

Abstract
Autocrine trophic functions of brain-derived neurotrophic factor (BDNF) have been proposed for many central neurons because this neurotrophin displays striking colocalization with its receptor trkB within the CNS. In the cortex, the distribution patterns of BDNF and trkB expression are almost identical. Corticospinal neurons (CSNs) are a major cortical long-distance projecting system. They are localized in layer V of the somatosensory cortex, and their axons project into the spinal cord where they contribute to the innervation of spinal motoneurons. We have shown recently that adult CSNs express trkB mRNA and are rescued from axotomy-induced death by BDNF treatment. Half of the axotomized CSNs survived without BDNF infusions. These findings raise the possibility that endogenous cortical BDNF is involved in the trophic support of this neuronal population. To test the hypothesis that endogenous cortical BDNF promotes survival of adult CSNs, we infused the BDNF-neutralizing affinity-purified antibody RAB to axotomized and unlesioned CSNs for 7 d. This treatment resulted in increased death of axotomized CSNs. Survival of unlesioned CSNs was not affected by RAB treatment. In situ hybridizations for BDNF and trkB mRNA revealed that virtually all CSNs express trkB, whereas only half of them express BDNF. Thus, autocrine/paracrine mechanisms are likely to contribute to the endogenous BDNF protection of axotomized CSNs. We have demonstrated previously that, in addition to BDNF, glial cell line-derived neurotrophic factor (GDNF) and neurotrophin 3 (NT-3) also rescue CSNs from axotomy-induced death. We now show that the rescuing by GDNF requires the presence of endogenous cortical BDNF, implicating a central role of this neurotrophin in the trophic support of axotomized CSNs and a trophic cross-talk between BDNF and GDNF regarding the maintenance of lesioned CSNs. In contrast, NT-3 promotes survival of axotomized CSNs even when endogenous cortical BDNF is neutralized by RAB, indicating a potential of compensatory mechanisms for the trophic support of CSNs.
Keywords: corticospinal neurons, axotomy, neurotrophin, BDNF, GDNF, autocrine, paracrine, gene expression, neuronal protection
Although survival of specific populations of neurons in the peripheral nervous system is frequently promoted by individual neurotrophins (Snider, 1994; Lindsay, 1996), the situation in the CNS seems to be much more complex. Neurons of the CNS are commonly supported by several neurotrophic factors (Barbacid, 1994; Snider, 1994; Lindsay, 1996), and knockouts of a particular neurotrophin rarely affect the survival of individual neuronal populations (Snider, 1994), suggesting a redundancy of trophic support for central neurons. This situation is further complicated by the fact that trophic support for neurons may be derived from different sources. According to the classical view of neurotrophic support, a neuron depends on a certain target-derived neurotrophic factor for survival (Barde, 1989). It is now well accepted, however, that this support can also be derived from factors along the axonal pathway of the neuron, from its immediate perikaryal surrounding (paracrine), or from the neuron itself (autocrine) (Korsching, 1993). In addition, trophic dependencies of adult CNS neurons may be altered by insults such as axonal injury (Sofroniew et al., 1990).
Support for the idea of trophic redundancy for central neurons is provided by the increased survival of axotomized corticospinal neurons (CSNs) by exogenously supplied brain-derived neurotrophic factor (BDNF) (Giehl and Tetzlaff, 1996), neurotrophin 3 (NT-3) (Giehl and Tetzlaff, 1996), ciliary neurotrophic factor (CNTF) (Dale et al., 1995), and glial cell line-derived neurotrophic factor (GDNF) (Giehl et al., 1997). CSNs are neocortical layer V neurons involved in motor control (Porter et al., 1987; Nudo and Masterton, 1988, 1990; Liang et al., 1991). An understanding of the endogenous support mechanisms for CSNs would be beneficial for the development of new therapeutic strategies for the treatment of amyotrophic lateral sclerosis, a fatal neurodegenerative disease displaying severe CSN and spinal motoneuron degeneration (Chou, 1995; Martin and Swash, 1995). However, little is known about the physiological trophic dependencies of CSNs. In the neurotrophin knockouts, CSNs do not seem to be severely affected because the cortex does not display gross morphological alterations (Ernfors et al., 1994a,b; Jones et al., 1994). Because more than half of the axotomized CSNs survive for at least 42 d without maintaining a connection to their original spinal cord targets (Giehl et al., 1997), we hypothesized that local cortical mechanisms exist to promote CSN survival in vivo.
BDNF seems to be a good candidate molecule for this support because it is expressed in developing and adult cortex (Ernfors et al., 1990;Maisonpierre et al., 1990; Kokaia et al., 1993a; Miranda et al., 1993; Altar et al., 1994; Conner et al., 1997; Yan et al., 1997a), and CSNs express full-length trkB mRNA (Giehl and Tetzlaff, 1996), which codes for the functional BDNF high-affinity receptor (Barbacid, 1994). In addition, endogenous BDNF has been established as a survival factor for embryonic cortical neurons (Ghosh et al., 1994), and BDNF treatment fully prevents axotomy-induced death of adult CSNs (Giehl and Tetzlaff, 1996).
We show here that endogenous BDNF is a survival factor for axotomized CSNs. Endogenous cortical BDNF was neutralized by infusing the BDNF-neutralizing affinity-purified antibody RAB (Yan et al., 1997a) to unlesioned or axotomized CSNs. This dramatically increased the number of CSNs that die after axotomy. Survival of unlesioned CSNs that are connected to their target was not affected by RAB infusion. RAB-induced death of CSNs could be counteracted by exogenously applied NT-3, which is consistent with the expression of trkC mRNA in CSNs (Giehl and Tetzlaff, 1996), but not by GDNF. Both factors have been shown previously to completely prevent axotomy-induced death of CSNs (Giehl and Tetzlaff, 1996; Giehl et al., 1997). Thus, we propose that BDNF is an autocrine/paracrine survival factor for axotomized CSNs and that the GDNF survival promotion on CSNs requires endogenous cortical BDNF.
MATERIALS AND METHODS
Operation procedure, antibody application, and neurotrophic factor application. The experimental procedures and maintenance of animals were approved by the local Animal Care Committee according to the German law regulating the experimental use of animals. Sixty-five male Sprague Dawley rats weighing 190–330 gm were used for this study. All surgery was performed under anesthesia with a combination of chloral hydrate (150 mg/kg) and sodium pentobarbital (32 mg/kg).
The operation procedure, neurotrophic factor application, and coordinates for stereotaxic lesion and intracortical antibody and/or neurotrophic factor delivery have been described in detail elsewhere (Giehl and Tetzlaff, 1996). In brief, to distinguish CSNs from other neurons in layer V of the cortex, they were retrogradely labeled by bilateral fast blue (FB) (2% in 0.2% DMSO) injections into both corticospinal tracts (CSTs) at cervical level C5 10 d before axotomy. The internal capsule lesion was performed using a stereotaxic wire knife. The axotomy was verified by injection of a 1:1 mixture of rhodamine dextran 10,000 (RDX) (20% in 0.2% DMSO) and rhodamine-b-isothiocyanate (RITC) (10% in 0.2% DMSO) into both CSTs at C3/C4 immediately after the lesion. Neurons doubly labeled with FB and RDX/RITC were regarded as unlesioned and those containing FB only as lesioned. This was the basis for the determination of the “lesion area” (see Analysis of Axotomy and Survival).
For antibody or neurotrophic factor application to axotomized CSNs, a 30 gauge steel cannula connected to an osmotic minipump (Alzet 2001) via a silicone tube was implanted intraparenchymally into the lesion area of the cortex on the lesion side. The pumps delivered either the rabbit affinity-purified BDNF-neutralizing rabbit antibody RAB [170 μg in sodium phosphate buffer (PBS)], vehicle solution (PBS), human recombinant GDNF (Amgen; 4 or 40 μg) in vehicle or RAB solution, human recombinant NT-3 (Regeneron Pharmaceuticals; 100 or 200 μg) in vehicle or RAB solution, or rabbit anti-turkey IgG [RIgG; Sigma (St. Louis, MO); 1 mg/ml in PBS] over 7 d. All solutions contained penicillin/streptomycin in a final concentration of 50 U/ml. The properties and specificity of the RAB antibody are described elsewhere (Yan et al., 1997a). The concentration of the respective neurotrophic factors was selected on the basis of the best survival promotion for axotomized CSNs (for NT-3, see Giehl and Tetzlaff, 1996; for GDNF, seeGiehl et al., 1997). The infusion rate was 1 μl/hr.
