

Abstract
In vivo microdialysis, radioimmunoassay, and HPLC with electrochemical or fluorometric detection were used to investigate the release of cholecystokinin (CCK), glutamate (Glu), and dopamine (DA) in nucleus accumbens septi (NAS) as a function of ipsilateral electrical stimulation of medial prefrontal cortex (mPFC). CCK was progressively elevated by mPFC stimulation at 50–200 Hz. Stimulation-induced CCK release was intensity-dependent at 250–700 μA. NAS Glu and DA levels were each elevated by stimulation at 25–400 Hz; the dopamine metabolites DOPAC and homovanillic acid were increased by stimulation at 100–400 Hz. When rats were trained to lever press for mPFC stimulation, the stimulation induced similar elevations of each of the three transmitters to those seen with experimenter-administered stimulation. Perfusion of 1 mm kynurenic acid (Kyn) into either the ventral tegmental area (VTA) or NAS blocked lever pressing for mPFC stimulation. VTA, but not NAS, perfusion of Kyn significantly attenuated the increases in NAS DA levels induced by mPFC stimulation. Kyn did not affect NAS CCK or Glu levels when perfused into either the VTA or NAS. The present results are consistent with histochemical evidence and provide the first in vivoevidence for the existence of a releasable pool of CCK in the NAS originating from the mPFC. Although dopamine is the transmitter most closely linked to reward function, it was CCK that showed frequency-dependent differences in release corresponding most closely to rewarding efficacy of the stimulation. Although not essential for the reward signal itself, coreleased CCK may modulate the impact of the glutamatergic action in this behavior.
Keywords: cholecystokinin, amino acids, dopamine, microdialysis, prefrontal cortex, nucleus accumbens, brain stimulation reward, rat
Cholecystokinin (CCK) is the most abundant peptide in the brain (Vanderhaegen et al., 1975; Rehfeld, 1985), occurring primarily as sulfated C-terminal octapeptide (Dockray et al., 1978; Larsson and Rehfeld, 1979; Dockray, 1980). CCK is synthesized at a high rate in the brain (Goltermann et al., 1980), is vesicularly located at synaptic terminals (Emson et al., 1980; Snyder et al., 1993; Morino et al., 1994a), and is released from synaptosomesin vitro (Dodd et al., 1980). Peptidase responsible for inactivation of endogenous CCK has been identified and purified from brain (Rose et al., 1996). CCK increases neuronal firing rates (Dodd and Kelly, 1981; Skirboll et al., 1981; Hommer et al., 1986) and excites patch-clamped neurons by opening nonselective cation channels via a G-protein-coupled mechanism (Wu and Wang, 1994, 1996). CCK exerts its function via CCKA and CCKB receptors, and each subtype has been cloned (Innis and Snyder, 1980; Van Dijk et al., 1984; Hill et al., 1987, 1990; Woodruff et al., 1991; Wank et al., 1992a,b). CCK receptor distribution matches the distribution of CCK immunoreactivity in most brain regions (Morency et al., 1994). Basal extracellular levels of CCK are in the low picomolar range and can be increased in a calcium-dependent manner after depolarization of cortex, neostriatum, or nucleus accumbens septi (NAS) (Butcher et al., 1989;Maidement et al., 1991; Meana et al., 1991; Vallebuona et al., 1993;You et al., 1994; Liu and Kato 1996; Nevo et al., 1996). Together, these findings suggest that CCK acts as a neurotransmitter or neuromodulator in the brain.
The medial prefrontal cortex (mPFC) projects heavily and topographically to the ventral striatum (Sesack et al., 1989;Berendse et al., 1992), using glutamate (Glu) or aspartate as its primary neurotransmitter (Fonnum et al., 1981; Druce et al., 1982;Sandberg et al., 1985; Christie et al., 1990). CCK is colocalized in this pathway (Meyer et al., 1982; Hökfelt et al., 1988; Burgunder and Young, 1990; Morino et al., 1994a,b). Pyramidal shaped CCK-containing cells of the mPFC project bilaterally (with an ipsilateral preference) to NAS (Morino et al., 1994b). Although this pathway appears to be a major source of NAS CCK, unilateral lesions of this pathway have little effect on NAS or striatal CCK (You et al., 1994; Kresse et al., 1995); this raises questions about the functional significance of the CCK localized to this pathway.
The NAS also receives a dense innervation of dopamine (DA) neurons from the ventral tegmental area (VTA); these neurons also colocalize CCK but here, too, unilateral lesions of the pathway have little effect on NAS or striatal CCK levels (Kresse et al., 1995; Maidment and Villafranca, 1997). DA function in the NAS is critical for goal-oriented behaviors, including those motivated by drug and brain stimulation reward (Wise, 1996a,b). Electrical stimulation of mPFC is rewarding and increases the release of DA in the NAS (Ferrer et al., 1988; Corbett, 1990; Taber and Fibiger, 1995). It has been suggested that the cortical projection to the VTA may contribute more importantly to the rewarding property of stimulation, because Glu antagonists infused into the VTA block the increase in DA release induced by mPFC stimulation (Taber et al., 1995). However, functional interactions between Glu and DA also occur at the level of NAS via metabotropic Glu receptors after mPFC stimulation (Taber and Fibiger, 1995). Thus, NAS may be an important site of CCK, Glu, and DA interactions. Indeed, CCK and glutamatergic drugs administrated into the NAS have been shown to modulate several DA-related behaviors, including cocaine self-administration (Ettenberg et al., 1982, Pulvirenti et al., 1992), expression of morphine-induced place preference (Higgins et al., 1992), locomotion (Crawley et al., 1985; Burns et al., 1994), and intracranial self-stimulation (Vaccarino and Vaccarino, 1989; Carlezon and Wise, 1996a).
The present study was designed to characterize the effects of electrical stimulation of mPFC on the release of CCK, Glu, and DA in the NAS. Transmitter release was determined as a function of stimulation frequency and stimulation intensity over the effective ranges of these parameters. The effect of stimulation of mPFC was also investigated in rats trained to lever press for the stimulation. Finally, the involvement of Glu receptors in the effect of mPFC stimulation on NAS transmitter release was investigated through perfusion of kynurenic acid (Kyn), a broad-spectrum antagonist at ionotropic Glu receptors, into the VTA or NAS.
MATERIALS AND METHODS
Animal preparation and testing
Subjects. All experiments were performed on 350–450 gm male Long–Evans rats (Charles-River, St. Constant, Quebec, Canada), which, except during testing, were housed individually under a 12 hr light/dark cycle (lights on from 8:00 A.M. to 8:00 P.M.) with access to food and water ad libitum. The experimental procedures were performed in accordance with the principles of animal care outlined by the Canadian Council on Animal Care and the National Institutes of Health.
Surgery. The rats were anesthetized for surgery with pentobarbital (65 mg/kg, i.p.) and then placed in a stereotaxic apparatus. A monopolar stimulating electrode and a guide cannula for microdialysis were stereotaxically implanted into the left mPFC and NAS, respectively, of each animal. Coordinates for the electrode implantation were as follows: anterioposterior (AP), 3.2; mediolateral (ML), 0.8; dorsoventral (DV), −4.4 relative to bregma and the skull surface (Paxinos and Watson, 1986). Coordinates for the guide cannula were as follows: AP 1.7; ML 1.5; DV −6.0. The anode was uninsulated stainless steel wire wrapped around one of four stainless steel screws that were threaded into the skull. For one series of experiments in which a Glu antagonist was infused into the VTA, a second guide cannula was implanted with coordinates of AP −5.0, ML 1.0, and DV −6.5. After the assemblies were secured to the skulls with dental cement, stainless steel blockers were inserted into the guide cannulas and fixed in place until the insertion of microdialysis probes. The animals were then returned to their home cages for at least 7 d recovery before receiving self-stimulation training or being subjected to microdialysis.
Forced stimulation. Thirty-one rats had dialysis probes inserted in preparation for forced stimulation on the following day (see below). Forced stimulation (0.5 sec trains of 0.1 msec pulses, with an intertrain interval of 2 sec) was given to five to eight animals at each of seven stimulation frequencies (6, 12, 25, 50, 100, 200, and 400 Hz, with a constant stimulation intensity at 500 μA) or of four stimulation intensities (50, 250, 500, and 700 μA, with a constant stimulation frequency at 100 Hz) for periods of 40 min during which microdialysis samples were taken (see below). Each animal was tested twice: once in the morning with stimulation at one frequency or intensity and once in the afternoon of the same day at a second frequency or intensity. Each stimulation frequency or intensity was presented in the morning session to half of the assigned animals and in the afternoon session to the other half of the assigned animals. The two stimulation periods for each animal were at least 3 hr apart.
Self-stimulation training. Thirteen rats were trained to lever press for mPFC stimulation. They were placed in operant chambers with a lever connected to a computer-controlled pulse generator (Mundl, 1980). Each lever press earned a 0.5 sec train of 0.1 msec rectangular cathodal pulses to the mPFC. The stimulation currents were adjusted to intensities at which no motor side effects were produced. The rats were subjected to the microdialysis experiment after a minimum of 5 d of stable responding was established. Seven rats were tested at each of three stimulation frequencies (25, 100, and 400 Hz); two frequencies were tested on the first microdialysis day, and one was tested on the following day. The order of testing of the three frequencies was counterbalanced across animals. Another six rats were used to investigate the effect of Kyn (Research Biochemicals International, Natick, MA) on the increases in NAS neurotransmitter levels induced by stimulation.
