

Abstract
Cell cultures were used to analyze the role of brain-derived neurotrophic factor (BDNF) and neurotrophin-3 (NT-3) in the development of synaptic transmission. Neurons obtained from embryonic day 18 (E18) rat hippocampus and cultured for 2 weeks exhibited extensive spontaneous synaptic activity. By comparison, neurons obtained from E16 hippocampus expressed very low levels of spontaneous or evoked synaptic activity. Neurotrophin treatment produced a sevenfold increase in the number of functional synaptic connections in the E16 cultures. BDNF induced formation of both excitatory and inhibitory synapses, whereas NT-3 induced formation of only excitatory synapses. These effects were independent of serum or the age of the glia bed used for the culture. They were not accompanied by significant changes in synaptic-vesicle-associated proteins or glutamate receptors. Treatment of the cultures with the neurotrophins for 3 d was sufficient to establish the maximal level of functional synapses. During this period, neurotrophins did not affect the viability or the morphology of the excitatory neurons, although they did produce an increase in the number and length of dendrites of the GABAergic neurons. Remarkably, only BDNF caused an increase in the number of axonal branches and in the total length of the axons of the GABAergic neurons. These results support a unique and differential role for neurotrophins in the formation of excitatory and inhibitory synapses in the developing hippocampus.
Keywords: synapses, hippocampus, culture, BDNF, NT-3, GABA
Since the pioneering work with nerve growth factor (NGF), it has been thought that soluble proteins regulate the initial steps in neuron target interactions (Levi-Montalcini, 1987). The neurotrophic factors, brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), neurotrophin-4/5 (NT-4/5), and ciliary neurotrophic factor (CNTF) modulate both neuromuscular and central synapses. Acutely applied BDNF, NT-3, and CNTF potentiate neuromuscular synaptic transmission in culture (Lohof et al., 1993; Stoop and Poo, 1996). Chronic exposure of Xenopus nerve–muscle cocultures to NT-3 and BDNF promoted the maturation of these synapses (Wang et al., 1995), probably in an activity-dependent manner (Xie et al., 1997). Synthesis of neurotrophins in the hippocampus is regulated by neuronal activity (Thoenen, 1995), and several groups have reported acute effects of these growth factors to enhance excitatory synaptic transmission (Lebmann et al., 1994; Kang and Schuman, 1995, 1996;Levine et al., 1995) and long-term potentiation (LTP) (Kang and Schuman, 1995; Figurov et al., 1996). BDNF knock-out mice show deficits in basal synaptic transmission in the Schaffer collateral CA1 pathway (Patterson et al., 1996) and in LTP (Korte et al., 1995; Patterson et al., 1996), supporting a role of neurotrophins in synaptic plasticity. These studies show that neurotrophins may strengthen ongoing synaptic transmission in the CNS, but they do not define a role for them in the induction of functional synaptic activity.
BDNF and NT-3 promote the differentiation of neurons derived from cultured neuroepithelial cells (Ghosh and Greenberg, 1995;Vicario-Abejón et al., 1995). These observations prompted us to examine a possible function of neurotrophins in the induction of synaptic transmission in cultures of neurons dissociated from rat embryonic day 16 (E16) hippocampus. At this embryonic age, the great majority of precursors are mitotically active, and synaptogenesis has barely begun in vivo (Altman and Bayer, 1990). Neurons derived from E18 hippocampus form functional synapses spontaneously (Fletcher et al., 1994; Matteoli et al., 1995). In contrast, E16-derived neurons establish functional synapses only when they are exposed to exogenous neurotrophins. The results reported here demonstrate that neurotrophins regulate synapse formation during CNS development.
MATERIALS AND METHODS
Hippocampal cultures. Reagents for tissue culture were purchased from Life Technologies (Grand Island, NY), Sigma (St. Louis, MO), and Intergen (Purchase, NY). Fetal bovine serum (FBS) was inactivated during 30 min at 56°C previous use. Neuronal cultures were prepared from rat embryonic hippocampus at E16 (the day when a copulation plug was found was considered day E1). The hippocampi were minced and trypsinized (Vicario-Abejón et al., 1995). Cells suspended in DMEM/F12/N2 and 10% FBS were plated on a monolayer of type I astrocytes at a density of 50,000–80,000 cells/cm2 and incubated at 37°C in 5% CO2 atmosphere. Medium was refreshed every 4–7 d by exchanging a third part of the medium with DMEM/N2 and 10% FBS. Neurotrophins (PreproTech, Rocky Hill, NJ) were added at different times in culture and replaced every 2 d at 20 ng/ml. Some experiments were conducted with the cells exposed to neurotrophins in serum-free medium. In these experiments, serum was removed at day 10, BDNF or NT-3 was added at day 11, and cells were recorded and fixed at day 14, as in other cultures.
To prepare type I astrocytes, hippocampal neuroepithelial cells were expanded and passaged in the presence of basic FGF (Vicario-Abejón et al., 1995). After three or four passages, cells were plated on glass coverslips, 4-well chamber slides, or 6 or 10 cm dishes coated with 15 μg/ml polyornithine and 1 μg/ml fibronectin. Cells were grown in DMEM/F12/N2 and 10% FBS and incubated at 37°C in 5% CO2. Under these conditions, a confluent monolayer of type I astrocytes is obtained after 8–10 d in vitro.
To prepare cultures from E18 hippocampus, cells were plated on glass coverslips coated with 15 μg/ml polyornithine and 1 μg/ml fibronectin at a density of 200,000 cells/cm2. After 4–5 d in culture, a third part of the medium was replaced by DMEM/N2 and 10% FBS and 6–8 μm cytosine β-d-arabinofuranoside (ARA-C) was added to halt glial proliferation. Further medium changes were done every 7–10 d. Neurons from E18 hippocampus were also plated on a monolayer of type I astrocytes as described above. In these experiments, the plating density of the E18 cells was 60,000 cells/cm2, and the rest of the treatments throughout the culture period were identical to that of the E16 cultures.
Electrophysiology. Electrophysiological experiments were performed in cells grown in culture for 13–15 d on glass coverslips. Patch-clamp recordings in the whole-cell configuration were used to record spontaneous and evoked synaptic activity. The recording medium contained (in mm): 130 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.4; osmolarity was adjusted to 310 mOsm with sucrose. The patch pipette contained (in mm): 130 Cs gluconate, 10 NaCl, 2 MgCl2, 0.2 EGTA, 1 NaATP, and 10 HEPES, pH 7.2; osmolarity was adjusted to 290 mOsm with sucrose. Patch pipettes were manufactured from 1.5 mm outer diameter thick glass (A & M Systems, Everett, WA) and had a resistance of 3–5 MΩ. Series resistance was <10 MΩ and was compensated by 50% in most experiments. Currents were recorded by an Axopatch 200B amplifier, filtered at 3 KHz, and digitized at 10 KHz using Axon Instruments (Foster City, CA) hardware and software. Afferent cells were stimulated with a 1 mmglutamate (10 psi, 5–10 msec) delivered by pressure through a glass micropipette (1 μm tip diameter). After whole-cell recording was established, cells were held at −60 mV. Single or clusters of afferent neurons were then stimulated, and the presence or absence of evoked postsynaptic currents was recorded. Typically 4–10 afferents were tested for each recorded cell. A measurement of synaptic connectivity was then obtained by calculating the ratio of afferents that elicited an evoked response over the total number of stimulated cells in the field surrounding the recorded cell. The ratio was then expressed as the percentage of afferents connected to each cell from which we recorded. Averages per each treatment were plotted, and statistical significance was evaluated with Student’s t test. We preferred to use the glutamate stimulation over electrical stimulation to maximize the chances to obtain a postsynaptic response. In some experiments, 1 μm tetrodotoxin (TTX) was added to the external medium to block action potentials. The presynaptic terminals were then stimulated with 50 μl of a hyperosmotic sucrose solution (300 mm added to the regular recording medium) delivered manually, whereas miniature postsynaptic currents were recorded during 10 sec intervals. In some experiments, miniature synaptic currents were elicited in the presence of CNQX (20 μm) or picrotoxin (100 μm). Data were analyzed using Axon Instruments software package. Each spontaneous event was also evaluated with specialized software (Jaejin Software, Leonia, NJ) that allowed for their interactive detection. The amplitudes and the time constant of decay were then measured to generate distribution histograms;t tests for independent groups were used to evaluate the treatment effects.
