

Abstract
The involvement of metabotropic glutamate receptors (mGluRs) in hippocampal long-term potentiation (LTP) is a matter of controversial debate. Using [Ca2+]imeasurements by confocal laser scanning microscopy and field recordings of EPSPs (fEPSPs) in the hippocampal CA1-region, we found that the efficacy of the broad-spectrum mGluR-antagonist (S)-α-methyl-4-carboxyphenylglycine (MCPG) and of (S)-4-carboxy-phenylglycine (4-CPG), a selective antagonist at class I mGluRs, in LTP is contingent on the tetanization strength and the resulting [Ca2+]iresponse. As indicated by experiments in which we blocked voltage-dependent calcium channels (VDCCs) and intracellular Ca2+ stores (ICSs), the functional significance of class I mGluRs in LTP is confined to certain types of potentiation, which are induced by weak tetanization protocols and require the release of Ca2+ from ICSs for induction. During strong tetanic stimulation, this Ca2+ source is functionally bypassed by activating VDCCs.
Keywords: class I metabotropic glutamate receptors, long-term potentiation, intracellular calcium concentration, intracellular calcium stores, voltage-dependent calcium channels, hippocampus
The induction of long-term potentiation (LTP), an activity-dependent form of synaptic plasticity, was proven to require the influx of Ca2+ through the NMDA type of glutamate receptors (NMDARs), under most experimental conditions (Bliss and Collingridge, 1993; Malenka, 1994). However, because an activation of NMDARs alone results in decremental short-term potentiation (Collingridge et al., 1983; Kauer et al., 1988), other mechanisms such as a coactivation of metabotropic glutamate receptors (mGluRs) have been suggested to be involved.
Until now, eight subtypes of mGluRs have been described and assigned to three different classes according to their sequence homology, pharmacological characterization, and coupling to second messenger pathways. While the activation of class I mGluRs (mGluR1, 5) stimulates phosphatidylinositol 4,5-bisphosphate hydrolysis [producing inositol 1,4,5-trisphosphate (IP3) and diacylglycerol], mGluRs of class II (mGluR2, 3) and class III (mGluR4, 6, 7, 8) are negatively coupled to adenylyl cyclase (Nakanishi, 1994; Conn and Pin, 1997).
The involvement of mGluRs in hippocampal synaptic plasticity has been a matter of controversial debate during the last few years. In particular, experiments using the class I/II specific antagonist (S)-α-methyl-4-carboxyphenylglycine (MCPG) yielded conflicting results. While some authors reported an inhibition of LTP (Bashir et al., 1993; Bortolotto et al., 1994; Brown et al., 1994;Richter-Levin et al., 1994; Little et al., 1995; Riedel et al., 1995), in other studies the MCPG actions could not be confirmed (Chinestra et al., 1993; Manzoni et al., 1994; Selig et al., 1995; Thomas and O’Dell, 1995). The issue of whether MCPG-sensitive mGluRs are involved in NMDAR-dependent synaptic plasticity was further confounded by the contrasting results of studies using mGluR1 and mGluR5 knock-out mice (Aiba et al., 1994; Conquet et al., 1994; Lu et al., 1997).
An intriguing possibility for resolving the controversy was provided by the “molecular switch hypothesis” (Bortolotto et al., 1994). According to this hypothesis, activation of mGluRs before LTP sets an input-specific molecular switch that then negates the necessity of further mGluR activation during LTP induction. However, other groups failed to confirm the existence of such a molecular switch (Selig et al., 1995; Thomas and O’Dell, 1995), giving a clear indication that the switch is not a general mechanism of synaptic plasticity but rather confined to certain experimental conditions.
Therefore, we pursued another idea to approach this question and focused on the effects of class I mGluRs on the IP3-mediated Ca2+ release from intracellular Ca2+ stores (ICSs) (Murphy and Miller, 1988; Berridge, 1993; Jaffe and Brown, 1994; Shirasaki et al., 1994;Phenna et al., 1995). Several laboratories have shown that the rise in intracellular Ca2+ concentration ([Ca2+]i) is a critical factor for LTP induction (Lynch et al., 1983; Malenka et al., 1988, 1992) and that the contribution of Ca2+ from ICSs may represent a critical factor for generating a long-lasting potentiation (Harvey and Collingridge, 1992; Behnisch and Reymann, 1995).
We suggest that this Ca2+ source should be particularly important under conditions where Ca2+entry through NMDARs and voltage-dependent calcium channels (VDCCs) does not attain the threshold concentration for triggering subsequent transduction pathways that are critical for LTP maintenance.
MATERIALS AND METHODS
Hippocampal slice preparation
Hippocampal slices were prepared from male rats (7–8 weeks old) of the Wistar outbred strain MOL: WIST (SHOE). After decapitation and dissection of the hippocampus, 400-μm-thick slices were cut in cold oxygenated physiological solution [artificial CSF (ACSF) (in mm): NaCl 124, KCl 4.9, MgSO4 1.3, CaCl2 2.5, KH2PO4 1.2, NaHCO3 25.6, d-glucose 10, saturated with 95% O2, 5% CO2, pH 7.4] using a tissue chopper. The slices were submerged and permanently perfused with 32°C ACSF.
Electrophysiological long-term recordings
Synaptic responses were elicited by stimulation of the Schaffer collateral–commissural fibers in stratum radiatum of the CA1 region using lacquer-coated stainless steel stimulating electrodes. Glass electrodes (filled with ACSF, 1–4 MΩ) were placed in the apical dendritic layer to record field EPSPs (fEPSPs). The initial slope of the fEPSP was used as a measure of this potential. The test stimulation strength was adjusted to 35% of the maximum. During baseline recording, four single stimuli (10 sec interval) were averaged every 5 min. After tetanization, recordings were taken every 10 min over a period of at least 120 min. Once a stable baseline had been established, LTP was induced by one of the following tetanization paradigms.
Strong tetanization. Three trains of 500 msec duration at 100 Hz and 0.2 msec pulse width, separated by 2 min intervals, induced a stable potentiation of fEPSP of at least 180 min in control experiments.
Weak tetanization. (1) Four paired pulses (10 msec interval, 0.2 msec pulse width) were applied at the theta frequency of 5 Hz. The tetanization was strengthened either by adding two more paired pulses up to eight × two pulses or further by increasing the number of pulses (eight × four). (2) A single train of 400 msec duration at 100 Hz and 0.2 msec pulse width was applied. These weak tetanization protocols triggered a potentiation that returned to baseline level within 180 min.
In some experiments, two independent pathways were stimulated in the same slice by placing the stimulation electrodes symmetrically to the recording electrode into the apical dendritic layer at a different distance from the pyramidal layer.
The mGluR antagonists MCPG and (S)-4-carboxyphenylglycine (4-CPG) (Tocris, Bristol, UK) were dissolved in ACSF and bath-applied from 10 min before until 5 min after tetanization. Thapsigargin (Calbiochem, Bad Soden, Germany), a potent and selective inhibitor of intracellular Ca2+ pumps, and the L-type VDCC blocker nimodipine (Sigma, Deisenhofen, Germany) were initially dissolved in dimethylsulfoxide (DMSO) and further diluted with ACSF (final concentration of DMSO <0.01%). Thapsigargin was added to the bath from 30 min before until 5 min after tetanus; nimodipine was added according to the same time schedule as MCPG and 4-CPG. The drugs were applied either alone or in combination, as indicated in the text. All solutions were adjusted to pH 7.4. For statistical analysis, the Mann–Whitney U test (independent samples) and the Wilcoxon matched pairs signed rank test were used with a significance level ofp < 0.05.
