant federal and local animal care guidelines. The Case Western Reserve University Animal Resource Center is fully accredited by the American Association for the Accreditation of Laboratory Animal Care.
Immunostaining. For all immunostaining protocols, antibodies were diluted in PBS/0.5% Tween and 5% (primary) or 20% (secondary) goat serum for antibody incubations. After permeabilization with HCl, BrdU was detected by rat anti-BrdU antibodies (Accurate Chemicals, Westbury, NY), diluted 1:6. The secondary antibody was an FITC-conjugated goat anti-rat IgG (Jackson ImmunoResearch, West Grove, PA) diluted 1:200. Anti-reelin antibody, G10, was diluted 1:1000 and visualized with RITC-conjugated goat anti-rabbit IgG (Cappell, West Chester, PA). Anti-chondroitin sulfate, CS56 (Sigma, St. Louis, MO) was diluted 1:100 and visualized with RITC-conjugated goat anti-mouse IgM (Cappell). Anti-L1CD, a rabbit polyclonal antiserum to the intracellular portion of the L1 protein (a generous gift of Dr. Vance Lemmon, Case Western Reserve University) was used at 1:1000 dilution. Secondary antibody was visualized by DAB reaction product as specified by Vectastain Elite protocol (Vector Laboratories, Burlingame, CA) and counterstained with hematoxylin. Anti-calretinin (Chemicon, Temecula, CA) was diluted 1:1000 and visualized with Cy3-conjugated anti-rabbit IgG (Jackson). All sections were examined by standard light or fluorescent microscopic techniques.
DiI experiments. Brains fixed with 4% paraformaldehyde from E18.5 embryos were bisected in the coronal plane caudal to the internal capsule, and solubilized DiI (3 mg/ml) was injected into thalamus. DiI was allowed to diffuse for 4–6 weeks while brains were stored in PBS with 0.8% paraformaldehyde at room temperature. Vibratome sections, 100 μm, were examined by fluorescent and confocal microscopic techniques.
Statistics. All SDs were obtained using Microsoft Excel 5.0 software.
RESULTS
Examination of cresyl violet-stained sections of embryonicCdk5−/− brains reveals a disruption in cerebral cortical morphology (Ohshima et al., 1996). To determine the layering pattern of cerebral cortical cells with respect to their final cell division or “birthday,” BrdU was injected into pregnant females at E12.5, to label the neurons of the deep cortical layers, or E15.5, to label the middle layers (Caviness, 1982). Because cdk5−/− mice die perinatally, embryos were killed for analysis at E18.5. In wild-type mice injected with BrdU at E12.5 (Fig.1A), BrdU-positive nuclei are located in the deep portions of cerebral cortex as expected (Caviness, 1982). In one experiment, the average distance of an E12.5 labeled cell from the pial surface was 313 ± 102 μm. By contrast, in littermate cdk5−/− mutant embryos, this same cohort is located much closer to the pial surface (Fig. 1B), at an average distance of 124 ± 59 μm. In wild-type embryos, the cells labeled by E15.5 BrdU injections are found in large numbers near the top of the developing cortical plate, although the 3 d survival after the BrdU injection is insufficient to allow many migratory cells to complete their transit of the intermediate zone (Fig. 1C). In mutant littermates, however, nearly all of the E15.5 BrdU-labeled nuclei were found in deeper locations (Fig. 1D).
Fig. 1.
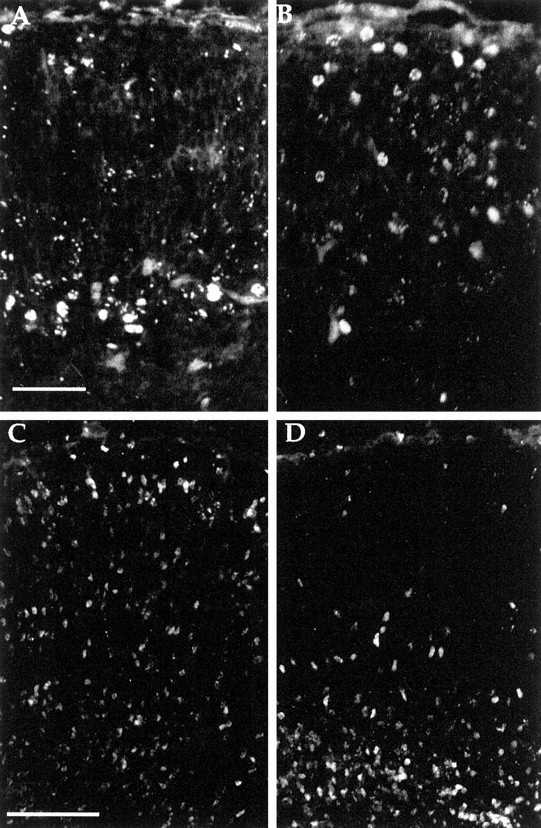
Coronal sections of cerebral cortex from E18.5 animals injected with BrdU at various gestational ages and stained with anti-BrdU antibody. A, B, Embryos from dams injected with BrdU at E12.5. Wild-type (A) cortex demonstrates expected pattern of labeling with BrdU-positive cells found in deeper portions of cerebral cortex. Thecdk5−/− (B) animals show BrdU-positive cells with a wider distribution including cells in positions near the pial surface. C, D, Embryos from dams injected with BrdU at E15.5. Cells in the wild type (C) were consistently found near the pial surface (and at lower positions, still in the process of migration). By contrast, in the cdk5−/− brain (D) very few cells were found near the pial surface. Indeed most appeared stalled in a layer below subplate. Scale bars: A, B, 50 μm; C, D, 100 μm.