Tissue processing. Seven days after the lesion, the animals were killed by an overdose of sodium pentobarbital and transcardially perfused with PBS followed by a 4% solution prepared from paraformaldehyde (PFA) in 0.1 m sodium phosphate, pH 7.2. The brains were post-fixed for 1 hr in 4% PFA, cryoprotected in 20% sucrose in PBS for 12 hr, and then frozen in dry ice-cooled isopentane. For cell counts, the brains containing the sensory motor cortex area were cut into 20 μm cryostat serial coronal sections.
For in situ hybridization, 20 μm cryostat serial coronal sections were collected. Every fifth section was collected separately to determine the localization of the lesion area based on the absence of the second tracer RDX/RITC. Only sections from the center of the lesion area, where all CSNs were lesioned on the lesion side, were used for in situ hybridization. It was necessary to determine the precise extent of the lesion area on separate sections because the second tracer does not withstand the in situ hybridization procedure.
Analysis of axotomy and survival. Every tenth 20 μm cryostat section was evaluated by cell counting performed blindly by a person who was unaware of the treatment given to the animal. The criterion for a CSN was an FB-filled pyramidal-shaped profile >4 μm in diameter. This criterion was used because cresyl violet counterstaining revealed that axotomized fast blue-filled profiles of this size but not of smaller size still contained nuclear condensation, indicating CSNs in a shrunken stage. Smaller fast blue-labeled profiles were regarded as dendritic processes from adjacent sections. The largest diameter of the neurons was determined by cross-sectional area measurement with the image analysis system Digger (MEDVIS-Medical Vision Systems, Homburg/Saar, Germany), which yields largest and smallest diameters that are correlated to a scale in the ocular to allow determination of cell size in critical cases during counting. The lesion area of the cortex on the lesion side was defined by the absence of the second tracer RDX/RITC. Cell counts from the areas anterior and posterior to the lesion area confirmed a well balanced labeling of CSNs. An area that spans the posterior two-thirds of the lesion area, minus 800 μm from the extreme posterior end of this area, was defined as “cell death area.” This cell death area is the area where dying CSNs were regularly observed in both lesion-only and lesion-plus-vehicle animals (“vehicle”). This area was used as an anatomical mask for the RAB, rabbit anti-turkey IgG, or neurotrophic factor-treated animals to obtain the respective survival data. Within the cell death area, percent survival is defined as “number of FB-labeled CSNs on the lesion side/number of FB-labeled CSNs contralateral to the lesion side × 100%.” The data are based on a total of >280,000 cells counted in all experiments. One-way ANOVA, which was followed by a post hoc Newman–Keuls test and a post hoc Fisher’s least significance difference test, was used to determine the statistical significance of differences in survival among the individual experimental groups.
In situ hybridizations. In situhybridizations were performed as described elsewhere (Giehl and Mestres, 1995). Oligonucleotide probes for the individual mRNAs were labeled with 35S-dATP (DuPont NEN, Wilmington, DE), using terminal deoxynucleotide transferase (Life Technologies, Gaithersburg, MD). The BDNF probe was a 40-mer oligonucleotide, complementary to the nucleotides 535–574 of the rat BDNF mRNA (Maisonpierre et al., 1991); the trkB probe was a 45-mer oligo (Giehl and Tetzlaff, 1996), complementary to nucleotides 1363–1407 of the rat trkB mRNA sequence (Middlemas et al., 1991), hence lying within the tyrosine kinase domain; and the GFR-α-1 probe was complementary to nucleotides 1109–1148 of the rat GFR-α-1 mRNA (Jing et al., 1996). Thus, at the high stringency used, the probes are specific for the BDNF, trkB–tyrosine kinase domain, or GFR-α-1 mRNA sequences, respectively. The specificity of all probes was confirmed by Northern blots (data not shown). The controls of the in situhybridization were performed by competition with an excess of cold probe, which resulted in grain densities below background staining (data not shown). In addition, each probe resulted in staining patterns consistent with the literature. Post-hybridization washes and autoradiography were performed as described elsewhere (Giehl and Mestres, 1995). A 3 week exposure time to the Kodak NTB 2 photoemulsion was used in these experiments.
Quantification of mRNA expression. To quantify BDNF, trkB, or GFR-α-1 mRNA expression in CSNs, the grain densities over fast blue-labeled CSNs were measured with the aid of the image analysis system DIGGER on unstained sections. The quantification was performed according to a standard procedure for autoradiographic in situ hybridization images (McCabe et al., 1989). For each quantification step, two images were loaded into two separate buffers of the image analysis system. The first buffer contained the fluorescent image of fast blue-labeled CSNs; the second buffer contained the dark-field image of the respective autoradiography at the same position of the section. The fast blue-labeled neurons were traced in the first buffer to create a mask that was automatically used for the quantification of grain densities in the second buffer at the corresponding position in the dark-field image. Hence, the user interaction was limited to tracing the fast blue-labeled CSNs without seeing the silver grains in the second image buffer. CSNs were traced along their perikaryal borders, excluding the apical dendrites. The linearity of grain measurements by the image analysis system used was verified by comparing the number of directly counted, not computer assisted, grains over CSNs with the grain density value of the same cell obtained by the image analysis system (McCabe et al., 1989). In various independent samples, the correlation coefficient of the two techniques was r = 0.96 (data not shown).
The grain densities of the CSNs were expressed as an x-fold of the mean background grain density. To account for unspecific and chemographic effects of the tissue whose background is always slightly higher than the grain density over the slide (Rogers, 1979; Harlan et al., 1987), background grain density was measured in an area of the section that did not contain positively labeled cells, i.e., cortical layer I for BDNF, trkB, and GFR-α-1 mRNA (Ernfors et al., 1990;Kokaia et al., 1993a; Miranda et al., 1993; Altar et al., 1994;Treanor et al., 1996; Conner et al., 1997). The mean background grain density value was determined for each specimen by 50 independent background measurements. Each measurement analyzed automatically the grain density in a circle 20 μm in diameter. A circle with this diameter was chosen because it has the average cross-sectional area of a CSN (Giehl et al., 1997). Care was taken that these circles did not overlap. The grain densities of each series of background measurement were normally distributed as determined by Kolmogorov–Smirnov test (with Lilliefors correction). Then, 3ς was calculated and expressed as a multiple of the respective mean background grain density to determine the expression level regarded as positive for CSNs. Among all background measurements, 3ς varied between 1.46- and 2.83-fold of the respective mean background grain densities. Thus, the criterion of a CSN expressing the respective mRNA was set as “more than threefold of the mean background grain density,” because this is outside the 3ς interval. A threshold of three times more than background has been well established for quantitative radioactive in situhybridization histochemistry as being stringent enough to eliminate unspecific labeling and sensitive enough to minimize false negative results (McCabe et al., 1989).
As determined by cresyl violet counterstaining of some sections, the grain clouds slightly exceeded the borders of neuronal cells, which is caused by the thickness of the sections. The section thickness of 20 μm was chosen to minimize the loss of tracer during the in situ hybridization procedure (Schwaber et al., 1989; Giehl and Mestres, 1995), which is important because the identification of a CSN is based solely on its fast blue content.
Six animals that received a unilateral CSN lesion at internal capsule level were used for the in situ hybridization analysis; five of them were analyzed for BDNF and GFR-α-1 mRNA expression, and all six were used for the determination of trkB mRNA expression. The data are based on a total of >7000 cells. One-way ANOVA, which was followed by a post hoc Newman–Keuls test and a post hocFisher’s least significance difference test, was used to determine the statistical significance of differences between lesion and control side.