In vivo microdialysis. Microdialysis probes with a concentric design were constructed as described previously (Wise et al., 1996), except that 22 gauge stainless steel outlet cannulas and a membrane with a larger molecular cutoff (molecular weight cutoff, 40 kDa) (AN69; Hospal-Gambro, Inc.) were used in the present study. The active membrane was ∼3 mm for NAS and 2 mm for VTA probes. Fifteen hours before the beginning of each microdialysis experiment, the rats were transported to the testing room. The blockers were removed, and the microdialysis probes were then fixed in position, connected to the microdialysis pump (Harvard), and perfused at a flow rate of 0.4 μl/min overnight. On the following day, the rats were transferred to the self-stimulation boxes, and the flow rate was increased to 2 μl/min until the end of the experiment. The microdialysis probes were perfused with a Krebs’–Ringer’s solution (in mm: 138 NaCl, 11 NaHCO3, 3 KCl, 1 CaCl2, 1 MgCl2, and 1 NaH2PO4), supplemented with 2 gm/l glucose, 2 gm/l bovine serum albumin, and 0.3 gm/l bacitracin (Sigma, St. Louis, MO). After four baseline samples were collected, the animals were either given 40 min of forced stimulation or allowed to access the lever to earn stimulation at assigned stimulation frequency and current intensity. Dialysis samples were collected at 40 min intervals and split for analysis of CCK (60 μl), DA (10 μl), and Glu (10 μl), respectively.
Biochemical analysis
CCK radioimmunoassay. The determination of CCK was performed by radioimmunoassay as reported previously (Meana et al., 1991; You et al., 1994). Briefly, samples (100 μl, containing 60 μl of perfusate) and standards diluted in the perfusion medium (100 μl) were incubated with the CCK antiserum (2609) (Rehfeld, 1978) in Eppendorf polyethylene tubes for 24 hr at 4°C. After subsequent addition of 125I-gastrin (1000 cpm/tube) (Milab, Malmo, Sweden), all samples were further incubated for 72 hr at 4°C. Samples without antiserum (to determine nonspecific binding) and samples without unlabeled peptide (to determine maximal tracer binding) were simultaneously incubated. The antiserum and labeled peptide used in the assay were diluted in barbital buffer to yield a final barbital concentration of 0.02 m in a total incubation volume of 150 μl. After incubation, the antibody-bound and free tracer were separated by the addition of anti-rabbit IgG coupled to Sepharose (Pharmacia Decanting Suspension 3; Pharmacia, Uppsala, Sweden) and centrifuged at 2000 × g for 15 min in a Beckman centrifuge. The bound fraction was counted in a gamma counter. The detection limit was 0.5 pm. The nonspecific binding of the tracer to the precipitated pellet in the absence of the antiserum was <2%.
Catecholamine determination. DA and its metabolites, dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA), were measured with HPLC coupled to a Coulochem II detector (5200; ESA, Inc., Bedford, MA) with two analytical electrodes and a dual-channel Spectra-Physics (Fremont, CA) SP4270 integrator connected to Spectra-Physics Winner software on an IBM computer. DA was quantified on a reducing electrode (−260 mV, 10 nA), and metabolites were quantified on an oxidizing electrode (340 mV, 500 nA). The limit of detection for DA was ∼0.3 pg/injection (Wise et al., 1996).
Glu determination. Glu was measured in 10 μl of perfusate using an HPLC system with a fluorescence detection and a precolumn derivatization method. The precolumn derivatization of Glu was performed with an o-phthalaldehyde–mercaptoethanol reagent. The derivatization and the injection were performed automatically by an autoinjector (AS3500; Spectra-Physics). The elution of Glu was achieved with a phosphate mobile phase (0.15 m phosphate, pH 6.8, adjusted with 1N NaOH containing 30% methanol and 25 mg/l EDTA) at a flow rate of 1 ml/min.
Histology
After the completion of the microdialysis experiment, the rats were anesthetized with pentobarbital, and a 250 μA 115 V current was passed through each stimulation electrode, producing a small lesion at the electrode tip. The rats were then decapitated, and the brains were removed and post-fixed as described previously (Bauco and Wise, 1994). Thirty micrometer coronal sections were sliced with a cryostat, and the probe and electrode locations were examined microscopically at low magnification. Only animals with correctly implanted probes and electrodes were included in the statistical comparisons. One animal was discarded because of an improperly placed probe, and one was discarded because of an improperly placed electrode. The histological localizations of the stimulating electrodes and of the microdialysis probes are shown in Figure 1.
Fig. 1.

Histological localizations of the stimulating electrode tips in the mPFC (left) and of the microdialysis membranes in the NAS (middle) and VTA (right). The number of circles or lines is less than the number of rats used in these studies because of overlapping placements among several animals. The numbers on each section indicate the distance from the bregma. Two rats were excluded from the data analysis because of the stimulating electrode located in the secondary motor cortex in one rat and the microdialysis membrane located dorsomedially to the NAS in another. The drawings were derived from the atlas of Paxinos and Watson (1986).
Statistics
The levels of the assayed substances were expressed as the concentrations found in the perfusates (mean ± SEM). Basal values refer to those obtained before each stimulation or before drug was added into the perfusion medium. When data were expressed as percentages of controls, the average concentration of the two samples preceding the stimulation were defined as 100%. Basal levels of the substances between trained and untrained rats were analyzed with Student’s t test. The stimulation effects on CCK, Glu, and DA and its metabolites were analyzed with one-way or two-way ANOVA with repeated measures over time. Comparisons with prestimulation baseline or between treatment groups were performed using Dunnett’s test or Fisher’s least significant difference (LSD) test. A level of p < 0.05 for a two-tailed test was considered critical for statistical significance.
RESULTS
Basal levels
Basal levels of CCK in the NAS were ∼2 pm, basal Glu levels were ∼12 μm, and basal levels of DA and its metabolites were ∼2 and 200 nm, respectively. After self-stimulation training, basal levels of NAS CCK were significantly elevated (t = 2.43; p < 0.02). Basal Glu levels showed a tendency to increase after self-stimulation training, but the increase was not statistically significant (t = 1.77; p = 0.09). Self-stimulation training did not change basal levels of either DA or its metabolite levels in the NAS (Table 1).
Table 1.
Basal extracellular levels of CCK, Glu, DA, DOPAC, HVA, and 5-HIAA
| Untrained | Trained | |
|---|---|---|
| CCK (pm) | 1.92 ± 0.26 | 3.03 ± 0.39* |
| Glu (μm) | 12.2 ± 1.7 | 17.3 ± 2.3 |
| DA (nm) | 2.07 ± 0.21 | 1.98 ± 0.38 |
| DOPAC (nm) | 207 ± 23 | 162 ± 26 |
| HVA (nm) | 181 ± 24 | 173 ± 27 |
| 5-HIAA (nm) | 66 ± 7 | 58 ± 13 |
Basal extracellular levels of CCK, Glu, DA, DOPAC, HVA, and 5-HIAA (mean ± SEM) measured by microdialysis in the nucleus accumbens of trained (n = 13) and untrained (n = 25–27) rats.
*p < 0.05, Student’s t test (two-tailed).
Effects of forced stimulation of mPFC on extracellular levels of CCK, Glu, DA, DOPAC, and HVA in the NAS
Effect of stimulation frequency
Experimenter-administered electrical stimulation of mPFC induced frequency-dependent increases in NAS CCK levels (Fig.2), with a significant increase obtained at 50 Hz and a maximal increase reached at 200 Hz. Extracellular levels of Glu were increased over a larger range of stimulation frequencies (25–400 Hz), but near-maximal elevations were seen with stimulation at 50 Hz and higher. DA levels in the NAS increased significantly in response to stimulation at 25 Hz and increased only slightly more with progressively higher stimulation frequencies. DOPAC and HVA levels were less sensitive than DA levels to stimulation at the lower frequencies; significant increases were seen only with stimulation frequencies of ≥100 Hz (Fig. 2). Electrical stimulation of mPFC at 6 or 12 Hz was ineffective in changing extracellular levels of any of the substances measured (Fig. 2).
Fig. 2.
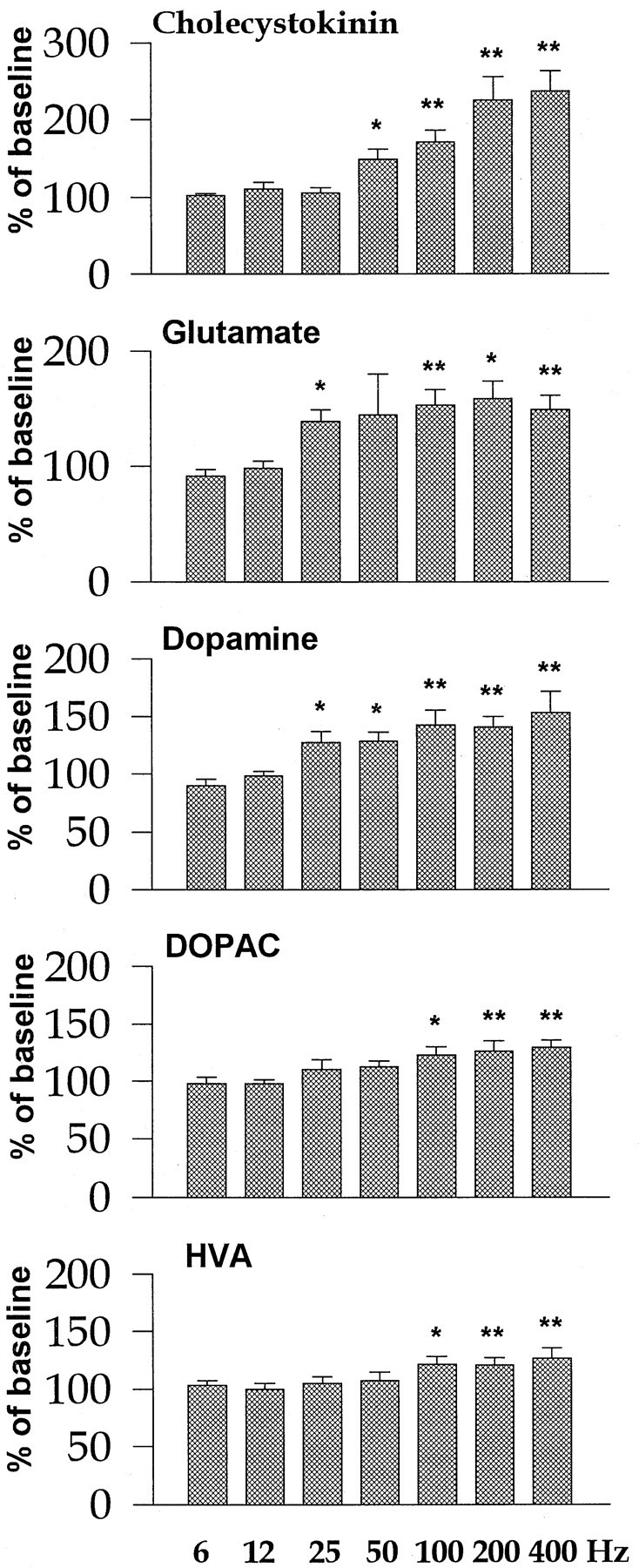
Effect of forced stimulation at various frequencies on extracellular CCK, Glu, DA, DOPAC, and HVA levels in the NAS. The stimulation parameters were set as follows: current intensity, 500 μA; train duration, 0.5 sec; pulse width, 0.1 msec; and interstimulation interval, 2 sec. The values are expressed as percentage of the basal values obtained before stimulation (mean ± SEM) (see also Materials and Methods). *p < 0.05; **p < 0.01, one-way ANOVA followed by Dunnett’s post hoc comparison; n = 5–8 per frequency.
