

Abstract
Spreading depression (SD) was analyzed in hippocampal and neocortical brain slices by imaging intrinsic optical signals in combination with either simultaneous electrophysiological recordings or imaging of intracellular calcium dynamics. The goal was to determine the roles of intracellular calcium (Ca2+int) waves in the generation and propagation of SD. Imaging of intrinsic optical signals in the hippocampus showed that ouabain consistently induced SD, which characteristically started in the CA1 region, propagated at 15–35 μm/sec, and traversed across the hippocampal fissure to the dentate gyrus. In the dendritic regions of both CA1 and the dentate gyrus, SD caused a transient increase in light transmittance, characterized by both a rapid onset and a rapid recovery. In contrast, in the cell body regions the transmittance increase was prolonged. Simultaneous imaging of intracellular calcium and intrinsic optical signals revealed that a slow Ca2+int increase preceded any change in transmittance. Additionally, a wave of increased Ca2+int typically propagated many seconds ahead of the change in transmittance. These calcium increases were also observed in individual astrocytes injected with calcium orange, indicating that Ca2+int waves were normally associated with SD. However, when hippocampal slices were incubated in calcium-free/EGTA external solutions, SD was still observed, although Ca2+int waves were completely abolished. Under these conditions SD had a comparable peak increase in transmittance but a slower onset and a faster recovery. These results demonstrate that although there are calcium dynamics associated with SD, these increases are not necessary for the initiation or propagation of spreading depression.
Keywords: calcium waves, astrocytes, spreading depression, intrinsic optical signals, ouabain, hippocampus, migraine
Spreading depression (SD) was first described in cortical tissue >50 years ago (Leao, 1944). SD is classically characterized as a slowly propagating wave of neuronal and astrocytic depolarization that results in a transient depression of synaptic transmission (Nicholson and Kraig, 1981; Somjen et al., 1992). SD propagates radially at 30–50 μm/sec from its initiation site and is associated with a redistribution of ions, shrinkage of the extracellular space, and an increase in energy metabolism (Nicholson and Kraig, 1981). Typically cells recover and are relatively unchanged after SD. However, ischemic insult can induce SD waves (Nedergaard and Hansen, 1993; Dietrich et al., 1994), and the extent of the resultant damage is increased with each incidence of SD (Mies et al., 1993). The underlying mechanisms of SD are still unknown.
Recent observations of the interactions between astrocytes and neurons in cell culture have suggested a novel explanation for the mechanism of SD initiation and propagation. Astrocytes exhibit waves of increased intracellular calcium that can propagate through gap junction-coupled networks of astrocytes. Several laboratories have shown that an increase in intracellular calcium in cultured glial cells can trigger an increase in intracellular calcium in neighboring neurons (Charles, 1994; Nedergaard, 1994; Parpura et al., 1994; Hassinger et al., 1995). These results suggest that complex and dynamic interactions can occur between glia and neurons. Furthermore, astrocytic calcium waves have many characteristics that are similar to SD (Nedergaard et al., 1995). For example, SD propagates at approximately the same rate as astrocytic calcium waves (Hansen, 1985; Cornell Bell et al., 1990), and both SD and astrocytic calcium waves appear to depend on functional gap junction communication (Finkbeiner, 1992; Martins-Ferreira and Ribeiro, 1995; Nedergaard et al., 1995). This has led to the hypothesis that astrocytic calcium waves play an integral role in SD (Cornell Bell and Finkbeiner, 1991; Nedergaard, 1994).
To address the role of intracellular calcium waves in the initiation and propagation of SD, we have examined SD in hippocampal and neocortical slices using a combination of electrophysiology, intrinsic optical imaging, and fluorescence microscopy. Imaging of intrinsic optical signals provides an excellent approach for examining SD because the large changes in light transmittance in brain slices caused by SD are easily detected, and the ability to visualize SD in an entire brain slice allows for the spatial and temporal mapping of SD propagation. We have designed a system for the simultaneous imaging of both intrinsic optical signals and Ca2+int dynamics. In the following study we have quantified the spatiotemporal intrinsic optical, electrophysiological, and Ca2+int events that occur during SD. We demonstrate that slow Ca2+int elevations precede a robust calcium wave that occurs concomitantly with SD. However, these calcium dynamics can be abolished by the removal of external calcium, without blocking SD. Therefore, although calcium waves are associated with SD, it is unlikely that a calcium wave in glial cells is critical for the successful initiation or propagation of SD.
MATERIALS AND METHODS
Slices. Hippocampal or neocortical slices (400 μm) were prepared from 15- to 25-d-old Sprague Dawley rats. Slices were maintained in artificial CSF (aCSF) aerated with 95% O2 and 5% CO2 for a minimum of 1 hr after preparation before experiments were performed. For calcium-imaging experiments, slices were placed on filter paper suspended over a solution of continuously stirring 10 μm fura-2 AM and aCSF in an enclosed interface chamber that was saturated with a humidified 95% O2 and 5% CO2 environment at a temperature of 25–30°C. To increase fura-2 AM loading efficiency, the aCSF was supplemented with an additional 10 mm glucose. The slices were maintained in fura-2 AM and aCSF for 2.5 hr and then transferred to room temperature (20–23°C) aCSF aerated with 95% O2and 5% CO2 until used. For all experiments the slices were transferred to a superfusion chamber mounted to the imaging setup described below. Slices were maintained at 33–34°C and held in place with platinum wires during the experiment. In experiments with zero-calcium aCSF (0-Ca2+ aCSF), slices were loaded with fura-2 AM in regular calcium aCSF as described above. Experiments were alternately performed on slices that were either in 0-Ca2+ aCSF or regular calcium aCSF. We only used data from slices in 0-Ca2+ aCSF when the matched controls showed robust Ca2+ waves in regular calcium aCSF. This ensured that any lack of a response in a 0-Ca2+ aCSF was not attributable to poor slice quality. We also ensured that there was adequate dye-loading of cells in each slice that was used. Before the onset of an experiment, slices were examined at both 365 and 380 nm fura-2 excitation to ensure that robust fura-2 dye-loading had occurred. This was evident by the easy identification of individually labeled fluorescent cell bodies. Any slice that did not show robust loading was discarded.