RESULTS
Endogenous BDNF supports survival of axotomized CSNsin vivo
Endogenous cortical BDNF may be a physiological survival factor for adult CSNs because BDNF mRNA and protein are expressed in the adult cortex (Ernfors et al., 1990; Kokaia et al., 1993a; Miranda et al., 1993; Ringstedt et al., 1993; Conner et al., 1997; Yan et al., 1997a) and intracortical administration of BDNF promotes survival of adult axotomized CSNs (Giehl and Tetzlaff, 1996). To test this hypothesis, the BDNF-neutralizing antibody RAB was intraparenchymally infused to the cell bodies of axotomized or unlesioned CSNs for 7 d. The rabbit affinity-purified anti-BDNF antibody RAB has recently been shown to be highly specific for BDNF and to potently neutralize the biological effects of BDNF in vitro (Yan et al., 1997a).
Application of RAB significantly decreased the survival of axotomized CSNs (39 ± 4%, mean survival ± SEM; n = 6) as compared with untreated axotomized CSNs (lesion only) (53 ± 3%; n = 8), vehicle-treated (69 ± 3%;n = 4), or RIgG-treated axotomized CSNs (65 ± 3%; n = 5) (Figs. 1, 2). Because the treatment of axotomized CSNs with vehicle or control IgG resulted in enhanced survival of CSNs, suggesting the release of endogenous survival-promoting factor(s) by the infusion, the effect of RAB treatment should be compared with vehicle or control IgG. The treatment of RAB resulted in an additional 26–30% of CSN degeneration after the axotomy. In contrast, RAB infused to unlesioned CSNs did not cause CSN death (97 ± 4%; n = 3) (Fig.2). Vehicle and RIgG treatment did not result in different survival of CSNs as determined by post hoc Fisher’s least significance difference test (p < 0.05). Thus, endogenous cortical BDNF is important for the survival of axotomized CSNs.
Fig. 1.

Neutralization of endogenous BDNF increases death of axotomized CSNs, which can be counteracted by simultaneously applied NT-3 but not by GDNF. Photomicrographs of FB-fluorescent CSNs at the lesion (A, C, E, G, I) and control side (B, D, F, H, J) of representative animals. Sections of (A, B) vehicle-, (C, D) RAB-, (E, F) RAB plus 200 μg NT-3-, (G, H) RAB plus 40 μg GDNF-, and (I, J) 40 μg GDNF-treated animal whose CSNs were unilaterally axotomized at internal capsule levels. Survival time after lesion and treatment period was 7 d. Treatment was given on the lesion side. Axotomy of CSNs at internal capsule levels (A) induces death of a substantial population of CSNs as compared with the control side (B).C, After treatment with RAB, the number of CSNs surviving their axotomy is markedly decreased (compare withA and D). E, A combination of RAB and 200 μg NT-3 completely counteracts RAB-induced CSN death and results in complete rescue of CSNs from axotomy-induced death (compare with F). In contrast, (G) 40 μg of GDNF was not able to prevent RAB-induced death, whereas (I) 40 μg of GDNF alone completely prevent axotomy-induced death, indicating that the survival promotion of GDNF is mediated by endogenous BDNF.
Fig. 2.

Quantification of CSN survival 7 d after axotomy (mean survival ± SEM are indicated). The differences of the mean values among the individual treatment groups were highly significant using one-way ANOVA (p < 0.001). Treatment of axotomized CSNs with BDNF-neutralizing RAB decreases [39 ± 4% survival, n = 6,p < 0.01 vs lesion only (l.o.), vehicle, and RIgG as determined by post hocNewman–Keuls test, **] their survival as compared with l.o. (53 ± 3%; n = 8), vehicle (69 ± 3%;n = 4), or rabbit anti-turkey IgG (RIgG) (65 ± 3%; n = 5). In contrast, RAB infused to unlesioned CSNs (RAB-only) did not cause CSN death (97 ± 4%; n = 3). Administration of vehicle was not significantly different (n.s.) from RIgG as determined with post hoc Fisher’s least significance difference test. NT-3 prevented RAB-induced death and promoted survival of axotomized CSNs in a dose-dependent manner at total doses of 100 μg (69 ± 2%, n = 4,p < 0.01 vs RAB and l.o. as determined bypost hoc Newman–Keuls test, **) and 200 μg (106 ± 10%, n = 2, p < 0.01 vs RAB, l.o., vehicle, and RIgG as determined with post hocNewman–Keuls test, **). In contrast, GDNF did not affect RAB-induced death at a total dose of 4 μg (n.s. vs RAB as determined withpost hoc Fisher’s least significance difference test,p < 0.01 vs vehicle or RIgG as determined bypost hoc Newman–Keuls test) and 40 μg (n.s. vs RAB as determined with post hoc Fisher’s least significance difference test, p < 0.05 vs vehicle or RIgG as determined by post hoc Newman–Keuls test) over 7 d. CSN survival in vehicle (n = 4) and RIgG (n = 5) animals was highly significantly increased as compared with lesion only (p < 0.01 as determined by post hoc Newman–Keuls test), confirming our previous finding of a vehicle effect on survival of axotomized CSNs (Giehl and Tetzlaff, 1996).
NT-3, but not GDNF, counteracts RAB-induced death of axotomized CSNs
Application of BDNF (Giehl and Tetzlaff, 1996), NT-3 (Giehl and Tetzlaff, 1996), or GDNF (Giehl et al., 1997) can completely prevent axotomy-induced death of CSNs in vivo. The question arises whether these factors can compensate for each other with regard to the promotion of the survival of CSNs. To test whether one neurotrophic factor may prevent the death of CSNs attributable to the neutralization of another, NT-3 or GDNF was infused simultaneously with RAB to axotomized CSNs.
For the combined application of NT-3 with RAB, a dose of 100 μg of NT-3 over 7 d was chosen because this dose had complete rescue effects on axotomized CSNs as demonstrated previously (Giehl and Tetzlaff, 1996). It has been shown before that RAB displays a very minor cross-reactivity to NT-3 (<0.5%) (Yan et al., 1997a). This may diminish the biological effectiveness of NT-3 within the cortical neuropil, which displays abundant trkC expression (Merlio et al., 1992;Kokaia et al., 1993a; Ringstedt et al., 1993; Altar et al., 1994). Therefore, we infused NT-3 at a dose of 200 μg in combination with RAB for 7 d in a different set of experiments. Application of NT-3 was able to counteract RAB-induced death of axotomized CSNs in a dose-dependent manner. Although 100 μg NT-3 increased CSN survival to 69 ± 2% (n = 4), the higher dose of 200 μg NT-3 resulted in complete CSN survival with 106 ± 10% (n = 2) (Figs. 1, 2). Because 100 or 200 μg NT-3, respectively, was infused simultaneously with 170 μg RAB, it is unlikely that NT-3 would significantly influence the biological activity of RAB at these doses. Thus, NT-3 is able to compensate for the neutralization of endogenous BDNF regarding the survival of axotomized CSNs. Because the lower dose of NT-3 did not rescue all CSNs in the presence of RAB, the biological activity of this NT-3 batch was assayed by infusing it alone to axotomized CSNs at a total dose of 100 μg for 7 d. After this treatment, 95 ± 4% (n = 4) of CSNs survived their axotomy. This confirmed the high biological potency of the NT-3 used for the present experiments.
The GDNF survival promotion of axotomized CSNs is maximal at doses of 4 or 40 μg over 7 d (Giehl et al., 1997). We therefore infused either 4 or 40 μg GDNF in combination with RAB to axotomized CSNs for 7 d. Doses of 100 μg or higher result in reduced CSN survival not distinguishable from vehicle and induce severe body weight reduction of the animals (Giehl et al., 1997). Animals that received GDNF intracortically at doses between 4 and 40 μg did not display body weight reductions (Giehl et al., 1997). Thus, we regarded higher doses of GDNF as unsuitable in this context. Neither 4 nor 40 μg GDNF had a statistically significant effect on RAB-induced death of CSNs with 47 ± 4% (n = 5) and 49 ± 5% (n = 4) CSN survival, respectively, as determined bypost hoc Fisher’s least significance difference test (p < 0.05) (Figs. 1, 2). In addition, the combination of GDNF with RAB was statistically different from vehicle, as determined by post hoc Newman–Keuls test at both the 4 μg (p < 0.01) and 40 μg (p < 0.05) GDNF doses. Because RAB does not display cross-reactivity to GDNF (Yan et al., 1997a), this finding is unlikely to be attributable to the interaction between GDNF and RAB. To verify the biological activity of the GDNF used for these experiments, GDNF from the same batch was infused intracortically to lesioned CSNs of a separate group of rats at a total dose of 40 μg for 7 d. This treatment completely rescued axotomized CSNs (97 ± 1%;n = 2), confirming the biological activity of the GDNF sample used in these experiments, as well as our previous observation that GDNF promotes CSN survival (Giehl et al., 1997). These data indicate that the rescue effect of GDNF on CSNs is mediated by endogenous BDNF.