Effects of current intensity
Levels of stimulation-induced CCK, Glu, and DA release were each intensity-dependent at 250–700 μA; 50 μA was without effect (Fig.3). The elevations in Glu levels were significant only at 500 and 700 μA, whereas the elevations of CCK and DA were significant at 250, 500, and 700 μA. DOPAC and HVA levels were significantly increased in response to stimulation at 500 and 700 μA but not at 50 or 250 μA.
Fig. 3.
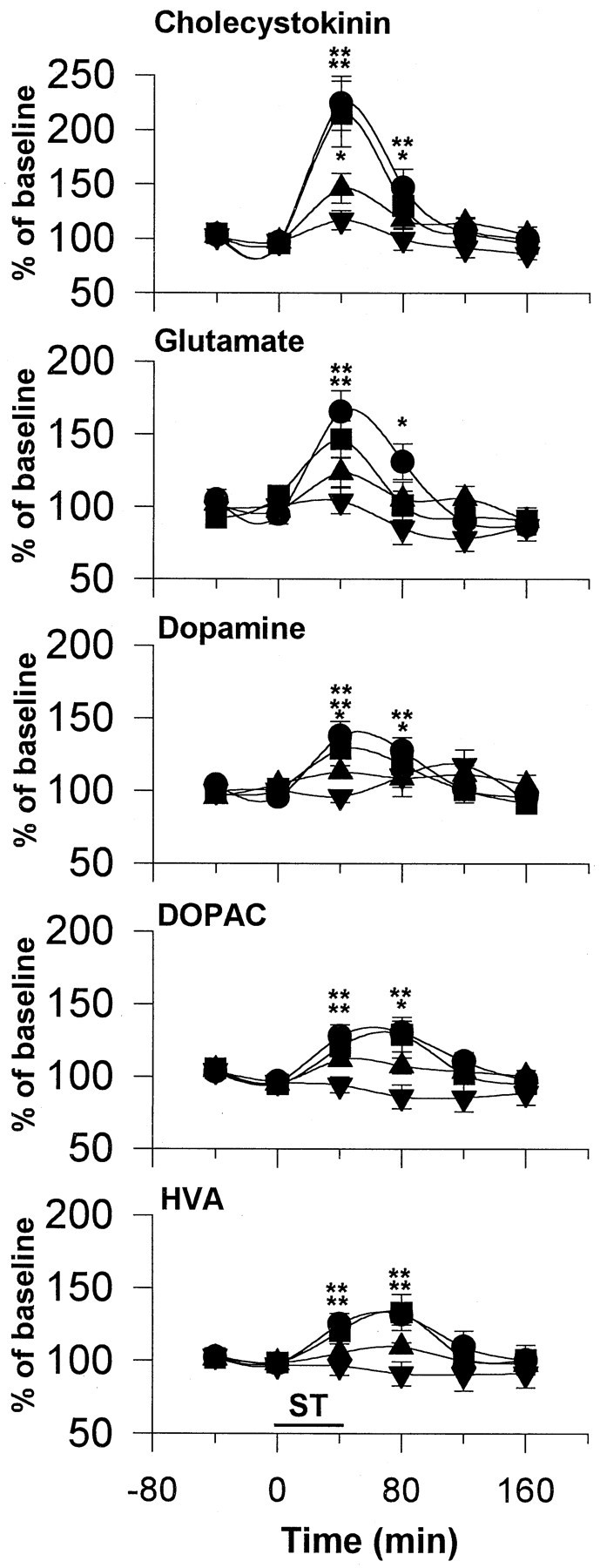
Effect of forced stimulation at various intensities on extracellular CCK, Glu, DA, DOPAC, and HVA levels in the NAS. The stimulation parameters were the same as in Figure 2, except that stimulation frequency was fixed at 100 Hz, whereas stimulation current was varied. Circles, 700 μA;squares, 500 μA; upward anddownward triangles, 250 and 50 μA, respectively. The data were analyzed using a two-way ANOVA with repeated measures over time, followed by Fisher’s LSD test. n = 6–7 in each group. For CCK: treatment, F(3,21) = 5.47; p < 0.01; time,F(5,105) = 30.58; p < 0.001; interaction,F(15,105) = 3.68; p < 0.001. For Glu: treatment, F(3,20) = 8.56;p < 0.001; time,F(5,100) = 13.49; p < 0.001; interaction, F(15,100) = 2.17;p < 0.01. For DA: treatment,F(3,21) = 0.80; p = 0.51; time, F(5,105) = 8.95;p < 0.001; interaction,F(15,105) = 2.44; p < 0.02. For DOPAC: treatment, F(3,21) = 4.59;p < 0.01; time,F(5,105) = 10.76; p < 0.001; interaction, F(15,105) = 3.224;p < 0.001. For HVA: treatment,F(3,21) = 2.65; p = 0.07; time, F(5,105) = 7.22;p < 0.001; interaction,F(15,105) = 1.91; p < 0.05. *p < 0.05; **p < 0.01, compared with respective baseline values.
Effects of self-stimulation of mPFC on extracellular levels of CCK, Glu, DA, DOPAC, and HVA in NAS
Most rats did not respond for 25 Hz stimulation beyond the first few minutes of their sessions; all animals readily responded for higher frequencies of stimulation. The mean lever-press rates were 26 ± 3 and 36 ± 5 responses/min at 100 and 400 Hz, respectively. Self-stimulation induced frequency-dependent increases in CCK levels, as found in forced-stimulated rats (Fig.4). Glu and DA levels were increased to similar extents by 100 and 400 Hz stimulation (Fig. 4). DOPAC and HVA levels were increased after 100 and 400 Hz self-stimulation to the extents found in forced-stimulated rats. No effects of 25 Hz self-stimulation were observed in any of the substances studied, but it must be remembered that the rats did not respond for this stimulation for more than a small fraction of the test period.
Fig. 4.
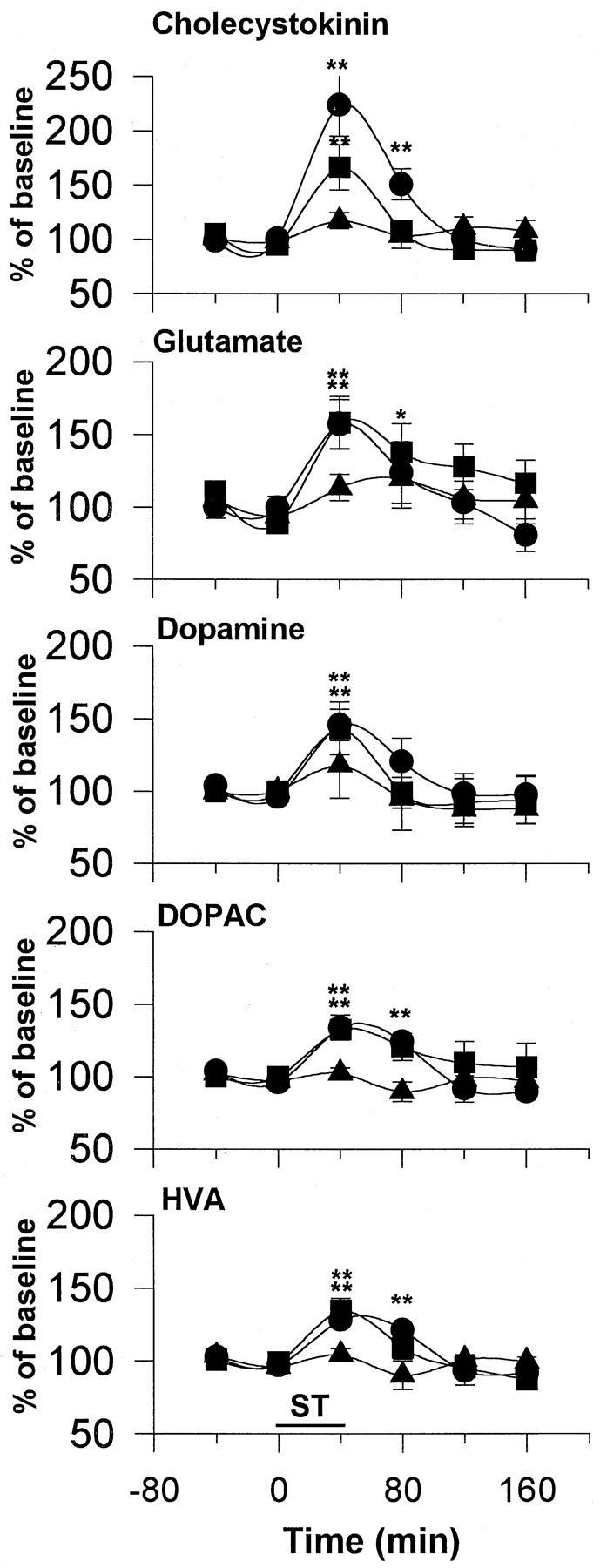
Effect of self-stimulation of mPFC on extracellular CCK, Glu, DA, DOPAC, and HVA levels in the NAS. The rats were allowed access to stimulation for 40 min at each of three frequencies: 400 Hz (circles), 100 Hz (squares), and 25 Hz (triangles) after the establishment of a stable baseline for DA. The mean lever-press rates were 36 ± 5, 26 ± 3, and 7 ± 3 responses/min, respectively. The current intensity was fixed at each animal’s training intensity (600–800 μA), whereas other stimulation parameters were the same as in Figure 2. The data were analyzed using a two-way ANOVA with repeated measures over time followed by Fisher’s LSD test. n = 6–7 in each group. For CCK: treatment, F(2, 18) = 4.11;p < 0.05; time, F(5, 90) = 21.08; p < 0.001; interaction,F(10,90) = 5.23; p < 0.001. For Glu: treatment, F(2,17) = 1.94; p = 0.17; time, F(5,85)= 4.75; p < 0.001; interaction,F(10,85) = 0.92; p = 0.52. For DA: treatment, F(2,18) = 0.45;p = 0.64; time, F(5,90)= 6.78; p < 0.001; interaction,F(10,90) = 0.48; p = 0.89. For DOPAC: treatment, F(2,18) = 2.20;p = 0.14; time, F(5,90)= 7.48; p < 0.001; interaction,F(10,90) = 2.76; p = 0.05. For HVA: treatment, F(2,18) = 1.39;p = 0.28; time, F(5,90)= 11.16; p < 0.001; interaction,F(10,90) = 3.25; p < 0.05. *p < 0.05; **p < 0.01, compared with respective baseline values.