Immunostaining of cultured cells. Cells were fixed with 4% paraformaldehyde and 0.1 m phosphate buffer, pH 7.4, for 30 min. After treatment with 0.1% Triton X-100/10% normal serum/PBS (Triton X-100 was avoided for some antigens), cells were incubated overnight at 4°C with the primary antibodies against MAP-2ab (1:200, Sigma); GABA (1:700, Sigma); glutamate (1:700, Sigma); GAD (1:100, Chemicon, Temecula, CA); synapsin-I (1:1000, from M. Kennedy); synaptophysin (1:4; Zymed, San Francisco, CA); SV2 (1:60); TrkB (1:50, from S. Feinstein and M. Radeke); TrkC (1:10–20; Santa Cruz Biotechnology, Santa Cruz, CA); and glutamate receptors (GluR1, 1:100; GluR2, 1:50; NMDAR1, 1:50, from R. Wenthold). The cells were then incubated with the corresponding fluorescein and/or rhodamine-conjugated secondary antibodies (1:100) (Jackson ImmunoResearch, West Grove, PA or Cappel, Durham, NC), or with a biotinylated secondary antibody (1:200) followed by avidin–biotin–horseradish peroxidase complex (Vectastain ABC kit; Vector Laboratories, Burlingame, CA) and developed using DAB. Coverslips were mounted in 1,4 diazabicyclo[2.2.2]-octane and glycerol.
The monoclonal antibody SV2 developed by K. M. Buckley was obtained from the Hybridoma Bank maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA, under contract NO1-HD-7–3263 from the National Institute of Child Health and Human Development. The anti-TrkB antibody recognizes the tyrosine kinase domain of TrkB and does not cross-react with rat-TrkC transfected human embryo kidney 293 cells (data not shown). According to the manufacturer, the anti-TrkC antibody used in this study (798; catalog #sc-117) does not cross-react with TrkA or TrkB. Preabsortion of the anti-TrkC antibody with a 10-fold (by weight) excess of peptide antigen abolished specific staining (data not shown).
Morphological analysis. To investigate the effects of neurotrophins on the morphology of excitatory and inhibitory neurons, E16 cells were double-immunostained for MAP2ab and GABA. Neurons were traced with a camera lucida using a 63× objective and rhodamine and fluorescein filters, and from these drawings the number of primary dendrites, dendritic branches, and axonal branches were directly counted. The total length of dendrites and axon was measured with a digital plan measure and the NIH Image program. For the GABAergic neurons, the dendrites (visualized with anti-MAP2ab and anti-GABA antibodies) and the axon (visualized with anti-GABA antibodies) were traced. For the rest of the neurons, i.e., those which were MAP2ab-positive and GABA-negative (i.e., excitatory neurons), only the dendritic tree could be traced. Drawings were digitally scanned and processed using Adobe Photoshop.
Western blotting. Cells from E16 hippocampus were plated on 6 or 10 cm dishes covered with a monolayer of type I astrocytes. Proteins were analyzed by SDS-PAGE 11–13 d later. Thirty to forty microliters of extracts (and standard for molecular weight) were separated by electrophoresis on a 8% polyacrylamide gel and electrotransferred to 0.2 μm nitrocellulose filters (Schleicher & Schuell, Keene, NH). Filters were treated with 2% BSA and TBS, pH 7.6, for 1 hr and incubated overnight at 4°C with the primary antibodies against synapsin-I (1:5,000), syntaxin (1:300; Chemicon), synaptotagmin (1:500, Stressgen, Victoria, BC), GluR1 (1:400), GluR2 (1:400), and NMDR1 (1:400). After incubation with peroxidase-conjugated secondary antibodies (Boehringer Mannheim, Indianapolis, IN), bands were developed on autoradiographic film (Eastman Kodak, Rochester, NY) by chemiluminescence using the ECL kit (Amersham, Arlington Heights, IL). The film signals were digitally scanned and then quantified using NIH Image software.
Electron microscopy. E16 cells, grown for 2 weeks in 4-well chambers, were fixed with 3.5% glutaraldehyde and 0.1 msodium cacodylate, pH 7.4, for 30 min. The cells were post-fixed with 1% OsO4, contrasted en bloc with uranyl acetate, dehydrated in ethanol, and embedded in Epon Araldite. Transverse sections of 800 Å were visualized and photographed under an electron microscope.
Confocal microscopic imaging. Cultures were fixed with 4% paraformaldehyde in PBS. Individual cells were stained with a microdrop of DiI dissolved in oil. The dye was allowed to diffuse across the cell surface for 4–8 hr. The cultures were visualized in a Zeiss confocal microscope. Images were reconstructed from serial optical sections and stored for off-line analysis using NIH Image software. Details of the procedures are presented elsewhere (Papa et al., 1995). For quantification of the number of synapsin-I-positive boutons, confocal images at 100× magnification were taken of E16 neurons immunostained for MAP2ab and synapsin-I. Boutons were counted using NIH Image software. Results are expressed as number of synapsin-I-positive boutons per 100× field.
RESULTS
Effects of neurotrophins on the formation of functional synapses in cultured hippocampal neurons
To measure the density of connections between neurons in culture, we studied their ability to respond to stimulation of afferent cells with EPSCs and IPSCs. Postsynaptic cells were patch clamped at resting potential (−60 mV), and 4–10 afferent cells in the field of view were stimulated by local application of brief pulses of glutamate (Fig. 1A); the glutamate-containing micropipette was placed near the somata of putative afferent cells to elicit a response. Neurons prepared from E18 hippocampus established extensive functional synaptic contacts within 2 weeks in culture and showed high levels of spontaneous excitatory and inhibitory currents (Fig. 1B,C). In current-clamped cells, this spontaneous activity was associated with action potential discharges (data not shown). Connections were found in ∼90% of E18 cells tested in the presence or absence of neurotrophins (Fig. 1D). Functional connections among E18 neurons were seen whether the cells were plated on coated coverslips at 200,000 cells/cm2 or on a monolayer of type I astrocytes at 60,000 cells/cm2 as used for the E16 cultures (Fig.1D). Exogenous neurotrophins did affect the magnitude of connections between E18 cells, however this was not analyzed systematically in the present study. In contrast, E16 cells grown in culture for 2 weeks did not express spontaneous synaptic activity (Fig.1C). Only 8.8% of afferent cells evoked PSCs in untreated E16 cultures, suggesting that the majority of these neurons had not formed functional synaptic connections (Fig. 1D). This conspicuous lack of spontaneous synaptic activity was ameliorated by growing the E16 cells in the presence of 20 ng/ml BDNF and/or 20 ng/ml NT-3 for the length of the culture. A highly significant, sevenfold increase in the proportion of active afferents (from 8.8% to 65%) (Fig. 1D) that evoked PSCs in postsynaptic cells after glutamate stimulation was found. BDNF and NT-3 produced similar increases in the proportion of cells that evoked postsynaptic currents in recorded neurons.
Fig. 1.

BDNF- and NT-3-induced functional synapses in hippocampal neurons in culture. A, An image of a field in E18 culture. One cell is patch clamped (right shadow), and an adjacent cell is approached with a glutamate-containing pipette (left shadow). Magnification, 40×. B, Trace 1, Illustration of the response of an afferent cell to the glutamate pulse with an action potential. Traces 2-5, Postsynaptic recordings in a single patch-clamped cell after stimulation of four afferent neurons to illustrate the variety of synaptic responses. C, Top trace, Spontaneous activity recorded from a cell taken from E16 and grown in culture for 2 weeks. Bottom trace, Recording from an E18 culture showing extensive EPSCs and IPSCs. D, Two week treatment of the E16 cultures with 20 ng/ml BDNF and/or 20 ng/ml NT-3 caused a marked increase in the levels of functional synaptic connectivity among neurons. In control cultures, only 8.8 ± 2.1% of all tested afferents were connected (n = 52 cells), however, the percentage of connected afferents reached 65.3 ± 5.2% (n = 36 cells), 56.4 ± 5.4% (n = 41 cells), or 58.6 ± 4.9% (n = 38 cells) in BDNF plus NT-3-treated (NT), BDNF-treated, or NT-3-treated cultures, respectively (Fig. 1D). The neurotrophins (NT) did not have a similar effect in the E18 cultures; in both control and NT-treated cultures the majority of cells (>90%, Fig. 1D) were connected. Control E18 neurons also form connections when they are plated at relatively low density on top of monolayer of type I astrocytes (Fig.1D, E18, shaded bar).
