

Abstract
A possible role for a protein kinase, PKN, a fatty acid-activated serine/threonine kinase with a catalytic domain homologous to the protein kinase C family and a direct target for Rho, was investigated in the pathology of Alzheimer’s disease (AD) using a sensitive immunocytochemistry on postmortem human brain tissues and a kinase assay for human tau protein. The present study provides evidences by light, electron, and confocal laser microscopy that in control human brains, PKN is enriched in neurons, where the kinase is concentrated in a subset of endoplasmic reticulum (ER) and ER-derived vesicles localized to the apical compartment of juxtanuclear cytoplasm, as well as late endosomes, multivesicular bodies, Golgi bodies, secretary vesicles, and nuclei. In AD-affected neurons, PKN was redistributed to the cortical cytoplasm and neurites and was closely associated with neurofibrillary tangles (NFTs) and their major constituent, abnormally modified tau. PKN was also found in degenerative neurites within senile plaques. In addition, we report that human tau protein is directly phosphorylated by PKN both in vitro and in vivo. Thus, our results suggest a specific role for PKN in NFT formation and neurodegeneration in AD damaged neurons.
Keywords: PKN, PRK1, kinase, phosphorylation, endoplasmic reticulum, neurofibrillary tangles, paired helical filaments, tau protein, Alzheimer’s disease, immunocytochemistry, kinase assay, human brain, neuron
Alzheimer’s disease (AD) pathology is characterized by intracellular neurofibrillary tangles (NFTs), extracellular senile plaques (SPs), neuronal loss, and activation of glia. The number of NFTs correlates directly with the severity of dementia (Alafuzoff et al., 1987). NFTs are composed of straight and paired helical filaments (PHFs), with a major component being an aberrantly hyperphosphorylated form of the microtubule-associated protein (MAP) tau (for review, see Billingsley and Kincaid, 1997). Such abnormal filaments accumulate in the cell bodies of diseased but surviving neurons, as well as in the neuropil threads, and dystrophic neurites in and around SPs. In normal brain, tau promotes the assembly and stabilization of microtubules (Drubin and Kirschner, 1986), one of the three major components of the cytoskeleton. The ability of tau to bind to microtubule is downregulated, however, after local phosphorylation, especially in the C-terminal repeats of microtubule-binding domain (Johnson, 1992; Drewes et al., 1997).In vitro, tau is a substrate for some proline-directed as well as nonproline-directed serine/threonine kinases (Morishima-Kawashima et al., 1995; Billingsley and Kincaid, 1997). The colocalization of NFTs with some proline-directed kinases, such as GSK3 (Yamaguchi et al., 1996; Pei et al., 1997), Cdc2-related kinase (Cdk5) (Liu et al., 1995; Yamaguchi et al., 1996), and Erk2 (Trojanowski et al., 1993), has been shown by immunohistochemical analysis, but it is not known whether other kinases are present. In addition, it remains to be elucidated which kinases modulate tau phosphorylation in vivo.
Recently we have found a novel 120 kDa lipid-activated serine/threonine kinase, PKN, which consists of a regulatory N-terminal region containing unique repeats of a leucine zipper-like motif and a C-terminal catalytic domain highly homologous to that of the protein kinase C (PKC) family (Mukai and Ono, 1994; Mukai et al., 1994;Kitagawa et al., 1995). In vitro, the amino-terminal region of PKN binds to GTP-Rho, activating PKN (Amano et al., 1996; Shibata et al., 1996; Watanabe et al., 1996), and to the head rod of intermediate filament proteins such as neurofilament, vimentin, and glial fibrillary acidic protein, leading to an inhibitory regulation of polymerization and fibril formation (Mukai et al., 1996a; Matsuzawa et al., 1997) into intermediate filament, a second major component of the cytoskeleton. PKN also interacts with α-actinin cross-linking actin, a third major component of the cytoskeleton (Mukai et al., 1997). Thus PKN may play important roles in the assembly or stabilization of various cytoskeletal proteins.
PKN is ubiquitously expressed but is enriched in brain (Kitagawa et al., 1995). To characterize the PKN pathway in AD-affected neurons, we have attempted to localize PKN immunocytochemically and to identify other target proteins interacting with PKN in human brain tissue. Here we report that PKN localizes in abnormal straight and paired helical filaments and in endoplasmic reticulum (ER)-derived vesicles closely associated with intracellular NFTs in damaged AD neurons, and that PKN directly phosphorylates human tau protein both in vitro and in vivo.
MATERIALS AND METHODS
Human brain tissue used. Twenty-five autopsied human brains were used in this study. Twelve of the brains came from cases without neurological disorders (seven males, five females; age range, 56–89 years; mean age ± SEM, 74.5 ± 11.8 years), and 13 came from sporadic AD cases (six males, seven females; age range: 58–90 years; mean age ± SEM, 76.8 ± 9.1 years). All experiments using human subjects were performed in compliance with the policies and principles contained in the Federal Policy for the Protection of Human Subjects [United States Office of Science and Technology Policy (1991)] and in the Declaration of Helsinki (1984).
Immunoblot analysis. A polyclonal antibody, αC6, was raised in rabbit against a recombinant C-terminus protein corresponding to residues 863–946 of rat PKN (Mukai et al., 1994). Cytoplasmic extracts were prepared from the appropriate postmortem brains of control and AD patients. The brain tissues were homogenized with a polytron in 10 vol of ice-cold 20 mm Tris-HCl, pH 7.4, containing 5 mm EDTA, 5 mm EGTA, 10 mg/ml leupeptin, 10 mg/ml pepstatin, and 2 mm phenylmethyl sulfonate, and mixed with SDS-PAGE sample buffer. The total brain homogenates were then transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA), which were incubated with αC6 diluted at 1:2000 and then soaked in 0.01 m PBS, pH 7.4, containing 0.3% Triton X-100 and HRP-conjugated donkey anti-rabbit IgG (Amersham, Buckinghamshire, UK). Immunoreactive bands were visualized by using the enhanced chemiluminescence method on a hyperfilm ECL (Amersham). Preincubation of the antibody either with the recombinant protein used as the immunogen or with the glutathioneS-transferase (GST)-full PKN fusion protein eliminated the bindings of αC6 both to human brain tissues and to the recombinant PKN protein on the membranes (data not shown). Such elimination of staining did not occur if the preincubation was with control peptides or with GST protein.