Imaging. Two independent imaging systems were used to simultaneously acquire intrinsic optical signals and fura-2 fluorescence signals (Fig. 1). The intrinsic optical system was composed of a charge-coupled device (CCD; model 4982, COHU) camera connected to an 8-bit frame grabber (model DT3155; Data Translations) that was driven by Axon Imaging Workbench (AIW, version 2.1; Axon Instruments, Foster City, CA). The illumination source was a standard Zeiss tungsten bulb whose output was directed through a 750DF20 discriminating filter. The fura-2 system used an image intensifier (VideoScope model VS-2525) coupled to a CCD camera (model COHU 4982) that was connected to an Axon Image Lightning 2000 frame grabber operating in 8-bit mode and driven by Axon Imaging Workbench. A lambda-10 filter wheel (Sutter Instruments) equipped with model 365HT15 and 380HT15 filters was used to provide alternating excitation for ratiometric fura-2 measurements. To maintain temporal synchronization between these two systems, a “master–slave” configuration was established using transistor–transistor logic (TTL) pulses generated by AIW and transmitted over the parallel ports of each computer. Typically four frame averages were acquired. This approach allowed the visualization of SD at a sampling frequency of 1 Hz, which was sufficiently fast given the relatively slow propagation rate of SD.
Fig. 1.

Schematic of the acquisition setup. Independent acquisition systems permitted simultaneous imaging of the intrinsic optical signals and fura-2-based intracellular calcium dynamics, as well as extracellular field potentials. Slices were continuously transilluminated with 750 nm light to monitor intrinsic optical signals and alternately epi-illuminated by 365 or 380 nm light when calcium images were acquired. The short-pass (<650 nm) dichroic and short-pass (<630 nm) barrier filter ensured that the intensified CCD only detected light from the fura-2 signal. An additional long-pass (>650 nm) filter on the intrinsic optical acquisition system ensured that there was no crossover of the fura-2 signal into the intrinsic optical channel. In some cases, extracellular field potentials were recorded. Typically the intrinsic optical computer provided the synchronizing trigger outputs to ensure temporal synchrony with the rest of the acquisition.
The optical path shown in Figure 1 demonstrates how the intrinsic optical signal was isolated from the fura-2 fluorescence signal. A 650 nm short-pass dichroic mirror reflected longer wavelengths to the intrinsic optical detection system and passed the shorter wavelengths to the fura-2 detection system in which an additional 630 nm short-pass barrier filter ensured no optical crossover from the intrinsic optical signals. The intrinsic optical signals were recorded and presented as subtracted images, with the first image acquired during acquisition serving as the reference image, which was then subtracted from all subsequent images during acquisition. The intrinsic optical signals were acquired at a frequency of 1 Hz but saved to disk at a variable frequency of between 0.008 and 1.0 Hz to reduce data storage requirements. Ratiometric images were initially acquired at 0.016 Hz, and this rate was increased to 1 Hz before the onset of SD. This configuration allowed for the continuous monitoring of changes in the intrinsic optical signal, yet minimized photobleaching and phototoxicity because synchronous ratiometric images were acquired at a higher rate only when necessary.
One caveat of using these two independent imaging systems was that the two images in the separate systems were not precisely aligned because of differences in the light path and detection systems. However, this was compensated for during analysis by aligning given regions of interest before intensity measurements were made. To achieve this alignment, an image that contained three easily identifiable reference points on the imaging coverslip was acquired on both imaging systems. Using these three reference points, custom software was used to mathematically zoom, rotate, and translate the coordinates of the individual regions of interest on the intrinsic optical signal into the appropriate coordinates for the fura-2 images.
In some experiments astrocytes were intracellularly injected with the long wavelength calcium indicator calcium orange, according to the methods described by Duffy and MacVicar (1995). It should be noted that when using either fura-2 or Calcium Orange, calcium measurements can only be obtained from cells close to the surface of the slice (∼75–100 μm deep) because of poor penetration of light at these excitation wavelengths. In contrast, intrinsic optical signals at 750 nm are sensitive to changes throughout the entire slice.
Electrophysiology. Standard extracellular recording techniques were used to measure the direct current field potentials during spreading depression. Microelectrodes were fabricated from 1.5 mm outer diameter borosilicate glass and typically had resistances of 15–20 MΩ when filled with 3 m NaCl. Potentials were measured with a dual channel NeuroData intracellular amplifier (model IR-283). To reduce noise, a differential recording configuration was used with one electrode in the bath and a second electrode embedded into the slice, and the preparation was grounded through a World Precision Instruments (Sarasota, FL) reference electrode (model DRIREF-2SH). Both electrode signals were fed into a Frequency Devices model 901F Bessel filter unit to generate a third differential signal that was filtered at 1 kHz. Both unfiltered electrode potentials and the filtered differential signal were then digitized at 16.67 Hz using Axoscope (version 1.1, Axon Instruments). To ensure spatial synchrony between the field potential recordings and the imaging measurements, a single frame was acquired at the end of each experiment to reveal the precise location of the electrode. This location was then used to define a region that was used for measurements of the acquired intrinsic optical signals. Temporal synchrony for the simultaneous electrophysiology and imaging was maintained with TTL triggers from the imaging system driving the electrophysiological acquisition.
Solutions. Regular aCSF contained (in mm) NaCl, 124; KCl, 5; MgCl2, 1.3; CaCl2, 2; glucose, 10; and NaHCO3, 26.2. For the zero-calcium aCSF, calcium was replaced with magnesium and 2 mm EGTA was added, yielding a 0-Ca2+aCSF that contained (in mm) NaCl, 114; KCl, 5; MgCl2, 3.3; glucose, 10; NaHCO3, 26.2, and EGTA, 2. The pH was adjusted to 7.37 for both of these solutions. For extracellular recordings the microelectrode was filled with 3 m NaCl. A stock solution of 10 mm fura-2 AM was prepared daily by sonication in a solution of pluronic F-127 acid (25% w/w), and DMSO and was used at a final fura-2 AM concentration of 10 μm.
Statistics. Unless otherwise stated, all statistics were performed with the Mann–Whitney U test. GB-STAT version 3.53 (Dynamic Microsystems) was used for all statistical calculations.