Confocal microscopy
Experiments were performed on ventral transverse hippocampal slices (250 μm thick) from 20-d-old male Wistar rats in the same solutions and conditions as the extracellular experiments.
Pyramidal CA1 neurons and their Ca2+ transients were visualized with a Odyssey XL confocal laser scanning microscope (Noran Instruments, Middleton, WI) mounted on an Axioskop-FS upright microscope (Zeiss, Jena, Germany). Image acquisition was performed using Intervision software (Noran Instruments) on a Silicon Graphics Indy workstation. All recordings were performed with a 488 nm excitation filter and a 515 nm long-pass emission filter. The slit aperture on the photomultipliers set was adjusted to 100 μm width. A 40× water-immersion objective (NA = 0.75) was used to visualize neurons.
CA1 pyramidal cells were impaled with potassium acetate (2m)-filled borosilicate glass microelectrodes (80–150 MΩ) (Clark, Pangbourne, UK). The tips of these sharp electrodes were filled with the Ca2+-sensitive dye Calcium Green-1 (2 mm; Molecular Probes, Leiden, Netherlands). For the experiments involving bath application of 4-CPG, and coapplication of 4-CPG and nimodipine, 30 mm QX-314 (Alomone Labs, Jerusalem, Israel) was added to the dye solution. A monopolar stimulating electrode was positioned in CA1 stratum radiatum. Only neurons with a membrane potential below −55 mV [recorded with an npi SEC 1L-amplifier (NPI Electronic, Tamm, Germany) at bridge-mode] were used. The electrophysiological properties of the neurons were controlled during the whole experiment. The dye was injected into the neurons by applying a steady-state hyperpolarizing current of 100–400 pA for 15–20 min. Well stained, in focus dendrite regions 80–200 μm from soma were chosen for recording the Ca2+responses to different tetani. Tetanizations were performed in a time interval of 2 min with biphasic pulses. The changes in fluorescence intensity were averaged over 66.7 msec (average of eight images). For data analysis, four regions of interest (ROI) were selected on the dendritic tree. The fluorescence intensities of these four ROI were averaged, and a background correction was performed, i.e., a nearby nonactive region of the same size as the recording region was measured in parallel and subtracted. The results were given asF/F0 wherebyF0 was the averaged intensity before the tetanization.
RESULTS
Because most of the studies that addressed the functional role of mGluRs in LTP were performed in the CA1 region of the hippocampus, we recorded fEPSPs in the apical dendritic layer of this area by stimulating the Schaffer collateral–commissural fibers. The initial slope of the fEPSPs was used as a measure of synaptic responses. In the first series of experiments we tested whether the ability of mGluR antagonists to block LTP depends on tetanization strength. In the strong tetanization paradigm (3 × 100 Hz, 500 msec, 2 min interval between trains), which led to a stable potentiation lasting >240 min under control conditions, the application of MCPG (400 μm) affected neither the initial magnitude (MCPG group: 213 ± 12%, n = 7; control: 220 ± 7%,n = 7) nor the time course of potentiation (after 240 min: 133 ± 6% and 137 ± 5%, respectively) (Fig.1A). Because MCPG is a broad-spectrum antagonist acting on class I mGluRs, and with a somewhat lower affinity at mGluRs of class II (Davies et al., 1995; Sekiyama et al., 1996), we repeated the same experiments with 4-CPG, which represents in the employed concentration range a specific antagonist toward class I mGluRs. 4-CPG (100 μm) had no effect on the initial potentiation (4-CPG group: 208 ± 9%,n = 7; control: 206 ± 10%, n = 7) and the maintenance of LTP (240 min after tetanus: 135 ± 8% and 132 ± 6%, respectively) (Fig. 1B). From these experiments, we concluded that activation of mGluR class I is not mandatory for the induction of a long-lasting potentiation by strong tetanization.
Fig. 1.

MCPG (400 μm; n= 7) (A) and 4-CPG (100 μm;n = 7) (B) did not influence LTP induced by a strong tetanization (3 × 100 Hz, 500 msec, 2 min interval between trains). The potentiation persisted for at least 240 min. The tetanus was applied at the time point 0. Horizontal bars under the time scale indicate the time of drug application. Analog traces represent typical recordings of single experiments taken 10 min before tetanization (1) and 120 min after tetanization (2). ○, Drug-treated groups; •, controls. Calibration: 2 mV, 3 msec.
To test whether these drugs can affect a potentiation induced by a weak tetanic stimulation, we conducted experiments in which we used tetanization paradigms consisting of either four paired pulses (10 msec interval, applied at the theta frequency of 5 Hz) or a single 100 Hz train of 400 msec duration.
“Paired-pulse tetanization” induced an LTP that lasted ∼180 min under control conditions. Application of 400 μm MCPG did not influence the initial magnitude of potentiation (MCPG group: 179 ± 6%; control: 190 ± 7%) but significantly impaired the maintenance of LTP beginning 50 min after the tetanus (n = 7; p < 0.05) (Fig.2A). After 120 min, the fEPSP potentiation of the MCPG-treated slices had returned to baseline values, whereas the control group was still above baseline (110 ± 2%; n = 9). Similarly, application of 50 μm 4-CPG (n = 6) led to a faster decline of LTP compared with control (from 65 min after tetanus) but left the initial potentiation untouched (181 ± 9% 4-CPG experiments; 192 ± 7% control) (Fig. 2B). The effect of 4-CPG on LTP induced by the weak 100 Hz tetanization resembled the effect observed in the previous tetanization paradigm, i.e., beginning 90 min post-tetanus, the potentiation of the 4-CPG group was significantly impaired (102 ± 6%; n = 6) as compared with control (122 ± 5%; n = 8) (Fig.2C).
Fig. 2.

LTP induced by a weak tetanization protocol was susceptible to the action of MCPG (A) or 4-CPG (B–D). The tetanization consisted of either four × two pulses (200 msec interval between pulse pairs) (A, B) or a single 100 Hz train, 400 msec duration (C). In the experiments depicted inD, two independent pathways in the same slice were used to allow a direct comparison of the effects of 4-CPG on LTP generated by induction protocols of different strength. The groups treated with mGluR antagonists are indicated by open symbols, and their respective controls are indicated by closed symbols. A, Application of 400 μmMCPG led to a significant reduction of LTP, starting at 50 min post-tetanus (n = 7, as compared with controls,n = 9; p < 0.05).B, 4-CPG (50 μm) caused a significant blockade of LTP from 65 min after tetanus (4-CPG groups:n = 6; controls: n = 8;p < 0.05). C, Similarly, an LTP induced by the weak 100 Hz tetanization decayed faster after application of 4-CPG (50 μm). D, 4-CPG (50 μm) significantly impaired a decremental LTP that was induced by a weak tetanization (single 100 Hz train, 400 msec duration;circles) of the first pathway, but had no significant effect on a robust potentiation (3 × 100 Hz, 500 msec duration, 2 min interval between trains; squares) generated 10 min afterward by strong tetanization of the second pathway. Note that inD the first sampling time after tetanus was 5 min, but it was 1 min in A–C. Analog traces represent typical recordings of single experiments taken 10 min before tetanization (1) and 60 min after tetanization (2). Calibration 2 mV, 3 msec.