The minimal duration of exposure to the neurotrophins required for the induction of synaptic currents was then studied (Fig.2A). E16 cells cultured for 2 weeks but exposed to neurotrophins for only 24 hr before recording showed a threefold enhancement in the levels of functional synaptic connections [from 6.7 ± 4.2%, n = 22 cells in control cultures to 23.1 ± 5.8%, n = 24 cells in the BDNF plus NT-3 (NT)-treated cultures, p < 0.001], but this was still lower than the proportion of active connections in the cells exposed for 2 weeks to the neurotrophins. When the cultures were exposed for 72 hr to both neurotrophins, the levels of functional connectivity were similar to the levels found in cultures treated with neurotrophins for the entire 2 weeks in vitro(65.9 ± 8.2%, n = 12 cells, p < 0.001). Treatment of the cultures separately with NT-3 or BDNF produced similar effects on connectivity (58.5 ± 9.3% in NT-3-treated cultures, n = 9 cells, p < 0.001; 64.6 ± 8.8% in BDNF-treated cultures, n = 21 cells, p < 0.001, compared with 12.2 ± 6.3% connections in the control, untreated cultures) (Table1). The effects of the neurotrophins were independent of the presence of serum in the growth medium. In the absence of serum, BDNF- or NT-3-treated cells expressed a high level of connectivity (BDNF, 80.6 ± 7.9%, n = 9 cells; NT-3, 76.9 ± 8.7%, n = 9 cells; control, 4.8 ± 5.7%, n = 7 cells). These results indicate that the neurotrophins regulate connectivity in the E16 cultures.
Fig. 2.

Time-dependent effects of neurotrophins on synaptic and glutamate responses. A, Synaptic responses in patch-clamped cells after exposure of a nearby neuron to glutamate. Culture of E16-derived neurons exposed to BDNF plus NT-3 (NT) for 24 hr produced a small but clear increase in the proportion of evoked responses, whereas exposure of these cultures for 72 hr produced an effect similar in magnitude to that of a chronic treatment with NT. B, Responses of E16 cells to topical application of glutamate does not differ in control, and in cultures exposed for 1, 2, or 3 d to the neurotrophins.Top, Sample records of the response to the application of glutamate. Bottom, Summary of peak inward current densities generated by exposure to glutamate.
Table 1.
Effect of serum on neurotrophin-induced synaptic connections among E16 hippocampal neurons
| % Connected afferents | ||
|---|---|---|
| Serum-containing medium | Serum-free medium | |
| Control | 12.2 ± 6.3 (n = 19 cells) | 4.8 ± 5.7 (n = 7 cells) |
| NT-3 | 58.5 ± 9.3 (n = 9 cells)** | 76.9 ± 8.7 (n = 9 cells)*** |
| BDNF | 64.6 ± 8.8 (n = 21 cells)** | 80.6 ± 7.9 (n = 9 cells)*** |
E16 hippocampal cells were grown for 14 d in serum-containing medium or initially in serum-containing medium and from day 10 to day 14 in serum-free medium. NT-3 or BDNF were added in both cases from day 11 to day 14 when recordings were done. Results are the mean ± SEM of connection ratios per cell. **p< 0.002; ***p < 0.001, t test.
The effects of the neurotrophins on connectivity in the E16 cultures could be attributable to an enhanced reactivity of the postsynaptic neurons to the neurotransmitter substance (i.e., glutamate or GABA), to a change in dendritic or axonal morphology, or to a change in the properties of the presynaptic neurons. A presynaptic change in release properties could be a consequence of an increase in the density of cells or synapses among them or an increase in release probability at individual synapses. As can be seen in Figure 2B, the responses to glutamate applied directly on the postsynaptic cells were the same in control and NT-treated E16 cells suggesting that neurotrophins do not exert their action by changing postsynaptic sensitivity to glutamate.
It has been suggested that synapses mature during development by converting from a silent state (i.e., a synapse containing only functional NMDA receptors) to a functional state (i.e., containing both NMDA and AMPA receptors; for review, see Ben-Ari et al., 1997). It is possible that the lack of synaptic responses in the control E16 cultures results from an immature synapse lacking AMPA receptors. In a subset of three cells from two separate cultures, we examined the possibility that the synapses are “silent” by depolarizing the postsynaptic cells from +20 to +40 mV, a condition that will reveal outward-going NMDA synaptic currents. Stimulation of afferent cells did not reveal a pure NMDA-mediated synaptic response in control cultures. In contrast, NMDA receptor-mediated outward synaptic currents (+20 mV) were seen in neurotrophin-treated cells (data not shown). These results show that NMDA receptors are only integrated in the membrane after neurotrophin treatment.
One possible site of presynaptic action is the spike-generating mechanism. As shown in Table 2, measurements of spike properties showed that the spikes of the neurotrophin-treated cultures were larger and had faster kinetics than those of the control cells. It was therefore necessary to study miniature synaptic currents in the presence of TTX to examine if the observed synaptic responses are of axonal or synaptic origin. Clear effects of the neurotrophins on synaptic currents in the presence of TTX were seen (below), indicating that the synaptic responses to neurotrophins are independent of changes in the properties of action potential discharges.
Table 2.
Properties of action potentials in E16 cultured hippocampal neurons
| Control | Neurotrophins | |
|---|---|---|
| Threshold (mV) | −29.40 ± 1.10 | −32.1 ± 1.16** |
| Rise time (msec) | 1.73 ± 0.10 | 1.46 ± 0.07** |
| Peak amp (mV) | 81.45 ± 6.50 | 94.01 ± 3.60** |
| Decay time (msec) | 3.90 ± 0.61 | 2.68 ± 0.22** |
| AP width (msec) | 5.63 ± 0.69 | 4.14 ± 0.19** |
| AHP amp (mV) | 10.90 ± 1.45 | 10.03 ± 0.91 |
Action potentials elicited in response to a glutamate pulse were recorded under current-clamp mode from E16 hippocampal neurons cultured for 14 d in the presence or absence of BDNF plus NT-3. Data are presented as mean ± SEM of 12 control neurons or 13 neurotrophin-treated neurons. **p < 0.01 vs control values using Student’s t test. amp, Amplitude; AP, action potential; AHP, afterhyperpolarization.
Glutamate stimulation of afferent cells elicited both EPSCs and IPSCs with properties that have been previously described (Segal, 1983;Basarsky et al., 1994). Three parameters were distinguished between EPSCs and IPSCs: the reversal potential, antagonist sensitivity, and the decay time constant of the PSC. The latter property was routinely used as a criterion to discriminate EPSCs from IPSCs (Fig.3A). Responses with a decay time constant of 3–6 msec were excitatory, whereas responses of 12–30 msec were inhibitory. The slow responses could be reversed at −20 to −30 mV (when a gluconate-containing pipette was used), and the fast events could be reversed at 0 mV. Therefore, the two types of events could be recorded simultaneously and clearly distinguished based on their time constant of decay, reversal potential, and pharmacology.
Fig. 3.

Pharmacology and kinetic characterization of excitatory and inhibitory synaptic events. A, Sample traces of evoked responses to glutamate application on afferent E16 neurons, exhibiting fast- and slow-decaying responses. When the membrane potential was clamped at −40 mV, the slow response was near its reversal potential. At 0 mV, the slow response was fully reversed and was outward, whereas the fast response was at its reversal potential. Right, Illustration of three afferents, stimulated for the same postsynaptic cell. In the top trace, there was no response of the postsynaptic cell, in themiddle trace stimulation of another afferent produced a fast response, and stimulation of yet another afferent,bottom, produced a slow IPSC. The decay time constant of the synaptic responses was determined by a best fit exponent from the peak with a time constant in these examples of 15.8 and 4.8 msec. This is illustrated in A in which the open symbol marks a slowly decaying response, and the closed symbol marks a response with a rapid time constant.B, Differential effects of NT-3 and BDNF on evoked EPSCs and IPSCs. The initial response to a single stimulation was measured, and the percentage of excitatory and inhibitory connections evoked by afferent stimuli were plotted. In control cultures, 86.7% of total PSCs were excitatory and 13.3% were inhibitory. In BDNF plus NT-3-treated cultures, 65.6% were excitatory and 34.4% were inhibitory. In BDNF-treated cultures 53.6% of the PSCs were excitatory and 46.4% were inhibitory. In NT3-treated cultures, 92.4% were excitatory and only 7.6% were inhibitory. Differences between the proportion of excitatory and inhibitory synapses were statistically significant (*p < 0.002, χ2 test) in BDNF or NT-3 plus BDNF-treated cultures compared with controls. In NT-3 alone-treated cultures, the proportion of excitatory and inhibitory synapses was not statistically different from controls.
As shown in Figure 3B, there was a marked difference in the synaptic effects exerted by each neurotrophin alone; 92.4% (157 of 170) of the PSCs elicited in the NT-3-treated cultures were EPSCs and only 7.6% were IPSCs, whereas in the BDNF-treated cultures 53.6% (113 of 211) of the responses were excitatory and 46.4% (98 out of 211) were inhibitory. In BDNF plus NT-3-treated cultures, 65.6% (128 of 198) of responses were excitatory and 34.4% inhibitory, whereas 86.7% (39 of 45) of PSCs were excitatory, and 13.3% were inhibitory in the untreated cultures. These results demonstrate that BDNF induces both EPSCs and IPSCs, whereas NT-3 induces predominantly EPSCs.