Expression of BDNF, trkB, and GFR-α-1 mRNA in CSNs
Endogenous BDNF may support survival of axotomized CSNs by an autocrine or paracrine mechanism. Assuming an autocrine support, one would expect that axotomized CSNs express BDNF mRNA and the trkB receptor. We thus analyzed BDNF mRNA and trkB mRNA expression in fast blue-labeled CSNs using quantitative in situ hybridization with radiolabeled oligonucleotides. Both lesioned and unlesioned CSNs were examined to reveal potential differences related to the axotomy. Many CSNs express BDNF mRNA, and almost all of them express trkB mRNA (Fig. 3). The quantification of thein situ hybridizations revealed that almost half of the CSNs express BDNF mRNA, and no differences exist between unlesioned (48.1 ± 6%; n = 5) and axotomized (48.8 ± 5%; n = 5) CSNs (Fig.4). Also, the percentage of trkB-expressing CSNs did not differ between unlesioned (88.7 ± 3%; n = 6) and lesioned (89.4 ± 2%;n = 6) CSNs (Fig. 4). The expression of BDNF mRNA seems to be most prominent in layer V cells adjoining CSNs, whereas trkB mRNA expression is very pronounced in CSNs (Fig. 3). The expression of both trkB and BDNF in the cortex suggests that both autocrine and paracrine mechanisms could be involved in the trophic influence on CSNs.
Fig. 3.
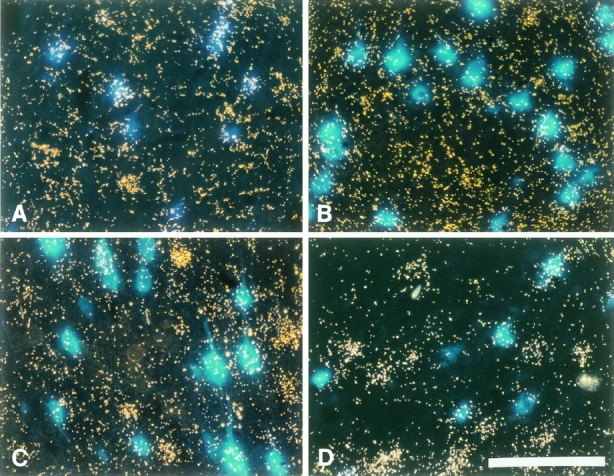
Expression of trkB, GFR-α-1, and BDNF mRNA in cortical layer V. Photomicrograph of FB-labeled CSNs under UV illumination in combination with dark field to simultaneously visualize FB-labeled CSNs and the silver grains produced in the photoemulsion after radioactive in situ hybridization for the respective mRNAs. CSNs appear as blue cells and those expressing a specific mRNA are covered by a cloud of silver grains. Survival time after lesion was 7 d. A, Full-length trkB mRNA is expressed in virtually all axotomized CSNs. Also many neighboring cells that do not contain FB express trkB mRNA. B, GFR-α-1 mRNA expression in several unlesioned CSNs and noncorticospinal cells within neocortical layer V. C, D, BDNF mRNA expression in several CSNs of the (C) control and (D) lesion side of representative lesion-only animals. Note that BDNF mRNA is most prominently expressed by noncorticospinal cells of layer V. Scale bar, 100 μm.
Fig. 4.

Quantification of BDNF, trkB, and GFR-α-1 mRNA expression in unlesioned and lesioned CSNs. Survival time after lesion was 7 d. Black bars represent axotomized CSNs;gray bars represent unlesioned CSNs. A, The percentage of BDNF-expressing CSNs was not altered by axotomy: 48 ± 6% (mean + SEM; n = 5) unlesioned and 49 ± 5% (n = 5) axotomized CSNs express BDNF mRNA. Virtually all CSNs express trkB mRNA regardless of whether they are unlesioned (89 ± 3%; n = 6) or axotomized (89 ± 2%; n = 6). In contrast, the percentage of GFR-α-1-expressing CSNs was higher in axotomized (65 ± 4%, n = 5, p < 0.05 as determined by post hoc Newman–Keuls test) than in unlesioned (41 ± 8%; n = 5) CSNs. (B) The mRNA levels in axotomized CSNs expressing the respective mRNAs are expressed as percent expression of unlesioned CSNs of the respective contralateral control sides. This analysis revealed that BDNF (114 ± 15%; n = 5) and GFR-α-1 mRNA (123 ± 12%; n = 5) levels in CSNs are slightly increased by axotomy, whereas trkB mRNA levels in lesioned CSNs are decreased to 81 ± 7% (n = 6) of unlesioned CSNs.
We further analyzed the in situ hybridization data for expression levels of each mRNA in axotomized CSNs as compared with their unlesioned contralateral counterparts. This revealed that BDNF expression in axotomized CSNs is slightly elevated to 114 ± 15% (n = 5) of the CSNs of the control side (Fig. 4). In contrast, trkB levels were decreased to 81 ± 7% (n = 6). The observation that the percentage of trkB-expressing CSNs did not increase after axotomy suggests that the endogenous BDNF is not the exclusive factor playing a role in the trophic support of axotomized CSNs.
We have shown above that the support of axotomized CSNs by exogenously applied GDNF depends critically on endogenous BDNF. To further examine the possibility that GDNF might interact directly with CSNs, the expression of GDNF receptor component α (GFR-α-1) mRNA (Jing et al., 1996; Treanor et al., 1996) was assessed in unlesioned and axotomized CSNs by radioactive oligonucleotide in situhybridization. Several cells in layer V, including a substantial population of CSNs, express mRNA for GFR-α-1 (Fig. 3). The percentage of CSNs expressing this mRNA was significantly increased by the axotomy: 41.3 ± 8% (n = 5) of unlesioned and 65.2 ± 5% (n = 5) of lesioned CSNs expressed GFR-α-1 mRNA (Fig. 4). Similarly, the levels of GFR-α-1 mRNA per cell were higher in axotomized CSNs (123 ± 13% of the unlesioned CSNs; n = 5) (Fig. 4). These results suggest that a subpopulation of CSNs could potentially respond directly to GDNF. However, because the expression of RET, the signal transduction component of the GDNF receptor complex (Jing et al., 1996; Treanor et al., 1996), was not examined in this study, it remains to be shown whether GDNF can directly influence CSNs.
DISCUSSION
We have shown here that endogenous cortical BDNF supports the survival of axotomized adult rat CSNs in vivo. The CSN death caused by the neutralization of endogenous BDNF was prevented by NT-3, indicating that NT-3 can compensate for BDNF regarding survival promotion of axotomized CSNs. In addition, we have shown that the previously described GDNF effect on CSN survival is mediated via endogenous BDNF.
Endogenous cortical BDNF supports survival of lesioned CSNsin vivo
The additional cell death of lesioned CSNs after intracortical infusion of RAB has been interpreted to reflect the neutralization of endogenous BDNF. Several lines of evidence exclude the theoretical possibility that this is the result of activation of immune cells or the complement system through RAB immunoglobulins. First, RAB did not induce death of unlesioned CSNs. Second, RAB antibody is affinity-purified and does not contain a serum complement system. Finally, infusion of normal rabbit IgG to axotomized CSNs did not result in enhanced death of CSNs as compared with vehicle treatment. We conclude that the RAB-induced death of axotomized CSNs is the direct result of the neutralization of endogenous cortical BDNF.