Effects of perfusion with Kyn on the increases in extracellular levels of CCK, Glu, DA, DOPAC, and HVA in the NAS induced by mPFC stimulation
In this experiment, 1 mm Kyn was perfused through the microdialysis probe into either the VTA or NAS; 40 min later the effect of stimulation was tested. Local administration of Kyn has been shown previously to inhibit burst firing of DA neurons in the VTA (Charlety et al., 1991), antagonize the increases in firing rates of DA neurons induced by both NMDA and AMPA application in the substantia nigra (Wu et al., 1994), and attenuate the increases in DA release induced by kainic acid in the striatum (Carrozza et al., 1991). Furthermore, local application of Kyn, but not the selective NMDA antagonist 2-AP7, inhibits the excitation of striatal neurons after stimulation of corticostriatal pathway (Herrling, 1985). At the concentration used here, Kyn blocks the changes in NAS DA release induced by NMDA perfusion but is below the threshold for altering basal NAS DA levels (Taber and Fibiger, 1995; Taber et al., 1996). In the present study, perfusion of Kyn into either the VTA or the NAS blocked lever pressing for mPFC stimulation.
Perfusion of Kyn into the VTA, but not into the NAS, significantly inhibited the increase in accumbens DA levels induced by forced stimulation. The effects of stimulation on DOPAC and HVA were also inhibited after the perfusion of Kyn into the VTA. Perfusion of Kyn into the VTA did not significantly affect stimulation-induced increases in either CCK or Glu levels (Fig. 5). Perfusion of Kyn into the NAS only slightly attenuated the increase in Glu levels induced by stimulation. NAS perfusion of Kyn contaminated the DOPAC peak; thus, the effect on DOPAC could not be determined (Fig.5).
Fig. 5.
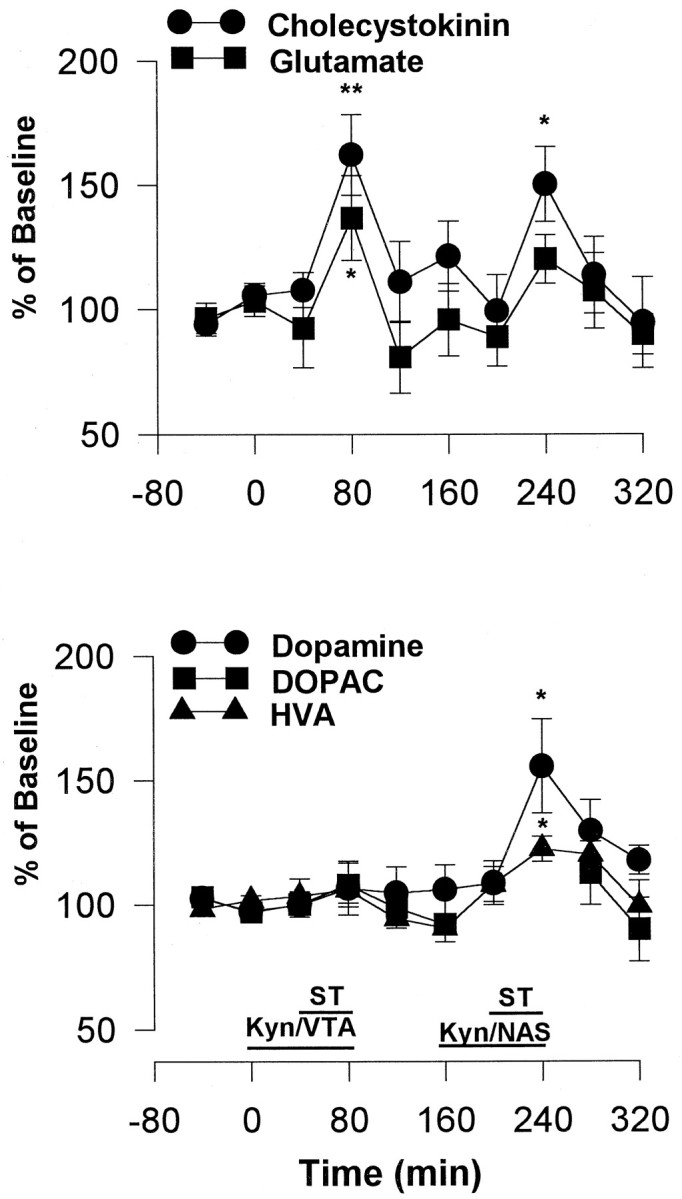
Effect of Kyn perfusion into the VTA (Kyn/VTA) or into the NAS (Kyn/NAS) on the stimulation-induced increases in extracellular CCK, Glu, DA, DOPAC, and HVA levels in the NAS. The stimulation frequency was set at 100 Hz, and the current intensity was set at each animal’s training intensity, whereas other stimulation parameters were the same as in Figure 2. *p < 0.05; **p < 0.01, one-way ANOVA followed by Dunnett’s post hoccomparison; n = 6.
DISCUSSION
The present study demonstrates for the first time that activation of mPFC neurons induces CCK release in the NAS. This finding strengthens the hypothesis that NAS levels of CCK reflect the neuronal input to this area. The possibility of direct depolarization of other NAS CCK afferents can be ruled out based on our previous finding showing that current spread at rewarding stimulation parameters is <0.5 mm in distance (Fouriezos and Wise, 1984), which is much shorter than the distance from the mPFC electrode to the NAS probe (>1.5 mm) in the present study. Supporting this, Taber and Fibiger (1995)have shown that NAS DA release after mPFC stimulation is completely TTX-dependent, indicating that stimulation does not have a direct effect on the mesoaccumbens DA pathway in which CCK is colocalized. Thus, the fact that the stimulation-induced increases in NAS CCK levels were both intensity- and frequency-dependent suggests that the CCK release depends on both the number of mPFC efferents activated and the rate of impulse flow in the activated fibers. This leaves unanswered the question of why unilateral lesions of this system have little effect on striatal or NAS CCK levels (You et al., 1994; Kresse et al., 1995; Maidment and Villafranca, 1997); perhaps there are compensatory changes in release or metabolism of CCK from the other CCK sources in NAS. In any case, the present study suggests that significant elevations of NAS CCK are caused by the activation of neuronal input to NAS from mPFC.
Of the various NAS afferents in which CCK is colocalized, the direct projection of glutamatergic pyramidal cells from mPFC is the most likely source of the stimulation-induced CCK release reported here. Both Glu and CCK were elevated by the stimulation, and the mPFC striatal fibers, presumed to colocalize the two substances (Morino et al., 1994a,b), offer the most direct link between the two structures. Still, the NAS receives other CCK-containing afferents. Cortical CCK-containing neurons innervate the whole rostrocaudal level of NAS, with the most dense innervation in the rostral part (Morino et al., 1994a,b). Mesoaccumbens DA neurons colocalize CCK and innervate most strongly the caudal NAS (Hökfelt et al., 1980; Seroogy et al., 1989). Other sources of CCK, although less pronounced, have been reported in both intrinsic medium spiny neurons and in aspiny neurons in NAS (Hökfelt et al., 1980; Schiffman and Vanderhaeghen, 1992) and also in extrinsic neurons originating from amygdala and nucleus tractus solitarius (Wang et al., 1992; Kresse et al., 1995). These multiple sources of NAS CCK offer several possible sources for the relatively normal basal levels of CCK found after unilateral lesions to the corticostriatal pathway.