The amplitudes of the evoked EPSCs and IPSCs were twofold greater in cultures treated with neurotrophins for 2 weeks. BDNF plus NT-3 added to the cultures increased the amplitude of the EPSCs from a mean of −21.3 ± 1.7 pA (n = 39 EPSCs) in the untreated cultures to −47.8 ± 4.4 pA (n = 128,p < 0.01). The amplitude of the IPSCs was enhanced from −41.2 ± 9.9 pA (n = 6 IPSCs) to −87.4 ± 13.4 pA (n = 67, p < 0.001). In the BDNF-treated cultures, the EPSC amplitude was −40.9 ± 3.5 pA (n = 113), and IPSC amplitude was −103.8 ± 10.5 pA (n = 98). In the NT3-treated cultures, EPSC amplitude was −37.3 ± 2.8 pA (n = 157), and IPSC was −107 ± 22 pA (n = 13). Short-term treatment of the cultures with BDNF plus NT-3 also enhanced the amplitudes of the evoked EPSCs (−36.0 ± 7.8 pA at 24 hr; −41.0 ± 5.7 pA at 72 hr; n = 48, 62) and IPSCs (−96.3 ± 22.6 pA at 24 hr; −71.0 ± 10.0 pA at 72 hr; n = 18, 19).
To further examine if the site of action of neurotrophins is presynaptic or postsynaptic, we studied the amplitudes and frequencies of miniature postsynaptic currents (mPSCs) generated in the presence of TTX by high osmotic stimulation (300 mm sucrose). Miniature events elicited by sucrose stimulation could also be isolated pharmacologically; i.e., in the presence of CNQX (to block AMPA/KA receptors) or picrotoxin (to block GABA-A receptors) (Fig.4). Almost all the events recorded in the presence of picrotoxin had decay time constants faster than 10 msec and were blocked by CNQX (Fig. 4A,B). Conversely, events elicited in the presence of CNQX had decay times longer than 10 msec and were completely blocked by the subsequent addition of picrotoxin (Fig. 4C,D). Treatment of the cultures with neurotrophins facilitated both types of mPSCs (Fig.5A,B). There was a 40% (p < 0.05) and a 16% (p < 0.01) increase in the amplitudes of mEPSCs and mIPSCs, respectively (Fig. 5C). However, neurotrophins enhanced the frequency of mEPSCs and mIPSCs elicited in a cell in a 10 sec measuring interval after sucrose application by threefold and fivefold, respectively (Fig. 5D). These results suggest that the neurotrophins enhance the probability of transmitter release or the number of releasing sites at the presynaptic terminal, but have little effect on the sensitivity of the postsynaptic cells to neurotransmitters.
Fig. 4.

Pharmacological and kinetic analysis of excitatory and inhibitory mPSCs. Miniature synaptic currents were elicited by exposure to 300 mm sucrose the presence of 1 μm TTX. In cultures treated with NT-3 for 72 hr, only fast decaying events were elicited, and are shown here in the presence of picrotoxin (A, B). These fast events were completely blocked by CNQX. In cultures treated for 5 d with BDNF, >60% of elicited events had long decay times. They are shown here in pharmacological isolation, after addition of CNQX, and were completely blocked by picrotoxin (C,D). Amplitude values shown in B andD are negative.
Fig. 5.

Neurotrophins enhanced the frequency of miniature synaptic events. mPSCs elicited by high osmotic stimulation in the presence of TTX were registered. Responses were recorded for 10 sec after stimulation. Excitatory events were defined as any mPSC with a decay time constant <10 msec. Inhibitory events were defined as any mPSC with a decay time constant >12 msec. A, E16 control cells present very few events after stimulation.B, The frequency of the mPSCs was markedly enhanced by BDNF plus NT-3 (NT) treatment. C, Plot of the mEPSC and mIPSC amplitudes show that neurotrophins only cause small increases in the amplitudes of the miniature events. In the neurotrophin-treated cultures, the amplitudes of mEPSCs and mIPSCs were 40% (p < 0.05) and 16% (p < 0.01) greater than in controls, respectively. The amplitudes of mEPSCs and mIPSCs in the untreated cultures were −11.9 ± 2.6 pA and −27.3 ± 3.3 pA, respectively (Fig. 5C). In the neurotrophin-treated cultures, the amplitudes of mEPSCs and mIPSCs were −16.7 ± 3.1 pA (40% above control levels; p < 0.05) and −31.6 ± 2.8 pA (16%; p < 0.01), respectively. D, Plot of the mean number of events in 10 sec in control and BDNF plus NT-3-treated cultures. Neurotrophin-treatment produced a threefold and fivefold enhancement in the number of mEPSC and mIPSC events, respectively. The frequency of mEPSCs and of mIPSCs in the untreated cultures was 46.0 ± 11 and 10.0 ± 3.9/10 sec (n = 10 cells), respectively. In the neurotrophin-treated cultures, the frequency of mEPSCs was 133.0 ± 38.0, and the frequency of mIPSCs was 48.0 ± 10.0 (n = 10 cells). E, Distribution of the frequency of mEPSCs amplitudes (left histogram) and mIPSCs amplitudes (right histogram) from BDNF plus NT-3-treated cultures.
Effects of neurotrophins on neuronal number, morphology, and differentiated state
To interpret these physiological results it is important to know whether the growth factors affected neuronal numbers, dendritic, axonal, and/or synaptic morphology or the expression of presynaptic and postsynaptic proteins. Addition of neurotrophins throughout the culture period did not change the total number of neurons expressing microtubule-associated protein-2ab (MAP-2ab) (Fig.6A), a general marker for neurons (Matus et al., 1986). The majority of neurons in culture stained for glutamate, and no differences were observed in the numbers of these cells between untreated and treated cultures (data not shown). Neurotrophins are known to influence the differentiation of striatal and hippocampal GABAergic neurons (Ventimiglia et al., 1995; Marty et al., 1996). In E16 cultures, the number of GABA-positive cells was similar in the presence or absence of BDNF plus NT-3 (Fig.7A). The proportion of GABA-positive neurons was ∼3% of total neuronal numbers. These results show that, under our conditions, neurotrophins do not change the survival of hippocampal neurons.
Fig. 6.

Effect of NT-3 and BDNF on total neuronal numbers and on dendritic morphology of excitatory neurons in E16 hippocampal cultures. A, At 14 d in vitro, cultures were fixed and immunostained for MAP2ab. To determine the number of cells, a total of 10 fields per coverslip were counted using 400× magnification under fluorescence filters or bright field (Zeiss Axioplan microscope). Results are expressed as the total number of cells staining for MAP2ab in 10 fields. Results are the mean ± SEM of data from seven cultures from four experiments.B–D, Cells were grown for 14 d in control conditions (CT) or from days 11–14 in the presence of NT-3 orBDNF. Cells were double immunostained for MAP2ab and GABA. MAP2ab-positive/GABA-negative neurons were traced with a camera lucida using a 63× objective, and from these drawings the number of primary dendrites and dendritic branches were directly counted. The total length of dendrites was measured with a digital plan measure and the NIH Image program. Results are the mean ± SEM of data from 10 cells per group from two experiments. CT, control;NT, NT-3 plus BDNF.
Fig. 7.

Effect of NT-3 and BDNF on numbers and morphology of inhibitory neurons in E16 hippocampal cultures. A, At 14 d in vitro, cultures were fixed and immunostained for GABA. To determine the number of cells, a total of 10 fields per coverslip were counted using 200× magnification under fluorescence filters. Results are expressed as the total number of cells staining for GABA in 10 fields. Results are the mean ± SEM of data from seven cultures from four experiments.B–F, Cells were grown for 14 d in control conditions (CT) or from days 11–14 in the presence of NT-3 orBDNF. Cells were double immunostained for MAP2ab and GABA. MAP2ab-positive/GABA-positive neurons were traced with a camera lucida using a 63× objective. Analyses were done as described in Figure 6. Results are the mean ± SEM of data from nine cells per group from three experiments. CT, control;NT, NT-3 plus BDNF. **p < 0.005; ***p < 0.001 versus control values.
To define the morphological effects of neurotrophins on excitatory and inhibitory neurons during synaptic activation, we traced cells double-immunostained for MAP2ab and GABA using camera lucida. As can be seen in Figures 6 and 8, treatment of the cultures with NT-3 or BDNF for 3 d, the time sufficient to express the effects of the factors on synaptic connectivity, did not change the number of primary dendrites, dendritic branching, or total dendritic length of MAP2ab-positive/GABA-negative neurons, i.e., of the excitatory neurons. These results suggest that NT-3 and BDNF induce functional excitatory transmission without altering the dendritic morphology of the excitatory cells.
Fig. 8.