Because not all of the axotomized CSNs die after RAB treatment, although almost all of them express trkB, endogenous NT-3, which is also expressed in the cortex (Ernfors et al., 1990; Maisonpierre et al., 1990; Miranda et al., 1993), or some other unidentified factors, could mediate this partial protection from RAB-induced death. Alternatively, the resistance of this CSN population to RAB treatment could be caused by incomplete neutralization of endogenous BDNF by RAB. Although RAB has been shown to potently neutralize BDNF in vitro (Yan et al., 1997a), it is hard to exclude the possibility that some endogenous BDNF is sequestered and not neutralizable by RAB infusions. It is unlikely, however, that a limited diffusion of RAB within the lesion area caused this phenomenon, because none of the sections displayed a gradient regarding survival of CSNs and the extension of the RAB infusion area was between 7 and 8 mm as determined by immunohistochemistry (data not shown). Finally, it is possible that the CSNs surviving RAB treatment receive BDNF support via long collaterals that reach cortical areas outside the RAB infusion area, because intracortical collaterals are spared by the internal capsule lesion.
Vehicle- or RIgG-treated animals displayed significantly more survival of axotomized CSNs than lesion-only animals, confirming our previous observation of a vehicle effect on CSN survival (Giehl and Tetzlaff, 1996; Giehl et al., 1997). This effect may be caused by increased CNTF expression around the intracortically implanted osmotic minipump applicator, because CNTF production is increased around stab wounds (Ip et al., 1993) and CNTF rescues axotomized CSNs in vivo (Dale et al., 1995). Because cortical BDNF expression is inducible by several depolarizing stimuli (Kokaia et al., 1993b, 1994), this survival may also be caused by upregulation of cortical BDNF through the continuous liquid flow of the pumps (Giehl and Teztlaff, 1996). The latter possibility is strongly supported by the fact that neutralization of endogenous cortical BDNF completely prevented the vehicle effect. This further emphasizes the importance of endogenous BDNF in supporting survival of axotomized CSNs. It is important to note that only 39% of axotomized CSNs do not depend on endogenous cortical BDNF for survival. Without treatment, 47% of CSNs die, and an additional 14% die if BDNF is neutralized. The survival of CSNs can be increased, e.g., by vehicle (an additional 16%) or GDNF treatment (an additional 44%, i.e., complete survival), which is completely prevented by RAB infusions. Thus, survival of at least 58% of axotomized CSNs can be promoted by endogenous cortical BDNF. This indicates that BDNF is a crucial endogenous survival factor for lesioned CSNs.
Neutralization of cortical BDNF did not affect the survival of unlesioned CSNs. RAB infusion also did not influence CSN cell size as determined by quantitative cross-sectional area measurements (data not shown). This shows that only acutely damaged CSNs critically depend on endogenous cortical BDNF. It is conceivable that unlesioned CSNs can receive sufficient trophic support from their brainstem or spinal cord targets, even when cortical BDNF is neutralized by RAB. This trophic support is not possible for axotomized CSNs because they are disconnected from their targets.
Although BDNF and several other neurotrophic factors can influence survival and differentiation of developing cortical neurons in vitro (Ghosh et al., 1994; Ghosh and Greenberg, 1995) and adult CSNs under lesion condition (Giehl and Tetzlaff, 1996; Giehl et al., 1997), it is still not clear whether these factors play a physiological role for CSNs in vivo during development and adulthood. For example, the BDNF (Ernfors et al., 1994a,b; Jones et al., 1994), GDNF (Moore et al., 1996; Pichel et al., 1996), or NT-3 (Ernfors et al., 1994a,b) knock-out mice do not display gross morphological alterations in the cortex. However, the cortical size is reduced and lamination less marked in BDNF and NT-3 knockout mice (Chen et al., 1997). Mice overexpressing BDNF under a nestin promotor display a deformed cortex and disturbance of the laminar architecture (Ringstedt et al., 1997). Because CSNs are a subpopulation of cortical neurons, a specific determination of CSN numbers and localization in the respective knock-out mouse and in mice that overexpress one of these factors would be important to elucidate the significance of these factors for CSN development.
BDNF influences CSNs through an autocrine/paracrine mechanism
The colocalization of BDNF and trkB mRNA in many areas of the CNS and peripheral nervous system during development and in adults leads to the hypothesis that this neurotrophic factor has autocrine or paracrine maintenance function for various neuronal populations in addition to its role as a target-derived neurotrophic factor (Schecterson and Bothwell, 1992; Miranda et al., 1993; Altar et al., 1994; Cohen-Cory and Fraser, 1994; Kokaia et al., 1995). This idea was strongly supported by the evidence that (1) a subpopulation of adult rat DRG depends on a BDNF autocrine loop in vitro(Acheson et al., 1995), (2) endogenous cortical BDNF supports survival of almost all cortical neurons in cultures from embryonic rats (Ghosh et al., 1994), and (3) autocrine/paracrine BDNF action promotes survival and differentiation of Xenopus retinal ganglion cells in vitro (Cohen-Cory et al., 1996). During development and in adults, BDNF and trkB mRNA are expressed in several neocortical layers, including layer V (Ernfors et al., 1990; Kokaia et al., 1993a; Miranda et al., 1993; Ringstedt et al., 1993; Altar et al., 1994; Conner et al., 1997; Yan et al., 1997a,b). We have shown previously that unlesioned adult CSNs express full-length trkB mRNA and intracortical infusion of BDNF prevents the axotomy-induced death of CSNs (Giehl and Tetzlaff, 1996). Thus, autocrine/paracrine BDNF mechanisms may play a role in the maintenance of adult CSNs. Our results in this study suggest that this is the case for axotomized CSNs. It is very difficult to distinguish between autocrine and paracrine action of BDNF. By definition, autocrine neurotrophic support can but may not involve protein secretion, whereas paracrine action requires secretion of the respective factor. Intracortical RAB infusions could thus neutralize autocrine and paracrine BDNF support to the CSNs. As shown here, expression of BDNF is most prominent in noncorticospinal cells of cortical layer V, indicating that there are potential sources of BDNF for a paracrine CSN support. Furthermore, because virtually all CSNs express trkB mRNA, whereas only half of them express BDNF, some of the CSNs must receive BDNF support through a paracrine mechanism. For further differentiation between paracrine and autocrine mechanisms, studies will be required that examine the sites of secretion and binding of endogenous cortical BDNF.
The effect of GDNF on lesioned CSNs requires the presence of endogenous BDNF
Although GDNF, a member of the TGFβ-1 family of growth factors (Lin et al., 1993), completely prevents axotomy-induced death of CSNs (Giehl et al., 1997), it did not prevent axotomy-induced CSN death in the presence of RAB. As shown by Western blotting, RAB does not display any cross-reactivity with GDNF (Yan et al., 1997a). Thus, RAB cannot interfere directly with the biological activity of GDNF. To further exclude any possibility that RAB might influence a GDNF survival promotion effect by a mechanism different from direct antibody/ligand interaction, we locally applied a combination of GDNF with RAB at ratios/concentrations identical to those above to the transected newborn facial nerve. Newborn facial motoneurons massively die after axotomy, which is completely prevented by GDNF (Yan et al., 1995). The combination of GDNF/RAB completely prevented lesion-induced death of newborn facial motoneurons to the same extend as GDNF alone (data not shown), indicating that GDNF is fully biologically active in the presence of RAB. By immunohistochemistry, we have determined that GDNF diffuses 2–3 mm in the cortex at a total dose of 4 μg and more than 5 mm at a total dose of 40 μg over 7 d [Giehl et al. (1997) and our unpublished data]. At least at the high GDNF concentrations, all CSNs subjected to axotomy were localized within the GDNF diffusion area. Thus, limited diffusion of GDNF also cannot account for the lack of GDNF survival promotion when RAB was applied simultaneously. Our data suggest, therefore, that the survival promotion of axotomized CSNs by GDNF requires the presence of endogenous cortical BDNF.
It will be important to determine whether GDNF stimulates cortical BDNF and/or trkB expression or enhances secretion of BDNF protein. Cortical BDNF expression is stimulated by several stimuli (Kokaia et al., 1993b,1994), and neurotrophin release can be mediated by neurotrophins in PC12 cells (Canossa et al., 1997; Krüttgen et al., 1997). However, an examination of the potential role of GDNF regarding these mechanisms is beyond the scope of this study. Alternatively, GDNF promotion of CSN survival may require a baseline activity of BDNF signaling pathways. Both GDNF (Creedon et al., 1997; Hiwasa et al., 1997; Ibanez and Trupp, 1997; Wang et al., 1998) and BDNF (Ip and Yancopoulos, 1996; Segal and Greenberg, 1996; Kaplan and Miller, 1997) can activate the Ras-MAP and PI3 kinase pathways. A convergence, interaction, or synergium of GDNF and BDNF signals at an intracellular level regarding the survival promotion of axotomized CSNs therefore is conceivable.