The inversion of the normal inside-out layering pattern of cortical neurons with respect to their birthday is strongly reminiscent of thereeler mouse mutant. To explore this resemblance further, we stained developing cdk5−/− embryos with an anti-reelin antibody. Reelin, the product of the gene disrupted inreeler mice, is a large extracellular glycoprotein normally located among the cells of the marginal zone. As shown in Figure2, A and B, the intensity and location of the reelin antigen was identical in mutant and wild-type embryos.
Fig. 2.
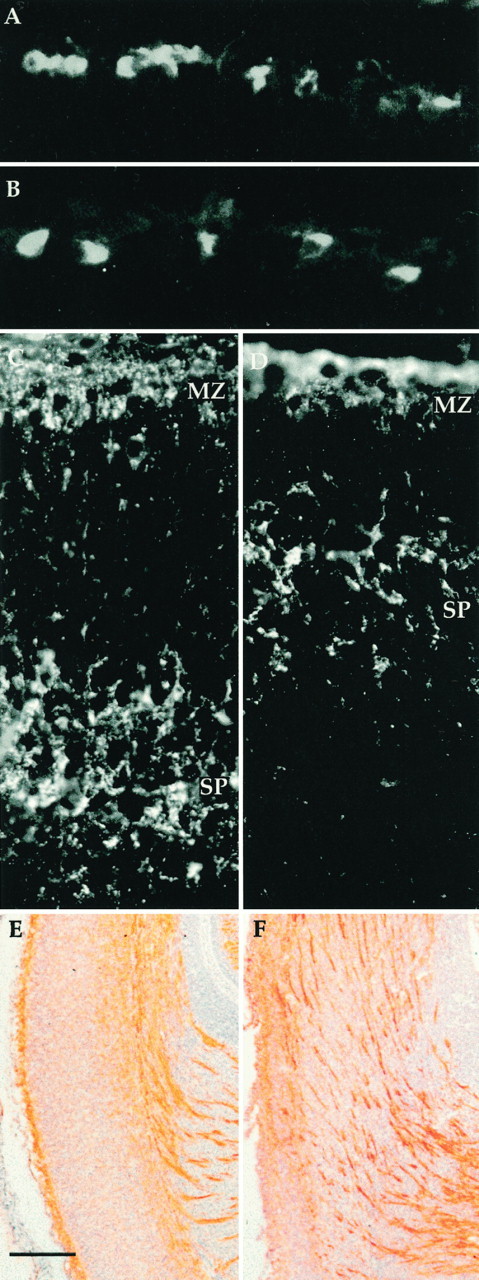
Anti-reelin antibody (G10) stains Cajal-Retzius cells in both wild-type (A) andcdk5−/− (B) E18.5 cerebral cortices. Anti-chondroitin sulfate antibody stains preplate derivatives in the marginal zone (MZ) and subplate (SP). Subplate is seen separated from marginal zone in both wild-type (C) andcdk5−/− (D) animals at E15.5. Note, however, the difference in the absolute distance of subplate separation. For full discussion see Results and Table 1. E, F, L1 staining of axons in wild-type (E) andcdk5−/− (F) embryonic cerebral cortex. Coronal sections of E18.5 embryos were stained with L1CD, an antibody to the cytoplasmic domain of L1 (a generous gift of Vance P. Lemon). Dorsal is toward thetop; the pial surface is to the left; the internal capsule can be seen in the bottom right. The wild-type section (E) shows the expected pattern of stained fascicles deep within cerebral cortex. In thecdk5−/− mutant (F) some fibers run deep, but many are found coursing obliquely through the cerebral cortex toward the pial surface. L1-positive bundles run parallel and near but not adjacent to the pial surface. Scale bars: E, F, 100 μm.