RESULTS
Imaging hippocampal spreading depression
Spreading depression was initiated by bath application of 100 μm ouabain, dihydro-ouabain, elevations in external potassium, temperature elevations, or through electrical stimulation. The most consistent stimulus for induction of SD was ouabain, and all results presented are from ouabain-induced SD. Figure2A illustrates the time course of changes in the intrinsic optical signal during a typical ouabain-induced SD. An increase in the intrinsic optical signal, measured as a change in transmittance (ΔT), represents an increase in tissue volume, or cell swelling (Holthoff and Witte, 1996). Therefore, SD is detected optically as a result of dynamic changes in cellular volume. Spreading depression typically initiated in area CA1 and propagated throughout this region at a velocity of 16.2 ± 2.9 μm/sec (n = 7, mean ± SEM) and through the dentate gyrus at 19.3 ± 2.4 μm/sec (n = 8) (Fig. 2). Changes in the extracellular field potential were consistent with classical SD (Nicholson and Kraig, 1981) and were tightly correlated with changes in the intrinsic optical signal (Fig.2C). In the hippocampus, the increase in transmittance in the stratum radiatum of CA1 and the molecular layer of the dentate gyrus was transient, whereas stratum pyramidale of the CA1 and the granule cell layer of the dentate gyrus showed a sustained increase.
Fig. 2.
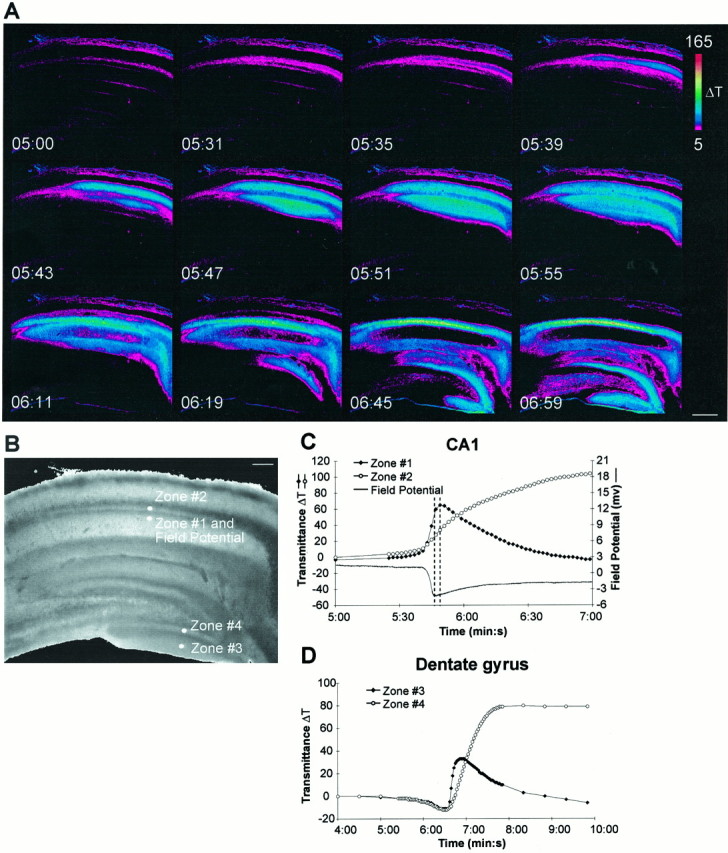
Imaging spreading depression in the hippocampus.A, Intrinsic optical signals during superfusion of regular ASCF containing 100 μm ouabain. Unless otherwise noted, ouabain addition was started at 1 min 30 sec in all experiments. Note the initial transient increase in stratum radiatum of CA1 that propagates throughout the entire slice. B, Demarcation of the areas that were used for measurements. C,D, Time course of the intrinsic optical signal and extracellular field potential. C, Responses in the stratum radiatum (zone 1) and stratum pyramidale (zone 2) of CA1.D, Responses in the molecular layer (zone 3) and granule cell layer (zone 4) of the dentate gyrus. The time course of the field potential in C resembles the time course of the intrinsic optical signal in the stratum radiatum. The dashed lines represent the peaks of the intrinsic optical signal and the field potential. The pseudocolor bar is a linear representation of the change in transmittance. Note that the time is given in minutes and seconds. Scale bars: A, 400 μm;B, 200 μm.
Typically SD originated in only one location, although there were a few slices in which SD initiated in multiple areas of CA1. Any slices with multiple initiation sites were excluded from our analysis. SD propagated radially from the initiation site and eventually crossed the hippocampal fissure to propagate throughout the dentate gyrus. The time course of the intrinsic optical signal in the dentate gyrus was very similar to that seen in CA1 (Fig. 2C,D). It was very rare to see SD initiate in areas outside of CA1. Furthermore, the wave typically slowed when entering area CA3, and the time course of recovery of the intrinsic optical signal was delayed in CA3. The features of SD in CA3 were not investigated further.
Quantification of spreading depression
To analyze such a wave-like event, it was necessary to define several features of the intrinsic optical signal (Fig.3). Several regions of interest were identified on each image. For each given region of interest the time course of the intrinsic optical signal was measured. To analyze the kinetics of the onset of SD, the time of peak, time to 20 or 40% of the peak, the slope between 20 and 40% of the peak, and the maximum slope between two measurements during the rising phase were determined. The kinetics of the recovery phase during SD was quantified by determining the extent of recovery toward initial transmittance levels during a fixed time period after the peak (30 sec, 1 min, or 2 min), and by measuring the time required to decay from the peak by a fixed percentage of the peak (25 or 50%). To measure the propagation velocity, three regions of interest were identified in either CA1 or the dentate gyrus. The time differential between the onset of SD in each of these zones was determined, and the distance between the zones was measured, enabling one to compute the velocity of the wave between any two regions of interest. The onset of the SD event was defined as the time to 20% of the peak amplitude because this point gave the most consistent velocity measurements. To alleviate any problems caused by an initial offset in the intrinsic optical signal, all values were baseline subtracted before any computations were performed.
Fig. 3.

Quantification of the intrinsic optical signal. Time course of the intrinsic optical signal from a single zone in the stratum radiatum of CA1. The onset of an event was defined by the time to reach 20% of the peak. The onset kinetics were characterized by determining the slope between 20 and 40% of the peak response, as well as the maximum slope during the onset phase. The recovery phase was defined by measuring the percentage decay at fixed time points (30 sec, 1 min, 2 min) after the peak response and by the time taken to decay a fixed percentage from the peak.