NT-3 can compensate the endogenous BDNF for lesioned CSNs
Consistent with the trkC expression in most CSNs (Giehl and Tetzlaff, 1996), NT-3 treatment was able to prevent the RAB-induced CSN death. However, a dose of NT-3 (100 μg over 7 d) that results in complete rescue of axotomized CSNs [Giehl and Tetzlaff (1996) and this study] was not able to exert the same quantity of biological effect in the presence of RAB. One possibility for this difference is that RAB has cross-reactivity to NT-3. However, as shown by in vitrostudies and in Western blots (Yan et al., 1997a), RAB displays only 0.5% cross-reactivity to NT-3. At the antibody and NT-3 concentrations used in the low- and high-dose NT-3/RAB combination experiments, the molar ratio was 1:3 and 1:6, respectively, i.e., maximum 1.5% and 3%, respectively, of RAB could theoretically have been sequestered by NT-3. Thus, a minimum of 97% of RAB was free to interact with endogenous BDNF. We further conducted RAB immunohistochemistry with NT-3-preincubated RAB at a molar ratio of 10:1 on forebrain sections, which yield a distribution of BDNF-like immunoreactivity as reported elsewhere (data not shown) (Yan et al., 1997a). This shows that there is sufficient RAB antibody left in the used combinations with NT-3 to recognize BDNF epitopes. In addition, it is most likely that a significant portion of NT-3, which displays much lower affinity to RAB than BDNF, was still free because the survival effects on axotomized CSNs were very pronounced. Another possibility for the reduced NT-3 effects observed in this study is that NT-3 may not reach all of the lesioned CSNs because its diffusion within the brain parenchyma is limited. It has been shown previously that the NT-3 diffusion area within the brain parenchyma has a diameter of ∼4 mm at comparable concentrations (Kobayashi et al., 1997). Thus, the access of NT-3 to lesioned CSNs should not be a problem. The likely explanation is that the complete rescue of axotomized CSNs by NT-3 alone is partially mediated via endogenous BDNF, which would not be possible in the presence of RAB. In addition, NT-3 might promote CSN survival directly via trkB because it also binds, although less avidly than BDNF, to trkB receptor (Davies et al., 1995; Ryden and Ibanez, 1996). The contribution of direct NT-3 action on trkB is supported by our observation that doubling the amount of NT-3 infused simultaneously with RAB resulted in complete rescue of axotomized CSNs.
The expression of BDNF, trkB, and GFR-α-1 mRNA in CSNs in response to axotomy
Almost all axotomized and unlesioned CSNs express trkB mRNA. However, the levels of trkB mRNA is decreased in axotomized CSNs. The percentage of BDNF-expressing CSNs was not changed after axotomy, and the level of BDNF mRNA was not significantly increased. This is in contrast to the situation in the peripheral nervous system. In peripheral BDNF-responsive neurons (Sendtner et al., 1992; Yan et al., 1992; Koliatsos et al., 1993; Acheson et al., 1995), trkB or BDNF mRNA is frequently strongly increased after axotomy (Ernfors et al., 1993;Sebert and Shooter, 1993; Piehl et al., 1994, Kobayashi et al., 1996). These neurons survive axotomy (Lowrie and Vrbova, 1992), and protection by endogenous BDNF is proposed to account for this survival (Ernfors et al., 1993; Sebert and Shooter, 1993; Piehl et al., 1994, Kobayashi et al., 1996). CSNs, however, like many other central neurons, die after axotomy (Aguayo et al., 1991; Tetzlaff et al., 1994; Giehl and Tetzlaff, 1996). This could be attributable to the fact that CSNs fail to upregulate BDNF or trkB expression in response to axotomy and therefore do not receive sufficient endogenous BDNF protection. In contrast, strong upregulation of trkB and BDNF mRNA in cortical and hippocampal areas is demonstrated after brain insults and seizures, and a role for BDNF in protecting neurons in these areas has been discussed extensively (Kokaia et al., 1993b, 1994, 1995; Acheson and Lindsay, 1996).
Interestingly, axotomy increased both the portion of GFR-α-1-expressing CSNs and the GFR-α-1 mRNA levels in these cells. Because GDNF is expressed in the neocortex (Schaar et al., 1993;Springer et al., 1994; Choi Lundberg and Bohn, 1995) and GDNF promotes survival of axotomized CSNs via BDNF, cortical GDNF could be involved in endogenous protection mechanisms for injured CSNs. GDNF-receptor expression in mouse spinal motoneurons (sMN) seems to be regulated differently after axotomy. Although GFR-α-1 expression remains constant in axotomized sMN, the expression of the GDNF receptor signal transducing component c-ret (Jing et al., 1996; Treanor et al., 1996) is upregulated (Naveilhan et al., 1997). Because the soluble, not GPI membrane-linked form of GFR-α-1 is also able to activate c-ret (Jing et al., 1996; Treanor et al., 1996) and GFR-α-1 is upregulated in the distal nerve stump after axotomy (Naveilhan et al., 1997), it is possible that binding of GDNF/soluble GFR-α-1-complexes to sMN c-ret reflects a potentially increased sensitivity of axotomized sMN to GDNF (Naveilhan et al., 1997). This spatially different expression of the individual GDNF receptor components is consistent with the role of GDNF in nerve regeneration (Naveilhan et al., 1997). It remains a matter of speculation whether the increase of GFR-α-1 expression in axotomized CSNs rather reflects different functions such as survival promotion of GDNF in this system or whether it is merely attributable to a species difference.
In conclusion, endogenous BDNF is an autocrine/paracrine survival factor for axotomized adult rat CSNs in vivo. Because this endogenous BDNF-mediated survival promotion of CSNs can be stimulated and compensated for by different trophic agents, these findings may be important for the development of new therapeutic strategies for the treatment of neurodegenerative diseases such as amyotrophic lateral sclerosis that aim at the stimulation of the endogenous survival potential of neurons.
Footnotes
This study was supported by grants from Amgen Inc. and the Zentrale Forschungskommission der Universität des Saarlandes to K.M.G. and the Deutsche Forschungsgemeinschaft to K.M.G. and P.M. The neurotrophic factors were generously supplied by Amgen Inc., Thousand Oaks, CA, and Regeneron Pharmaceuticals Inc., Tarrytown, NY. We thank Britta Leiner for skillful technical assistance and Holger Summa for help with the figures. We also thank Ann Soether for proofreading this manuscript and Dr. H. M. Cooper for helpful discussion and critical proofreading of this manuscript.
Correspondence should be addressed to Dr. Klaus Giehl, Anatomisches Institut, Universität des Saarlandes, D-66421 Homburg/Saar, Germany.