We next examined the condition of the subplate incdk5−/− mice. The proteoglycan chondroitin sulfate labels the preplate and its derivatives, the marginal zone and subplate (Sheppard et al., 1991; Bicknese et al., 1994). We used an antibody to chondroitin sulfate, CS56, to immunostain E15.5 mice that were either wild type (Fig. 2C) orcdk5−/− (Fig. 2D). Mutant cdk5−/− mice have two layers of immunostaining, one above and one below the developing cortical plate, suggesting that the preplate has been split into marginal zone and subplate. This finding is substantiated by the observation that L1 staining axons are found within the chondroitin sulfate staining portions of both mutant and wild-type subplate (data not shown). The splitting of the preplate into marginal zone and subplate represents a significant divergence between thecdk−/− and reelerphenotypes. Recall that in reeler the preplate remains as a single “superplate” layer (Goffinet, 1984; Sheppard and Pearlman, 1997). The arrest of cortical development incdk5−/− mice thus occurs at a stage after that described for reeler.
A series of late-gestation (E18.5) wild-type andcdk5−/− brains were stained with an antibody to L1, a marker of embryonic axons. In wild-type animals, as expected, axon bundles are found within the presumptive white matter tracts underlying the developing cerebral cortex with only thin single fibers penetrating the cortical plate (Fig. 2E). Incdk5−/− deficient mice, however, many broad axon fascicles have oblique trajectories across cerebral cortex ending in a noncontiguous band, parallel to the pial surface but not adjacent to it (Fig. 2F). These oblique axon bundles are reminiscent of the axon tracts in reeler mice with the exception that the reeler axons run immediately adjacent to the pial surface, rather than below it (Godfraind et al., 1988).
As the cell bodies of cortical neurons assume their adult location, the efferent and afferent axon populations follow a stereotyped growth pattern. To determine whether neurons that split the preplate were connected to thalamus, solubilized carbocyanine dye (DiI) was injected into the thalamus of E18.5 wild-type andcdk5−/− animals. Such an injection would be expected to label cell bodies of any corticothalamic neurons as well as the thalamocortical axons that project into the cerebral cortex. In wild-type embryos, as expected, DiI injections label cell bodies within the cortical plate and subplate as well as axons in the thalamocortical tracts (Fig.3A,C). Significantly, identical DiI injections in cdk5−/−mice also label cell bodies with a pyramidal morphology. These cells are found in a location above the subplate and below the marginal zone with an axon projecting toward the ventricular zone and a pyramidal morphology (Fig. 3B,D). However, most of the labeled cells within the mutant are within the subplate region or below. Although clearly projecting to the thalamus, cells below the cortical plate never have a pyramidal morphology.
Fig. 3.
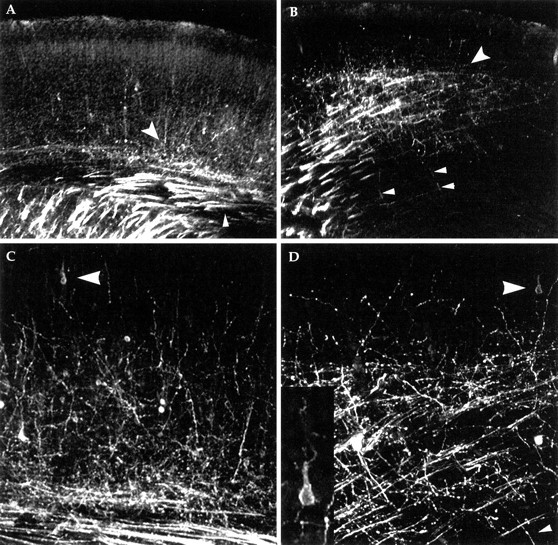
Thalamic injections of DiI reveal corticothalamic and thalamocortical axons in E18.5 wild-type (A, C) andcdk5−/− mutant (B, D) cerebral cortex. In A and B, the heavily labeled internal capsule is seen in the bottom left corner. The small arrowhead in Aindicates the location of the tract of cortical efferents (running in the presumptive subcortical white matter). In mutant (B), note the sparse horizontal stained fibers, near arrowheads, which are the corticothalamic tract. The large arrowheads in A andB mark the plexus of afferent fibers (the endings of the thalamocortical tract axons). In both genotypes, this plexus is found within the subplate. Above this, small fibers ascend into the developing cortical plate in both wild type and mutant. In wild type, these fibers can often be traced to a cell body of origin, superficial to the subplate, with a clear pyramidal morphology. In thecdk5−/− cerebral cortex (B), a number of thin fibers (B, D, small arrowheads), nearly perpendicular to the larger mass of fibers, can be traced to cell bodies deep to subplate, indicating they too are cortical efferents (connections not seen because of limitations in plane of focus). In the cdk5−/−mutant no labeled cell bodies located above the subplate had an abnormal morphology, whereas no cells in or below the subplate had the normal pyramidal morphology. Thalamic DiI injections also backfill neuronal cell bodies in or below the afferent plexus. Many of these are likely to be subplate cells but may also include aberrantly located and shaped layer VI neurons. The inset in Dshows a higher magnification of the DiI-filled cell, indicated by thelarge arrowhead.