Cortical spreading depression
In addition to examining SD in hippocampal slices, SD was induced by ouabain in neocortical slices (Fig.4). Spreading depression propagation was measured in two regions that were defined as the outermost one-third and innermost one-third of the distance from the neocortical surface to the white matter. At the outermost layer, cortical SD propagated at 33.8 ± 1.9 μm/sec (n = 5, SEM), whereas propagation was significantly slower in the innermost layer of the cortex (27.5 ± 2.4 μm/sec; p < 0.05, Mann–Whitney U test). These values are comparable but slightly faster than the velocities determined in hippocampal slices. The SD response was observed as a uniform wave propagating throughout the entire slice with all regions demonstrating a transient increase in cell swelling, in contrast to the regional differences observed between stratum radiatum and stratum pyramidale of the hippocampus. In general, the kinetics of the responses were similar between hippocampal and neocortical slices (Fig. 4C)
Fig. 4.

Imaging spreading depression in neocortical slices. A, Intrinsic optical signals during superfusion of aCSF containing 100 μm ouabain. In this case, SD was initiated at the leftmost visible portion of the slice and propagated uniformly across the entire slice. B, Description of the areas that were used for measurements. C, Time course of the intrinsic optical signal and extracellular field potential for one region. Note the similar time course of the field potential and associated intrinsic optical signal. The dashed linesrepresent the peaks of the intrinsic optical signal and the field potential. Scale bars: A, 400 μm; B, 200 μm.
Intracellular calcium dynamics during spreading depression
To determine what role intracellular calcium waves play in the initiation and propagation of SD, we examined intracellular calcium dynamics during SD (Fig. 5). To increase spatial resolution we used a higher power objective (25× compared with 5×), and limited our imaging to the infrapyramidal blade (free blade) of the dentate gyrus. A number of technical reasons favored this imaging location: (1) the dentate gyrus demonstrated the most robust loading of fura-2 AM; (2) SD always propagated through the dentate gyrus from the suprapyramidal blade (enclosed blade) into the infrapyramidal blade, and thus, the direction of wave propagation was known in advance; and (3) because SD propagated into the dentate gyrus, complications caused by multiple initiation sites were avoided. As shown in Figure 2D the intrinsic optical signal response in the dentate gyrus was comparable to the response seen in CA1.
Fig. 5.
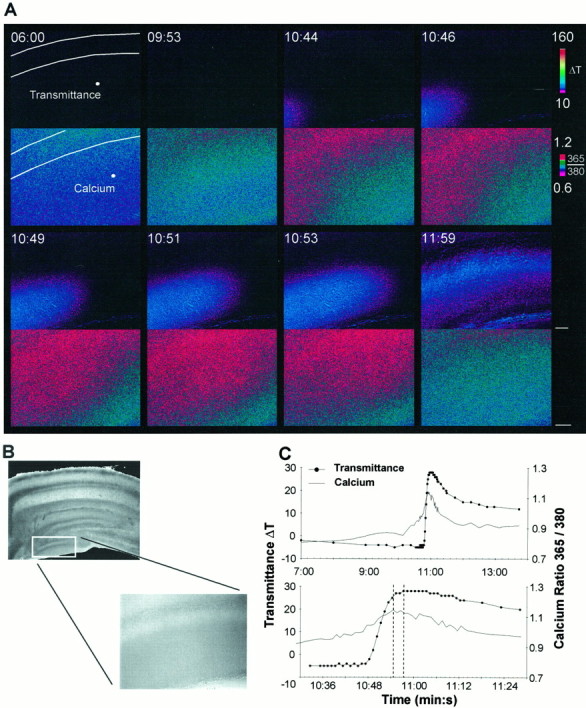
Simultaneous imaging of intracellular calcium and intrinsic optical signals in the dentate gyrus. A, Intrinsic optical signals (top) and intracellular calcium (bottom) during superfusion of aCSF containing 100 μm ouabain. A slow rise in calcium precedes any detectable change in the intrinsic optical signal, and a calcium wave preceded the intrinsic optical signal wave. The white lines denote the regions corresponding to the cell body layer in the dentate gyrus. The white dots denote the areas that were used for measurements. B, Because a higher power objective was required for enhanced spatial resolution, only a portion of the dentate gyrus was imaged (see Results for details).C, Time course of the intrinsic optical signal and intracellular calcium levels during SD. The same data with an expanded time scale are shown in the bottom panel. Thedashed lines represent the peaks of the intrinsic optical signal and the calcium ratio. Scale bar in A, 50 μm for both intrinsic and calcium images.
Using two independent imaging systems it was not possible to ensure complete spatial registry of the individual images between the two setups. However, it was possible to ensure spatial registry on given regions of interest before measurements were made (see Materials and Methods). This ensured that the extracted intrinsic and calcium measurements originated from precisely the same slice location. Coupling this with the synchronizing triggers between the two systems ensured tight spatial and temporal synchrony for analysis purposes. It must be noted that for display purposes, the raw images were not translated, and instead, the regions corresponding to the cell body layer in the dentate gyrus are outlined on both the intrinsic optical and fura-2 signals (Fig. 5A, white lines).
In all cases, an increase in intracellular calcium was detected during SD (n = 14). Typically, a slow calcium rise preceded any increase in the intrinsic optical signal and, subsequently, a calcium wave propagated through the dentate gyrus, ahead of the intrinsic optical signal (Fig.5A,C) Because the calcium signal was composed of a slow rise and a subsequent wave, three different onset parameters were used to demonstrate that the calcium signal preceded the intrinsic optical signal (Fig.6). The calcium signal peaked 15.8 ± 2.2 sec ahead of the intrinsic optical signal and propagated through the slice at a velocity of 21.1 ± 6.3 μm/sec. In comparison, the propagation velocity of the associated intrinsic optical signal was 24.8 ± 2.7 μm/sec, comparable to the velocity of 19.3 ± 2.4 μm/sec that had been measured at lower magnification (p > 0.05, Mann–Whitney U test). On occasion only a slow elevation in calcium without the calcium wave was observed, or only a wave occurred without the slow rise.
Fig. 6.

Intracellular calcium elevations preceded the onset of the intrinsic optical signal. The time differential between intracellular calcium elevations and the intrinsic optical signal is shown. The δ time is given for three different onset parameters, 20% of the peak, 40% of the peak, and the time of the peak. These three parameters are described in detail in Figure 3. In all measurements the calcium signal preceded the intrinsic optical signal.
It has been established that calcium waves occur in cultured astrocytes (Cornell Bell et al., 1990). To determine whether glia cells contributed to the observed Ca2+intdynamics, astrocytes were injected intracellularly with the calcium indicator calcium orange (Fig. 7). Under these conditions, any detectable change in Ca2+int would be attributable to astrocytic Ca2+int dynamics. Associated with spreading depression, an increase in astrocytic Ca2+int was observed, and the peak of these increases preceded the intrinsic optical signal of SD (n = 4). These results demonstrate that an increase in Ca2+int precedes spreading depression and suggest that a calcium wave is involved in the initiation or propagation of spreading depression. Furthermore, such calcium dynamics are attributable in part to astrocytic contributions.