REFERENCES
- 1.Acheson A, Lindsay RM. Non-target-derived roles of the neurotrophins. Philos Trans R Soc Lond B Biol Sci. 1996;351:417–422. doi: 10.1098/rstb.1996.0037. [DOI] [PubMed] [Google Scholar]
- 2.Acheson A, Conover JC, Fandl JP, DeChiara TM, Russell M, Thadani A, Squinto SP, Yancopoulos GD, Lindsay RM. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374:450–453. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- 3.Aguayo AJ, Rasminsky M, Bray GM, Carbonetto S, McKerracher L, Villegas Perez MP, Vidal Sanz M, Carter DA. Degenerative and regenerative responses of injured neurons in the central nervous system of adult mammals. Philos Trans R Soc Lond B Biol Sci. 1991;331:337–343. doi: 10.1098/rstb.1991.0025. [DOI] [PubMed] [Google Scholar]
- 4.Altar CA, Siuciak JA, Wright P, Ip NY, Lindsay RM, Wiegand SJ. In situ hybridization of trkB and trkC receptor mRNA in rat forebrain and association with high-affinity binding of [125I]BDNF, [125I]NT-4/5 and [125I]NT-3. Eur J Neurosci. 1994;6:1389–1405. doi: 10.1111/j.1460-9568.1994.tb01001.x. [DOI] [PubMed] [Google Scholar]
- 5.Barbacid M. The Trk family of neurotrophin receptors. J Neurobiol. 1994;25:1386–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- 6.Barde YA. Trophic factors and neuronal survival. Neuron. 1989;2:1525–1534. doi: 10.1016/0896-6273(89)90040-8. [DOI] [PubMed] [Google Scholar]
- 7.Canossa M, Griesbeck O, Berninger B, Campana G, Kolbeck R, Thoenen H. Neurotrophin release by neurotrophins: implications for activity-dependent neuronal plasticity. Proc Natl Acad Sci USA. 1997;94:13279–13286. doi: 10.1073/pnas.94.24.13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen S, Liebl DJ, Parada LF, Llinas R, Hillman D. Marked developmental impairment in the forebrain and diencephalon of BDNF and NT-3 knockout mice. Soc Neurosci Abstr. 1997;23:564.5. [Google Scholar]
- 9.Choi Lundberg DL, Bohn MC. Ontogeny and distribution of glial cell line-derived neurotrophic factor (GDNF) mRNA in rat. Brain Res Dev Brain Res. 1995;85:80–88. doi: 10.1016/0165-3806(94)00197-8. [DOI] [PubMed] [Google Scholar]
- 10.Chou SM. Pathology of motor system disorders. In: Leigh PN, Swash M, editors. Motor neuron disease, biology and management. Springer; New York: 1995. pp. 53–92. [Google Scholar]
- 11.Cohen-Cory S, Fraser SE. BDNF in the development of the visual system of Xenopus. Neuron. 1994;12:747–761. doi: 10.1016/0896-6273(94)90328-x. [DOI] [PubMed] [Google Scholar]
- 12.Cohen-Cory S, Escandon E, Fraser SE. The cellular patterns of BDNF and trkB expression suggest multiple roles for BDNF during Xenopus visual system development. Dev Biol. 1996;179:102–115. doi: 10.1006/dbio.1996.0244. [DOI] [PubMed] [Google Scholar]
- 13.Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Creedon DJ, Tansey MG, Baloh RH, Osborne PA, Lampe PA, Fahrner TJ, Heuckeroth RO, Milbrandt J, Johnson EM., Jr Neurturin shares receptors and signal transduction pathways with glial cell line-derived neurotrophic factor in sympathetic neurons. Proc Natl Acad Sci USA. 1997;94:7018–7023. doi: 10.1073/pnas.94.13.7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dale SM, Kuang RZ, Wei X, Varon S. Corticospinal motor neurons in the adult rat: degeneration after intracortical axotomy and protection by ciliary neurotrophic factor (CNTF). Exp Neurol. 1995;135:67–73. doi: 10.1006/exnr.1995.1066. [DOI] [PubMed] [Google Scholar]
- 16.Davies AM, Minichiello L, Klein R. Developmental changes in NT3 signalling via TrkA and TrkB in embryonic neurons. EMBO J. 1995;14:4482–4489. doi: 10.1002/j.1460-2075.1995.tb00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ernfors P, Wetmore C, Olson L, Persson H. Identification of cells in rat brain and peripheral tissues expressing mRNA for members of the nerve growth factor family. Neuron. 1990;5:511–526. doi: 10.1016/0896-6273(90)90090-3. [DOI] [PubMed] [Google Scholar]
- 18.Ernfors P, Rosario CM, Merlio JP, Grant G, Aldskogius H, Persson H. Expression of mRNAs for neurotrophin receptors in the dorsal root ganglion and spinal cord during development and following peripheral or central axotomy. Brain Res Mol Brain Res. 1993;17:217–226. doi: 10.1016/0169-328x(93)90005-a. [DOI] [PubMed] [Google Scholar]
- 19.Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994a;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- 20.Ernfors P, Lee KF, Kucera J, Jaenisch R. Lack of neurotrophin-3 leads to deficiencies in the peripheral nervous system and loss of limb proprioceptive afferents. Cell. 1994b;77:503–512. doi: 10.1016/0092-8674(94)90213-5. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh A, Greenberg ME. Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron. 1995;15:89–103. doi: 10.1016/0896-6273(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- 23.Giehl K, Mestres P. Somatostatin-mRNA expression in brainstem projections into the medial preoptic nucleus. Exp Brain Res. 1995;103:344–354. doi: 10.1007/BF00241494. [DOI] [PubMed] [Google Scholar]
- 24.Giehl KM, Tetzlaff W. BDNF and NT-3, but not NGF, prevent axotomy induced death of rat corticospinal neurons in vivo. Eur J Neurosci. 1996;8:1167–1175. doi: 10.1111/j.1460-9568.1996.tb01284.x. [DOI] [PubMed] [Google Scholar]
- 25.Giehl KM, Schacht CM, Yan Q, Mestres P. GDNF is a trophic factor for adult rat corticospinal neurons and promotes their long-term survival after axotomy in vivo. Eur J Neurosci. 1997;9:2479–2489. doi: 10.1111/j.1460-9568.1997.tb01665.x. [DOI] [PubMed] [Google Scholar]
- 26.Harlan RE, Shivers BD, Romano GJ, Howells RD, Pfaff DW. Localization of preproenkephalin mRNA in the rat brain and spinal cord by in situ hybridization. J Comp Neurol. 1987;258:159–184. doi: 10.1002/cne.902580202. [DOI] [PubMed] [Google Scholar]
- 27.Hiwasa T, Kondo K, Hishiki T, Koshizawa S, Umezawa K, Nakagawara A. GDNF-induced neurite formation was stimulated by protein kinase inhibitors and suppressed by Ras inhibitors. Neurosci Lett. 1997;238:115–118. doi: 10.1016/s0304-3940(97)00861-6. [DOI] [PubMed] [Google Scholar]
- 28.Ibanez CF, Trupp M. On the signalling mechanisms of GDNF receptors. Soc Neurosci Abstr. 1997;23:249.3. [Google Scholar]
- 29.Ip NY, Yancopoulos GD. The neurotrophins and CNTF: two families of collaborative neurotrophic factors. Annu Rev Neurosci. 1996;19:491–515. doi: 10.1146/annurev.ne.19.030196.002423. [DOI] [PubMed] [Google Scholar]
- 30.Ip NY, Wiegand SJ, Morse J, Rudge JS. Injury-induced regulation of ciliary neurotrophic factor mRNA in the adult rat brain. Eur J Neurosci. 1993;5:25–33. doi: 10.1111/j.1460-9568.1993.tb00201.x. [DOI] [PubMed] [Google Scholar]
- 31.Jing S, Wen D, Yu Y, Holst PL, Luo Y, Fang M, Tamir R, Antonio L, Hu Z, Cupples R, Louis J-C, Hu S, Altrock BW, Fox GM. GDNF-induced activation of the Ret protein tyrosine kinase is mediated by GFR-α-1, a novel receptor for GDNF. Cell. 1996;85:1113–1124. doi: 10.1016/s0092-8674(00)81311-2. [DOI] [PubMed] [Google Scholar]