Immunocytochemistry. The fixation and processing of human brain tissues, including cerebellum, brainstem, thalamus, striatum, basal forebrain, hippocampus, entorhinal cortex, amygdala, midfrontal gyrus, precentral and postcentral gyri, angular gyrus, primary visual cortex, and superior temporal gyrus, and the immunohistochemical examinations were performed as described previously (Kawamata et al., 1993).
For immunoelectron microscopic study, the stained sections were post-fixed in 1% osmium tetroxide, dehydrated in graded alcohol, and flat-embedded in Epon WE812 (Wako, Osaka, Japan). Ultrathin sections were examined by using a Hitachi H-7100 electron microscope at 75 kV.
Colocalization of PKN-positive structures with such organelles as ER, endosome–lysosome, and the cytoskeleton, and with AD pathological markers, was analyzed by double immunofluorescence staining. After immunostaining followed by incubation with appropriate secondary antibodies coupled either to fluorescein or rhodamine isothiocyanate (Chemicon, Temecula, CA), sections were analyzed by a confocal scanning laser microscope (Bio-Rad MRC1024, Bio-Rad, Hercules, CA). The monoclonal primary antibodies used in this study were against microtubule-associated protein (MAP2; Sigma, St. Louis, MO), nonphosphorylated neurofilament (SMI-32; Sternberger, Baltimore, MD), phosphorylated neurofilament (SMI-31; Sternberger), an Ig-heavy chain-binding protein BiP (StressGen, Victoria, British Columbia), cathepsin-D (Chemicon), β-amyloid precursor protein Aβ1–17 (Senetek, Maryland Heights, MD), complement protein C4d (Quidel, San Diego, CA), ubiquitin (Senetek), nonphosphorylated tau [tau-1 (Boehringer Mannheim, Mannheim, Germany) diluted at 1:1000], phosphorylated tau [AT8, AT180, and AT270 (Innogenetics, Zwijndrecht, Belgium) diluted 1:10000], and tau with abnormal conformation [Alz50 (generously provided by P. Davies, Albert Einstein College of Medicine) diluted 1:400].
Preparation of recombinant tau, recombinant PKN, and expression plasmids. cDNA encoding the human brain tau (383 residues) was cloned from human brain cDNA library (clone T9) (Mori et al., 1989). The full-length coding region ligated to pGEX4T vector (Pharmacia, Uppsala, Sweden) was transformed into DH5α cells. Expression and purification of GST or GST-tau fusion protein were performed according to the manufacturer’s instruction (Pharmacia). Lysates were cleared by centrifugation, filtered, and loaded overnight at 4°C onto a glutathione–Sepharose 4B column (Sigma). GST-tau fusion protein was eluted by adding 15 mm reduced glutathione (Sigma) to the buffer and appeared homogenous on Coomassie blue-stained SDS-PAGE.
pGFP-N3/GST vector was constructed by subcloning the cDNA fragment for the coding region of GST prepared by PCR from a pGEX4T vector intoXhoI site of pGFP-N3 vector (Clontech, Palo Alto, CA). phPKN-H4 (Mukai and Ono, 1994) was digested with SalI, and the resultant insert fragment was subcloned into the SalI site of the pGFP-N3/GST vector. The insert fragment for GST-tagged full length of human PKN was obtained by digesting the resultant vector withKpnI, blunting with T4 DNA polymerase, ligating withNcoI linker, and digesting withBglII/NcoI. pBlueBacHis/GST/PKNf for expression of 6× His- and GST-tagged full length of human PKN in Sf9 cells was constructed by subcloning the insert fragment into pBlueBacHis-C (Invitrogen, Carlsbad, CA). To generate the recombinant baculovirus, pBlueBacHis/GST/PKNf was cotransfected with BacVector-2000 Triple Cut Virus DNA into Sf9 cells. Isolation, amplification, and expression of baculovirus were performed according to the manufacturer’s instruction (Novagen, Madison, WI). Recombinant PKN was first purified with Ni-NTA agarose from lysates of infected Sf9 cells according to the protocols for purification under native conditions (Qiagen, Chatsworth, CA) and was further purified by loading the eluate from Ni-NTA agarose onto glutathione-Sepharose 4B (Pharmacia).
pTB701/PKN/AF3 was constructed by insertion of the cDNA fragment encoding amino acids 561–942 of PKN into mammalian expression vector pTB701 (Ono et al., 1988). pTB701/PKN/AF3(K644E) was generated by Quik Change Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) from pTB701/PKN/AF3 to mutate the lysine residue (amino acids 644) at the ATP binding site of the protein kinase domain to glutamic acid.
Kinase assay. The phosphorylation of tau by PKN was assayed at 30°C in a reaction mixture containing 20 mm Tris/HCl, pH 7.5, 4 mm MgCl2, 100 μmATP, 185 kBq of [γ-32P] ATP, recombinant tau protein, and 50 ng of recombinant PKN, with or without 40 μmarachidonic acid, as indicated in each experiment. The reaction was terminated by adding an equal volume of Laemmli’s sample buffer after the indicated time in each experiment, followed by separation on SDS-PAGE. The gel was dried under vacuum, and the phosphorylation was visualized and quantified by an imaging analyzer, FUJI BAS2000.
Human neuroblastoma SK-N-MC cells transfected with pTB701 vector, pTB701/PKN/AF3 (catalytically active variant of PKN), and pTB701/PKN/AF3(K644E) (catalytically inactive variant of PKN) were pulse-labeled in medium supplemented with 50 μCi/ml carrier-free [32P] orthophosphate for 1 hr. The [32P]-labeled proteins were purified with tau-1 antibody (Boehringer Mannheim), separated by SDS-PAGE, and visualized by autoradiography.
RESULTS
Characterization of PKN antibody
Immunoblot analysis revealed that the αC6 antibody against PKN detected a single band in total brain homogenates from human control and AD cases at 120 kDa (Fig. 1), which corresponds to the full-size PKN molecule as noted in rat brain (Kitagawa et al., 1995). There was not much difference in PKN immunoreactivity between human control and AD brains.
Fig. 1.