The identity of the abnormal axon bundles described above are revealed through these studies as well. DiI stained fibers with abnormal oblique projections through the cerebral cortex are never connected to neuronal cell bodies, indicating that they are the axons of the thalamocortical tract. Single DiI-labeled axons extend from deep portions of cerebral cortex toward pial surface and are often connected to cell bodies (Fig.3B,D). These data indicate that corticothalamic tract neurons that reach thalamus have the correct projection pattern within deep white matter tracts while the fibers of the thalamic projection neurons appear at first to be abnormal, but are in reality within their correct substrate, the subplate.
We examined the possibility that the absence of Cdk5 function merely slows cortical development. We estimated the maturity of the developing cortical plate at various embryonic ages, using the distance from the pial surface to the top of the subplate as a reflection of the maturation process. The location of the subplate can be seen by staining with anti-calretinin antibody (Fig.4A–D) (Vogt-Weisenhorn et al., 1994; Fonseca et al., 1995). Normally, the cortex in lateral regions develops in advance of more medial regions, and, as would be predicted, the subplate to pial distance increases from medial to lateral in any one coronal section. As illustrated in Table1, this gradient is apparent at E15.5 in both mutant and wild-type cortex. Incdk5−/− mice, however, neither the absolute distances nor the mediolateral differences are as great as in wild-type. By E18.5, as cortical development continues in wild-type animals, the absolute distances between subplate and pial surface increase, and the mediolateral differential in cortical plate thickness is maintained. Strikingly, however, in the cortical plate of E18.5cdk5−/− mice, subplate is nearly equidistant from pia in all mediolateral regions. Furthermore, there is little additional thickening of the cortical plate in any region (beyond the values seen at E15.5). These observations are strong evidence in support of a block in cortical development that occurs at a stage shortly after the splitting of the preplate. After this occurs, the neurons that would subsequently migrate into the cortical plate to form the adult cortex end up in an ectopic position beneath the subplate.
Fig. 4.
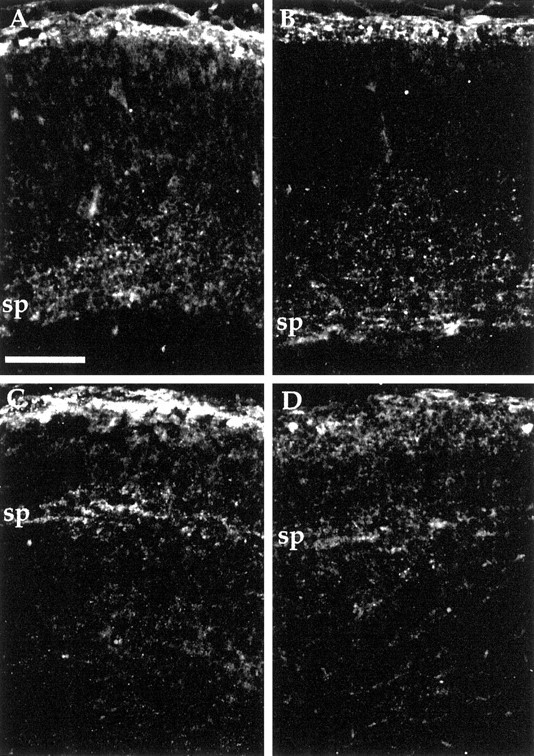
Anti-calretinin immunostaining of wild-type andCdk5−/− cerebral cortex at E18.5. Coronal sections of medial (A, C) and lateral (B, D) cerebral cortex indicate location of subplate cells and fibers. Subplate (sp) is located deep within cerebral cortex of wild-type (A, B) but near the pial surface ofCdk5−/− mice (C, D). Because A and B and C andD are from the same coronal section, direct comparisons can be made concerning their degree of development. Separation of subplate away from pial surface was used as a marker for development of the cortex and quantified in Table 1. Scale bar, 100 μm.
Table 1.
Cortical thickness as a measure of maturation in wild-type and cdk5−/− mutant
| Age/genotype | Lateral (μm) | Medial (μm) | Lateral − medial |
|---|---|---|---|
| E15.5/wt | 184 ± 13 | 74 ± 13 | 110 ± 18 |
| E18.5/wt | 381 ± 51 | 293 ± 77 | 88 ± 34 |
| E15.5/Cdk5−/− | 90 ± 11 | 55 ± 6 | 35 ± 13 |
| E18.5/Cdk5−/− | 102 ± 7 | 107 ± 13 | −5 ± 19 |
The distance between the top of the subplate and the pial surface was measured at two different ages in both wild-type (wt) andcdk5−/− mice. At E15.5, both L1 and CS56 immunoreactivity was used to define the superficial portion of subplate. At E18.5, L1 immunoreactivity alone was used to determine the upper boundary of subplate, because chondroitin sulfate immunoreactivity is no longer confined to preplate derivatives. Calretinin immunoreactivity (Fonseca et al., 1995) and thalamic DiI injection were used to confirm the location of subplate.