Fig. 7.

Imaging of astrocytic intracellular calcium and intrinsic optical signals in the hippocampus. A, Intrinsic optical signals (top) and intracellular calcium (bottom) during bath application of 100 μm ouabain, which was started at 2 min. Individual astrocytes were intracellularly injected with calcium orange. Thedotted lines represent the onset of the calcium signal, the peak of the calcium signal, and the peak of the intrinsic optical signal. Note that the peak of the calcium signal precedes the peak of the intrinsic optical signal.
Spreading depression in the absence of external calcium
To determine whether the SD-associated increases in Ca2+int are necessary for the initiation and propagation of SD, we evoked SD in the absence of external calcium. After incubation of the slices in 0-Ca2+ aCSF with 2 mm EGTA (0-Ca2+ aCSF) for >20 min, SD initiation and propagation was still induced by superfusion of ouabain (Fig.8A,C). However, Ca2+int increases during SD were completely inhibited (Fig.8D,E). In regular calcium aCSF, the baseline fura-2 ratio was 0.77 ± 0.02 and peaked at 1.12 ± 0.10 (p < 0.001, Student’s t test) during SD. In 0-Ca2+ aCSF, the baseline was 0.73 ± 0.01 and during SD reached 0.76 ± 0.01 (p > 0.05, Student’s t test). The inhibition of Ca2+int dynamics without a concomitant inhibition of SD indicates that the increase in Ca2+int observed in regular aCSF is not critical for the initiation or propagation of SD.
Fig. 8.
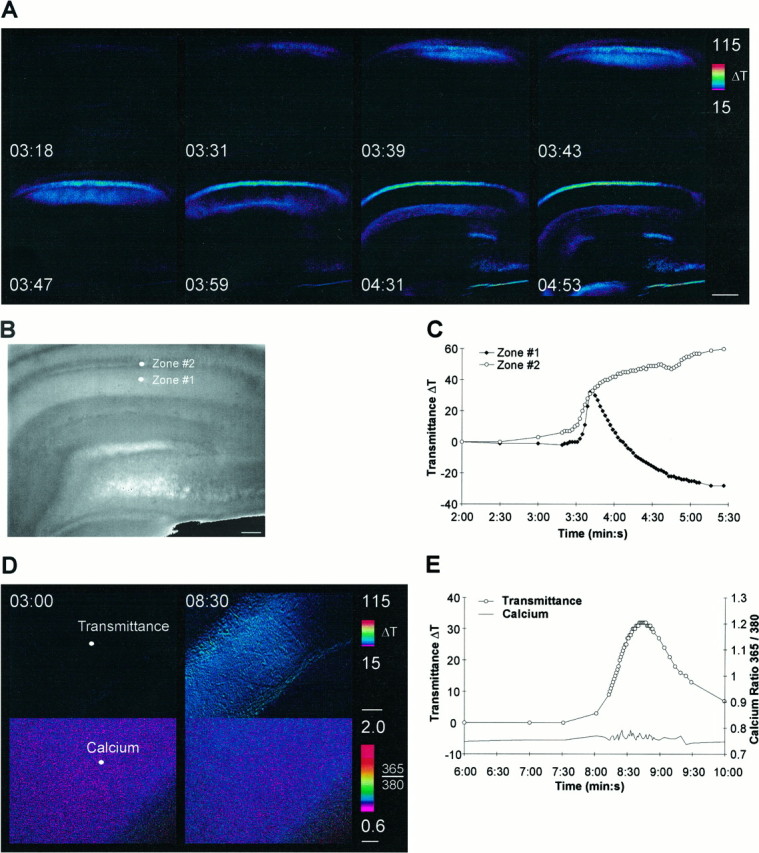
Spreading depression in the absence of external calcium. A, Intrinsic optical signals in 0-Ca2+ aCSF during bath application of 100 μm ouabain. B, Description of the areas that were used for measurements. C, Time course of the intrinsic optical signal in stratum radiatum and stratum pyramidale of CA1. D, Intracellular calcium increases are absent in 0-Ca2+ aCSF. Left, Before spreading depression. Right, Peak of spreading depression. Thewhite dots denote the areas of measurement shown inE. E, Time course of the intrinsic optical signal and intracellular calcium levels in 0-Ca2+ aCSF. Note the complete absence of any calcium increase, although SD still occurred. Scale bars:A, 400 μm; B, 200 μm;D, 50 μm (top andbottom).
The peak amplitudes of the intrinsic optical signal were similar in regular and 0-Ca2+ aCSF (Fig.9A), indicating that the extent of swelling was comparable in both conditions. However, SD in 0-Ca2+ aCSF was not identical to SD in regular aCSF. In 0-Ca2+ aCSF, SD had a smaller onset slope and lower maximum onset slope, indicative of a slower rise time (Fig.9B,C). In contrast, the kinetics of recovery were faster in 0-Ca2+ aCSF, as shown by the faster rate of decay and decreased time to a fixed decay percentage (Fig. 9D,E).
Fig. 9.

Analysis of spreading depression in the presence and absence of external calcium. The amplitudes of the peak response are shown in A. Note that the peak responses are expressed as δT/T, where T was defined as the initial transmittance level for each zone. The rising phase of SD was decreased in the absence of calcium. The onset slope, measured as either the slope between 20 and 40% of the peak (B), or the maximum slope from onset to peak (C), was smaller in the absence of calcium. The recovery phase is faster in 0-Ca2+ aCSF (D, E). The time to decay 25 or 50% from the peak is shown inD, whereas the rate of decay defined as the percentage decay over a fixed time interval is given in E.
DISCUSSION
We have used a combination of imaging and electrophysiological techniques to record the propagation of SD in the rat hippocampus. Imaging of intrinsic optical signals permitted us to map the propagating wave front and to correlate it with the Ca2+int changes monitored with fura-2 fluorescence. In normal solutions, SD was associated with increases in Ca2+int that had a wave-like appearance, and part of this Ca2+int increase was of astrocytic origin. However, calcium waves were not critical for SD, because SD was still observed in the absence of external calcium and any detectable increase in Ca2+int.