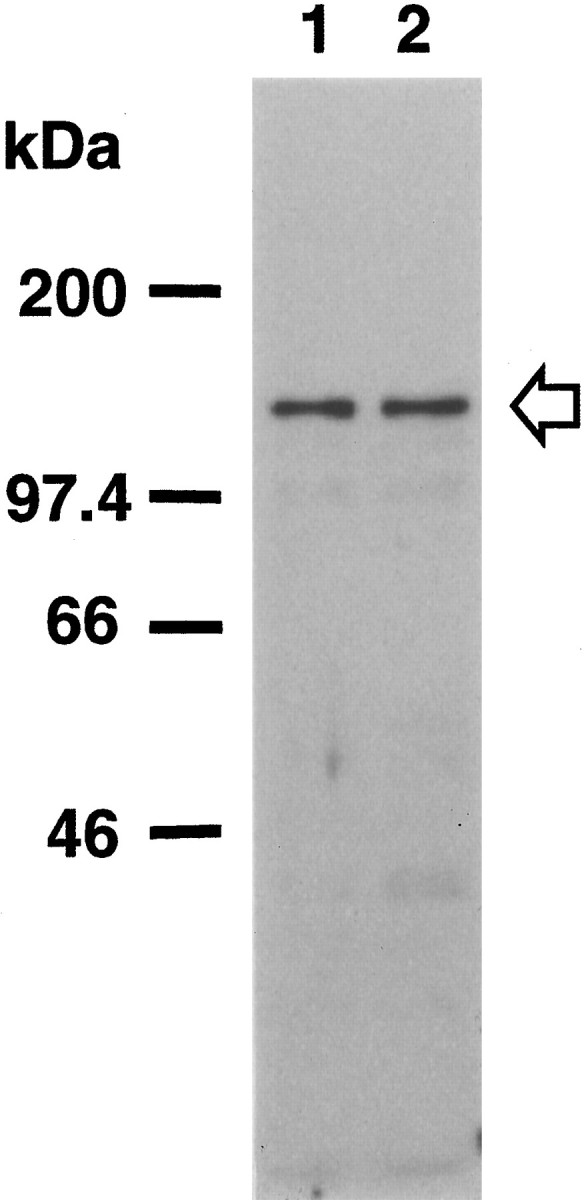
PKN expression in human brain. Total homogenates (50 μg of protein) of human brain tissue from the temporal or occipital cortices of control (lane 1) and AD cases (lane 2) were subjected to SDS-PAGE followed by immunoblotting. Immunostaining was performed with the antisera αC6 against PKN. The positions of marker proteins are indicated inkDa, and the position of PKN is indicated by anarrow.
Cellular localization of PKN in human control and AD brains
PKN was localized by immunocytochemistry at the light and electron microscopic levels. The expression of PKN was mostly ubiquitous, appearing in all layers of the neocortex, all subfields of hippocampus, and all brain regions examined. PKN immunoreactivity in control brains was virtually restricted to neurons, whereas some glia-like cells were immunostained in addition to surviving neurons in AD brains. In control brains, large neurons, such as pyramidal cells, were stained more intensely than small neurons, such as granule cells. In normal neurons, many coarse or fine granules positive for PKN were located in or around the nucleus, with a small number of the granules distributed in the proximal portion of dendrites or axons (Fig.2A).
Fig. 2.
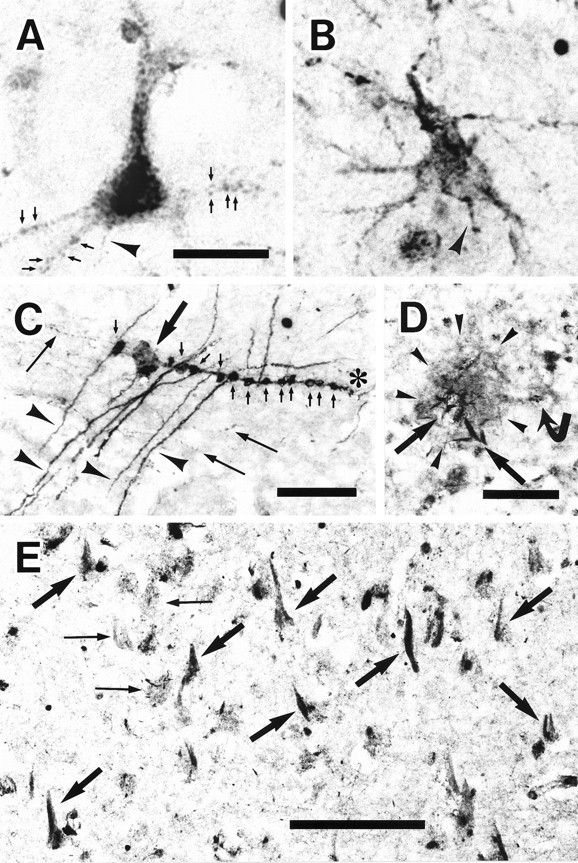
PKN distribution in human brain tissues.A, In a representative neuron from the angular cortex of a control case, PKN was concentrated in many small or large vesicles accumulating in the proximal dendrites and cell bodies, especially the nucleus and perinuclear region. The axon (arrowhead) was weakly stained. Some immunoreactive dots looked like synaptic boutons (arrows) on dendrites. B, Dendrites, axons, and the cytoplasm just under cell membranes were strongly immunostained in a surviving neuron in AD angular gyrus, whereas PKN immunoreactivity was slightly decreased in or around the nucleus. Axonal immunoreactivity (arrowhead) was greater than that in control neuron. C, In the molecular layer (Figure legend continues) of the dentate gyrus from AD hippocampus, some apical dendrites (arrowheads) positive for PKN made synapses (small arrows) on immunoreactive varicose fibers (asterisk). Many varicose and thin fibers (long arrows) were also stained for PKN, which was accumulated in the dystrophic axon (thick arrow). D, A senile plaque (SP) (demarcated by arrowheads) was weakly stained for PKN, but PKN was highly enriched in the degenerative neurites (thick arrows). Faintly stained cells with glial profiles surrounded the SP (curved arrow).E, Intracellular NFTs (thick arrows) were strongly immunolabeled for PKN, as were degenerative neurites and neuropil threads in the subiculum of AD hippocampus. Extracellular NFTs were not labeled (thin arrows). A andB are at the same magnification. Scale bars:A, 25 μm; C, D, 50 μm;E, 100 μm.
By contrast, additional or predominant redistribution of PKN to the periphery was seen in degenerating neurons in AD hippocampus and angular cortex (Fig. 2B). Because of such redistribution, PKN immunoreactivity in the distal portions of dendrites and axons was markedly upregulated in AD hippocampus (Fig.2C). PKN was vigorously accumulated in synaptic boutons and dystrophic axons. Degenerating neurites were intensely labeled for PKN within weakly positive SPs (Fig. 2D). Neuritic pathologies such as intracellular NFTs and degenerative neurites were decorated with PKN in AD hippocampus (Fig. 2E). No extracellular tangles were immunoreactive for PKN.