To verify the actions of 4-CPG with a different experimental approach, we used two independent stimulation pathways in the same slice (n = 6). A weak tetanization was applied to the first pathway (single 100 Hz train, 400 msec duration) followed 10 min afterward by a strong tetanization (3 × 500 msec, 100 Hz, 2 min interval between trains) of the second pathway. 4-CPG (50 μm) significantly impaired the decremental LTP that was induced by the weak tetanization (4-CPG group: 109 ± 4%; control: 124 ± 6%, at 100 min; p < 0.05) but had no significant effect on the robust potentiation generated by the strong tetanization paradigm (4-CPG group: 161 ± 15%; control: 174 ± 14%, at 100 min) (Fig. 2D).
Because MCPG and 4-CPG were effective only in the weak tetanization paradigm, we investigated whether the efficiency of class I antagonists is dependent on the tetanization strength. To test this, the tetanization strength of the weak tetanization paradigm of four bursts of two pulses at 5 Hz was gradually increased by adding more bursts or by increasing the number of pulses per burst. The resulting fEPSP slope potentiation (normalized to controls 90 min after tetanization) was compared with the fEPSP slope potentiation that was obtained with the other tetanization protocols used in this study. The results depicted in Figure 3 (columns 1–5, left) clearly indicate that the 4-CPG effect is contingent on tetanization strength. The fEPSP slope potentiation was mostly impaired using tetanization protocols of four × two pulses (82.4 ± 2.5%, n = 7; p < 0.05; left column) and 100 Hz, 400 msec duration (83.4 ± 5.7%,n = 5; p < 0.05). The effect decreased if six instead of four paired pulses were applied in the paired-pulse protocol (89.5 ± 2.9%, n = 5; p< 0.05). Enhancing the tetanus strength either by adding two more paired pulses (eight × two) (data not shown) and further by increasing the number of pulses (eight × four) or by applying a strong tetanization of 3 × 500 msec at 100 Hz (2 min interval between trains) abolished the effect of 4-CPG on LTP (eight × four pulses: 103.9 ± 10.6%, n = 5; 3 × 500 msec: 100.4 ± 2.3%, n = 6).
Fig. 3.
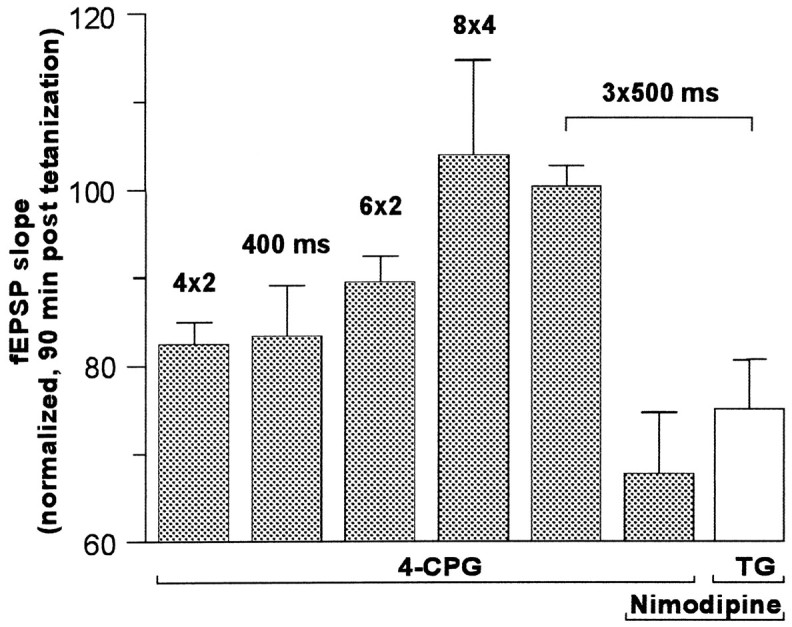
The effectiveness of 4-CPG on LTP was contingent on the strength of tetanization. The fEPSP slope potentiation (normalized to controls 90 min after tetanization) was mostly impaired using tetanization protocols of four × two pulses (82.4 ± 2.5%, n = 7; p < 0.05;left column) and 100 Hz, 400 msec duration (83.4 ± 5.7%, n = 5, p < 0.05). The effect decreased if six instead of four paired pulses were applied in the paired-pulse protocol (89.5 ± 2.9%, n = 5; p < 0.05). Enhancement of the tetanus strength, by adding two more pulses (8 × 2) (data not shown) or by increasing the number of pulses (8 × 4) or by applying a strong tetanization of 3 × 100 Hz (500 msec, 2 min interval between trains) abolished the effect of 4-CPG on LTP (103.9 ± 10.6%,n = 5, and 100.4 ± 2.3%,n = 6, respectively). Coapplication of 4-CPG (100 μm) and nimodipine (10 μm) resulted in a clear decrease of potentiation to 67.8 ± 6.9% (n = 7). This effect could be mimicked by coapplication of thapsigargin (TG) and nimodipine (n = 7).
The experiments presented above showed that the effectiveness of MCPG and 4-CPG on LTP is confined to weak tetanization paradigms. Because it has been demonstrated that the magnitude of Ca2+influx through NMDARs may be a critical factor in determining whether a robust or decremental potentiation is induced (Malenka et al., 1992), we suggested that the involvement of class I mGluRs in LTP may depend on the level and dynamics of [Ca2+]iduring tetanization. To examine how an increment of the duration of tetanization affects the rise of [Ca2+]i in dendrites, pyramidal CA1 neurons were loaded with the Ca2+-sensitive dye Calcium Green-1 and stimulated by a monopolar stimulating electrode positioned 80 μm from soma in CA1 stratum radiatum. Tetanizations of different durations (and pulse widths) were applied in a time interval of 2 min, and the Ca2+ responses of selected dendritic regions were visualized by confocal laser scanning microscopy and subsequently analyzed. As exemplified in Figure4A, weak 100 Hz tetanization protocols (e.g., 200 msec, pulse width 0.1 msec) induced a submaximal, short-lasting rise of [Ca2+]i. Augmenting the pulse width or increasing the duration of tetanization advanced the [Ca2+]i response toward maximum values. After approaching these peak Ca2+concentrations, any further extension of tetanization broadened the peak and slowed down the decay. For example, a 1 sec train of 100 Hz caused an elevation of [Ca2+]i that lasted for more than 2 sec. Application of a weak tetanization paradigm of four × two pulses at 100 Hz (200 msec interburst interval) triggered a [Ca2+]i rise that resembled the [Ca2+]i responses obtained with the common 100 Hz protocols but was superimposed by steep pinnacles that were synchronized with the interburst interval of 200 msec (5 Hz) (Fig. 4A, shaded area). It is important to note that the Ca2+ transients to different tetanization protocols were found to be independent of the sequence of application. As depicted in Figure 4D, the averaged areas of the fluorescence intensity changes (AreaF/F0) were correlated linearly, to both the duration of tetanic 100 Hz stimulation and the pulse width that we used [correlation coefficients of 0.99; data of pulse width 0.2 msec (data not shown)]. The AreaF/F0 of the tetanus of four × two pulses was comparable to the 400 msec tetanus (100 Hz). This finding corresponds very well with the duration of potentiation obtained with these stimulation protocols in the extracellular experiments (Fig. 2A–D) (additional data not shown).