Intrinsic optical signals
These experiments demonstrate that imaging intrinsic optical signals can be used to monitor slow activity-dependent changes in brain tissue and to provide novel insights into the cellular mechanisms of SD. Intrinsic optical signals develop over seconds (MacVicar and Hochman, 1991) and reflect the cellular changes in volume that occur in response to activity (Andrew and MacVicar, 1994). Changes in light transmittance have been shown to be directly correlated with extracellular space changes (Holthoff and Witte, 1996). Cellular swelling causes a decrease in extracellular space and an increase in light transmittance, whereas cell shrinkage causes the reverse. It should be noted that the slices we imaged were composed of both neurons and glia and that the intrinsic optical signal could represent volume changes in both cell types.
The maintained light transmittance increase in the cell body region versus the transient increase in the dendritic region during SD may reflect different regional and cellular patterns of volume changes. The maintained increase in stratum pyramidale during SD likely indicates that neurons underwent swelling. Previous reports have shown that neurons swell (Van Harreveld and Khattab, 1967) and that the extracellular space shrinks during SD (Nicholson and Kraig, 1981; Jing et al., 1994), consistent with our observations. The transient increase in light transmittance in stratum radiatum is more difficult to interpret. A previous study suggested that the increase in light transmittance in hippocampal slices during synaptic activity results from K+ accumulation and swelling in astrocytes (MacVicar and Hochman, 1991). It is possible that the subsequent decreases in the dendritic region reflect compensatory astrocytic regulatory volume decreases. Regulatory volume decrease has been observed in astrocytes in cell culture (Pasantes-Morales et al., 1994) but not in the CNS to date (Krizaj et al., 1996; Andrew et al., 1997). Alternatively, the decrease in light transmittance in the dendritic region could represent neuronal dendritic shrinkage. In support, it recently has been shown that neurons are capable of undergoing regulatory volume decrease (Churchwell et al., 1996), although this was under conditions in which NMDA receptors were inhibited. Therefore, our study illustrates the utility of imaging intrinsic optical signals in mapping SD. Further studies are in progress to differentiate the underlying neuronal and astrocytic cellular changes.
Ouabain induction of spreading depression
Although it has long been known that ouabain can induce SD by inhibiting Na+,K+ ATPases, the sequence of events that leads to SD is unknown (Nicholson and Kraig, 1981; Haglund and Schwartzkroin, 1990). At the concentrations that we used ouabain will inhibit both the α2 and α3 isoforms of Na+,K+ ATPase that are thought to be expressed mainly in astrocytes and neurons, respectively (Watts et al., 1991; Sweadner, 1992). Inhibiting Na+,K+ ATPases would increase intracellular Na+ and extracellular K+ concentrations, leading to cellular depolarization, which could contribute to the triggering of SD. It has been suggested that the sensitivity of the CA1 region to the induction of SD by ouabain is attributable to the reduced amounts of Na+,K+ ATPases in CA1 compared with CA3 (Haglund et al., 1985; Haglund and Schwartzkroin, 1990). Our observation that SD was typically initiated in the CA1 region is consistent with this.
Although ouabain can induce cell death (Lees et al., 1990; Lees, 1991) our observations of fura-2-loaded cells suggest that this was not a significant problem during the time course of our experiments. We did not observe maintained calcium elevations that typically indicate cell death. Furthermore, we did not observe any decrease in the intensity of fura-2 fluorescence at the near isosbestic wavelength of 365 nm that would occur if there was any dye loss because of cell lysis.
Astrocyte calcium waves and spreading depression
Our studies were designed to test the hypothesis that Ca2+int waves in glial cells are important in SD (Cornell Bell and Finkbeiner, 1991; Nedergaard, 1994). There is increasing evidence that astrocytes and neurons are capable of bidirectional communication. For example, it has been shown that neuronal activity can induce elevations in Ca2+int in neighboring astrocytes (Dani et al., 1992). Conversely, there is evidence that elevations in Ca2+int in astrocytes can induce both neuronal depolarization and elevations of neuronal Ca2+int (Charles, 1994; Nedergaard, 1994; Parpura et al., 1994; Hassinger et al., 1995). Because SD is characterized as a slowly propagating wave of neuronal and glial depolarization, it has been postulated that such complex interactions between neurons and glia are necessary for SD. In fact, several features of SD and astrocytic calcium waves, such as velocity of propagation (Hansen, 1985; Cornell Bell et al., 1990) and sensitivity to gap junction inhibitors, (Finkbeiner, 1992; Martins-Ferreira and Ribeiro, 1995; Nedergaard et al., 1995) are very similar (Nedergaard et al., 1995). Therefore, it has been proposed that astrocytic calcium waves play a role in the initiation and propagation of SD (Cornell Bell and Finkbeiner, 1991; Nedergaard, 1994).
How might an astrocytic calcium wave be involved in spreading depression? It has been demonstrated that elevations in astrocytic calcium result in the subsequent depolarization and elevation of Ca2+int in neighboring neurons (Charles, 1994; Nedergaard, 1994; Parpura et al., 1994; Hassinger et al., 1995). Two mechanisms have been proposed to explain such glia–neuron signaling. Elevations of Ca2+int in astrocytes causes the release of glutamate, which depolarizes adjacent neurons and activates neuronal NMDA receptors (Parpura et al., 1994;Hassinger et al., 1995). Astrocytes have also been postulated to be transiently coupled to neurons through gap junctions, allowing for the propagation of a calcium wave between the two cell types (Nedergaard, 1994). Both of these mechanisms are consistent with the ability of a propagating astrocytic calcium wave to induce propagating neuronal depolarization, similar to that which is observed in SD. Such a mechanism would imply that Ca2+intchanges should precede SD. We typically observed slow Ca2+int elevations minutes before the onset of SD, and a calcium wave typically preceded SD by many seconds. Experiments performed with individual astrocytes loaded with fura-2 revealed that astrocytic Ca2+int changes partially account for the observed Ca2+int dynamics. Taken together, these data are consistent with the idea that astrocytic calcium waves are normally associated with SD.