Subcellular localization of PKN in human control and AD neurons
Electron microscopic analysis of PKN-positive control neurons revealed that strong immunoreactivity was present mainly in the juxtanuclear, cytoplasmic, and dendroplasmic compartments, and in presynapses and postsynapses. Dense labeling was associated with ER, multivesicular bodies, microtubules, and many small vesicular structures, whereas weak immunoreaction was seen in the nucleoplasm (Fig. 3A). Dense PKN immunoreactivity was also detected in ribosomes and late endosomes and along the Golgi-secretory pathway (Fig. 3B). The membranes of these vesicles appeared highly immunoreactive for PKN. In synapses, the ER-related spine apparatus in the postsynapse was clearly labeled for PKN, as were many synaptic vesicles in the presynaptic bouton (Fig.3C). In a damaged AD neuron bearing a small NFT, straight or twisted tubules in the tangle were strongly stained for PKN and were decorated with numerous small vesicles intensely labeled for PKN (Fig.3D).
Fig. 3.
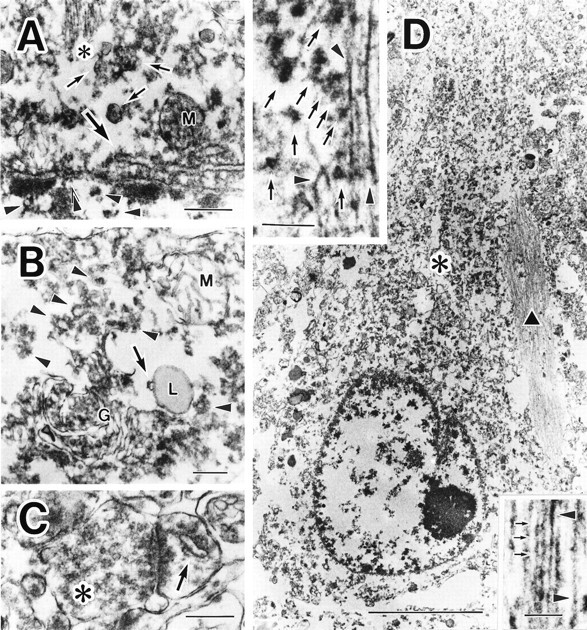
Subcellular localization of PKN in control and AD neurons. A, In control neuron, the antibody to PKN labeled nucleoplasm weakly (arrowheads) at high magnification. The nuclear envelope (double arrowheads), endoplasmic reticulum (thick arrow), multivesicular bodies (arrows), microtubules (asterisk), and small vesicles were moderately to intensely immunostained. Mitochondrial labeling (M) was not consistent: the mitochondria (M) inB was not immunoreactive for PKN. B,cis-/medial/trans-Golgi compartments (G) and trans-Golgi network, including secretory vesicles (arrowheads), were all strongly immunopositive. PKN was also found on many ribosomes. Note that a late endosome (arrow), fusing to primary lysosome (L), was immunolabeled for PKN. Lipofuscins were negative. C, In an asymmetric synapse, many synaptic vesicles in a presynaptic terminal (asterisk) were weakly immunoreactive; a spine apparatus (arrow) in the postsynapse was strongly stained for PKN. D, At lower magnification, many fine or coarse granular structures at the apical part of the perinuclear compartment (asterisk) and an intracellular NFT (black triangle) were intensely immunolabeled in a hippocampal pyramidal neuron. Top inset, Some of the abnormal filaments were markedly immunopositive for PKN (arrowheads), as were numerous vesicles 40–80 nm in diameter (arrows) surrounding the filamentous structures in a NFT. Bottom inset, Such filaments were composed of two types of tubules: straight tubules (arrowheads) and twisted tubules with a periodic constriction (small arrows) occurring every ∼80 nm, corresponding to paired helical filaments. Scale bars:A, 400 nm; B, C, 300 nm;D, 5 μm; top and bottom insets of D, 200 nm.
Intraneuronal accumulation of PKN in specific organelles
The intraneuronal colocalization of PKN with selected organelles and PHF pathology was also investigated with confocal laser microscopy. PKN immunoreactivity was distributed in a subset of ER, which in normal neurons appeared in the nuclear or perinuclear compartments and in the apical part of cell soma (Fig.4A). In degenerating AD neurons, these vesicles were redistributed in association with intracellular NFTs, whereas other ER-related vesicles negative for PKN showed no change in cellular distribution (Fig. 4B). PKN was also concentrated in fine or coarse vesicles labeled for cathepsin D around the nuclei (Fig. 4C). These results indicate that most of these vesicles are ER, late endosomes, or intermediates such as multivesicular bodies. Nonphosphorylated neurofilaments, phosphorylated neurofilaments, and MAP2 displayed no close association with PKN in AD neurons (data not shown).
Fig. 4.