Fig. 4.

Ca2+ imaging of the rise of [Ca2+]i in the dendritic tree of CA1 neurons (filled with the Ca2+-sensitive dye Calcium Green-1) on stimulation with different tetanization protocols and bath application of the mGluR class I antagonist 4-CPG (50 μm) and the L-type VDCC antagonist nimodipine (10 μm). A, Averaged Ca2+ response curves (transients) of seven neurons to a single set of tetanization paradigms. The Ca2+ transients of four dendritic regions were averaged. The tetanization protocols corresponding to the curves are indicated by an arrow. An increasing duration of the 100 Hz stimulation led initially to an increment of the peak amplitude, but after reaching a maximum amplitude the high Ca2+ level is maintained, followed by a slower decay (1 sec train). Application of the standard weak tetanization paradigm of four bursts of two pulses at 100 Hz (200 msec interburst interval) triggered a [Ca2+]i rise that resembled the [Ca2+]i responses obtained with the common 100 Hz protocols but was superimposed by steep pinnacles that were synchronized with the interburst interval of 200 msec (5 Hz) (shaded area). B, C, Representative images of the Ca2+ response of one neuron. The Ca2+ response to a weak (200 msec) (B) and a strong tetanization (1 sec) (C) is illustrated. The examples were taken at the onset of tetanization (top images), at the maximum of [Ca2+]i elevation (middle), and during the decay phase of the response (bottom). Note the clear difference in the Ca2+ level during the decay of response at 933 msec (bottom traces). D, As indicated by the averaged areas of the fluorescence intensity changes (AreaF/F0), the rise of [Ca2+]i was closely correlated to both the duration of tetanic 100 Hz stimulation and the pulse width that was used (correlation coefficients of 0.99; data not shown of pulse width 0.2 msec). The area below theF/F0 curves was calculated asAreaF/F0 = ∫B(F/F0 − 1) dt;B = [0 sec, 8 sec]. E, F, After a tetanization of four × two pulses, bath application of 4-CPG (50 μm) led to a significant reduction (p < 0.05) of AreaF/F0 to 67% (F), which was caused predominantly by a slower rise and earlier decay of [Ca2+]i. Note that the effect of 4-CPG was reversible (wash outline in E). G, Only the coapplication of 4-CPG and nimodipine caused a significant reduction ofAreaF/F0 (78%;p < 0.05) during strong tetanization (1 sec, 100 Hz). The application of 4-CPG alone had no effect.
Although these data provide only a momentary and localized image of the [Ca2+]i dynamics occurring in the dendritic regions, they support the hypothesis that the intradendritic level of Ca2+ is closely correlated to the type and duration of tetanization. Next, we examined by confocal laser scanning microscopy whether the elevation of [Ca2+]i after weak tetanic stimulation can be impaired by an inhibition of class I mGluRs, as implied by the coupling of class I mGluRs to IP3-sensitive ICSs and suggested by the action of 4-CPG in the weak potentiation paradigms (Fig. 2B–D). As shown in Figure4F, bath application of the same concentration of 4-CPG (50 μm) led to a significant reduction of AreaF/F0 to 67% of control (n= 6; p < 0.05). This reduction was predominantly caused by a slower rise and earlier decay of [Ca2+]i (Fig. 4E). The effect of 4-CPG was reversible in all neurons tested (Fig.4E, wash out).
The results of the Ca2+ imaging studies led us to conclude that weak tetanization paradigms that evoked a weak or moderate [Ca2+]i response could be affected by a mGluR class I antagonist. We thus investigated whether a blockade of other Ca2+ sources during strong tetanization, such as the entry of Ca2+ through VDCCs, could enable class I mGluRs to affect LTP. To examine this hypothesis, we coapplied the L-type VDCC antagonist nimodipine together with the mGluR antagonists MCPG and 4-CPG during strong tetanization. Nimodipine (10 μm; n = 8) applied on its own during tetanization did not modify the potentiation in comparison to control experiments (n = 6; data not shown). However, the coapplication of nimodipine (10 μm) and MCPG (400 μm) led to a marked reduction of LTP (Fig.5A). The impairment of LTP became significant 150 min after tetanization and resulted in a diminished potentiation of 127 ± 8% 240 min after LTP induction (n = 7) as compared with 179 ± 15% in the control group (n = 7; p < 0.05). Similar to the experiments with the weak tetanization, the initial potentiation was unaffected, showing that post-tetanic potentiation was not modified. The effects of 100 μm 4-CPG coapplied with nimodipine (10 μm) (n = 7) resembled the MCPG/nimodipine experiments. Although the initial potentiation was not influenced, the 4-CPG/nimodipine group displayed a faster rundown of potentiation, which became statistically distinguishable from controls 80 min after tetanus (Fig. 5B). After 240 min, the potentiation had nearly declined to baseline (108 ± 12%) as compared with controls (152 ± 7%).
Fig. 5.
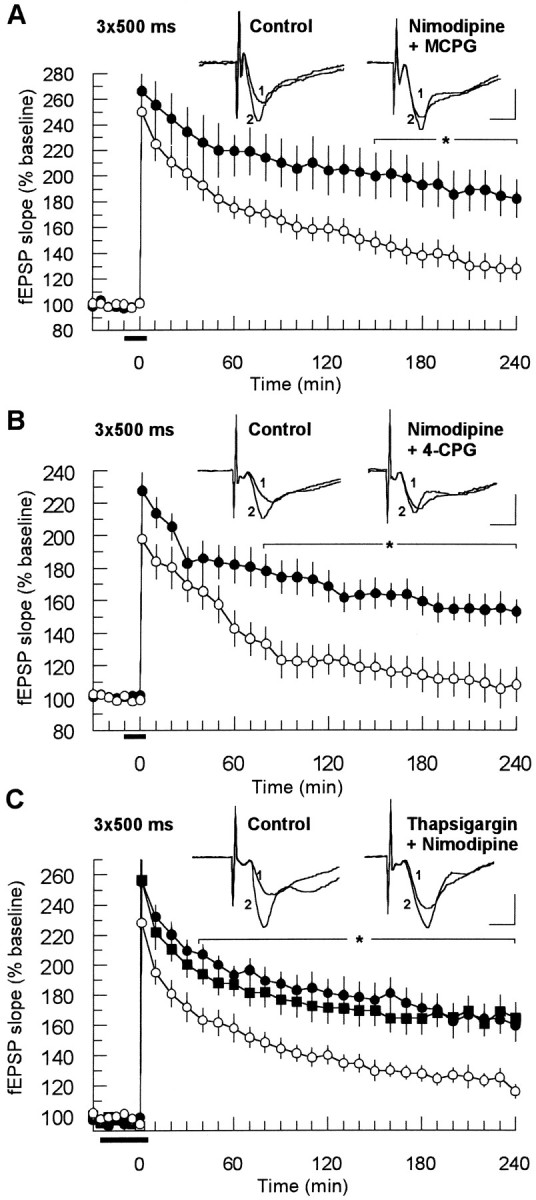
Coapplication of mGluR class I antagonists and the L-type calcium channel antagonist nimodipine impaired LTP evoked by a strong tetanization protocol. A, MCPG (400 μm) affected the potentiation when nimodipine (10 μm) was coapplied. The reduction became significant 150 min after tetanization (p < 0.05;n = 7). The control application of nimodipine alone (n = 8) had no effect on LTP. B, Similarly, coapplication of 4-CPG (100 μm) and nimodipine resulted in a significant impairment of LTP starting 80 min after tetanization (p < 0.05;n = 7), whereas nimodipine by itself was not effective (n = 8). C, Application of thapsigargin (6 μm) had the same effect as mGluR class I antagonists if coapplied with nimodipine (○, p < 0.05; n = 7). The application of nimodipine (•,n = 7) and thapsigargin (▪, n= 6) alone had no influence on LTP. See Figure 2 for further explanation.