One possibility can be ruled out based on the present evidence. Although there is a glutamatergic projection from mPFC to the DA CCK neurons of the VTA (which project to the NAS), it seems clear that mPFC stimulation did not elevate NAS CCK significantly by transsynaptically activating the mesolimbic DA CCK projection. Stimulation of mPFC does activate glutamatergic projections to the VTA, and such activation is capable of activating the mesolimbic DA system and elevating NAS DA (Taber and Fibiger, 1995; Rossetti et al., 1998). Such activation is blocked by coapplication of AP-5 and CNQX into the VTA (Taber et al., 1995) and was confirmed by Kyn application in the present study. However, in the present experiment, the same VTA Kyn treatment that reduced stimulation-induced DA release in the NAS failed to reduce stimulation-induced CCK release in the same perfusate. Thus, it seems likely that the mPFC stimulation in the present study was sufficient to cause Glu release in both the NAS and VTA. It is also likely that the Glu release in the VTA was sufficient to cause DA release and the accumulation of DA metabolites in NAS, but that this activation of mesolimbic DA fibers was insufficient to cause significant corelease of CCK. This reasoning would leave the direct Glu–CCK pathway from mPFC to NAS as the major source of the stimulation-induced CCK release in NAS.
It is interesting to note that stimulation-induced release of Glu and DA were each near the maximal release with lower stimulation frequencies than were necessary to cause maximal CCK release. This is consistent with what is known about classic and peptide cotransmitter release in general; amino acid transmitters tend to be released by stimulation at lower frequencies than are needed to drive the release of peptide cotransmitters (Bartfai et al., 1988; Lundberg et al., 1994). Presumably, this means that peptide cotransmitters are preferentially released by rapidly firing or bursting of neurons. Such preferential release offers a possible explanation for how mPFC stimulation might be sufficient to transsynaptically activate DA fibers and cause DA release but be insufficient to release significant amounts of the peptide cotransmitter CCK.
The frequency response of the self-stimulation behavior more closely paralleled the frequency response of CCK release than it did the frequency response of Glu or DA release. Although near-maximal release of Glu and DA was seen with 25 Hz stimulation, the animals were minimally interested in stimulation at this frequency; although the animals became progressively more engaged by stimulation at increasing frequencies >50 Hz, increasing stimulation frequency >50 Hz caused minimal increases in Glu or DA release. Meanwhile, stimulation at 50, 100, and 200 Hz caused progressively stronger release of CCK, just as it caused progressively stronger motivation of behavior. These data are of considerable interest; the discrepancy between the frequency responses of DA release and the frequency responses of the behavior have been of long-standing concern (Wise, 1978).
The relatively high frequency required for mPFC self-stimulation (>25 Hz) raises the possibility that although Glu from the mPFC projection is essential for the behavior, CCK or some other peptide cotransmitter plays a significant modulatory role, determining the motivational impact of the basic Glu signal. Electrophysiological and pharmacological studies have shown that CCK and its analogs are involved in memory processes (Itoh et al., 1988), induction of long-term potentiation (Rich-Bennett et al., 1993), and potentiation of extinction of a learned task (Itoh and Katsura, 1987). CCK perfused into the NAS increases DA release (Ruggeri et al., 1987), at least in the posterior NAS (Ladurelle et al., 1993). However, it has been shown that NAS CCK administration inhibits the rewarding efficacy of VTA stimulation (Vaccarino and Koob, 1984). CCK injected directly into the cortex has been reported to have no effect on mPFC self-stimulation (Ferrer et al., 1988), but we know of no studies of the effect on mPFC self-stimulation of CCK injected into the NAS or VTA. Thus, further study is needed to elucidate the function of NAS CCK on mPFC self-stimulation.
Perfusion of Kyn into either the NAS or VTA, without significant affect on CCK or Glu release in the NAS, totally blocked lever pressing for mPFC stimulation. Kyn into the VTA blocked the increase in NAS DA induced by stimulation, confirming reports that cortical input to the VTA contributes more importantly in regulation of DA function than does cortical input to the NAS (Taber et al., 1995; Karreman et al., 1996). In agreement with the present finding, Glu agonists into the VTA have been shown to increase the firing rate and cause bursting in VTA DA neurons (Johnson et al., 1992; Chergui et al., 1993), as well as the release of DA in NAS (Karreman et al., 1996; Westerink et al., 1996). Thus, perhaps the inhibiting effect of VTA Kyn on self-stimulation can be partially attributable to blockade of the effect of Glu on DA function in this region. The blocking effect of Kyn in the NAS is unexpected and difficult to interpret, because most NMDA antagonists in the NAS have been shown to have rewarding and reward-enhancing effects (Carlezon and Wise, 1996a,b; Olds, 1996; Wise 1996a,b). Unlike the substrates of lateral hypothalamus and VTA self-stimulation (Gallistel et al., 1981), which are critically dependent on mesolimbic DA functions, the substrate of mPFC self-stimulation has not been very well characterized. Although it has been shown to be relatively insensitive to some DA antagonists (Corbett, 1990) and to DA depletion (Phillips and Fibiger, 1978; Simon et al., 1979), chronic cocaine administration does enhance the reward value of mPFC stimulation, as revealed by increases in response rate and decreases in reward threshold (Corbett, 1991). Thus, although the details of mechanism are not known, it seems possible that, in addition to some contribution of the mesolimbic DA pathway, mPFC brain stimulation reward involves circuitry in which Glu and CCK interact or make series (transsynaptic) contributions.
Despite a dense projection of glutamatergic fibers to the NAS (Sesack et al., 1989; Berendse et al., 1992), stimulation in the present study caused much weaker elevations of Glu than of CCK. Numerous studies have characterized the origin of extracellular Glu levels as measured by microdialysis. The insensitivity of Glu concentration to both TTX perfusion and calcium depletion, along with its weak responsiveness to potassium depolarization, has been considered an indication that basal Glu levels are more closely related to metabolic events than to neuronal impulse flow (Westerink et al., 1987; Moghaddam, 1993; Morari et al., 1993; You et al., 1994; Herrera-Marschitz et al., 1996). Thus, the contribution of neuronally released Glu to the pool of Glu sampled by microdialysis has been difficult to characterize. On the basis of inclusion of high Mg2+ in a Ca2+-free perfusion medium or of administration of the gliotoxin fluorocitrate, it has been estimated that perhaps only 10–30% of extracellular levels of this transmitter are of immediate neuronal origin (Fonnum, 1984; Paulsen and Fonnum, 1989). Basal levels of Glu are thought to reflect a net sum of release from the neuronal pool and secretion from the metabolic pool on the one hand and uptake by neuronal terminals and astrocytes on the other.
However, although it has been difficult to confirm the neuronal origin of basal Glu levels, TTX-dependent elevations in VTA and NAS Glu levels have been found to result from electrical stimulation of mPFC (Rossetti et al., 1998) and stress (Moghaddam, 1993). Because a major portion of basal Glu is unresponsive to neuronal firing, the percentage increase in Glu levels induced by neuronal stimulation must necessarily give an underestimate of the effectiveness of the stimulation. Thus, a less than twofold increase in extracellular Glu levels may actually represent a much higher and very significant increase in stimulation-induced Glu release. Perfusion of Kyn into the NAS slightly attenuated the increase in Glu levels induced by stimulation; one possibility is that this contribution could reflect an axon–axonal interaction between cortical Glu neurons and Glu afferents to NAS. Glutamatergic afferents to NAS originate in the amygdala, hippocampus, and thalamus (Walaas and Fonnum, 1980; Sesack et al., 1989), and these afferents are each likely to contribute, along with afferents from mPFC, to NAS Glu levels.
In summary, the present study demonstrates that phasic activation of mPFC by electrical stimulation increases CCK and Glu release in the ipsilateral NAS. The increases are mainly of cortical origin, because blockade of Glu receptors in either the VTA or the NAS failed to block the stimulation-induced increases. The effects were seen at levels of stimulation that were self-selected by the animal, suggesting the possible relevance of these transmitters to the fact that such stimulation can be rewarding. Although the attenuation of mPFC self-stimulation after Kyn perfusion into either the VTA or NAS suggests an essential role of postsynaptic Glu receptor activation, the corelation of increasing CCK release with increasingly preferred stimulation frequencies indicates this coreleased peptide may modulate the impact of the glutamatergic action in this behavior.
Footnotes
This study was supported by National Institute on Drug Abuse Grant DA01720 and Swedish Medical Research Council Grant 6836. T.M.T. was the recipient of a Deutscher Akademischer Austauschdienst scholarship (Doktorandenstipendium HSP3). We gratefully acknowledge the excellent assistance of Annika Olsson. We thank Dr. J. Rehfeld (Rigshospitalet, University of Copenhagen, Copenhagen, Denmark) for the generous supply of gastrin/CCK antiserum.
Correspondance should be addressed to Dr. Zhi-Bing You, Center for Studies in Behavioral Neurobiology, Department of Psychology, Concordia University, 1455 Boulevard De Maisonneuve W, Montreal, Quebec, Canada H3G 1M8.
Dr. Tzschentke’s present address: Department of Neuropharmacology, Zoological Institute, University of Tubingen, Mohlstrasse 54/1, 72074 Tubingen, Germany.