DISCUSSION
In this paper we present data describing the cerebral cortical phenotype of mice deficient of Cdk5. Abnormal cell movements lead us to propose that the block in cdk5−/−development defines a heretofore unexpected stage in the maturation of neocortex. Previous chemical or genetic manipulations have disrupted individual components of cortical development (cell migration, cell survival, or formation of entire cortical plate), but none has led to a block at a clear intermediate point in the acquisition of the stereotyped six-layered neocortex. The results of our studies document that, in the absence of Cdk5 function, cortical development is arrested at a stage after the preplate stage, but before completion of the cortical plate stage. The result is a four-tiered structure consisting of marginal zone (MZ), early cortical plate (ECP), subplate (SP), and a deeper layer of postmitotic neurons that we call the underplate (UP). In both wild-type (Fig. 5A) and cdk5−/− mice (Fig. 5B), marginal zone separates from subplate by the interstitial migration of layer VI cells. In wild-type cortex, neuronal migration past subplate to the marginal zone continues and the cortical plate thickens as illustrated in Table 1. By contrast, incdk5−/− mice, cortical plate formation stops at the newly defined ECP stage (and in keeping with this, the pial to subplate distance fails to increase in magnitude). Cells born after the earliest cortical plate neurons seem able to migrate away from ventricular zone but are incapable of passing subplate or the previously born neurons and thus pile up both in intermediate zone and underplate. This forms a stratum of seemingly postmigratory cells, trapped between ventricular zone and the bottom of subplate (Fig.5B).
Fig. 5.

Comparison of the cytoarchitectonics of wild-type (A) and mutant (B) E18.5 cerebral cortex. In the wild type, the marginal zone (MZ) is adjacent to the pial surface. The layer of differentiating cells marking the cortical plate (CP) is easily distinguished from the cells of the subplate (SP), which in turn are found above the relatively cell-sparse intermediate zone (IZ). The ventricular zone (VZ) is still present at this age. In thecdk5−/− mutant (B) the MZ is present as is a thin layer with some cells. Deep to this lie the layer of neurons of the early cortical plate (ECP). Below these cells is a layer resembling subplate, and deep to these cells is a thick tier of cells, the underplate (UP), that contains the later-generated cortical plate neurons. The IZ is present in its normal position but appears to have a higher density of cells, as if the backup in the underplate has extended to this depth. Scale bar, 100 μm.
Figure 6 is a diagram of the differences between wild-type, reeler, andcdk5−/− mice. Similarities among the phenotypes of reeler, reeler-like mutants (scrambler, yotari, andmdab-1−/−) (Howell et al., 1997;Sheldon et al., 1997; Ware et al., 1997), andcdk5−/− mice have opened the possibility that mDab-1 and Cdk5 may be in the same biochemical pathway as the reelin receptor. We feel that although the same cell types may be affected in both mdab−/− andcdk5−/− cerebral cortices, the differences in phenotypes make it unlikely that they are in a single biochemical pathway. An important conclusion illustrated by this summary is that the phenotypes ofcdk5−/− andp35−/− cerebral cortices have a significant difference despite the fact that p35 is an activator of Cdk5. The location of subplate incdk5−/− and p35−/−mice at E15.5 is similar. However, by P0, the subplate inp35−/− mice has fully separated from near marginal zone to the bottom of cortical plate (Kwon and Tsai, 1998). The cause of this difference in phenotype remains unknown. It may be attributable to delayed activation of other activators of Cdk5, such as p39, which is present in cerebral cortex by birth in rat (Cai et al., 1997). This would imply first that these other gene products are present in embryonic cortex and second that the blockage in neuronal positioning of subplate caused by the absence of Cdk5 activity is at least partly reversible.
Fig. 6.

Models of the proposed phenotypic differences of the early cortical plate stage in wild-type, reeler, andcdk5−/− mouse cerebral cortices. Phenotypic differences between wild-type and reelerdeveloping cortical plate have been reviewed elsewhere (Caviness and Rakic, 1978; Goffinet, 1984; Gilmore and Herrup, 1997). Ventricular zone cells (open ovals) lie at the bottom of the cerebral wall with radial glia (darkly stippled) running the length of the wall. Cells derived from the preplate, Cajal–Retzius and subplate cells, are lightly stippled, whereas reelin protein is represented by the speckles around the Cajal–Retzius cells (absent in reeler). Cells of the early cortical plate are cross-hatched, whereas later born cells are crisscrossed. Cortical efferents are represented by dark lines, whereas cortical afferents are light gray. Shape of cortical plate neurons is a representation of final phenotype rather than morphology at the stage depicted. See Discussion for details.