Because the electrophysiological studies showed that nimodipine may enable 4-CPG to affect LTP induced by strong tetanization, we tested the coapplication of 4-CPG and nimodipine in confocal imaging experiments. 4-CPG, bath-applied in the strong tetanization protocol (1 sec, 100 Hz), had no effect (Fig. 4G). However, coapplication of 4-CPG with nimodipine (10 μm) caused a significant reduction of AreaF/F0 to 78% of control (n = 5; p < 0.05).
These results support our hypothesis that the efficiency of mGluR class I antagonists depends on the level of [Ca2+]i. During strong tetanization, the Ca2+ influx through NMDARs is significantly augmented by Ca2+ entering the neuron through L-type VDCCs. The high Ca2+ level originating from these two sources overrides the functional role of Ca2+release from IP3-sensitive ICSs. If MCPG and 4-CPG block release of Ca2+ from ICSs, then depleting these stores by the Ca2+-ATPase inhibitor thapsigargin should lead to a similar effect in the nimodipine/strong tetanization paradigm.
As shown in Figure 5C, the application of thapsigargin (6 μm; n = 6) or nimodipine (10 μm; n = 7) had no independent effect on robust LTP, which was induced by strong tetanization (initial values: 256 ± 12% and 255 ± 8%, respectively; 240 min values: 164 ± 10% and 159 ± 11%). However, coapplication of the two drugs (n = 7) caused a severe impairment of potentiation that declined from 228 ± 12% initially to 115 ± 4% after 240 min (controls: 256 ± 12% and 164 ± 10%, respectively; p < 0.05).
Therefore, the mGluR antagonists MCPG and 4-CPG and the Ca2+ store depletor thapsigargin only affected a robust LTP if the intracellular Ca2+ level during the tetanus was artificially lowered by blocking the Ca2+ entry through L-type VDCCs.
DISCUSSION
The presented results demonstrate that the role of mGluRs in LTP is contingent on the tetanization paradigm that is used. The stronger the tetanization, the less potent were the mGluR antagonists MCPG and 4-CPG in affecting the time course of the resulting potentiation. MCPG has been characterized as a competitive antagonist of class I and class II mGluRs, whereas 4-CPG displays a selectivity toward class I mGluRs and only weak activity on mGluRs of class II (Davies et al., 1995;Sekiyama et al., 1996). Because in most of our experiments the results obtained with the two antagonists were very similar, the effects of these compounds on LTP may be assigned to an action on class I mGluRs. This is consistent with the findings of other laboratories that excitatory postsynaptic mGluR actions in the CA1 region are mediated by activation of class I receptors (Davies et al., 1995;Gereau and Conn, 1995) and that agonists of class I mGluRs may facilitate the induction of LTP or induce a potentiation on their own [McGuinness et al., 1991; Otani and Ben-Ari, 1991; Ben-Ari et al., 1992; Bortolotto and Collingridge, 1992, 1995; Manahan-Vaughan and Reymann, 1995, 1996; Breakwell et al., 1996; O’Leary and O’Connor, 1997 (but see Brown and Reymann, 1995)]. Both subtypes of class I receptors, mGluR1 and mGluR5, are found in the CA1 subfield as shown byin situ hybridization and immunohistochemical studies [Petralia et al., 1997 (for further references, see Luján et al., 1996)].
Our findings shed new light on the controversy concerning the functional role of mGluRs in LTP and the contradictory data that have been obtained with MCPG. If the studies that dealt with the action of MCPG on hippocampal LTP are classified according to the type and strength of tetanization applied, then the resulting picture is largely consistent with the conclusions drawn from our experiments. Whereas in all studies using a “strong” tetanization (theta-burst stimulation or repetitive 100 Hz/1 sec protocols) MCPG failed to block LTP (Manzoni et al., 1994; Selig et al., 1995; Thomas and O’Dell, 1995), a single 100 Hz stimulation of 1 sec duration appears to mark a critical threshold where the involvement of mGluRs depends on certain (as yet unknown) experimental conditions (Bashir et al., 1993; Chinestra et al., 1993; Bortolotto et al., 1994; Thomas and O’Dell, 1995). In contrast, all tetanization paradigms that used single 100 Hz protocols <1 sec resulted in a potentiation that was dependent on a mGluR class I activation. In our experiments, the potentiation became susceptible to the action of MCPG and 4-CPG as soon the duration of the tetanus was reduced to 400 msec.
In our search for the basic mechanism that may underlie the tetanization strength-dependent efficiency of MCPG and 4-CPG, we focused on changes of the intracellular Ca2+ level after activation of class I mGluRs during tetanization. It is generally accepted that the induction of LTP in the CA1 area requires an elevation of the free, intracellular Ca2+concentration in the postsynaptic neuron (Lynch et al., 1983; Malenka et al., 1988). In addition to Ca2+ influx through NMDARs (Collingridge et al., 1983; Malenka, 1994), two other important Ca2+ sources can feed the rise of [Ca2+]i during tetanization: Ca2+ influx through VDCCs and the release of Ca2+ from IP3- and ryanodine-sensitive ICSs (Thastrup et al., 1990; Berridge, 1993; Frenguelli et al., 1996).
Activation of VDCCs appears to be critically involved in the generation of LTP by strong tetanization at 200 Hz and higher frequencies (Grover and Teyler, 1990). In contrast, the widely used strong 100 Hz paradigms evoke a potentiation that contains a smaller, inconspicuous VDCC-dependent component, as shown by Grover and Teyler (1994), and confirmed in our experiments by the lack of significant effects of nimodipine on potentiation.
Ca2+ release from ICSs (Thastrup et al., 1990;Berridge, 1993) seems to play a decisive role in LTP induction under certain conditions. Harvey and Collingridge (1992) reported a block of LTP if the selective Ca2+-ATPase inhibitor thapsigargin was applied during tetanization, but not if given 30 min afterward. In previous experiments in our laboratory, we found that bath application of thapsigargin during tetanization did not affect a robust LTP generated by a triple 100 Hz tetanus, but impaired a weaker potentiation induced by a single 100 Hz train of 400 msec duration (Behnisch and Reymann, 1995).