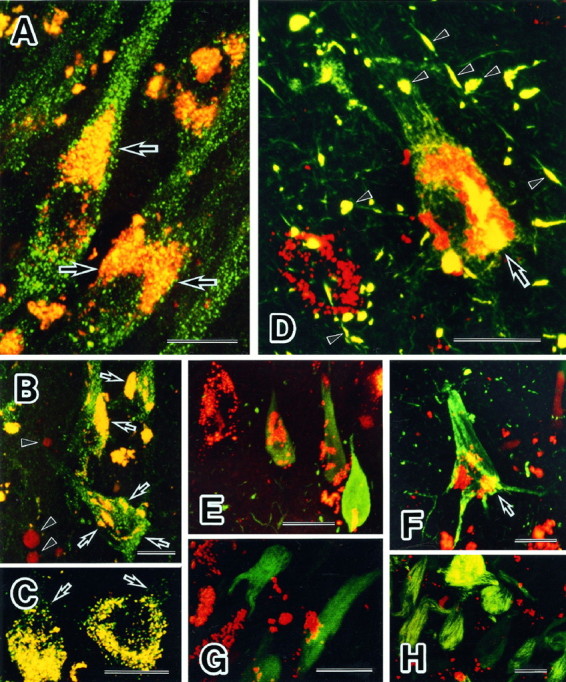
Colocalization of PKN with specific organelles and AD pathology in neurons. A, B, Double-labeling immunofluorescence microscopy demonstrating colocalization of PKN (red) and BiP (Grp-78, green). Numerous BiP-positive ER were scattered in cell bodies and apical dendrites of pyramidal neurons from a control hippocampus (A,green). A subset of these vesicles residing in the juxtanucleus and apical part of cell body and the proximal portion of apical dendrite was stained simultaneously for PKN and BiP (yellow, arrow inA). In tangle-bearing neurons in AD hippocampus, such doubly labeled vesicles were translocated and associated with intracellular tangles (arrows in B). Nuclear translocation of PKN was occasionally seen in AD degenerating neurons (arrowheads in B).C, Double immunolabeling for PKN (red) and cathepsin D, a candidate tau protease or APP secretase (green). Many small vesicles were doubly stained in the cytoplasm, especially in the perinuclear compartment of pyramidal neurons from AD hippocampus. Note that vesicles reactive for cathepsin D alone were near the cytoplasmic membrane (arrows), consistent with the report that cathepsin D is in early to late endosomes as well as in lysosomes in AD brain (Cataldo et al., 1997). D–H, Double immunofluorescent labeling of tangles for PKN (red) and NFT constituents (green). The AT8 monoclonal antibody recognized phosphoepitopes on tau in neuropil threads, degenerative neurites, and intracellular NFTs (D, green), which were decorated with many vesicles labeled for PKN (D, red). PKN and AT8 phosphoepitope were colocalized in filamentous structures within the cell soma (arrow) or within degenerative neurites (arrowheads). A similar pattern of filamentous structures within cell soma or degenerative neurites was seen in the staining with AT180 or AT270. PKN was localized in small vesicles within neurons overexpressing tau-1-positive nonphosphorylated tau (E, green). In F, PKN-containing vesicles (red) were seen within a vulnerable neuron overexpressing the Alz50 epitope (green). Some PKN-positive vesicles were seen very close to tau having an abnormal conformation (arrow in F). Few PKN-positive vesicles (G, red) were found on ubiquitinated intracellular NFTs (G, green). Extracellular NFTs labeled for complement protein C4d (H, green) were not associated with vesicles containing PKN (H, red). Scale bar: 20 μm in all panels.
Colocalization of PKN with abnormal tau in NFT
The association of PKN with developing NFTs was examined in AD hippocampus. In tangle-bearing neurons, PKN was localized in small vesicles clustering in association with the NFTs (Fig.4D,F). Moreover, PKN was colocalized within NFTs in fibrillar structures with abnormal tau having either the phosphoepitopes detected by AT8/180/270 antibodies (Fig.1D) or the intramolecular linkage recognized by antibody Alz50 (Carmel et al., 1996) (Fig. 4F). A tau-1-positive nonphosphorylated tau at Ser199 or Ser202 was highly expressed in a homogenous pattern in a subset of affected neurons, which were called pretangled neurons. In such damaged neurons, at the early stage of degeneration before NFT formation, PKN was densely packed in small vesicles (Fig. 4E), as it is in normal neurons (Fig. 4A). Ubiquitination of tau protein appears to be a late event in NFT development, because ubiquitination of PHFs occurs after the formation of NFTs (Bancher et al., 1991). At such a late stage, fewer PKN-containing vesicles were associated with the ubiquitinated form of NFTs (Fig. 4G). Extracellular ghost tangles, found after neuronal death, were decorated with the complement protein C4d (McGeer et al., 1989) but were not labeled for PKN (Fig. 4H).
Phosphorylation of tau protein by PKN
Recombinant PKN directly phosphorylated recombinant human tauin vitro, even in the absence of activator (Fig.5A,B). The phosphorylation was significantly potentiated by arachidonic acid, resulting in a decrease in the mobility on SDS-PAGE. The level of tau phosphorylation increased in a time-dependent manner up to ∼2.5 or 4.5 mol of Pi per monomer of tau protein in the absence or presence of 40 μmarachidonic acid (Fig. 5B). A large molecular weight shift took place concomitantly with the phosphorylation. In vivophosphorylation of tau was also found in the SK-N-MC human neuroblastoma cells expressing active forms of PKN, but not in the cells expressing an inactive variant of PKN (Fig. 5C). The tau phosphorylated in vitro by PKN was immunoreactive with tau-1 but not with AT8, AT180, or AT270 antibodies on immunoblot analysis (Fig. 5D), indicating that PKN did not phosphorylate the proline-directed phosphorylation sites recognized by these antibodies (Fig. 5E).
Fig. 5.

Phosphorylation of human tau protein by PKN. A, Phosphorylation was detected using autoradiography and SDS-PAGE. The white arrowheadindicates the position of autophosphorylation of recombinant PKN. One hundred nanograms of recombinant human tau were incubated with assay mixture without (lane 1) or with (lanes 2 and 3) recombinant PKN in the absence (lanes 1 and 2) or presence (lane 3) of 40 μm arachidonic acid. The reaction was terminated after 5 min, followed by separation on SDS-PAGE. Theblack arrowhead indicates the position of GST-tau fusion protein. Note the upper band of tau in lane 3, indicating molecular weight shift on more phosphorylation than inlane 2. B, Time course of tau phosphorylation by PKN. One hundred nanograms of GST-tau protein were incubated with recombinant PKN in the absence (closed circles or −AA) or presence (open circles or +AA) of arachidonic acid for 5 min (lane 1), 10 min (lane 2), 30 min (lane 3), 1 hr (lane 4), 2 hr (lane 5), 4 hr (lane 6), 6 hr (lane 7), 8 hr (lane 8), and 10 hr (lane 10). Mol of Pi incorporated into a mol of tau was calculated from the radioactivity quantified with an image analyzer.Arrows indicate the time point when 50 ng of additional PKN was applied in the assay mixture. C, In vivo phosphorylation of tau in human neuroblastoma SK-N-MC cells transfected with vector only or PKN transgenes. After immunoprecipitation with tau-1 antibody, intense phosphorylation signal was seen in the cells expressing active PKN (+aPKN, lane 2), but not in those expressing no (V, lane 1) or inactive PKN (+iPKN, lane 3). D, Immunoblot analysis with tau-1 (lanes 1 and2), AT8 (lane 3), AT180 (lane 4), and AT270 (lane 5) antibodies of recombinant tau phosphorylated by PKN in the absence (lane 1) or presence (lanes 2–5) of ATP. Note that tau-1 recognizes PKN-phosphorylated tau (lane 2), which does not react with AT8, AT180, or AT270 (lanes 3–5).E, Diagram of human tau isoform used. The four repeats of microtubule-binding domain in the C terminus are numbered 1–4 (closed circle). Epitopes of a couple of phosphorylation-dependent antibodies (open circles), a dephosphorylation-dependent antibody (open rectangle), and a conformation-dependent antibody (solid line) are depicted with their names initalic.