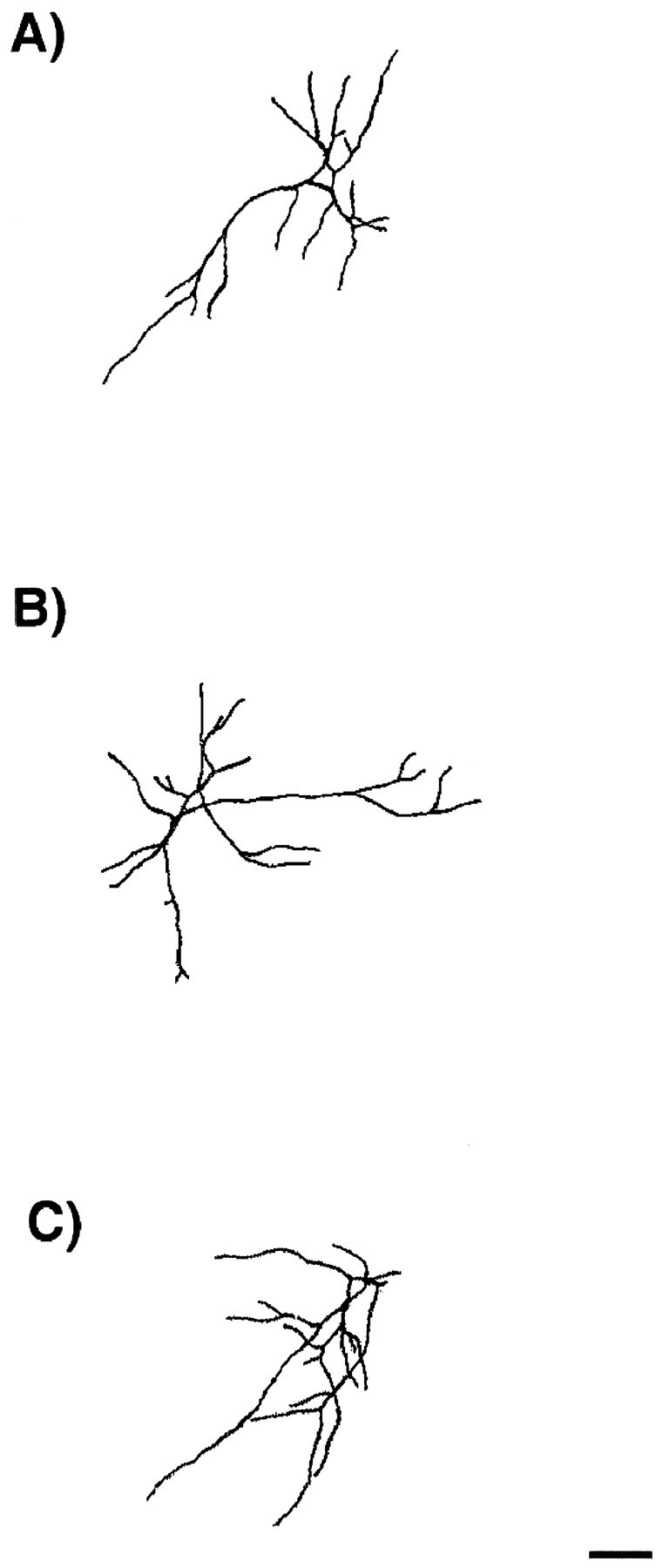
Morphology of excitatory E16 neurons in culture. Cells were grown for 14 d in control cultures (A) or in the presence of NT-3 (B) or BDNF (C) from days 11–14. They were fixed and double immunostained for the expression of MAP2ab and GABA. Camera lucida drawings of dendrites and cell bodies were done on MAP2ab-positive/GABA-negative neurons using a 63× objective. Drawings were digitally scanned and processed using Adobe Photoshop. Neurotrophins did not cause a change in dendritic morphology of the excitatory neurons. See Figure 6 for quantitative analysis of morphology. The thickness of the dendrites is represented arbitrarily. Scale bar, 50 μm.
In contrast, NT-3 and BDNF increased the number of dendritic branches of the GABAergic neurons by 50% (p < 0.001) and 71% (p < 0.001), respectively, and the total dendritic length by 48% (p < 0.005) and 68% (p < 0.001), respectively (Figs. 7,9). Notably, only BDNF caused a marked increase in the number of axonal branches (85%; p < 0.001) and in the total length of the axons (60%; p < 0.001). The morphological effects elicited by the neurotrophins were seen in serum-containing or in serum-free medium. These results suggest that the selective effect of BDNF on inhibitory transmission may be related to an increase in the length of the axons and number of terminals of the inhibitory neurons.
Fig. 9.

Morphology of inhibitory E16 neurons in culture. Cells were grown for 14 d in control cultures (A) or in the presence of NT-3 (B) or BDNF (C) from days 11–14. They were fixed and double immunostained for the expression of MAP2ab and GABA. Camera lucida drawings of dendrites, axon, and cell body were done on MAP2ab-positive/GABA-positive neurons using a 63× objective. Drawings were digitally scanned and processed using Adobe Photoshop. For clarity, the dendrites are represented inred and the axon in black. BDNF simulated the growth of dendrites and axon, whereas NT-3 only promoted dendritic growth. See Figure 7 for quantitative analysis of morphology. The thickness of the dendrites and axons is represented arbitrarily. Scale bar, 150 μm.
Electron microscopy revealed the presence of synaptic structures both in untreated (Fig.10A) and neurotrophin-treated neurons (Fig. 10B); there were no apparent differences between the characteristics of the synapses in the two groups. The number of synaptic vesicles was lower, and the postsynaptic membrane and synaptic cleft were less electron dense in comparison to synapses in the adult rat brain (Vaughn, 1989; Peters et al., 1991; Burns and Augustine, 1995; Sudhof, 1995), but the neurotrophins did not seem to affect these properties in the cultured neurons.
Fig. 10.
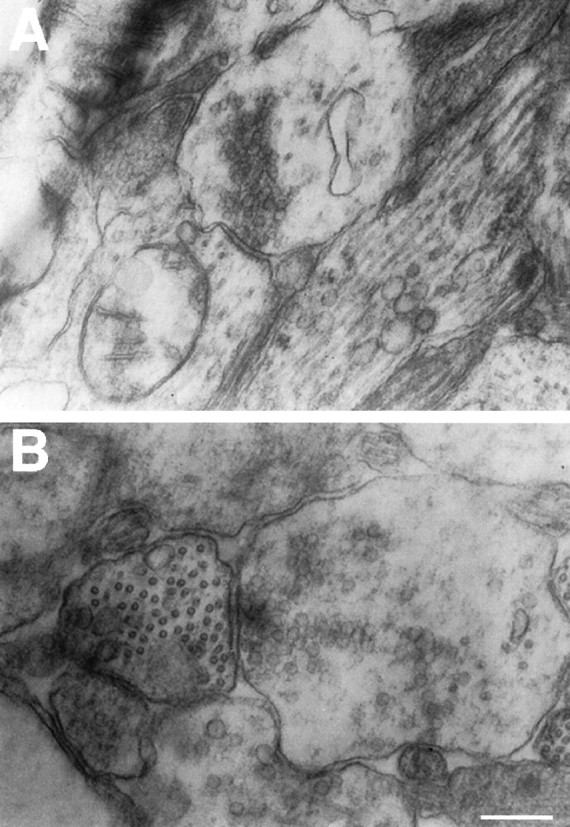
Synapses are present in control and neurotrophin-treated cultures. Transverse sections were visualized and photographed under 20,000× magnification. E16 control culture (A) and BDNF plus NT-3-treated culture (B) present synaptic structures. The number of synaptic vesicles was low, and the postsynaptic membrane and synaptic cleft were less electron dense in comparison to synapses in the adult rat brain. Scale bar, 200 nm.
When E16 cultures were stained for the synaptic-vesicle-associated protein, synapsin-I (Fig.11A,B), we found that the number of synapsin-I-positive boutons per 100× field was similar in control (211.1 ± 11.1) and in neurotrophin-treated cultures (196.8 ± 11.5). Because 97% of MAP2ab-positive neurons are excitatory, the lack of change of synapsin-I-positive boutons suggests that neurotrophins do not significantly modify the number of excitatory presynaptic structures. Staining of the E16 cultures for synaptophysin (Fig. 11C,D) and SV2 (Fig.11E,F) supports this conclusion. Double staining for synapsin-I and SV2 showed an overlap of synapsin-I-positive boutons and SV2-positive boutons (data not shown).
Fig. 11.