In this study, we did not see an effect of MCPG or 4-CPG on potentiation induced by a strong 100 Hz protocol, which is in accordance with the findings of other groups (Chinestra et al., 1993;Izumi and Zorumski, 1994; Manzoni et al., 1994; Thomas and O’Dell, 1995; Selig et al., 1995). However, coapplication of MCPG or 4-CPG with nimodipine had detrimental consequences on potentiation. Thus, inhibition of the Ca2+ influx through VDCCs during tetanization did not result in overt changes of LTP, but the potentiation became dependent on activation of class I mGluRs. Because thapsigargin mimicked exactly the action of MCPG and 4-CPG in impairing a robust LTP only when L-type VDCCs were additionally inhibited by nimodipine, we concluded that both antagonists acted via an inhibition of the IP3-mediated Ca2+-release from ICSs (Thastrup et al., 1990; Berridge, 1993). These findings indicated that the involvement of class I mGluRs in synaptic plasticity depends on [Ca2+]i attained during tetanization and the interaction between VDCCs and ICSs.
Although the confocal [Ca2+]imeasurements did not resolve the contribution of the different Ca2+ sources to the tetanic [Ca2+]i rise, they gave clear evidence of a tight correlation between [Ca2+]iand the strength of tetanization. In addition, they supported a different functional role for mGluR-triggered Ca2+release from IP3-sensitive ICSs during weak and strong tetanic stimulation. Under our experimental conditions, AreaF/F0 was not only monotonic but linearly correlated to the pulse width and duration of tetanization. However, the shape of [Ca2+]i responses and the resulting AreaF/F0 might have been influenced by methodical constraints such as saturation of the Ca2+ dye Calcium Green-1 (Kd 189 nm). The Ca2+ kinetics that we obtained after tetanic stimulation of increasing duration and pulse width extend the previous findings of Regehr and Tank (1992), who described Ca2+ accumulations in response to tetanization (100 Hz for 1 sec) with increasing stimulation intensity. Malenka et al. (1992) reported that the [Ca2+]i surge within the initial 2 sec after onset of potentiation determines the resulting type and the properties of plasticity. In our study, the often used 100 Hz/1 sec stimulation led to intradendritic [Ca2+]i kinetics with a steep increase, a maximum of ∼1000 msec and an exponential decay resulting in [Ca2+]i, which was still elevated after 2 sec. The finding that 4-CPG attenuates the tetanus-induced [Ca2+]i surge is consistent with the studies of Frenguelli et al. (1993), which described a similar effect for MCPG, and in accordance with Alford et al. (1993), who reported that blockade of Ca2+release from ICSs may cause a considerable reduction of [Ca2+]i in CA1 pyramidal neurons. However, in our hands, 4-CPG reduced only the [Ca2+]i elevation induced by weak but not by strong tetanization.
Putting all of these facts together, we hypothesize that the three main Ca2+ sources interfere with each other during LTP induction, as schematically outlined in Figure6. During weak tetanization (Fig.6A1), the characteristics of the attained depolarization do not allow the L-type VDCCs to contribute significantly to the Ca2+ influx required for LTP induction. The Ca2+ entry is therefore accomplished predominantly via NMDARs. The moderate intracellular Ca2+ level and IP3 liberated by activation of class I mGluRs trigger the release of Ca2+ from IP3-sensitive stores resulting in an amplification and prolongation of the Ca2+signal beyond the threshold for LTP induction, as supported by computational studies (Schiegg et al., 1995).
Fig. 6.

Scheme of the role of the three main Ca2+ sources during LTP induction in dependence of the tetanization strength. The final [Ca2+]i necessary for induction of LTP is determined by two factors, the NMDARs and an additional source provided by either VDCCs or the Ca2+ release from ICSs after activation of class I mGluRs. Top scheme, A1, During a weak, single tetanization the rise of [Ca2+]i is fed by the activation of NMDARs and class I mGluRs, whereas the contribution of L-type VDCCs is negligible under these conditions. Thus, application of class I antagonists causes an impairment of LTP (A2).Bottom scheme, B1, A strong tetanization paradigm leads to a sustained depolarization enabling the Ca2+ entry via NMDARs and VDCCs, as well as to the release of Ca2+ from ICSs.B2, The additional Ca2+ that is provided by the release from ICSs via liberation of IP3on class I mGluR activation is not required for LTP induction.B3, Blockade of VDCCs is counterbalanced by the class I mGluR-triggered Ca2+ release from ICSs.B4, Concomitant inhibition of class I mGluRs and VDCCs results in a decremental potentiation.
In contrast, during strong tetanization (Fig.6B1) leading to a sustained depolarization, the contribution of VDCCs to the [Ca2+]i surge is increased, i.e., Ca2+ enters the neuron via NMDARs and VDCCs. The corresponding high [Ca2+]i may interfere via two different mechanisms with the class I-triggered Ca2+ release. (1) The high [Ca2+]i could functionally override the contribution of the surplus Ca2+ from ICSs, i.e., the threshold for LTP induction (Lisman, 1989; Artola and Singer, 1993; Cummings et al., 1996; Neveu and Zucker, 1996; Tsumoto and Yasuda, 1996) can be achieved solely by activation of NMDARs and VDCCs. (2) The high cytosolic Ca2+ concentration can decrease the Ca2+ release from IP3-sensitive stores because of the bell-shaped Ca2+ sensitivity of IP3 channels (Bootman and Berridge, 1995).
The findings of the present study and the available experimental evidence lead us to hypothesize that class I mGluRs have a particular function in LTP. During weak and moderate tetanization, they amplify the initial [Ca2+]i surge that originates from Ca2+ influx through NMDARs, beyond the threshold for LTP induction; i.e., by instigating the Ca2+ release from IP3-sensitive ICSs they enable an NMDA-dependent input-specific LTP. Therefore, at moderate levels of synaptic activation, class I mGluRs may function as threshold boosters in input-specific Hebb-type plasticity.
Footnotes
This research was supported by the Deutsche Forschungsgemeinschaft Sonderforschungsbereich 426. We thank Dr. Denise Manahan-Vaughan and Dr. Ritchie Brown for critical comments on this manuscript, and Ms. Katrin Böhm for excellent technical assistance.
Correspondence should be addressed to Detlef Balschun, Leibniz Institute for Neurobiology, Department of Neurophysiology, P.O. Box 1860, D-39008 Magdeburg, Germany.