The normal, pyramidal morphology of some of the neurons incdk5−/− deserves further comment. Analysis of the reeler mutation raised the possibility that disruption of pyramidal morphology and cortical inversion are directly linked. In the cdk5−/− cerebral cortex, however, the earliest cortical plate neurons are superficial to all later born cells yet still have a pyramidal morphology. One important difference between early cortical plate neurons in reelerand cdk5−/− mice that may explain this disparity is the normal location of some layer VI cells between the subplate and marginal zone in cdk5−/−mice. Perhaps only within the environment between subplate and marginal zone can cortical neurons achieve a normal pyramidal morphology. In this position, young neurons would be able to interact with Cajal–Retzius cells of the marginal zone and their secreted factor, reelin. This interaction might then serve as a mechanism to confer a pyramidal morphology.
A persistent question raised by these and previous findings is the identity of substrate(s) for Cdk5 kinase and the role that the phosphorylation state of these molecules might have in the process of cerebral cortical development. Previous in vitro studies have shown that Cdk5 can phosphorylate neurofillament as well as other proteins, but no gross alterations have yet been found in the pattern of protein phosphorylation in thecdk5−/− mice (H.C. Pant, Veeranna, N.D. Amin, and A.B. Kulkarni, unpublished observations). The findings ofNikolic et al. (1996) suggest that one function of Cdk5 activity may be to assist in the process of axon elongation. Although thecdk5 mutant has abundant fiber tracts (Fig.2F), we consistently observed that DiI labeling intensity was reduced compared with wild-type (Fig. 3), suggesting possible delays or disruptions in axon growth to thalamus. However, because we did not perform quantitative injections of DiI into thalamus, no firm conclusions can be drawn from this study.
Our analysis of Cdk5−/− mice suggests that there are intermediate steps in the development of the cortical plate. It is intriguing to consider this conclusion in the light of the work of McConnell and collaborators (McConnell, 1995; Franz and McConnell, 1996), which shows that neurons have differing potentials depending on their developmental stage. It has also been proposed that the earliest born neurons in reeler mice are functionally different from later neurons (Hoffrarth et al., 1995). Taken together with our results, we believe the data suggest that the cellular migration mechanism of the early neurons is Cdk5 independent and distinct from that of later born cells. This observation is consistent with the growing concept that the progenitors of adult cerebral cortical neurons are not a uniform population, differing only in the time at which they exit the cell cycle and in their adult phenotype. Rather, the behavior of the precursor cells suggests that they also have different genetic requirements for proper migration. Among the questions posed by this interpretation is why all but the earliest born cortical plate cells are located under the subplate. One possibility is that early and late migrating neurons use different mechanisms to move from the ventricular zone to the marginal zone. Perhaps the earlier born cells move by a Cdk5-independent process while the later cell movements require the kinase. Although most CNS migration is accomplished by active movement along a radial glial cell, there is evidence that developing neurons can also move by translocation of the nucleus inside a preexisting radial process of the neuron itself (Morest, 1970; Shoukimas and Hinds, 1978; Nowakowski and Rakic, 1979; Brittis et al., 1995). Early cortical neurons might use this method of moving their cell body to cortical plate. Because it would represent a totally different cell biological process, it might be shown to be Cdk5-independent.
Footnotes
This work was supported by National Institutes of Health Grant NS20591 to K.H. and by National Institute of Dental Research, and National Institute of Neurological Diseases and Stroke, DIR, to A.K.
E.C.G. and T.O. contributed equally to this work.
Correspondence should be addressed to Dr. Edward C. Gilmore, E504 School of Medicine, Case Western Reserve Medical School, 10900 Euclid Avenue, Cleveland, OH 44106.