REFERENCES
- 1.Bartfai T, Iverfeldt K, Fisone G, Serfozo P. Regulation of the release of coexisting neurotransmitters. Annu Rev Pharmacol Toxicol. 1988;28:285–310. doi: 10.1146/annurev.pa.28.040188.001441. [DOI] [PubMed] [Google Scholar]
- 2.Bauco P, Wise RA. Potentiation of lateral hypothalamus and midline mesencephalic brain stimulation reinforcement by nicotine: examination of repeated treatment. J Pharmacol Exp Ther. 1994;271:294–301. [PubMed] [Google Scholar]
- 3.Berendse HW, Galis-de Graaf Y, Groenewegen HJ. Topographical organization and relationship with ventral striatal compartments of prefrontal cortical striatal projections in the rat. J Comp Neurol. 1992;316:314–347. doi: 10.1002/cne.903160305. [DOI] [PubMed] [Google Scholar]
- 4.Burgunder J-M, Young WS. Cortical neurons expressing the cholecystokinin gene in the rat: distribution in the adult brain, ontogeny, and some of their projections. J Comp Neurol. 1990;300:26–46. doi: 10.1002/cne.903000104. [DOI] [PubMed] [Google Scholar]
- 5.Burns LH, Everitt BJ, Kelley AE, Robbins TW. Glutamate–dopamine interactions in the ventral striatum: role in locomotor activity and responding with conditioned reinforcement. Psychopharmacology. 1994;115:516–528. doi: 10.1007/BF02245576. [DOI] [PubMed] [Google Scholar]
- 6.Butcher SP, Varro A, Kelly JS, Dockray GJ. In vivo studies on the enhancement of cholecystokinin release in the rat striatum by dopamine depletion. Brain Res. 1989;505:119–122. doi: 10.1016/0006-8993(89)90121-2. [DOI] [PubMed] [Google Scholar]
- 7.Carlezon WA, Wise RA. Microinjections of phencyclidine and related drugs into nucleus accumbens shell potentiate medial forebrain bundle brain stimulation reward. Psychopharmacology. 1996a;128:413–420. doi: 10.1007/s002130050151. [DOI] [PubMed] [Google Scholar]
- 8.Carlezon WA, Wise RA. Rewarding actions of phencyclidine and related drugs in nucleus accumbens shell and frontal cortex. J Neurosci. 1996b;16:3112–3122. doi: 10.1523/JNEUROSCI.16-09-03112.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carrozza DP, Ferraro TN, Golden GT, Reyes PF, Hare TA. Partial characterization of kainic acid-induced striatal dopamine release using in vivo microdialysis. Brain Res. 1991;543:69–76. doi: 10.1016/0006-8993(91)91049-7. [DOI] [PubMed] [Google Scholar]
- 10.Charlety PJ, Grenhoff J, Chergui K, De la Chapelle B, Buda M, Svensson TH, Chouvet Q. Burst firing of mesencephalic dopamine neurons is inhibited by somatodendritic application of kynurenate. Acta Physiol Scand. 1991;142:105–112. doi: 10.1111/j.1748-1716.1991.tb09134.x. [DOI] [PubMed] [Google Scholar]
- 11.Chergui K, Charlety PJ, Akaoka H, Saunier CF, Brunet J-L, Buda M, Svensson TH, Chouvet G. Tonic activation of NMDA receptors causes spontaneous burst discharge of rat midbrain dopamine neurons in vivo. Eur J Neurosci. 1993;5:137–144. doi: 10.1111/j.1460-9568.1993.tb00479.x. [DOI] [PubMed] [Google Scholar]
- 12.Christie MJ, Summers RJ, Stephenson JA, Cook CJ, Beart PM. Excitatory amino acid projections to the nucleus accumbens septi in the rat: a retrograde transport study utilizing D[3H]aspartate and [3H]GABA. J Neurosurg. 1990;72:231–244. doi: 10.1016/0306-4522(87)90345-9. [DOI] [PubMed] [Google Scholar]
- 13.Corbett D. Differences in sensitivity to neuroleptic blockade: medial forebrain bundle versus frontal cortex self-stimulation. Behav Brain Res. 1990;36:91–96. doi: 10.1016/0166-4328(90)90163-9. [DOI] [PubMed] [Google Scholar]
- 14.Corbett D. Cocaine enhances the reward value of medial prefrontal cortex self-stimulation. NeuroReport. 1991;2:805–808. doi: 10.1097/00001756-199112000-00019. [DOI] [PubMed] [Google Scholar]
- 15.Crawley JN, Hommer DW, Skirboll LR. Topographical analysis of nucleus accumbens sites at which cholecystokinin potentiates dopamine induced hyperlocomotion in the rat. Brain Res. 1985;335:337–341. doi: 10.1016/0006-8993(85)90489-5. [DOI] [PubMed] [Google Scholar]
- 16.Dockray GJ. Cholecystokinin in rat cerebral cortex: identification, purification and characterization by immunochemical methods. Brain Res. 1980;188:155–165. doi: 10.1016/0006-8993(80)90564-8. [DOI] [PubMed] [Google Scholar]
- 17.Dockray GJ, Gregory RA, Hutchinson JB, Harris JI, Runswick MJ. Isolation, structure and biological activity of two cholecystokinin octapeptides from sheep brain. Nature. 1978;274:711–713. doi: 10.1038/274711a0. [DOI] [PubMed] [Google Scholar]
- 18.Dodd J, Kelly JS. The actions of cholecystokinin and related peptides on pyramidal neurones of the mammalian hippocampus. Brain Res. 1981;205:337–350. doi: 10.1016/0006-8993(81)90344-9. [DOI] [PubMed] [Google Scholar]
- 19.Dodd PR, Edwardson JA, Dockray GJ. The depolarisation-induced release of CCK C-terminal octapeptide (CCK-8) from rat synaptosomes and brain slices. Regul Pept. 1980;1:17–29. [Google Scholar]
- 20.Druce D, Peterson D, De Belleroche J, Bradford HF. Differential amino acid neurotransmitter release in rat neostriatum following lesioning of the cortico-striatal pathway. Brain Res. 1982;247:303–307. doi: 10.1016/0006-8993(82)91254-9. [DOI] [PubMed] [Google Scholar]
- 21.Emson PC, Rehfeld JF, Langevin H, Rossor MN. Reduction in cholecystokinin-like immunoreactivity in the basal ganglia in Huntington’s disease. Brain Res. 1980;198:497–500. doi: 10.1016/0006-8993(80)90767-2. [DOI] [PubMed] [Google Scholar]
- 22.Ettenberg A, Pettit HO, Bloom FE, Koob GF. Heroin and cocaine intravenous self-administration in rats: mediation by separate neural mechanisms. Psychopharmacology. 1982;78:204–209. doi: 10.1007/BF00428151. [DOI] [PubMed] [Google Scholar]
- 23.Ferrer JM, Cobo M, Mora F. Peptides and self-stimulation of the medial prefrontal cortex in the rat: effects of intracerebral microinjections of substance P and cholecystokinin. Peptides. 1988;9:937–943. doi: 10.1016/0196-9781(88)90071-x. [DOI] [PubMed] [Google Scholar]
- 24.Fonnum F. Glutamate: a neurotransmitter in mammalian brain. J Neurochem. 1984;42:1–10. doi: 10.1111/j.1471-4159.1984.tb09689.x. [DOI] [PubMed] [Google Scholar]
- 25.Fonnum F, Storm-Mathisen J, Divac I. Biochemical evidence for glutamate as neurotransmitter in the cortico-striatal and cortico-thalamic fibres in rat brain. Neuroscience. 1981;6:863–875. doi: 10.1016/0306-4522(81)90168-8. [DOI] [PubMed] [Google Scholar]
- 26.Fouriezos G, Wise RA. Current-distance relation for rewarding brain stimulation. Behav Brain Res. 1984;14:85–89. doi: 10.1016/0166-4328(84)90023-8. [DOI] [PubMed] [Google Scholar]
- 27.Gallistel CR, Shizgal P, Yeomans J. A portrait of the substrate for self-stimulation. Psychol Rev. 1981;88:228–273. [PubMed] [Google Scholar]
- 28.Goltermann NR, Rehfeld JF, Roigaard-Petersen H. In vivo biosynthesis of cholecystokinin in rat cerebral cortex. J Biol Chem. 1980;255:6181–6185. [PubMed] [Google Scholar]
- 29.Herrera-Marschitz M, You Z-B, Goiny M, Meana JJ, Silveira R, Godukhin OV, Chen Y, Espinoza S, Pettersson E, Loidl CF, Lubec G, Andersson K, Nylander I, Terenius L, Ungerstedt U. On the origin of extracellular glutamate levels monitored in the basal ganglia of the rat by in vivo microdialysis. J Neurochem. 1996;66:1726–1735. doi: 10.1046/j.1471-4159.1996.66041726.x. [DOI] [PubMed] [Google Scholar]
- 30.Herrling PL. Pharmacology of the corticocaudate excitatory postsynaptic potential in the cat: evidence for its mediation by quisqualate- or kainate-receptors. Neuroscience. 1985;14:417–426. doi: 10.1016/0306-4522(85)90301-x. [DOI] [PubMed] [Google Scholar]
- 31.Higgins GA, Nguyen P, Sellers EM. Morphine place conditioning is differentially affected by CCKA and CCKB receptor antagonists. Brain Res. 1992;572:208–215. doi: 10.1016/0006-8993(92)90471-k. [DOI] [PubMed] [Google Scholar]
- 32.Hill DR, Campell NJ, Shaw TM, Woodruff GN. Autoradiographic localization and biochemical characterization of peripheral type CCK receptors in rat CNS using highly selective nonpeptide CCK antagonists. J Neurosci. 1987;7:2967–2977. doi: 10.1523/JNEUROSCI.07-09-02967.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hill DR, Shaw TM, Graham W, Woodruff GN. Autoradiographical detection of cholecystokinin-A receptors in primate brain using 125I-Bolton Hunter CCK-8 and 3H-MK-329. J Neurosci. 1990;10:1070–1081. doi: 10.1523/JNEUROSCI.10-04-01070.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hökfelt T, Skirboll L, Rehfeld JF, Goldstein M, Markey K, Dann O. A subpopulation of mesencephalic dopamine neurons projecting to limbic areas contains a cholecystokinin-like peptide: evidence from immunohistochemistry combined with retrograde tracing. Neuroscience. 1980;5:2093–2124. doi: 10.1016/0306-4522(80)90127-x. [DOI] [PubMed] [Google Scholar]