REFERENCES
- 1.Aiba A, Chen C, Herrup K, Rosenmund C, Stevens CF, Tonegawa S. Reduced hippocampal long-term potentiation and context-specific deficit in associative learning in mGluR1 mutant mice. Cell. 1994;79:365–375. doi: 10.1016/0092-8674(94)90204-6. [DOI] [PubMed] [Google Scholar]
- 2.Alford S, Frenguelli BG, Schofield JG, Collingridge GL. Characterization of Ca2+ signals induced in hippocampal CA1 neurones by the synaptic activation of NMDA receptors. J Physiol (Lond) 1993;469:693–716. doi: 10.1113/jphysiol.1993.sp019838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Artola A, Singer W. Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci. 1993;16:480–487. doi: 10.1016/0166-2236(93)90081-v. [DOI] [PubMed] [Google Scholar]
- 4.Bashir ZI, Bortolotto ZA, Davies CH, Beretta N, Irving AJ, Sea AJ, Henley JM, Jane DE, Watkins JC, Collingridge GL. Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature. 1993;363:347–350. doi: 10.1038/363347a0. [DOI] [PubMed] [Google Scholar]
- 5.Behnisch T, Reymann KG. Thapsigargin blocks long-term potentiation induced by weak, but not strong tetanization in rat hippocampal CA1 neurons. Neurosci Lett. 1995;192:185–188. doi: 10.1016/0304-3940(95)11641-9. [DOI] [PubMed] [Google Scholar]
- 6.Ben-Ari Y, Aniksztejn L, Bregestowski P. Protein kinase-C modulation of NMDA currents: an important link for LTP induction. Trends Neurosci. 1992;15:333–339. doi: 10.1016/0166-2236(92)90049-e. [DOI] [PubMed] [Google Scholar]
- 7.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 8.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 9.Bootman MD, Berridge MJ. The elemental principles of calcium signaling. Cell. 1995;83:675–678. doi: 10.1016/0092-8674(95)90179-5. [DOI] [PubMed] [Google Scholar]
- 10.Bortolotto ZA, Collingridge GL. Activation of glutamate metabotropic receptors induces long-term potentiation. Eur J Pharmacol. 1992;214:297–298. doi: 10.1016/0014-2999(92)90135-q. [DOI] [PubMed] [Google Scholar]
- 11.Bortolotto ZA, Collingridge GL. On the mechanism of long-term potentiation induced by (1S,3R)-1-aminocyclopentane-1,3-dicarboxylic acid (ACPD) in rat hippocampal slices. Neuropharmacology. 1995;34:1003–1014. doi: 10.1016/0028-3908(95)00054-a. [DOI] [PubMed] [Google Scholar]
- 12.Bortolotto ZA, Bashir ZI, Davies CH, Collingridge GL. A molecular switch activated by metabotropic glutamate receptors regulates induction of long-term potentiation. Nature. 1994;368:740–743. doi: 10.1038/368740a0. [DOI] [PubMed] [Google Scholar]
- 13.Breakwell NA, Rowan MJ, Anwyl R. Metabotropic glutamate receptor dependent EPSP and EPSP-spike potentiation in area CA1 of the submerged rat hippocampal slice. J Neurophysiol. 1996;76:3126–3135. doi: 10.1152/jn.1996.76.5.3126. [DOI] [PubMed] [Google Scholar]
- 14.Brown RE, Reymann KG. Class I metabotropic glutamate receptor agonists do not facilitate the induction of long-term potentiation in the dentate gyrus of the rat in vitro. Neurosci Lett. 1995;202:73–76. doi: 10.1016/0304-3940(95)12202-8. [DOI] [PubMed] [Google Scholar]
- 15.Brown RE, Rabe H, Reymann KG. (RS)-α-methyl-4-carboxyphenylglycine (MCPG) does not block theta burst-induced long-term potentiation in area CA1 of rat hippocampal slices. Neurosci Lett. 1994;170:17–21. doi: 10.1016/0304-3940(94)90228-3. [DOI] [PubMed] [Google Scholar]
- 16.Chinestra P, Aniksztejn L, Diabira D, Ben-Ari Y. (RS)-alpha-methyl-4-carboxyphenylglycine neither prevents induction of LTP nor antagonizes metabotropic glutamate receptors in CA1 hippocampal neurons. J Neurophysiol. 1993;70:2684–2689. doi: 10.1152/jn.1993.70.6.2684. [DOI] [PubMed] [Google Scholar]
- 17.Collingridge GL, Kehl SJ, McLennon H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol (Lond) 1983;334:33–46. doi: 10.1113/jphysiol.1983.sp014478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conn PJ, Pin J-P. Pharmacology and function of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 19.Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franzbacon K, Reggiani A, Matarese V, Conde F, Collingridge GL, Crepel F. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–243. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- 20.Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca2+ signaling requirements for long-term depression in the hippocampus. Neuron. 1996;16:825–833. doi: 10.1016/s0896-6273(00)80102-6. [DOI] [PubMed] [Google Scholar]
- 21.Davies CH, Clarke VRJ, Jane DE, Collingridge GL. Pharmacology of postsynaptic metabotropic glutamate receptors in rat hippocampal CA1 pyramidal neurons. Br J Pharmacol. 1995;116:1859–1869. doi: 10.1111/j.1476-5381.1995.tb16674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frenguelli BG, Potier B, Slater NT, Alford S, Collingridge GL. Metabotropic glutamate receptors and calcium signalling in dendrites of hippocampal CA1 neurones. Neuropharmacology. 1993;32:1229–1237. doi: 10.1016/0028-3908(93)90017-w. [DOI] [PubMed] [Google Scholar]
- 23.Frenguelli BG, Irving AJ, Collingridge GL. Ca2+ stores and hippocampal synaptic plasticity. Semin Neurosci. 1996;8:301–309. [Google Scholar]
- 24.Gereau RW, Conn PJ. Roles of metabotropic glutamate receptor subtypes in regulation of hippocampal CA1 pyramidal cell excitability. J Neurophysiol. 1995;74:122–129. doi: 10.1152/jn.1995.74.1.122. [DOI] [PubMed] [Google Scholar]
- 25.Grover LM, Teyler TJ. Two components of long-term potentiation induced by different patterns of activation. Nature. 1990;347:477–479. doi: 10.1038/347477a0. [DOI] [PubMed] [Google Scholar]
- 26.Grover LM, Teyler TJ. Activation of NMDA receptors by low and high frequency orthodromic stimulation, and their contribution to induction of long-term potentiation. Synapse. 1994;16:66–75. doi: 10.1002/syn.890160108. [DOI] [PubMed] [Google Scholar]
- 27.Harvey J, Collingridge GL. Thapsigargin blocks the induction of long-term potentiation in rat hippocampal slices. Neurosci Lett. 1992;139:197–200. doi: 10.1016/0304-3940(92)90551-h. [DOI] [PubMed] [Google Scholar]
- 28.Izumi Y, Zorumski CF. Developmental changes in the effects of metabotropic glutamate receptor antagonists on CA1 long-term potentiation in rat hippocampal slices. Neurosci Lett. 1994;176:89–92. doi: 10.1016/0304-3940(94)90878-8. [DOI] [PubMed] [Google Scholar]
- 29.Jaffe DB, Brown TH. Metabotropic glutamate receptor activation induces calcium waves within hippocampal dendrites. J Neurophysiol. 1994;72:471–474. doi: 10.1152/jn.1994.72.1.471. [DOI] [PubMed] [Google Scholar]
- 30.Kauer JA, Malenka RC, Nicoll RA. NMDA application potentiates synaptic transmission in the hippocampus. Nature. 1988;334:250–252. doi: 10.1038/334250a0. [DOI] [PubMed] [Google Scholar]