Zero-calcium and spreading depression
To directly test the involvement of calcium waves in SD, we initiated SD in slices incubated in 0-Ca2+ aCSF. In regular aCSF the calcium waves that we observed are most likely attributable to calcium influx or calcium release from internal calcium stores. Therefore, we expected that the magnitude of the calcium wave would be smaller in 0-Ca2+ aCSF. For example, calcium waves in astrocytes in cell culture and in retinal glia (Newman and Zahs, 1997) are primarily attributable to IP3-mediated release of calcium from intracellular stores (Charles et al., 1993), and these waves exist in the absence of external calcium (Newman and Zahs, 1997). Our observations that there was no detectable increase in [Ca2+]int during SD in 0-Ca2+ aCSF suggests that release from internal stores does not contribute to the calcium waves we observed in SD. However, preincubating slices for 20 min in 0-Ca2+aCSF could have depleted calcium stores before SD was initiated, thus accounting for the lack of any calcium signals generated by release from internal stores. In agreement, Venance et al. (1997) have demonstrated that internal calcium stores in cultured astrocytes are depleted within 10 min after incubation in a 0-Ca and 2 mmEGTA external solution. An alternative explanation for the lack of any detectable calcium dynamic during SD in 0-Ca2+ aCSF is that the calcium increase is attributable to influx through voltage-gated calcium channels. In addition to neuronal calcium channels, it has been reported that astrocytic calcium channels are activated when extracellular K+ exceeds 25 mm (Duffy and MacVicar, 1994) within the 25–80 mm range reported during SD (Vyskocil et al., 1972). Our observation that SD still occurred in the absence of any detectable Ca2+int response is consistent with previous reports that SD in the hippocampus is not inhibited by calcium channel antagonists (Jing et al., 1993; Herreras et al., 1994). However, it should be noted that in the retina, SD is inhibited in the absence of external calcium (Martins-Ferreira et al., 1974; Nedergaard et al., 1995).
Taken together, these data demonstrate that although calcium elevations are associated with SD, such calcium dynamics are not critical for SD, suggesting that SD propagation can occur by a mechanism independent of calcium wave propagation. Our results demonstrate that SD can occur in the hippocampus with no detectable increase in Ca2+int. The underlying mechanisms of SD remain to be determined.
Footnotes
This work was supported by the Medical Research Council (MRC). We thank J. Armstrong, D. Doll, and V. Parpura for helpful comments on this manuscript. We also thank A. Salkauskas and S. Borg for helpful discussions on analysis software development. T.A.B. is a postdoctoral fellow of the Alberta Heritage Foundation for Medical Research (AHFMR). B.A.M. is an MRC Senior Scientist and an AHFMR Scientist.
Correspondence should be addressed to Dr. Brian MacVicar, Department of Physiology and Biophysics, University of Calgary, 3330 Hospital Drive NW, Calgary, Alberta Canada T2N 4N1.
REFERENCES
- 1.Andrew RD, MacVicar BA. Imaging cell volume changes and neuronal excitation in the hippocampal slice. Neuroscience. 1994;62:371–383. doi: 10.1016/0306-4522(94)90372-7. [DOI] [PubMed] [Google Scholar]
- 2.Andrew RD, Lobinowich ME, Osehobo EP. Evidence against volume regulation by cortical brain cells during acute osmotic stress. Exp Neurol. 1997;143:300–312. doi: 10.1006/exnr.1996.6375. [DOI] [PubMed] [Google Scholar]
- 3.Charles AC. Glia-neuron intercellular calcium signalling. Dev Neurosci. 1994;16:196–206. doi: 10.1159/000112107. [DOI] [PubMed] [Google Scholar]
- 4.Charles AC, Dirksen ER, Merrill JE, Sanderson MJ. Mechanisms of intracellular calcium signalling in glial cells studied with dantrolene and thapsigargin. Glia. 1993;7:134–145. doi: 10.1002/glia.440070203. [DOI] [PubMed] [Google Scholar]
- 5.Churchwell KB, Wright SH, Emma F, Rosenberg PA, Strange K. NMDA receptor activation inhibits neuronal volume regulation after swelling induced by veratridine-stimulated Na+ influx in rat cortical cultures. J Neurosci. 1996;16:7447–7457. doi: 10.1523/JNEUROSCI.16-23-07447.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cornell Bell AH, Finkbeiner SM. Ca2+ waves in astrocytes. Cell Calcium. 1991;12:185–204. doi: 10.1016/0143-4160(91)90020-f. [DOI] [PubMed] [Google Scholar]
- 7.Cornell Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990;247:470–473. doi: 10.1126/science.1967852. [DOI] [PubMed] [Google Scholar]
- 8.Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron. 1992;8:429–440. doi: 10.1016/0896-6273(92)90271-e. [DOI] [PubMed] [Google Scholar]
- 9.Dietrich WD, Feng ZC, Leistra H, Watson BD, Rosenthal M. Photothrombotic infarction triggers multiple episodes of cortical spreading depression in distant brain regions. J Cereb Blood Flow Metab. 1994;14:20–28. doi: 10.1038/jcbfm.1994.4. [DOI] [PubMed] [Google Scholar]
- 10.Duffy S, MacVicar BA. Potassium-dependent calcium influx in acutely isolated hippocampal astrocytes. Neuroscience. 1994;61:51–61. doi: 10.1016/0306-4522(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 11.Duffy S, MacVicar BA. Adrenergic calcium signaling in astrocyte networks within the hippocampal slice. J Neurosci. 1995;15:5535–5550. doi: 10.1523/JNEUROSCI.15-08-05535.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finkbeiner S. Calcium waves in astrocytes-filling in the gaps. Neuron. 1992;8:1101–1108. doi: 10.1016/0896-6273(92)90131-v. [DOI] [PubMed] [Google Scholar]
- 13.Haglund MM, Schwartzkroin PA. Role of Na-K pump potassium regulation and IPSPs in seizures and spreading depression in immature rabbit hippocampal slices. J Neurophysiol. 1990;63:225–239. doi: 10.1152/jn.1990.63.2.225. [DOI] [PubMed] [Google Scholar]
- 14.Haglund MM, Stahl WL, Kunkel DD, Schwartzkroin PA. Developmental and regional differences in the localization of Na,K-ATPase activity in the rabbit hippocampus. Brain Res. 1985;343:198–203. doi: 10.1016/0006-8993(85)91180-1. [DOI] [PubMed] [Google Scholar]