DISCUSSION
The present study demonstrates that PKN phosphorylates tau protein and that neuronal PKN is highly enriched in a specific organelle, ER, around nucleus and is redistributed to ER-derived vesicles associated with Alzheimer NFTs in AD-affected neurons.
PKN directly phosphorylates tau
PKN phosphorylates human tau protein both in vitro andin vivo. PKC, which has a catalytic domain homologous to the corresponding region of PKN and has been implicated in a broad spectrum of neuronal functions (Tanaka and Nishizuka, 1994), phosphorylates tau at Ser324 in the third repeat of the microtubule-binding domainin vitro (Correas et al., 1992). p110mark(MARK), one of the other serine/threonine kinases phosphorylating tau, does so in vitro and in vivo at Ser262 in the first repeat (Drewes et al., 1995, 1997), resulting in disruption of the microtubule array followed by morphological changes and cell death (Drewes et al., 1997). The sites of phosphorylation by PKN remain be determined. Phosphorylation of the microtubule-binding region of tau may play a pivotal role in the aggregation of tau into PHFs, because tau not bound to microtubules may be sequestered and polymerized and the microtubule-binding domains compose the framework of PHFs (Ksiezak-Reding and Yen, 1991; Wille et al., 1992). PKN, a PKC-related kinase 1 (PRK1), located around NFTs (Figs. 2-4) may inhibit the interaction of tau with microtubules and give rise to monomeric or polymeric tau molecules available for PHF formation.
PKN is enriched in a subset of granular ER in human neurons
Recent studies have indicated that ER may be particularly important in AD pathology (Cook et al., 1997; Hartmann et al., 1997;Yan et al., 1997). Presenilins 1 and 2, whose mutations cause most early onset familial AD, are localized to the ER or early Golgi compartments (Kovacs et al., 1996) and have been suggested to interact directly with amyloid precursor protein (APP) in the ER (Xia et al., 1997). The ER-derived vesicles reactive for PKN were localized in the juxtanuclear compartment in control neurons, whereas they were mainly redistributed to the periphery of the cytoplasm or to neurites and were associated with intracellular NFTs in damaged AD neurons (Figs.2A,B, 4A,B). This is the first evidence for the possible involvement of selected ER containing PKN in NFT formation.
The sorting or processing of APP and Aβ generation have been suggested to occur not only in the ER but also in the Golgi body, secretary pathway, synaptic vesicles, and endosomal–lysosomal system. Most of these organelles were also immunolabeled for PKN (Figs.3B,C, 4C). Thus, PKN might be involved in the dysfunctions of various organelles within degenerating AD neurons.
PKN is closely associated with NFTs at an early stage of their development
Tau protein, normally distributed in axons, shows translocation to cell bodies and dendrites in AD-affected neurons (Kowall and Kosik, 1987). NFTs are frequently seen in the perinuclear region, closely encircling the nuclei, and in proximal dendrites in tangle-bearing neurons. An inverse change was observed in the distribution of PKN. PKN, normally concentrated in the juxtanuclear compartment, was redistributed predominantly to distal compartments or NFTs in AD degenerating neurons, indicating likely sites for PKN to interact with tau, possibly leading to PHF formation (Figs. 2B–E,3D, 4B–F). Colocalization of PKN with abnormal tau in NFT at an early stage provides the first evidence for the association with tangles in AD neurons of a nonproline-directed kinase that is directly activated by lipid messengers, including arachidonic acid. Although PKC has a homologous catalytic domain to PKN, no NFT has been noted to be reactive for PKC. The structural difference in the regulatory domain between PKN and PKC may account for such distinct localization in AD brains. The colocalization of PKN with phosphorylated tau at proline-directed sites may indicate its involvement in the regulation of proline-directed kinases working on tau (Fig. 4D). The normal distribution of PKN in the perikarya and dendrites (Fig. 2A) may contribute to the sorting of nonphosphorylated tau into axons by a regulation of its ability to bind to microtubules. The altered localization of PKN in AD axons (Fig. 2B,C) may affect microtubule dynamics, leading to dysfunctions in axonal transport and synaptic transmission and inevitably to neuritic degeneration.
Redistribution of PKN may also affect the sequential or multiple phosphorylation of tau by other kinases, leading to the PHF formation, because it has been suggested that the hyperphosphorylation of tau precedes its polymerization into PHF (Bancher et al., 1991). Recently a conformational alteration of tau, an intramolecular linkage detected by Alz50 (Wolozin et al., 1986) between the N terminus and the third microtubule-binding domain of tau, has also been found to precede the progressive modification and polymerization into abnormal filaments (Carmel et al., 1996; Jicha et al., 1997). PKN was closely associated with abnormal tau immunoreactive with Alz50 (Fig.4F), indicating the possible involvement of PKN in this conformational change of tau.
PHFs are seen not only in NFTs but also inside the nuclei (Metuzals et al., 1988), where tau exits in normal neurons, indicating a role for tau in the production or function of ribosomes (Loomis et al., 1990). PKN is also found in the neuronal nuclei in postmortem human brains (Figs. 2A,B, 3A,D, 4B), which is consistent with the translocation of PKN under stress from the cytosol to the nucleus in cultured fibroblasts (Mukai et al., 1996b). These findings may indicate that PKN participates in signal transduction to the nucleus in neurons.
Remaining neurons were less in number but displayed higher immunoreactivity for PKN in AD than in control brains (Fig. 2). Indeed, the protein level of PKN was approximately the same in control and AD brain tissues on immunoblot analysis (Fig. 1), suggesting that PKN accumulates in surviving AD neurons. Although most members of the PKC family are activated by arachidonic acid at the high concentration of 100–600 μm (Kitagawa et al., 1995), the activity of PKN is increased 2- to 18-fold by the lower, possibly physiological concentration of arachidonate (7–40 μm), release of which is induced by phospholipase A2 during phospholipid hydrolysis. Excess release of arachidonic acid might occur in AD brain, resulting in PKN activation, because marked increases have been reported in the amount or activity of phospholipase A2 and in the levels of prostaglandins, which are arachidonate metabolites (Farooqui et al., 1997). It is possible that overstimulation of PKN contributes to neurodegeneration in AD brains.
Thus, the phosphorylation of tau by PKN may play crucial roles in AD pathology by regulating the biological functions of tau in modulating neurite outgrowth and ribosomal function, as well as by modulating various cellular functions based on cytoskeletal responses to extracellular signals. The translocation of PKN associated with a subset of ER-derived vesicles toward NFTs may provide a molecular linkage between the ER, an organelle increasingly recognized as a key regulator in AD pathology, and PHF formation in degenerating AD neurons.