- 31.Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Little Z, Grover LM, Teyler TJ. Metabotropic glutamate receptor antagonist, (R,S)-alpha-methyl-4-carboxyphenylglycine, blocks two distinct forms of long-term potentiation in area CA1 of rat hippocampus. Neurosci Lett. 1995;201:73–76. doi: 10.1016/0304-3940(95)12141-p. [DOI] [PubMed] [Google Scholar]
- 33.Lu YM, Jia Z, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, Roder JC. Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J Neurosci. 1997;17:5196–5205. doi: 10.1523/JNEUROSCI.17-13-05196.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luján R, Nusser Z, Roberts JDB, Shigemoto R, Somogyi P. Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci. 1996;8:1488–1500. doi: 10.1111/j.1460-9568.1996.tb01611.x. [DOI] [PubMed] [Google Scholar]
- 35.Lynch GS, Larson J, Kelso S, Barrionuevo G, Schottler F. Intracellular injections of EGTA block the induction of hippocampal long-term potentiation. Nature. 1983;305:719–721. doi: 10.1038/305719a0. [DOI] [PubMed] [Google Scholar]
- 36.Malenka RC. Synaptic plasticity in the hippocampus, LTP and LTD. Cell. 1994;78:535–538. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- 37.Malenka RC, Kauer JA, Zucker RJ, Nicoll RJ. Post-synaptic calcium is sufficient for potentiation of hippocampal synaptic transmission. Science. 1988;242:81–84. doi: 10.1126/science.2845577. [DOI] [PubMed] [Google Scholar]
- 38.Malenka RC, Lancaster B, Zucker RS. Temporal limits on the rise in postsynaptic calcium required for the induction of long-term potentiation. Neuron. 1992;9:121–128. doi: 10.1016/0896-6273(92)90227-5. [DOI] [PubMed] [Google Scholar]
- 39.Manahan-Vaughan D, Reymann KG. 1S,3R-ACPD dose-dependently induces a slow-onset potentiation in the rat hippocampal CA1 region in vivo. Neuropharmacology. 1995;34:1103–1105. doi: 10.1016/0028-3908(95)00108-i. [DOI] [PubMed] [Google Scholar]
- 40.Manahan-Vaughan D, Reymann KG. Metabotropic glutamate receptor subtype agonists facilitate LTP within a distinct time window in the dentate gyrus in vivo. Neuroscience. 1996;74:723–731. doi: 10.1016/0306-4522(96)00162-5. [DOI] [PubMed] [Google Scholar]
- 41.Manzoni OJ, Weisskopf MG, Nicoll RA. MCPG antagonizes metabotropic glutamate receptors but not long-term potentiation in the hippocampus. Eur J Neurosci. 1994;6:1050–1054. doi: 10.1111/j.1460-9568.1994.tb00599.x. [DOI] [PubMed] [Google Scholar]
- 42.McGuinness N, Anwyl R, Rowan M. Trans-ACPD enhances long-term potentiation in the hippocampus. Eur J Pharmacol. 1991;19:231–232. doi: 10.1016/0014-2999(91)90529-y. [DOI] [PubMed] [Google Scholar]
- 43.Murphy SN, Miller RJ. A glutamate receptor regulates Ca2+ mobilisation in hippocampal neurones. Proc Natl Acad Sci USA. 1988;85:8737–8741. doi: 10.1073/pnas.85.22.8737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakanishi S. Metabotropic glutamate receptors: synaptic transmission, modulation, and plasticity. Neuron. 1994;13:1031–1037. doi: 10.1016/0896-6273(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 45.Neveu D, Zucker RS. Postsynaptic levels of [Ca2+]i needed to trigger LTD and LTP. Neuron. 1996;16:619–629. doi: 10.1016/s0896-6273(00)80081-1. [DOI] [PubMed] [Google Scholar]
- 46.O’Leary DM, O’Connor JJ. Potentiation of synaptic transmission in the rat dentate gyrus in vitro by (S)-3,5-dihydroxyphenylglycine ((S)-DHPG). Neurosci Lett. 1997;229:29–32. doi: 10.1016/s0304-3940(97)00404-7. [DOI] [PubMed] [Google Scholar]
- 47.Otani D, Ben-Ari Y. Metabotropic receptor mediated long-term potentiation in rat hippocampal slices. Eur J Pharmacol. 1991;205:325–326. doi: 10.1016/0014-2999(91)90920-l. [DOI] [PubMed] [Google Scholar]
- 48.Petralia RS, Wang Y-X, Singh S, Wu C, Shi L, Wenthold RJ. A monoclonal antibody shows discrete cellular and subcellular localizations of mGluR1α metabotropic glutamate receptors. J Chem Neuroanat. 1997;13:77–93. doi: 10.1016/s0891-0618(97)00023-9. [DOI] [PubMed] [Google Scholar]
- 49.Phenna S, Jane SD, Chad JE. Increased perinuclear Ca2+ activity evoked by metabotropic glutamate receptor activation in rat hippocampal neurones. J Physiol (Lond) 1995;486:149–161. doi: 10.1113/jphysiol.1995.sp020799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Regehr WG, Tank DW. Calcium concentration dynamics produced by synaptic activation of CA1 hippocampal pyramidal cells. J Neurosci. 1992;12:4202–4223. doi: 10.1523/JNEUROSCI.12-11-04202.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Richter-Levin G, Errington ML, Maegawa H, Bliss TVP. Activation of metabotropic glutamate receptors is necessary for long-term potentiation in the dentate gyrus and for spatial learning. Neuropharmacology. 1994;33:853–857. doi: 10.1016/0028-3908(94)90181-3. [DOI] [PubMed] [Google Scholar]
- 52.Riedel G, Casabona G, Reymann KG. Inhibition of long-term potentiation in the dentate gyrus of freely moving rats by the metabotropic glutamate receptor antagonist MCPG. J Neurosci. 1995;15:87–98. doi: 10.1523/JNEUROSCI.15-01-00087.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schiegg A, Gerstner W, Ritz R, van Hemmen JL. Intracellular Ca2+ stores can account for the time course of LTP induction: a model of Ca2+ dynamics in dendritic spines. J Neurophysiol. 1995;74:1046–1055. doi: 10.1152/jn.1995.74.3.1046. [DOI] [PubMed] [Google Scholar]
- 54.Sekiyama N, Hayashi Y, Nakanishi S, Jane DE, Tse H-W, Birse EF, Watkins JC. Structure-activity relationships of new agonists and antagonists of different metabotropic glutamate receptor subtypes. Br J Pharmacol. 1996;117:1493–1503. doi: 10.1111/j.1476-5381.1996.tb15312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Selig DK, Lee HK, Bear MF, Malenka RC. Re-examination of the effects of MCPG on hippocampal LTP, LTD and depotentiation. J Neurophysiol. 1995;74:1075–1082. doi: 10.1152/jn.1995.74.3.1075. [DOI] [PubMed] [Google Scholar]
- 56.Shirasaki T, Harata N, Akaike N. Metabotropic glutamate response in acutely dissociated hippocampal CA1 pyramidal neurones of the rat. J Physiol (Lond) 1994;475:439–453. doi: 10.1113/jphysiol.1994.sp020084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promotor, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmatic reticulum Ca2(+)ATPase. Proc Natl Acad Sci USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thomas MJ, O’Dell TJ. The molecular switch hypothesis fails to explain the inconsistent effects of the metabotropic glutamate receptor antagonist MCPG on long-term potentiation. Brain Res. 1995;695:45–52. doi: 10.1016/0006-8993(95)00757-h. [DOI] [PubMed] [Google Scholar]
- 59.Tsumoto T, Yasuda H. A switching role of postsynaptic calcium in the induction of long-term potentiation or long-term depression in visual cortex. Semin Neurosci. 1996;8:311–319. doi: 10.1016/0168-0102(95)01001-7. [DOI] [PubMed] [Google Scholar]