REFERENCES
- 1.Allendoerfer KL, Shatz CJ. The subplate, a transient neocortical structure: its role in the development of connections between thalamus and cortex. Annu Rev Neurosci. 1994;17:185–218. doi: 10.1146/annurev.ne.17.030194.001153. [DOI] [PubMed] [Google Scholar]
- 2.Angevine JB, Sidman RL. Autoradiographic study of cell migration during histogenesis of cerebral cortex in mice. Nature. 1961;192:766–768. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- 3.Bicknese AR, Sheppard AM, O’Leary DDM, Pearlman AL. Thalamocortical axons extend along a chondroitin sulfate proteoglycan-enriched pathway coincident with the neocortical subplate and distinct from the efferent path. J Neurosci. 1994;14:3500–3510. doi: 10.1523/JNEUROSCI.14-06-03500.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boulder-Committee. Embryonic vertebrate CNS: revised terminology. Anat Rec. 1970;166:257–261. doi: 10.1002/ar.1091660214. [DOI] [PubMed] [Google Scholar]
- 5.Brittis P, Meiri K, Dent E, Silver J. The earliest patterns of neuronal differentiation and migration in the mammalian central nervous system. Exp Neurol. 1995;134:1–12. doi: 10.1006/exnr.1995.1031. [DOI] [PubMed] [Google Scholar]
- 6.Cai X-H, Tomizama K, Tang D, Lu Y-F, Moriwaki A, Tokuda M, Nagahata S, Hatase O, Matsui H. Changes in the expression of novel Cdk5 activator messenger RNA (p39nck5ai mRNA) during rat brain development. Neurosci Res. 1997;28:355–360. doi: 10.1016/s0168-0102(97)00063-1. [DOI] [PubMed] [Google Scholar]
- 7.Caviness VS., Jr Neocortical histogenesis in normal and reeler mice: a developmental study based upon [3H]thymidine autoradiography. Dev Brain Res. 1982;4:293–302. doi: 10.1016/0165-3806(82)90141-9. [DOI] [PubMed] [Google Scholar]
- 8.Caviness VS, Jr, Rakic P. Mechanisms of cortical development: a view from mutations in mice. Annu Rev Neurosci. 1978;1:297–326. doi: 10.1146/annurev.ne.01.030178.001501. [DOI] [PubMed] [Google Scholar]
- 9.Chae T, Kwon YT, Bronson R, Dikkes P, Li E, Tsai L-H. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- 10.D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 11.des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrié A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Ctala M, Kahn A, Beldjord C, Chelly J. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- 12.Fonseca M, del Rio JA, Martinez A, Gomez S, Soriano E. Development of calretinin immunoreactivity in the neocortex of the rat. J Comp Neurol. 1995;361:177–192. doi: 10.1002/cne.903610114. [DOI] [PubMed] [Google Scholar]
- 13.Franz GD, McConnell SK. Restrictioni of late cerebral cortical progenitors to an upper-layer fate. Neuron. 1996;17:55–61. doi: 10.1016/s0896-6273(00)80280-9. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh A, Shatz CJ. A role for subplate neurons in the patterning of connections from thalamus to neocortex. Development. 1993;117:1031–1047. doi: 10.1242/dev.117.3.1031. [DOI] [PubMed] [Google Scholar]
- 15.Gilmore EC, Herrup K. Cortical development: layers of complexity. Curr Biol. 1997;7:R231–R234. doi: 10.1016/s0960-9822(06)00108-4. [DOI] [PubMed] [Google Scholar]
- 16.Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signalling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 17.Godfraind C, Schachner M, Goffinet AM. Immunohistological localization of cell adhesion molecules L1, J1, N-CAM, and their common carbohydrate L2 in the embryonic cortex of normal and reeler mice. Dev Brain Res. 1988;42:99–111. doi: 10.1016/0165-3806(88)90205-2. [DOI] [PubMed] [Google Scholar]
- 18.Goffinet AM. Events governing organization of postmigratory neurons: studies on brain development in normal and reeler mice. Brain Res. 1984;319:261–296. doi: 10.1016/0165-0173(84)90013-4. [DOI] [PubMed] [Google Scholar]
- 19.Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. Miller-Dieker lissencephaly gene encodes a subunit of brain platelet-activating factor acetylhydrolase. Nature. 1994;370:216–218. doi: 10.1038/370216a0. [DOI] [PubMed] [Google Scholar]
- 20.Hellmich MR, Pant HC, Wada E, Batty JF. Neuronal cdc2-like kinase: a cdc-related protein kinase with predominately neuronal expression. Proc Natl Acad Sci USA. 1992;89:10867–10871. doi: 10.1073/pnas.89.22.10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoffrarth RM, Johnston GJ, Krushel LA, van der Kooy D. The mouse mutantion reeler causes increased adhesion within a subpopulation of early postmitotic cortical neurons. J Neurosci. 1995;15:4838–4850. doi: 10.1523/JNEUROSCI.15-07-04838.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;289:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- 23.Kwon Y, Tsai LW. A novel disruption of cortical development in p35−/− mice distinct from reeler. J Comp Neurol. 1998;395:510–522. doi: 10.1002/(sici)1096-9861(19980615)395:4<510::aid-cne7>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 24.Lew J, Winkfein RJ, Paudel HK, Wang JH. Brain proline-directed protein kinase is a neurofilament kinase which displays high sequence homology to p34cdc2. J Biol Chem. 1992;267:25922–25926. [PubMed] [Google Scholar]