- 35.Hökfelt T, Herrera-Marschitz M, Seroogy K, Ju G, Staines WA, Holets V, Schalling M, Ungerstedt U, Post C, Rehfeld JF, Frey P, Fischer J, Dockray GJ, Hamaoka T, Walsh JH, Goldstein M. Immunohistochemical studies on cholecystokinin (CCK)-immunoreactive neurons in the rat using sequence specific antisera and with special reference to the caudate nucleus and primary sensory neurons. J Chem Neuroanat. 1988;1:11–52. [PubMed] [Google Scholar]
- 36.Hommer D, Stoner G, Crawley J, Paul S, Skirboll L. CCK-dopamine coexistence: electrophysiological actions corresponding to CCK receptor subtype. J Neurosci. 1986;6:3039–3043. doi: 10.1523/JNEUROSCI.06-10-03039.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Innis RB, Snyder S. Distinct cholecystokinin receptors in brain and pancreas. Proc Natl Acad Sci USA. 1980;77:6917–6921. doi: 10.1073/pnas.77.11.6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Itoh S, Katsuura G. Cholecystokinin octapeptide prevents extinction of active avoidance behavior in the rat. Drug Dev Res. 1987;10:171–175. [Google Scholar]
- 39.Itoh S, Takashima A, Katsuura G. Preventive effect of cholecystokinin octapeptide on scopolamine-induced memory impairment in the rat. Drug Dev Res. 1988;12:63–70. [Google Scholar]
- 40.Johnson SW, Seutin V, North RA. Burst firing in dopamine neurons induced by N-methyl-d-aspartate: role of electrogenic sodium pump. Science. 1992;258:665–667. doi: 10.1126/science.1329209. [DOI] [PubMed] [Google Scholar]
- 41.Karreman M, Westerink BHC, Moghaddam B. Excitatory amino acid receptors in the ventral tegmental area regulate dopamine release in the ventral striatum. J Neurochem. 1996;67:601–607. doi: 10.1046/j.1471-4159.1996.67020601.x. [DOI] [PubMed] [Google Scholar]
- 42.Kresse AE, Reyes A, Micevych P, Maidment NT. The origin of CCK in caudate and nucleus accumbens: combined microdialysis, retrograde tracing and in situ hybridization analysis. Soc Neurosci Abstr. 1995;21:2050. [Google Scholar]
- 43.Ladurelle N, Keller G, Roques BP, Dauge V. Effects of CCK8 and of CCKB-selective agonist BC264 on extracellular dopamine content in the anterior and posterior nucleus accumbens: a microdialysis in freely moving rats. Brain Res. 1993;628:254–262. doi: 10.1016/0006-8993(93)90962-m. [DOI] [PubMed] [Google Scholar]
- 44.Larsson L-I, Rehfeld J. Localization and molecular heterogeneity of cholecystokinin in the central and peripheral nervous system. Brain Res. 1979;165:201–218. doi: 10.1016/0006-8993(79)90554-7. [DOI] [PubMed] [Google Scholar]
- 45.Liu JK, Kato T. Simultaneous determination of cholecystokinin-like immunoreactivity and dopamine release after treatment with veratrine, NMDA, scopolamine and SCH23390 in rat medial frontal cortex: a brain microdialysis study. Brain Res. 1996;735:30–35. doi: 10.1016/s0006-8993(96)00577-x. [DOI] [PubMed] [Google Scholar]
- 46.Lundberg JM, Franco-Cereceda A, Lou YP, Modin A, Pernow J. Differential release of classical transmitters and peptides. Adv Second Messenger Phosphoprotein Res. 1994;29:223–234. doi: 10.1016/s1040-7952(06)80018-2. [DOI] [PubMed] [Google Scholar]
- 47.Maidment NT, Villafranca NP. Persistence of the releasable pool of CCK in the rat nucleus accumbens and caudate-putamen following lesions of midbrain. Brain Res. 1997;747:290–296. doi: 10.1016/s0006-8993(96)01238-3. [DOI] [PubMed] [Google Scholar]
- 48.Maidment NT, Siddall BJ, Rudolph VD, Erdelyi E, Evans CJ. Dual determination of extracellular cholecystokinin and neurotensin fragments in rat forebrain: microdialysis combined with a sequential multiple antigen radioimmunoassay. Neuroscience. 1991;45:81–93. doi: 10.1016/0306-4522(91)90105-w. [DOI] [PubMed] [Google Scholar]
- 49.Meana JJ, Herrera-Marschitz M, Brodin E, Hökfelt T, Ungerstedt U. Simultaneous determination of cholecystokinin, glutamate and aspartate in cortex and striatum of the rat using in vivo microdialysis. Amino Acids. 1991;1:365–373. doi: 10.1007/BF00814005. [DOI] [PubMed] [Google Scholar]
- 50.Meyer DK, Beinfeld MC, Oertel WH, Brownstein MJ. Origin of the cholecystokinin containing fibers in the caudate-putamen. Science. 1982;215:187–188. doi: 10.1126/science.7053570. [DOI] [PubMed] [Google Scholar]
- 51.Moghaddam B. Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: comparison to hippocampus and basal ganglia. J Neurochem. 1993;60:1650–1657. doi: 10.1111/j.1471-4159.1993.tb13387.x. [DOI] [PubMed] [Google Scholar]
- 52.Morari M, O’Connor WT, Ungerstedt U, Fuxe K. N-methyl-d-aspartic acid differentially regulates extracellular dopamine, GABA, and glutamate levels in the dorsolateral neostriatum of the halothane-anesthetized rat: an in vivo microdialysis study. J Neurochem. 1993;60:1884–1893. doi: 10.1111/j.1471-4159.1993.tb13416.x. [DOI] [PubMed] [Google Scholar]
- 53.Morency MA, Quirion R, Misra RK. Distribution of cholecystokinin receptors in the bovine brain: a quantitative autoradiographic study. Neuroscience. 1994;62:307–316. doi: 10.1016/0306-4522(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 54.Morino P, Herrera-Marschitz M, Castel MN, Ungerstedt U, Varro A, Dockray G, Hökfelt T. Cholecystokinin in cortico-striatal neurons in the rat: immunohistochemical studies at the light and electron microscopical level. Eur J Neurosci. 1994a;6:681–692. doi: 10.1111/j.1460-9568.1994.tb00980.x. [DOI] [PubMed] [Google Scholar]
- 55.Morino P, Mascagni F, McDonald A, Hökfelt T. Cholecystokinin corticostriatal pathway in the rat: evidence for bilateral origin from medial prefrontal cortical areas. Neuroscience. 1994b;59:939–952. doi: 10.1016/0306-4522(94)90297-6. [DOI] [PubMed] [Google Scholar]
- 56.Mundl WJ. A constant-current stimulator. Physiol Behav. 1980;24:991–993. doi: 10.1016/0031-9384(80)90162-6. [DOI] [PubMed] [Google Scholar]
- 57.Nevo I, Becker C, Hamon M, Benoliel J-J. Stress- and yohimbine-induced release of cholecystokinin in the frontal cortex of the freely moving rat: prevention by diazepam but not ondansetron. J Neurochem. 1996;66:2041–2049. doi: 10.1046/j.1471-4159.1996.66052041.x. [DOI] [PubMed] [Google Scholar]
- 58.Olds ME. Dopaminergic basis for the facilitation of brain stimulation reward by the NMDA receptor antagonist, MK-801. Eur J Pharmacol. 1996;306:23–32. doi: 10.1016/0014-2999(96)00217-8. [DOI] [PubMed] [Google Scholar]
- 59.Paulsen RE, Fonnum F. Role of glial cells for the basal and Ca2+-dependent K+-evoked release of transmitter amino acids investigated by microdialysis. J Neurochem. 1989;52:1823–1829. doi: 10.1111/j.1471-4159.1989.tb07263.x. [DOI] [PubMed] [Google Scholar]
- 60.Paxinos G, Watson C. The rat brain in stereotaxic coordinates, Ed 2. Academic; San Diego: 1986. [DOI] [PubMed] [Google Scholar]
- 61.Phillips AG, Fibiger HC. The role of dopamine in maintaining intracranial self-stimulation in the ventral tegmentum, nucleus accumbens and medial prefrontal cortex. Can J Psychol. 1978;32:58–66. doi: 10.1037/h0081676. [DOI] [PubMed] [Google Scholar]
- 62.Pulvirenti L, Maldonado-Lopez R, Koob GF. NMDA receptors in the nucleus accumbens modulate intravenous cocaine but not heroin self-administration in the rat. Brain Res. 1992;594:327–330. doi: 10.1016/0006-8993(92)91145-5. [DOI] [PubMed] [Google Scholar]
- 63.Rehfeld JF. Immunohistochemical studies on cholecystokinin. II. Distribution and molecular heterogeneity in the central nervous system and small intestine of man and hog. J Biol Chem. 1978;253:4022–4030. [PubMed] [Google Scholar]
- 64.Rehfeld JF. Neuronal cholecystokinin: one or multiple transmitters. J Neurochem. 1985;44:1–10. doi: 10.1111/j.1471-4159.1985.tb07105.x. [DOI] [PubMed] [Google Scholar]
- 65.Rich-Bennett E, Dahl D, Lecompte BB., III Modulation of paired-pulse activation in the hippocampal dentate gyrus by cholecystokinin, baclofen and bicuculline. Neuropeptides. 1993;24:263–270. doi: 10.1016/0143-4179(93)90014-2. [DOI] [PubMed] [Google Scholar]
- 66.Rose C, Vargas F, Facchinetti P, Bourgeat P, Bambal RB, Bishop PB, Chan SM, Moore AN, Ganellin CR, Schwartz JC. Characterization and inhibition of a cholecystokinin-inactivating serine peptidase. Nature. 1996;380:403–409. doi: 10.1038/380403a0. [DOI] [PubMed] [Google Scholar]