Footnotes
We are grateful to Dr. Hiroshi Mori and Dr. Peter Davies for the generous gifts of tau cDNA and Alz50 monoclonal antibody, respectively. We also thank A. Hori, Y. Tokunaga, and M. Sumida for excellent technical assistance.
Correspondence should be addressed to Dr. Toshio Kawamata, Hyogo Institute for Aging Brain and Cognitive Disorders, 520 Saisho-Koh, Himeji 670, Japan.
REFERENCES
- 1.Alafuzoff I, Iqbal K, Friden H, Adolfsson R, Winblad B. Histopathological criteria for progressive dementia disorders: clinico-pathological correlation and classification by multivariate data analysis. Acta Neuropathol (Berl) 1987;74:209–225. doi: 10.1007/BF00688184. [DOI] [PubMed] [Google Scholar]
- 2.Amano M, Mukai H, Ono Y, Chihara K, Matsui T, Hamajima Y, Okawa K, Iwamatsu A, Kaibuchi K. Identification of a putative target for Rho as the serine-threonine kinase protein kinase N. Science. 1996;271:648–650. doi: 10.1126/science.271.5249.648. [DOI] [PubMed] [Google Scholar]
- 3.Bancher C, Grundke-Iqbal I, Iqbal K, Fried VA, Smith HT, Wisniewski HM. Abnormal phosphorylation of tau preceded ubiquitination in neurofibrillary pathology of Alzheimer disease. Brain Res. 1991;539:11–18. doi: 10.1016/0006-8993(91)90681-k. [DOI] [PubMed] [Google Scholar]
- 4.Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997;323:577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz50’s selectivity for Alzheimer’s disease pathology. J Biol Chem. 1996;271:32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- 6.Cataldo AM, Barnett JL, Pieroni C, Nixon RA. Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer’s disease: neuropathologic evidence for a mechanism of increased β-amyloidogenesis. J Neurosci. 1997;17:6142–6151. doi: 10.1523/JNEUROSCI.17-16-06142.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM-Y, Doms RW. Alzheimer’s Aβ(1–42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med. 1997;3:1021–1023. doi: 10.1038/nm0997-1021. [DOI] [PubMed] [Google Scholar]
- 8.Correas I, Diaz-Nido J, Avila J. Microtubule-associated protein tau is phosphorylated by protein kinase C on its tubulin binding domain. J Biol Chem. 1992;267:15721–15728. [PubMed] [Google Scholar]
- 9.Declaration of Helsinki. (1984) The main issue in bioethics, revised edition (Varga AC, ed). New York: Paulist.
- 10.Drewes G, Trinczek B, Illenberger S, Biernat J, Schmitt-Ulms G, Meyer HE, Mandelkow E-M, Mandelkow E. Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark): a novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J Biol Chem. 1995;270:7679–7688. doi: 10.1074/jbc.270.13.7679. [DOI] [PubMed] [Google Scholar]
- 11.Drewes G, Ebneth A, Preuss U, Mandelkow E-M, Mandelkow E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell. 1997;89:297–308. doi: 10.1016/s0092-8674(00)80208-1. [DOI] [PubMed] [Google Scholar]
- 12.Drubin DG, Kirschner MW. Tau protein function in living cells. J Cell Biol. 1986;103:2739–2746. doi: 10.1083/jcb.103.6.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Farooqui AA, Yang H-C, Rosenberger TA, Horrocks LA. Phospholipase A2 and its role in brain tissue. J Neurochem. 1997;69:889–901. doi: 10.1046/j.1471-4159.1997.69030889.x. [DOI] [PubMed] [Google Scholar]
- 14.Federal policy for the protection of human subjects: notices and rules. (1991) Federal Register, Vol 56, No. 117, pp 28002–28007. [PubMed]
- 15.Hartmann T, Bieger SC, Brühl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K. Distinct sites of intracellular production for Alzheimer’s disease Aβ40/42 amyloid peptides. Nat Med. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- 16.Jicha GA, Lane E, Vincent I, Otvos L, Jr, Hoffmann R, Davies P. A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer’s disease. J Neurochem. 1997;69:2087–2095. doi: 10.1046/j.1471-4159.1997.69052087.x. [DOI] [PubMed] [Google Scholar]
- 17.Johnson GV. Differential phosphorylation of tau by cyclic AMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase II: metabolic and functional consequences. J Neurochem. 1992;59:2056–2062. doi: 10.1111/j.1471-4159.1992.tb10094.x. [DOI] [PubMed] [Google Scholar]
- 18.Kawamata T, Tooyama I, Yamada T, Walker DG, McGeer PL. Lactotransferrin immunocytochemistry in Alzheimer and normal human brain. Am J Pathol. 1993;142:1574–1585. [PMC free article] [PubMed] [Google Scholar]
- 19.Kitagawa M, Mukai H, Shibata H, Ono Y. Purification and characterization of a fatty acid-activated protein kinase (PKN) from rat testis. Biochem J. 1995;310:657–664. doi: 10.1042/bj3100657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kovacs DM, Fausett HJ, Page KJ, Kim TW, Moir RD, Merriam DE, Hollister RD, Hallmark OG, Mancini R, Felsenstein KM, Hyman BT, Tanzi RE, Wasco W. Alzheimer-associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- 21.Kowall NW, Kosik KS. Axonal disruption and aberrant localization of tau protein characterize the neuropil pathology of Alzheimer’s disease. Ann Neurol. 1987;22:639–643. doi: 10.1002/ana.410220514. [DOI] [PubMed] [Google Scholar]
- 22.Ksiezak-Reding H, Yen S-H. Structural stability of paired helical filaments requires microtubule-binding domains of tau: a model for self-association. Neuron. 1991;6:717–728. doi: 10.1016/0896-6273(91)90169-z. [DOI] [PubMed] [Google Scholar]
- 23.Liu WK, Williams RT, Hall FL, Dickson DW, Yen SH. Detection of a Cdc2-related kinase associated with Alzheimer paired helical filaments. Am J Pathol. 1995;146:228–238. [PMC free article] [PubMed] [Google Scholar]