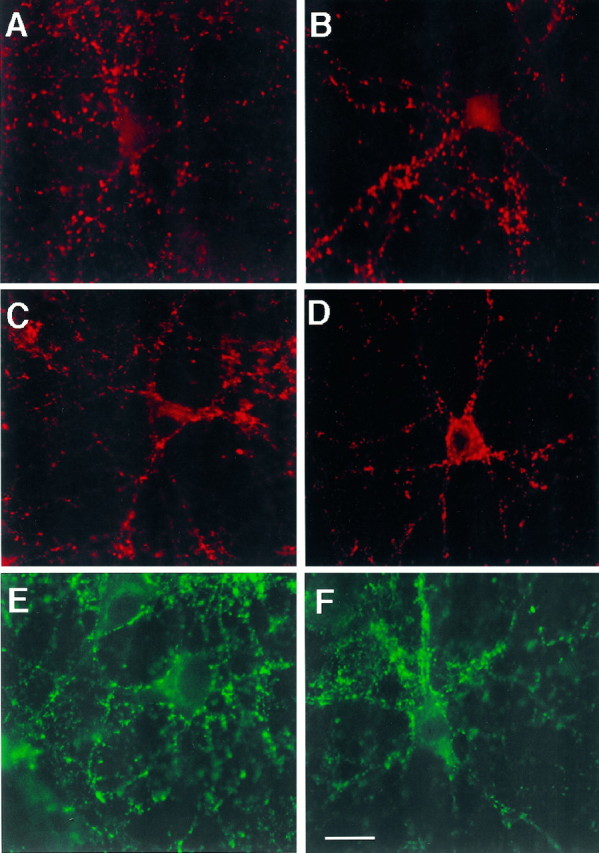
Expression of synaptic-vesicle proteins by hippocampal neurons in culture. Cells were grown for 14 d in control (A, C, E) or BDNF plus NT-3 conditions (B, D,F). Then, they were fixed and immunostained for the expression of synapsin-I (A,B). Different cultures were immunostained for the expression of synaptophysin (C, D), and SV2 (E, F). Magnification, 63×. No appreciable differences in the expression of the synaptic proteins were produced by the addition of BDNF plus NT-3 to the cultures. The same pattern was obtained after staining cultures from three experiments. Scale bar, 15 μm.
DiI labeling of individual neurons was also used to obtain quantitative measurements of detailed dendritic morphology of the glutamatergic neurons (Papa et al., 1995). DiI-labeled neurons from E16 and E18 cultures were analyzed under a confocal laser-scanning microscope (Fig.12A–D). The total number of spines (including mature spines with heads and filopodia) was similar in control and neurotrophin-treated neurons (Fig. 12E). Spine density was greater in the E18-derived neurons than in neurons derived from E16 hippocampus (Fig.12E), but the neurotrophins did not change the density or patterns of spines in either age group.
Fig. 12.
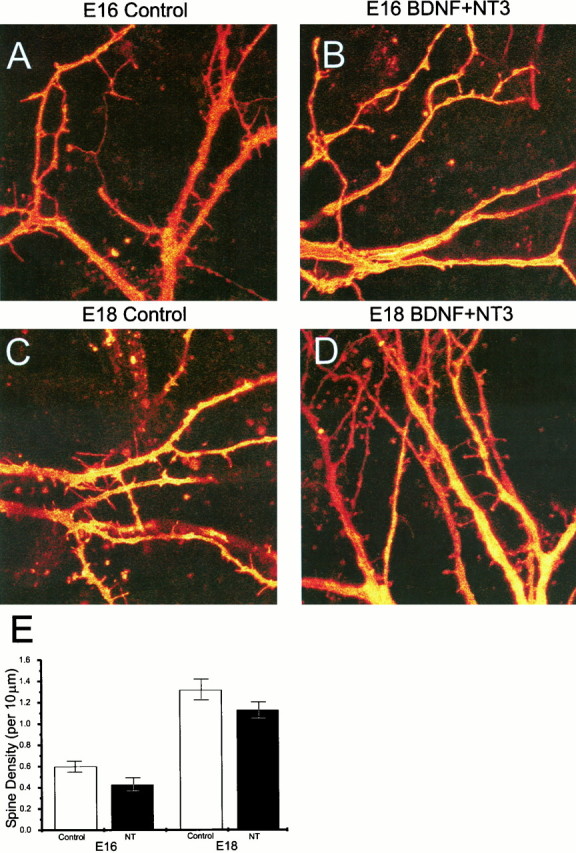
Dendritic spines are present in control and neurotrophin-treated cultures. Cells from E16 (A,B) and E18 (C, D) hippocampus were fixed and stained with DiI. Images were taken in a confocal microscope from control (A, C) and BDNF plus NT-3-treated (B, D) cultures. E, Number of spines per unit length (10 μm) was analyzed as described elsewhere (Papa et al., 1995). The density of spines slightly decreased in neurotrophin-treated cultures. There was a marked difference in spine density between neurons derived from E16 and E18 hippocampus.
E16 cultures were immunostained to reveal the presence of TrkB and TrkC, the receptors of BDNF and NT-3 (Klein, 1994) (Fig.13). Double staining for MAP2ab and TrkB or TrkC showed that these receptors were present in the majority of neurons in culture (data not shown), in agreement with the expression of these receptors in the glutamatergic neurons of the hippocampal pyramidal layer and dentate gyrus (Lamballe et al., 1994;Fryer et al., 1996). A double staining for TrkB and GAD (Fig.13A,B) and TrkC and GAD (Fig.13C,D) showed that GAD-positive neurons express TrkB and TrkC. These results indicate that both excitatory and inhibitory neurons express Trk receptors for BDNF and NT3.
Fig. 13.

Expression of TrkB and TrkC by E16 hippocampal neurons in culture. E16 cells grown for 14 d were fixed and immunostained for the expression of TrkB (A), TrkC (C), and GAD (B,D). Photographs were taken using a 40× objective. At least 95% of the neurons showed expression of TrkB and TrkC. TrkB and TrkC were present in both excitatory and inhibitory neurons. The same patterns were obtained after staining cultures from three experiments. Scale bars: A, B, 20 μm;C, D, 15 μm.
The levels of expression of glutamate receptors 1 and 2 (GluR1, GluR2) and NMDAR1 (data not shown) were not modified by neurotrophin application to the cultures (Fig. 14). In the presence of neurotrophins, the relative amounts of these proteins versus control values (100%) were 108.7 ± 22.3% for GluR1 and 96.1 ± 9.3% for GluR2. Postsynaptic clustering of glutamate receptors has been correlated to the development of excitatory synapses in cultured hippocampal and spinal neurons (Craig et al., 1993; O’Brien et al., 1997). Under our conditions, we did not observe an appreciable change in GluR1 or GluR2 clustering during the time required for synaptic activation by NT-3 or BDNF in the E16 cultures (data not shown). The amounts of the synaptic-vesicle proteins synapsin-I, synaptotagmin, and syntaxin (data not shown) were also similar in both groups of neurons (relative amounts vs control values were 102.2 ± 2.3% for synapsin-I and 103.1 ± 17.8% for synaptotagmin) (Fig.14). These biochemical results indicate that the increase in the probability of neurotransmitter release and/or in the releasing units caused by neurotrophins is not attributable to a change in the levels of these synaptic proteins.
Fig. 14.

Expression of synaptic-vesicle proteins and glutamate receptors by hippocampal neurons in culture. E16 hippocampal cells were grown on 6 or 10 cm dishes. After 11–13 d in vitro, proteins were analyzed by SDS-PAGE. Extracts were separated by electrophoresis on a 8% polyacrylamide gel and electrotransferred to nitrocellulose filters. Filters were immunoblotted with antibodies against synapsin-I (80–83 kDa), synaptotagmin (65 kDa), GluR2 (100 kDa), and GluR1 (100 kDa). The same pattern was obtained after analyzing extracts from two or three different cultures. Lane 1, Control; lane 2, chronic addition of BDNF and NT-3. There were not significant differences in the amount of these proteins between control and neurotrophin-treated cells (see Results).
DISCUSSION
Competition between neurons for target-derived factors is thought to be a central part of an activity-dependent program that regulates synapse formation and neuronal death. The classical evidence for this possibility was provided by experiments showing that neuronal numbers were a function of target size in the periphery, and that NGF actedin vivo as a neurotrophic factor for sympathetic and sensory neurons (Levi-Montalcini, 1987). Gene-targeting experiments confirm that many peripheral neurons require neurotrophins for their survival and differentiation (Snider, 1994). However, only some central neurons, including hippocampal dentate, cerebellar granule neurons, and Purkinje cells are affected on deletion of TrkB and TrkC (Minichiello and Klein, 1996; Alcántara et al., 1997; Schwartz et al., 1997). A role for BDNF in the development of PNS synapses has been proposed based on studies in transgenic mice (Causing et al., 1997). In the CNS, agonists and antagonists of neurotrophins perturb ocular dominance column formation (Cabelli et al., 1995, 1997), and different neurotrophins have specific and opposite effects on neuronal morphology (McAllister et al., 1997). Despite the theoretical importance of activity-dependent competition in models of CNS function, there are few studies on the role of neurotrophic factors in the formation of functional synaptic interactions between central neurons.