- 25.Lew J, Winkfein RJ, Huang QQ, Qi Z, Aebersold R, Hunt T, Wang JH. Neuronal cdc2-like kinase is a complex of cyclin-dependent kinase 5 and a novel brain-specific regulatory subunit. Nature. 1994;371:423–426. doi: 10.1038/371423a0. [DOI] [PubMed] [Google Scholar]
- 26.Marin-Padilla M. Dual origin of the mammalian neocortex and evolution of the cortical plate. Anat Embryol. 1978;152:109–126. doi: 10.1007/BF00315920. [DOI] [PubMed] [Google Scholar]
- 27.McConnell SK. Constructing the cerebral cortex: neurogenesis and fate determination. Neuron. 1995;15:761–768. doi: 10.1016/0896-6273(95)90168-x. [DOI] [PubMed] [Google Scholar]
- 28.Meyerson M, Enders GH, Wu CL, Su LK, Gorka C, Nelson C, Harlow E, Tsai LH. A family of human cdc2-related protein kinases. EMBO J. 1992;11:2909–2917. doi: 10.1002/j.1460-2075.1992.tb05360.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Molnar Z, Blakemore C. How do thalamic axons find their way to the cortex? Trends Neurosci. 1995;18:389–397. doi: 10.1016/0166-2236(95)93935-q. [DOI] [PubMed] [Google Scholar]
- 30.Morest DK. A study of neurogenesis in the forebrain of the opossum pouch young. Z Anat Entwicklungsgesch. 1970;130:265–305. doi: 10.1007/BF00520999. [DOI] [PubMed] [Google Scholar]
- 31.Nikolic M, Dudek H, Kwon YT, Ramos YFM, Tsai LH. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996;10:816–825. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- 32.Nowakowski RS, Rakic P. The mode of migration of neurons to the hippocampus: a golgi and electon microscopic analysis in foetal rhesus monkey. J Neurocytol. 1979;8:697–718. doi: 10.1007/BF01206671. [DOI] [PubMed] [Google Scholar]
- 33.Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- 34.Ohshima T, Ward JM, Huh CG, Longnecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH. Isolation of a Miller-Dieker lissencephaly gene containing G-protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 36.Sheldon M, Rice DS, D’arcangelo G, Yoneshima H, Nakajima K, Mikoshiba K, Howell BW, Cooper JA, Goldowitz D, Curran T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature. 1997;389:730–733. doi: 10.1038/39601. [DOI] [PubMed] [Google Scholar]
- 37.Sheppard AM, Pearlman AL. Abnormal reorganization of preplate neurons and their associated extracellular matrix: an early manifestation of altered neocortical development in the reeler mutant mouse. J Comp Neurol. 1997;378:173–179. doi: 10.1002/(sici)1096-9861(19970210)378:2<173::aid-cne2>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 38.Sheppard AM, Hamilton SK, Pearlman AL. Changes in the distribution of extracellular matrix components accompany early morphological events of mammalian cortical development. J Neurosci. 1991;11:3928–3942. doi: 10.1523/JNEUROSCI.11-12-03928.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shoukimas GM, Hinds JW. The development of the cerebral cortex in the embryonic mouse: an electron microscopic serial section analysis. J Comp Neurol. 1978;179:795–830. doi: 10.1002/cne.901790407. [DOI] [PubMed] [Google Scholar]
- 40.Tang D, Yeung J, Li KY, Matsushita M, Tomizama K, Matsui H, Hatase O, Wang JH. An isoform of neuronal cyclin dependent kinase 5. J Biol Chem. 1995;270:26897–26903. doi: 10.1074/jbc.270.45.26897. [DOI] [PubMed] [Google Scholar]
- 41.Tsai LH, Delalle I, Caviness VS, Jr, Chae R, Harlow E. Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development. 1993;119:1029–1040. doi: 10.1242/dev.119.4.1029. [DOI] [PubMed] [Google Scholar]
- 42.Tsai LH, Delalle I, Caviness VS, Jr, Chae T, Harlow E. p35 is a neuronal-specific regulatory subunit of cyclin-dependent kinase 5. Nature. 1994;271:419–423. doi: 10.1038/371419a0. [DOI] [PubMed] [Google Scholar]
- 43.Vogt-Weisenhorn DM, Weruaga-Prieto E, Celio MR. Localization of calretinin in cells of layer I (Cajal-Retzius cells) of the developing cortex of the rat. Dev Brain Res. 1994;82:293–297. doi: 10.1016/0165-3806(94)90171-6. [DOI] [PubMed] [Google Scholar]
- 44.Ware ML, Fox JW, Gonzalez JL, Davis NM, Lambert de Rouvroit C, Russo CJ, Chua SC, Goffinet AM, Walsh CA. Aberrant splicing of a mouse disabled homolog, mdab1, in the scrambler mouse. Neuron. 1997;19:239–249. doi: 10.1016/s0896-6273(00)80936-8. [DOI] [PubMed] [Google Scholar]