- 24.Loomis PA, Howard TH, Castleberry RP, Binder LI. Identification of nuclear tau isoforms in human neuroblastoma cells. Proc Natl Acad Sci USA. 1990;87:8422–8426. doi: 10.1073/pnas.87.21.8422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsuzawa K, Kosako H, Inagaki N, Shibata H, Mukai H, Ono Y, Amano M, Kaibuchi K, Matsuura Y, Azuma I, Inagaki M. Domain-specific phosphorylation of vimentin and glial fibrillary acidic protein by PKN. Biochem Biophys Res Commun. 1997;234:621–625. doi: 10.1006/bbrc.1997.6669. [DOI] [PubMed] [Google Scholar]
- 26.McGeer PL, Akiyama H, Itagaki S, McGeer EG. Immune system response in Alzheimer’s disease. Can J Neurol Sci. 1989;16:516–527. doi: 10.1017/s0317167100029863. [DOI] [PubMed] [Google Scholar]
- 27.Metuzals J, Robitaille Y, Susan H, Gauthier S, Leblanc R. Paired helical filaments and the cytoplasmic-nuclear interface in Alzheimer’s disease. J Neurocytol. 1988;17:827–833. doi: 10.1007/BF01216709. [DOI] [PubMed] [Google Scholar]
- 28.Mori H, Hamada Y, Kawaguchi M, Honda T, Kondo J, Ihara Y. A distinct form of tau is selectively incorporated into Alzheimer’s paired helical filaments. Biochem Biophys Res Commun. 1989;159:1221–1226. doi: 10.1016/0006-291x(89)92240-7. [DOI] [PubMed] [Google Scholar]
- 29.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, Ihara Y. Proline-directed and non-proline directed phosphorylation of PHF-tau. J Biol Chem. 1995;270:823–829. doi: 10.1074/jbc.270.2.823. [DOI] [PubMed] [Google Scholar]
- 30.Mukai H, Ono Y. A novel protein kinase with leucine zipper-like sequences: its catalytic domain is highly homologous to that of protein kinase C. Biochem Biophys Res Commun. 1994;199:897–904. doi: 10.1006/bbrc.1994.1313. [DOI] [PubMed] [Google Scholar]
- 31.Mukai H, Kitagawa M, Shibata H, Takanaga H, Mori K, Shimakawa M, Miyahara M, Hirao K, Ono Y. Activation of PKN, a novel 120-kDa protein kinase with leucine zipper-like sequences, by unsaturated fatty acids and by limited proteolysis. Biochem Biophys Res Commun. 1994;204:348–356. doi: 10.1006/bbrc.1994.2466. [DOI] [PubMed] [Google Scholar]
- 32.Mukai H, Toshimori M, Shibata H, Kitagawa M, Shimakawa M, Miyahara M, Sunakawa H, Ono Y. PKN associates and phosphorylates the head-rod domain of neurofilament protein. J Biol Chem. 1996a;271:9816–9822. doi: 10.1074/jbc.271.16.9816. [DOI] [PubMed] [Google Scholar]
- 33.Mukai H, Miyahara M, Sunakawa H, Shibata H, Toshimori M, Kitagawa M, Shimakawa M, Takanaga H, Ono Y. Translocation of PKN from the cytosol to the nucleus induced by stresses. Proc Natl Acad Sci USA. 1996b;93:10195–10199. doi: 10.1073/pnas.93.19.10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mukai H, Toshimori M, Shibata H, Takanaga H, Kitagawa M, Miyahara M, Shimakawa M, Ono Y. Interaction of PKN with α-actinin. J Biol Chem. 1997;272:4740–4746. doi: 10.1074/jbc.272.8.4740. [DOI] [PubMed] [Google Scholar]
- 35.Ono Y, Fujii T, Ogita K, Kikkawa U, Igarashi K, Nishizuka Y. The structure, expression, and properties of additional members of the protein kinase C family. J Biol Chem. 1988;263:6927–6932. [PubMed] [Google Scholar]
- 36.Pei J-J, Tanaka T, Tung Y-C, Braak E, Iqbal K, Grundke-Iqbal I. Distribution, levels, and activity of glycogen synthase kinase-3 in the Alzheimer disease brain. J Neuropathol Exp Neurol. 1997;56:70–78. doi: 10.1097/00005072-199701000-00007. [DOI] [PubMed] [Google Scholar]
- 37.Shibata H, Mukai H, Inagaki Y, Homma Y, Kimura K, Kaibuchi K, Narumiya S, Ono Y. Characterization of the interaction between RhoA and the amino-terminal region of PKN. FEBS Lett. 1996;385:221–224. doi: 10.1016/0014-5793(96)00385-7. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka C, Nishizuka Y. The protein kinase C family for neuronal signaling. Annu Rev Neurosci. 1994;17:551–567. doi: 10.1146/annurev.ne.17.030194.003003. [DOI] [PubMed] [Google Scholar]
- 39.Trojanowski JQ, Mawal-Dewan M, Schmidt ML, Martin J, Lee VM. Localization of the mitogen activated protein kinase ERK2. Brain Res. 1993;618:333–337. doi: 10.1016/0006-8993(93)91286-2. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe G, Saito Y, Madaule P, Ishizaki T, Fujisawa K, Morii N, Mukai H, Ono Y, Kakizuka A, Narumiya S. Protein kinase N (PKN) and PKN-related protein rhophilin as targets of small GTPase Rho. Science. 1996;271:645–648. doi: 10.1126/science.271.5249.645. [DOI] [PubMed] [Google Scholar]
- 41.Wille H, Drewes G, Biernat J, Mandelkow E-M, Mandelkow E. Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J Cell Biol. 1992;118:573–584. doi: 10.1083/jcb.118.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolozin BL, Pruchnicki A, Dickson DW, Davies P. A neuronal antigen in the brains of patients with Alzheimer’s disease. Science. 1986;232:648–650. doi: 10.1126/science.3083509. [DOI] [PubMed] [Google Scholar]
- 43.Xia W, Zhang J, Perez R, Koo EH, Selkoe DJ. Interaction between amyloid precursor protein and presenilins in mammalian cells: implications for the pathogenesis of Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:8208–8213. doi: 10.1073/pnas.94.15.8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamaguchi H, Ishiguro K, Uchida T, Takashima A, Lemere CA, Imahori K. Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3β and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol. 1996;92:232–241. doi: 10.1007/s004010050513. [DOI] [PubMed] [Google Scholar]
- 45.Yan SD, Fu J, Soto C, Chen X, Zhu H, Al-Mohanna F, Collison K, Zhu A, Stern E, Saido T, Tohyama M, Ogawa S, Roher A, Stern D. An intracellular protein that binds amyloid-β peptide and mediates neurotoxicity in Alzheimer’s disease. Nature. 1997;389:689–695. doi: 10.1038/39522. [DOI] [PubMed] [Google Scholar]