Neurotrophins activate excitatory synapses without changing the morphology of glutamatergic neurons
The neurotrophins BDNF and NT-3 promote early steps in the differentiation and morphological maturation of hippocampal neurons (Collazo et al., 1992; Vicario-Abejón et al., 1995; Ghosh and Greenberg, 1995; Marty et al., 1996). To test whether the neurotrophins also play a role in the formation of functional synaptic connections, we established a tissue culture system of hippocampal neurons to study synaptogenesis. After 2 weeks in culture, neurons prepared from E16 hippocampus showed very low levels of functional synaptic connectivity. Exposure to BDNF or NT-3 for 24–72 hr induced synaptic responses. It is important to note that no neurotrophins were present in the medium when the electrophysiological recordings were made. Thus the presence of synaptic currents did not require the continuous exposure to the growth factors. The time course of neurotrophin action and the fact that the untreated cultures do not possess functional synapses suggests that the major effect elicited by these factors is not to acutely strengthen synaptic transmission, as previously reported (Lebmann et al., 1994; Kang and Schuman, 1995, 1996; Levine et al., 1995), but to create functional synapses where they did not previously exist.
The majority of neurons in the system are glutamatergic, and they express TrkB and TrkC receptors. Both BDNF and NT-3 induce excitatory synaptic transmission. The relatively slow kinetics for synaptic activation by neurotrophins suggests that these factors could promote structural changes in synapses. Dendritic spines are known to be a major site for the establishment of excitatory synapses (Harris and Kater, 1994). However, the number of dendritic spines were not modified by the presence of neurotrophins. Similarly, the number of synapsin-I-positive boutons was not changed by the neurotrophins. In another series of experiments, the number of dendritic branches and the total dendritic length of excitatory neurons was measured after 72 hr of exposure to NT-3 or BDNF. There was no noticeable morphological effect, yet the connectivity was established within 3 d of treatment with neurotrophins. These results suggest that neurotrophins can activate excitatory synapses by mechanisms that are independent of gross changes in neuronal morphology.
The enhancement of the frequency of mPSCs elicited by hyperosmotic stimulation in the presence of TTX suggests that the neurotrophins act presynaptically to promote the maturation of neurotransmitter release mechanisms. Whereas this is likely to be the major effect of neurotrophins in the E16 cultures, they also could affect the postsynaptic receptors as suggested by the small, yet significant effect on the amplitudes of the mPSCs. A predominantly presynaptic action is also believed to underlie the potentiation of hippocampal and neuromuscular synapses by neurotrophins (Lohof et al., 1993; Lebmann et al., 1994; Wang et al., 1995), although postsynaptic effects have also been reported (Levine et al., 1995).
Spontaneous release of neurotransmitter before neurons contact synaptic targets has been shown (Hume et al., 1983; Hall and Sanes, 1993; Young and Poo, 1983; Basarsky et al., 1994). The data presented here suggest that when a neuron first contacts its target, a further maturation step is required for synaptic function. The biochemistry and molecular genetics of the presynaptic machinery have been extensively studied recently, defining roles for synaptic-vesicle proteins such as synapsin-I, syntaxin, synaptotagmin, and Rab3A in the release of neurotransmitters (Scheller, 1995; Sudhof, 1995; Geppert et al., 1997). Postranslational mechanisms such as phosphorylation of synapsin-I, syntaxin, SNAP-25, or synaptobrevin have been implicated in the modulation of synaptic strength (Knipper et al., 1994; Jovanovic et al., 1996; Hirling and Scheller, 1996). Indeed, overexpression of synaptic-vesicle proteins has been reported to increase synaptic connectivity in vitro (Greengard et al., 1993). In E16 cultures, the lack of correlation between the levels of synapsin-I, synaptotagmin, or syntaxin and the strong increase in transmitter release caused by neurotrophins suggests that the amount of these proteins is not a critical control point in synaptic activation. However, neurotrophins could regulate the expression of other synaptic-vesicle proteins and/or their phosphorylation state.
Regulation of inhibitory synapses by BDNF correlates with a specific change in axonal morphology
The number of functional glutamatergic synapses was enhanced by treatment of the cells with BDNF or NT-3. However, BDNF also promoted the formation of inhibitory synaptic connections. Both NT-3 and BDNF increase the dendritic complexity of the GABAergic neurons. BDNF alone had a marked effect on axonal morphology. The increase in inhibitory transmission seen with BDNF could therefore be a consequence of the promotion of axonal growth and branching in the inhibitory cells.
In hippocampal slices, both BDNF and NT-3 act acutely to strengthen glutamatergic synapses (Kang and Schuman, 1995; Levine et al., 1995). Potentiation of excitatory transmission could also be achieved by depression of inhibitory inputs by neurotrophins, as has been reported for both NT-3 and BDNF (Kim et al., 1994; Tanaka et al., 1997). The results presented here suggest that BDNF acts to promote inhibitory synaptic transmission, an extension of previous reports showing neurotrophic effects of BDNF on GABAergic neurons (Ventimiglia et al., 1995; Marty et al., 1996). Thus, in early development, BDNF appears to be critical in regulating GABAergic synaptic function.
The distinct physiological and morphological effects of NT-3 and BDNF reported here are in agreement with previous results that showed that neurotrophins elicited specific morphological effects on different neuronal classes (McAllister et al., 1997). Our results suggest that the levels of BDNF and NT-3 may regulate the balance between excitatory and inhibitory transmission during early stages of circuit formation in the hippocampus. BDNF produced by hippocampal glutamatergic neurons may promote the differentiation of GABAergic neurons (Marty et al., 1996). The results presented here extend this model of interaction between the excitatory and inhibitory systems as they suggest that excitatory neurons can use the activity-dependent release of BDNF to regulate the number of inhibitory connections they receive.
Footnotes
We thank Drs. S. Okabe and M. Nguyen for their help with the Western blotting. We also thank Dr. M. B. Kennedy (Caltech) for the gift of anti-synapsin-I; Dr. R. Wenthold [National Institutes of Health (NIH)] for the antibodies against glutamate receptors; Drs. S. Feinstein and M. Radeke (University of California) for antibodies against TrkB; Drs. S. Landis (NIH), W. Catterall (University of Washington), T. Reese (NIH), E. Stanley (NIH), J. Auerbach (NIH), and D. Panchision (NIH) for valuable comments; Dr. C. Smith (NIH) for advice with video and confocal microscopy; and Drs. S. Cheng, P. Zerfas, and V. Tanner (NIH) for help with EM analysis. Part of this work was conducted while M.S. was a Scholar in Residence at the Fogarty International Center for Advanced Study in the Health Sciences at NIH.
Correspondence should be addressed to Dr. Ronald D. G. McKay, National Institutes of Health, National Institute of Neurological Disorders and Stroke, Laboratory of Molecular Biology, Building 36, Room 3DO2, Bethesda, MD 20892-4092.