- 15.Hansen AJ. Effect of anoxia on ion distribution in the brain. Physiol Rev. 1985;65:101–148. doi: 10.1152/physrev.1985.65.1.101. [DOI] [PubMed] [Google Scholar]
- 16.Hassinger TD, Atkinson PB, Strecker GJ, Whalen LR, Dudek FE, Kossel AH, Kater SB. Evidence for glutamate-mediated activation of hippocampal neurons by glial calcium waves. J Neurobiol. 1995;28:159–170. doi: 10.1002/neu.480280204. [DOI] [PubMed] [Google Scholar]
- 17.Herreras O, Largo C, Ibarz JM, Somjen GG, Martin del Rio R. Role of neuronal synchronizing mechanisms in the propagation of spreading depression in the in vivo hippocampus. J Neurosci. 1994;14:7087–7098. doi: 10.1523/JNEUROSCI.14-11-07087.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holthoff K, Witte OW. Intrinsic optical signals in rat neocortical slices measured with near-infrared dark-field microscopy reveal changes in extracellular space. J Neurosci. 1996;16:2740–2749. doi: 10.1523/JNEUROSCI.16-08-02740.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jing J, Aitken PG, Somjen GG. Role of calcium channels in spreading depression in rat hippocampal slices. Brain Res. 1993;604:251–259. doi: 10.1016/0006-8993(93)90376-x. [DOI] [PubMed] [Google Scholar]
- 20.Jing J, Aitken PG, Somjen GG. Interstitial volume changes during spreading depression (SD) and SD-like hypoxic depolarization in hippocampal tissue slices. J Neurophysiol. 1994;71:2548–2551. doi: 10.1152/jn.1994.71.6.2548. [DOI] [PubMed] [Google Scholar]
- 21.Krizaj D, Rice ME, Wardle RA, Nicholson C. Water compartmentalization and extracellular tortuosity after osmotic changes in cerebellum of Trachemys scripta. J Physiol (Lond) 1996;492:887–896. doi: 10.1113/jphysiol.1996.sp021354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leao AAP. Spreading depression of activity in the cerebral cortex. J Neurophysiol. 1944;7:359–390. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- 23.Lees GJ. Inhibition of sodium-potassium-ATPase: a potentially ubiquitous mechanism contributing to central nervous system neuropathology. Brain Res Rev. 1991;16:283–300. doi: 10.1016/0165-0173(91)90011-v. [DOI] [PubMed] [Google Scholar]
- 24.Lees GJ, Lehmann A, Sandberg M, Hamberger A. The neurotoxicity of ouabain, a sodium-potassium ATPase inhibitor, in the rat hippocampus. Neurosci Lett. 1990;120:159–162. doi: 10.1016/0304-3940(90)90027-7. [DOI] [PubMed] [Google Scholar]
- 25.MacVicar BA, Hochman D. Imaging of synaptically evoked intrinsic optical signals in hippocampal slices. J Neurosci. 1991;11:1458–1469. doi: 10.1523/JNEUROSCI.11-05-01458.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martins-Ferreira H, Ribeiro LJ. Biphasic effects of gap junctional uncoupling agents on the propagation of retinal spreading depression. Braz J Med Biol Res. 1995;28:991–994. [PubMed] [Google Scholar]
- 27.Martins-Ferreira H, De Oliveira Castro G, Struchiner CJ, Rodrigues PS. Circling spreading depression in isolated chick retina. J Neurophysiol. 1974;37:773–784. doi: 10.1152/jn.1974.37.4.773. [DOI] [PubMed] [Google Scholar]
- 28.Mies G, Iijima T, Hossmann KA. Correlation between peri-infarct DC shifts and ischaemic neuronal damage in rat. NeuroReport. 1993;4:709–711. doi: 10.1097/00001756-199306000-00027. [DOI] [PubMed] [Google Scholar]
- 29.Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- 30.Nedergaard M, Hansen AJ. Characterization of cortical depolarizations evoked in focal cerebral ischemia. J Cereb Blood Flow Metab. 1993;13:568–574. doi: 10.1038/jcbfm.1993.74. [DOI] [PubMed] [Google Scholar]
- 31.Nedergaard M, Cooper AJ, Goldman SA. Gap junctions are required for the propagation of spreading depression. J Neurobiol. 1995;28:433–444. doi: 10.1002/neu.480280404. [DOI] [PubMed] [Google Scholar]
- 32.Newman EA, Zahs KR. Calcium waves in retinal glial cells. Science. 1997;275:844–847. doi: 10.1126/science.275.5301.844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicholson C, Kraig RP. The behaviour of extracellular ions during spreading depression. In: Zeuthen T, editor. The application of ion-selective electrodes. Elsevier; Amsterdam: 1981. pp. 217–238. [Google Scholar]
- 34.Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 35.Pasantes-Morales H, Murray RA, Lilja L, Moran J. Regulatory volume decrease in cultured astrocytes. I. Potassium- and chloride-activated permeability. Am J Physiol. 1994;266:C165–C171. doi: 10.1152/ajpcell.1994.266.1.C165. [DOI] [PubMed] [Google Scholar]
- 36.Somjen GG, Aitken PG, Czeh GL, Herreras O, Jing J, Young JN. Mechanism of spreading depression: a review of recent findings and a hypothesis. Can J Physiol Pharmacol. 1992;70:S248–S254. doi: 10.1139/y92-268. [DOI] [PubMed] [Google Scholar]
- 37.Sweadner KJ. Overlapping and diverse distribution of Na-K ATPase isozymes in neurons and glia. Can J Physiol Pharmacol. 1992;70:S255–S259. doi: 10.1139/y92-269. [DOI] [PubMed] [Google Scholar]
- 38.Van Harreveld A, Khattab FI. Changes in cortical extracellular space during spreading depression investigated with the electron microscope. J Neurophysiol. 1967;30:911–929. doi: 10.1152/jn.1967.30.4.911. [DOI] [PubMed] [Google Scholar]
- 39.Venance L, Stella N, Glowinski J, Giaume C. Mechanism involved in initiation and propagation of receptor-induced intercellular calcium signaling in cultured rat astrocytes. J Neurosci. 1997;17:1981–1992. doi: 10.1523/JNEUROSCI.17-06-01981.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vyskocil F, Kritz N, Bures J. Potassium-selective microelectrodes used for measuring the extracellular brain potassium during spreading depression and anoxic depolarization in rats. Brain Res. 1972;39:255–259. doi: 10.1016/0006-8993(72)90802-5. [DOI] [PubMed] [Google Scholar]
- 41.Watts AG, Sanchez-Watts G, Emanuel JR, Levenson R. Cell-specific expression of mRNAs encoding Na+,K(+)-ATPase alpha- and beta-subunit isoforms within the rat central nervous system. Proc Natl Acad Sci USA. 1991;88:7425–7429. doi: 10.1073/pnas.88.16.7425. [DOI] [PMC free article] [PubMed] [Google Scholar]