- 67.Rossetti Z, Marcangione C, Wise RA. Increase of extracellular glutamate and expression of fos-like immunoreactivity in the ventral tegmental area in response to electrical stimulation of the prefrontal cortex. J Neurochem. 1998;70:1503–1512. doi: 10.1046/j.1471-4159.1998.70041503.x. [DOI] [PubMed] [Google Scholar]
- 68.Ruggeri M, Ungerstedt U, Agnati LF, Mutt V, Hörfstrand A, Fuxe K. Effect of cholecystokinin peptides and neurotensin on dopamine release and metabolism in the rostral and caudal part of the nucleus accumbens using intracerebral dialysis in the anesthetized rat. Neurochem Int. 1987;10:509–520. doi: 10.1016/0197-0186(87)90077-5. [DOI] [PubMed] [Google Scholar]
- 69.Sandberg M, Ward HK, Bradford HF. Effect of cortico-striatal pathway lesion on the activities of enzymes involved in synthesis and metabolism of amino acid neurotransmitters in the striatum. J Neurochem. 1985;44:42–47. doi: 10.1111/j.1471-4159.1985.tb07110.x. [DOI] [PubMed] [Google Scholar]
- 70.Schiffmann SN, Vanderhaeghen J-J. Lesion of the nigrostriatal pathway induces cholecystokinin messenger RNA expression in the striatum. An in situ hybridization histochemistry study. Neuroscience. 1992;50:551–557. doi: 10.1016/0306-4522(92)90446-9. [DOI] [PubMed] [Google Scholar]
- 71.Seroogy KB, Dangaran K, Lim S, Haycock JW, Fallon JH. Ventral mesencephalic neurons containing both cholecystokinin- and tyrosine hydroxylase-like immunoreactivities project to forebrain regions. J Comp Neurol. 1989;279:397–414. doi: 10.1002/cne.902790306. [DOI] [PubMed] [Google Scholar]
- 72.Sesack SR, Deutch AY, Roth RH, Bunney BS. Topographical organization of the efferent projections of the medial prefrontal cortex in the rat: an anterograde tract-tracing study with Phaseolus vulgaris leucoagglutinin. J Comp Neurol. 1989;290:213–242. doi: 10.1002/cne.902900205. [DOI] [PubMed] [Google Scholar]
- 73.Simon H, Stinus L, Tassin JP, Lavielle S, Blanc G, Thierry AM, Glowinski J, Le Moal M. Is the dopaminergic mesocorticolimbic system necessary for intracranial self-stimulation? Behav Neural Biol. 1979;27:125–145. doi: 10.1016/s0163-1047(79)91745-x. [DOI] [PubMed] [Google Scholar]
- 74.Skirboll LR, Grace AA, Hommer DW, Rehfeld J, Goldstein M, Hökfelt T, Bunney BS. Peptide-monoamine coexistence: studies on the actions of cholecystokinin-like peptide on the electrical activity of midbrain dopamine neurons. Neuroscience. 1981;6:2111–2124. doi: 10.1016/0306-4522(81)90002-6. [DOI] [PubMed] [Google Scholar]
- 75.Snyder GL, Fisone G, Morino P, Gundersen V, Ottersen OP, Hökfelt T, Greengard P. Regulation by the neuropeptide cholecystokinin (CCK-8S) of protein phosphorylation in the neostriatum. Proc Natl Acad Sci USA. 1993;90:11277–11281. doi: 10.1073/pnas.90.23.11277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taber MT, Fibiger HC. Electrical stimulation of the medial prefrontal cortex increases dopamine release in the nucleus accumbens of the rat: modulation by metabotropic glutamate receptors. J Neurosci. 1995;15:3896–3904. doi: 10.1523/JNEUROSCI.15-05-03896.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Taber MT, Das S, Fibiger HC. Cortical regulation of subcortical dopamine release: mediation via the ventral tegmental area. J Neurochem. 1995;65:1407–1410. doi: 10.1046/j.1471-4159.1995.65031407.x. [DOI] [PubMed] [Google Scholar]
- 78.Taber MT, Baker GB, Fibiger HC. Glutamate receptor agonists decrease extracellular dopamine in the rat nucleus accumbens in vivo. Synapse. 1996;24:165–172. doi: 10.1002/(SICI)1098-2396(199610)24:2<165::AID-SYN8>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 79.Vaccarino FJ, Koob GF. Microinjections of nanogram amounts of sulfated cholecystokinin octapeptide into the rat nucleus accumbens attenuates brain stimulation reward. Neurosci Lett. 1984;52:61–66. doi: 10.1016/0304-3940(84)90351-3. [DOI] [PubMed] [Google Scholar]
- 80.Vaccarino FJ, Vaccarino AL. Antagonism of cholecystokinin function in the rostral and caudal nucleus accumbens: differential effects on brain stimulation reward. Neurosci Lett. 1989;97:151–156. doi: 10.1016/0304-3940(89)90155-9. [DOI] [PubMed] [Google Scholar]
- 81.Vallebuona F, Paudice P, Raiteri M. Release of cholecystokinin-like immunoreactivity in the frontal cortex of conscious rats as assessed by transcerebral microdialysis: effects of different depolarizing stimuli. J Neurochem. 1993;61:490–495. doi: 10.1111/j.1471-4159.1993.tb02150.x. [DOI] [PubMed] [Google Scholar]
- 82.Van Dijk A, Richards JG, Trzeciak A, Gillessen D, Mohler H. Cholecystokinin receptors: biochemical demonstrations and autoradiographical localization in rat brain and pancreas using 3H cholecystokinin as radioligand. J Neurosci. 1984;4:1021–1033. doi: 10.1523/JNEUROSCI.04-04-01021.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vanderhaegen JJ, Signeau JC, Gepts W. New peptide in the vertebrate CNS reacting with antigastrin antibodies. Nature. 1975;257:604–605. doi: 10.1038/257604a0. [DOI] [PubMed] [Google Scholar]
- 84.Walaas I, Fonnum F. Biochemical evidence for glutamate as a transmitter in hippocampal efferents to the basal forebrain and hypothalamus in rat brain. Neuroscience. 1980;5:1691–1698. doi: 10.1016/0306-4522(80)90088-3. [DOI] [PubMed] [Google Scholar]
- 85.Wang Z, Rao Z, Shi J. Tyrosine hydroxylase-, neurotensin- and cholecystokinin-containing neurons in the nucleus tractus solitarii send projection fibers to the nucleus accumbens in the rat. Brain Res. 1992;578:347–350. doi: 10.1016/0006-8993(92)90269-f. [DOI] [PubMed] [Google Scholar]
- 86.Wank SA, Harkins R, Jensen R, Shapira H, de Weerth A, Slattery T. Purification, molecular cloning, and functional expression of the cholecystokinin receptor from rat pancreas. Proc Natl Acad Sci USA. 1992a;89:3125–3129. doi: 10.1073/pnas.89.7.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wank SA, Pisegna IR, de Weerth A. Brain and gastrointestinal cholecystokinin receptor family: structure and functional expression. Proc Natl Acad Sci USA. 1992b;89:8691–8695. doi: 10.1073/pnas.89.18.8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Westerink BHC, Damsma G, Rollema H, de Vries JB, Horn AS. Scope and limitations of in vivo brain dialysis: a comparison of its application to various neurotransmitter systems. Life Sci. 1987;41:1763–1766. doi: 10.1016/0024-3205(87)90695-3. [DOI] [PubMed] [Google Scholar]
- 89.Westerink BHC, Kwint H-F, de Vries JB. The pharmacology of mesolimbic dopamine neurons: a dual-probe microdialysis study in the ventral tegmental area and nucleus accumbens of the rat brain. J Neurosci. 1996;16:2605–2611. doi: 10.1523/JNEUROSCI.16-08-02605.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wise RA. Catecholamine theories of reward: a critical review. Brain Res. 1978;152:215–247. doi: 10.1016/0006-8993(78)90253-6. [DOI] [PubMed] [Google Scholar]
- 91.Wise RA. Addictive drugs and brain stimulation reward. Annu Rev Neurosci. 1996a;19:319–340. doi: 10.1146/annurev.ne.19.030196.001535. [DOI] [PubMed] [Google Scholar]
- 92.Wise RA. Neurobiology of addiction. Curr Opin Neurobiol. 1996b;6:243–251. doi: 10.1016/s0959-4388(96)80079-1. [DOI] [PubMed] [Google Scholar]
- 93.Wise RA, Leone P, Rivest R, Leeb K. Elevations of nucleus accumbens dopamine and DOPAC levels during intravenous heroin self-administration. Synapse. 1996;21:140–148. doi: 10.1002/syn.890210207. [DOI] [PubMed] [Google Scholar]
- 94.Woodruff GN, Hill DR, Boden P, Pinnock R, Singh L, Hughes J. Functional role of brain CCK receptors. Neuropeptides [Suppl] 1991;19:45–56. doi: 10.1016/0143-4179(91)90082-t. [DOI] [PubMed] [Google Scholar]
- 95.Wu HQ, Schwarcz R, Shepard PD. Excitatory amino acid-induced excitation of dopamine-containing neurons in the rat substantia nigra: modulation by kynurenic acid. Synapse. 1994;16:219–230. doi: 10.1002/syn.890160307. [DOI] [PubMed] [Google Scholar]
- 96.Wu T, Wang H-L. CCK-8 excites substantia nigra dopaminergic neurons by increasing a cationic conductance. Neurosci Lett. 1994;170:229–232. doi: 10.1016/0304-3940(94)90325-5. [DOI] [PubMed] [Google Scholar]
- 97.Wu T, Wang H-L. The excitatory effect of cholecystokinin on rat neostriatal neurons: ionic and molecular mechanisms. Eur J Pharmacol. 1996;307:125–132. doi: 10.1016/0014-2999(96)00213-0. [DOI] [PubMed] [Google Scholar]
- 98.You Z-B, Herrera-Marschitz M, Brodin E, Meana JJ, Morino P, Hökfelt T, Silveira R, Goiny M, Ungerstedt U. On the origin of striatal cholecystokinin release: studies with in vivo microdialysis. J Neurochem. 1994;62:76–85. doi: 10.1046/j.1471-4159.1994.62010076.x. [DOI] [PubMed] [Google Scholar]