REFERENCES
- 1.Alcántara S, Frisén J, del Río J, Soriano E, Barbacid M, Silos-Santiago I. TrkB signalling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell dead. J Neurosci. 1997;15:3623–3633. doi: 10.1523/JNEUROSCI.17-10-03623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altman J, Bayer S. Prolonged sojourn of developing pyramidal cells in the intermediate zone of the hippocampus and their settling in the stratum pyramidale. J Comp Neurol. 1990;301:343–364. doi: 10.1002/cne.903010303. [DOI] [PubMed] [Google Scholar]
- 3.Basarsky T, Parpura V, Haydon P. Hippocampal synaptogenesis in cell culture: developmental time course of synapse formation, calcium influx, and synaptic protein distribution. J Neurosci. 1994;14:6402–6411. doi: 10.1523/JNEUROSCI.14-11-06402.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ben-Ari Y, Khazipov R, Leinekugel X, Caillard O, Gaiarsa J-L. GABAA, NMDA and AMPA receptors: a developmentally regulated “ménage a trois.”. Trends Neurosci. 1997;20:523–529. doi: 10.1016/s0166-2236(97)01147-8. [DOI] [PubMed] [Google Scholar]
- 5.Burns M, Augustine G. Synaptic structure and function: dynamic organization yields architectural precision. Cell. 1995;83:187–194. doi: 10.1016/0092-8674(95)90160-4. [DOI] [PubMed] [Google Scholar]
- 6.Cabelli R, Hohn A, Shatz C. Inhibition of ocular dominance column formation by infusion of NT-4/5 or BDNF. Science. 1995;267:1662–1666. doi: 10.1126/science.7886458. [DOI] [PubMed] [Google Scholar]
- 7.Cabelli R, Shelton D, Segal R, Shatz C. Blockade of endogenous ligands of TrkB inhibits formation of ocular dominance columns. Neuron. 1997;19:63–76. doi: 10.1016/s0896-6273(00)80348-7. [DOI] [PubMed] [Google Scholar]
- 8.Causing CG, Gloster A, Aloyz R, Bamji SX, Chang E, Fawcett J, Kuchel G, Miller FD. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron. 1997;18:257–267. doi: 10.1016/s0896-6273(00)80266-4. [DOI] [PubMed] [Google Scholar]
- 9.Collazo D, Takahashi H, McKay RDG. Cellular targets and trophic functions of neurotrophin-3 in the developing rat hippocampus. Neuron. 1992;9:643–656. doi: 10.1016/0896-6273(92)90028-c. [DOI] [PubMed] [Google Scholar]
- 10.Craig AM, Blackstone CD, Huganir RL, Banker G. The distribution of glutamate receptors in cultured rat hippocampal neurons: postsynaptic clustering of AMPA-selective subunits. Neuron. 1993;10:1055–1068. doi: 10.1016/0896-6273(93)90054-u. [DOI] [PubMed] [Google Scholar]
- 11.Figurov A, Pozzo-Miller L, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- 12.Fletcher T, De Camilli P, Banker G. Synaptogenesis in hippocampal cultures: evidence indicating that axons and dendrites become competent to form synapses at different stages of neuronal development. J Neurosci. 1994;14:6695–6706. doi: 10.1523/JNEUROSCI.14-11-06695.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fryer R, Kaplan D, Feinstein S, Radeke M, Grayson D, Kromer L. Developmental and mature expression of full-length and truncated trkB receptors in the rat forebrain. J Comp Neurol. 1996;374:21–40. doi: 10.1002/(SICI)1096-9861(19961007)374:1<21::AID-CNE2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 14.Geppert M, Goda Y, Stevens C, Sudhof T. The small GTP-binding protein Rab3A regulates a late step in synaptic vesicle fusion. Nature. 1997;387:810–814. doi: 10.1038/42954. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh A, Greenberg M. Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron. 1995;15:89–103. doi: 10.1016/0896-6273(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 16.Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- 17.Hall Z, Sanes J. Synaptic structure and development: the neuromuscular junction. Cell. 1993;72:99–121. doi: 10.1016/s0092-8674(05)80031-5. [DOI] [PubMed] [Google Scholar]
- 18.Harris K, Kater S. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- 19.Hirling H, Scheller R. Phosphorylation of synaptic vesicle proteins: modulation of the aSNAP interaction with the core complex. Proc Natl Acad Sci USA. 1996;93:11945–11949. doi: 10.1073/pnas.93.21.11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hume R, Role L, Fischbach G. Acetylcholine release from growth cones detected with patches of acetylcholine receptor-rich membranes. Nature. 1983;305:632–634. doi: 10.1038/305632a0. [DOI] [PubMed] [Google Scholar]
- 21.Jovanovic J, Benfenati F, Siow Y, Sihra T, Sanghera J, Pelech S, Greengard P, Czernik A. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc Natl Acad Sci USA. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang H, Schuman E. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 23.Kang H, Schuman E. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- 24.Kim H, Wang T, Olafsson P, Lu B. Neurotrophin 3 potentiates neuronal activity and inhibits g-aminobutyratergic synaptic transmission in cortical neurons. Proc Natl Acad Sci USA. 1994;91:12341–12345. doi: 10.1073/pnas.91.25.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klein R. Role of neurotrophins in mouse neuronal development. FASEB J. 1994;8:738–744. doi: 10.1096/fasebj.8.10.8050673. [DOI] [PubMed] [Google Scholar]
- 26.Knipper M, Leung L, Zhao D, Rylett R. Short-term modulation of glutamatergic synapsis in adult rat hippocampus by NGF. NeuroReport. 1994;5:2433–2436. doi: 10.1097/00001756-199412000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamballe F, Smeyne R, Barbacid M. Developmental expression of trkC, the neurotrophin-3 receptor, in the mammalian nervous system. J Neurosci. 1994;14:14–28. doi: 10.1523/JNEUROSCI.14-01-00014.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lebmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurons. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- 30.Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 31.Levine E, Dreyfus C, Black I, Plummer M. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lohof A, Ip N, Poo M. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 33.Marty S, Berninger B, Carroll P, Thoenen H. GABAergic stimulation regulates the phenotype of hippocampal interneurons through the regulation of Brain-derived neurotrophic factor. Neuron. 1996;16:565–570. doi: 10.1016/s0896-6273(00)80075-6. [DOI] [PubMed] [Google Scholar]
- 34.Matteoli M, Verderio C, Krawzeski K, Mundigl O, Coco S, Fumagalli G, De Camilli P. Mechanisms of synaptogenesis in hippocampal neurons in primary culture. J Physiol (Paris) 1995;89:51–55. doi: 10.1016/0928-4257(96)80551-1. [DOI] [PubMed] [Google Scholar]
- 35.Matus A, Bernhardt R, Bodmer R, Alaimos D. Microtubule-associated protein-2 and tubulin are differently distributed in the dendrites of the developing neurons. Neuroscience. 1986;17:371–389. doi: 10.1016/0306-4522(86)90253-8. [DOI] [PubMed] [Google Scholar]
- 36.McAllister A, Katz L, Lo D. Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron. 1997;18:767–778. doi: 10.1016/s0896-6273(00)80316-5. [DOI] [PubMed] [Google Scholar]
- 37.Minichiello L, Klein R. TrKB and TrkC neurotrophin receptors cooperate in promoting survival of hippocampal and cerebellar granule neurons. Genes Dev. 1996;10:2849–2858. doi: 10.1101/gad.10.22.2849. [DOI] [PubMed] [Google Scholar]
- 38.O’Brien RJ, Mammen AL, Blackshaw S, Ehlers MD, Rothstein JD, Huganir RL. The development of excitatory synapses in cultured spinal neurons. J Neurosci. 1997;17:7339–7350. doi: 10.1523/JNEUROSCI.17-19-07339.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papa M, Bundman M, Greenberger V, Segal M. Morphological analysis of dendritic spine development in primary cultures of hippocampal neurons. J Neurosci. 1995;15:1–11. doi: 10.1523/JNEUROSCI.15-01-00001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Patterson S, Abel T, Deuel T, Martin K, Rose J, Kandel E. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 41.Peters A, Palay S, Webster HD. The fine structure of the nervous system. Neurons and their supporting cells. Oxford UP; New York: 1991. [Google Scholar]
- 42.Scheller R. Membrane trafficking in the presynaptic nerve terminal. Neuron. 1995;14:893–897. doi: 10.1016/0896-6273(95)90328-3. [DOI] [PubMed] [Google Scholar]
- 43.Schwartz PM, Borghesian PR, Levy RL, Poeroy SL, Segal RA. Abnormal cerebellar development and foliation in BDNF−/− mice reveals a role for neurotrophins in CNS patterning. Neuron. 1997;19:269–281. doi: 10.1016/s0896-6273(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 44.Segal M. GABA induces a unique rise of [Ca]i in cultured rat hippocampal neurons. J Neurophysiol. 1983;50:1249–1264. [Google Scholar]
- 45.Snider W. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 46.Stoop R, Poo MM. Synaptic modulation by neurotrophic factors: differential and synergistic effects of brain-derived neurotrophic factor and ciliary neurotrophic factor. J Neurosci. 1996;16:3256–3264. doi: 10.1523/JNEUROSCI.16-10-03256.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sudhof T. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 48.Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 50.Vaughn J. Review: fine structure of synaptogenesis in the vertebrate central nervous system. Synapse. 1989;3:255–285. doi: 10.1002/syn.890030312. [DOI] [PubMed] [Google Scholar]
- 51.Ventimiglia R, Mather P, Jones B, Lindsay R. The neurotrophins BDNF, NT-3 and NT-4/5 promote survival and morphological and biochemical differentiation of striatal neurons in vitro. Eur J Neurosci. 1995;7:213–222. doi: 10.1111/j.1460-9568.1995.tb01057.x. [DOI] [PubMed] [Google Scholar]
- 52.Vicario-Abejón C, Johe K, Hazel T, Collazo D, McKay R. Functions of basic-fibroblast growth factor and neurotrophins in the differentiation of hippocampal neurons. Neuron. 1995;15:105–114. doi: 10.1016/0896-6273(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 53.Wang T, Xie K, Lu B. Neurotrophins promote maturation of developing neuromuscular synapses. J Neurosci. 1995;15:4796–4805. doi: 10.1523/JNEUROSCI.15-07-04796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xie K, Wang T, Olafsson P, Mizuno K, Lu B. Activity-dependent expression of NT-3 in muscle cells in culture: implications in the development of the neuromuscular junctions. J Neurosci. 1997;17:2947–2958. doi: 10.1523/JNEUROSCI.17-09-02947.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Young S, Poo M. Spontaneous release of transmitter from growth cones of embryonic neurones. Nature. 1983;305:634–637. doi: 10.1038/305634a0. [DOI] [PubMed] [Google Scholar]