- 32.Jones KR, Farinas I, Backus C, Reichardt LF. Targeted disruption of the brain-derived neurotrophic factor gene perturbs brain and sensory neuron but not motor neuron development. Cell. 1994;76:989–1000. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaplan DR, Miller FD. Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol. 1997;9:213–221. doi: 10.1016/s0955-0674(97)80065-8. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi NR, Bedard AM, Hincke MT, Tetzlaff W. Increased expression of BDNF and trkB mRNA in rat facial motoneurons after axotomy. Eur J Neurosci. 1996;8:1018–1029. doi: 10.1111/j.1460-9568.1996.tb01588.x. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi NR, Fan D-P, Giehl KM, Bedard AM, Wiegand SJ, Tetzlaff W. BDNF and NT-4/5 prevent atrophy of rat rubrospinal neurons after cervical axotomy, stimulate GAP-43 and Tα1-tubulin mRNA expression, and promote axonal regeneration. J Neurosci. 1997;17:9583–9595. doi: 10.1523/JNEUROSCI.17-24-09583.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kokaia Z, Bengzon J, Metsis M, Kokaia M, Persson H, Lindvall O. Coexpression of neurotrophins and their receptors in neurons of the central nervous system. Proc Natl Acad Sci USA. 1993a;90:6711–6715. doi: 10.1073/pnas.90.14.6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kokaia Z, Gido G, Ringstedt T, Bengzon J, Kokaia M, Siesjo BK, Persson H, Lindvall O. Rapid increase of BDNF mRNA levels in cortical neurons following spreading depression: regulation by glutamatergic mechanisms independent of seizure activity. Brain Res Mol Brain Res. 1993b;19:277–286. doi: 10.1016/0169-328x(93)90126-a. [DOI] [PubMed] [Google Scholar]
- 38.Kokaia Z, Metsis M, Kokaia M, Bengzon J, Elmer E, Smith ML, Timmusk T, Siesjo BK, Persson H, Lindvall O. Brain insults in rats induce increased expression of the BDNF gene through differential use of multiple promoters. Eur J Neurosci. 1994;6:587–596. doi: 10.1111/j.1460-9568.1994.tb00303.x. [DOI] [PubMed] [Google Scholar]
- 39.Kokaia Z, Metsis M, Kokaia M, Elmer E, Lindvall O. Co-expression of TrkB and TrkC receptors in CNS neurones suggests regulation by multiple neurotrophins. NeuroReport. 1995;6:769–772. doi: 10.1097/00001756-199503270-00016. [DOI] [PubMed] [Google Scholar]
- 40.Koliatsos VE, Clatterbuck RE, Winslow JW, Cayouette MH, Price DL. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron. 1993;10:359–367. doi: 10.1016/0896-6273(93)90326-m. [DOI] [PubMed] [Google Scholar]
- 41.Korsching S. The neurotrophic factor concept: a reexamination. J Neurosci. 1993;13:2739–2748. doi: 10.1523/JNEUROSCI.13-07-02739.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krüttgen A, Möller JC, Heymach JV, Jr, Shooter EM. NGF and BDNF induce regulated secretion of NT-3 in transfected PC12 cells. Soc Neurosci Abstr. 1997;23:667.20. [Google Scholar]
- 43.Liang FY, Moret V, Wiesendanger M, Rouiller EM. Corticomotoneuronal connections in the rat: evidence from double-labeling of motoneurons and corticospinal axon arborizations. J Comp Neurol. 1991;311:356–366. doi: 10.1002/cne.903110306. [DOI] [PubMed] [Google Scholar]
- 44.Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- 45.Lindsay RM. Role of neurotrophins and trk receptors in the development and maintenance of sensory neurons: an overview. Philos Trans R Soc Lond B Biol Sci. 1996;351:365–373. doi: 10.1098/rstb.1996.0030. [DOI] [PubMed] [Google Scholar]
- 46.Lowrie MB, Vrbova G. Dependence of postnatal motoneurons on their targets: review and hypothesis. Trends Neurosci. 1992;15:80–84. doi: 10.1016/0166-2236(92)90014-y. [DOI] [PubMed] [Google Scholar]
- 47.Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- 48.Maisonpierre PC, Le Beau MM, Espinosa RI, Ip NY, Belluscio L, de la Monte SM, Squinto S, Furth ME, Yancopoulos GD. Human and rat brain-derived neurotrophic factor and neurotrophin-3: gene structures, distributions, and chromosomal localizations. Genomics. 1991;10:558–568. doi: 10.1016/0888-7543(91)90436-i. [DOI] [PubMed] [Google Scholar]
- 49.Martin JE, Swash M. The pathology of motor neuron disease. In: Leigh PN, Swash M, editors. Motor neuron disease, biology and management. Springer; New York: 1995. pp. 288–321. [Google Scholar]
- 50.McCabe JT, Desharnais RA, Pfaff DW. Graphical and statistical approaches to data analysis for in situ hybridization. Methods Enzymol. 1989;168:822–848. doi: 10.1016/0076-6879(89)68061-5. [DOI] [PubMed] [Google Scholar]
- 51.Merlio JP, Ernfors P, Jaber M, Persson H. Molecular cloning of rat trkC and distribution of cells expressing messenger RNAs for members of the trk family in the rat central nervous system. Neuroscience. 1992;51:513–532. doi: 10.1016/0306-4522(92)90292-a. [DOI] [PubMed] [Google Scholar]
- 52.Middlemas DS, Lindberg RA, Hunter T. trkB, a neuronal receptor protein-tyrosine kinase: evidence for a full-length and two truncated receptors. Mol Cell Biol. 1991;11:143–153. doi: 10.1128/mcb.11.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miranda RC, Sohrabji F, Toran Allerand CD. Neuronal colocalization of mRNAs for neurotrophins and their receptors in the developing central nervous system suggests a potential for autocrine interactions. Proc Natl Acad Sci USA. 1993;90:6439–6443. doi: 10.1073/pnas.90.14.6439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moore MW, Klein RD, Farinas I, Sauer H, Armanini M, Philipps H, Reichardt LF, Ryan AM, Carver-Moore K, Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- 55. Naveilhan P, El Shamy WM, Ernfors P. Differential regulation of mRNAs for GDNF and its receptors ret and GDNFRα after sciatic nerve lesion in the mouse. Eur J Neurosci 9 1997. 1450 1460. [DOI] [PubMed] [Google Scholar]
- 56.Nudo RJ, Masterton RB. Descending pathways to the spinal cord: a comparative study of 22 mammals. J Comp Neurol. 1988;277:53–79. doi: 10.1002/cne.902770105. [DOI] [PubMed] [Google Scholar]
- 57.Nudo RJ, Masterton RB. Descending pathways to the spinal cord. III: Sites of origin of the corticospinal tract. J Comp Neurol. 1990;296:559–583. doi: 10.1002/cne.902960405. [DOI] [PubMed] [Google Scholar]
- 58.Pichel JG, Shen L, Sheng HZ, Granholm A-C, Drago J, Grinberg A, Lee EJ, Huang SP, Saarma M, Hoffer BJ, Sariola H, Westphal H. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- 59.Piehl F, Frisen J, Risling M, Hokfelt T, Cullheim S. Increased trkB mRNA expression by axotomized motoneurones. NeuroReport. 1994;5:697–700. doi: 10.1097/00001756-199402000-00009. [DOI] [PubMed] [Google Scholar]
- 60.Porter LL, Cedarbaum JM, O’Leary DDM, Stanfield BB, Asanuma H. The physiological identification of pyramidal tract neurons within transplants in the rostral cortex taken from the occipital cortex during development. Brain Res. 1987;436:136–142. doi: 10.1016/0006-8993(87)91565-4. [DOI] [PubMed] [Google Scholar]
- 61.Ringstedt T, Lagercrantz H, Persson H. Expression of members of the trk family in the developing postnatal rat brain. Dev Brain Res. 1993;72:119–131. doi: 10.1016/0165-3806(93)90165-7. [DOI] [PubMed] [Google Scholar]
- 62.Ringstedt T, Arenas E, Lendahl U, Ibanez CF. Neuronal heterotopias in neocortex and hippocampus of transgenic mice overexpressing BDNF in the developing nervous system. Soc Neurosci Abstr. 1997;23:564.3. [Google Scholar]
- 63.Rogers AW. Techniques of autoradiography. Elsevier; Amsterdam: 1979. [Google Scholar]
- 64.Ryden M, Ibanez CF. Binding of neurotrophin-3 to p75LNGFR, TrkA, and TrkB mediated by a single functional epitope distinct from that recognized by trkC. J Biol Chem. 1996;271:5623–5627. doi: 10.1074/jbc.271.10.5623. [DOI] [PubMed] [Google Scholar]
- 65.Schaar DG, Sieber BA, Dreyfus CF, Black IB. Regional and cell-specific expression of GDNF in rat brain. Exp Neurol. 1993;124:368–371. doi: 10.1006/exnr.1993.1207. [DOI] [PubMed] [Google Scholar]
- 66.Schecterson LC, Bothwell M. Novel roles for neurotrophins are suggested by BDNF and NT-3 mRNA expression in developing neurons. Neuron. 1992;9:449–463. doi: 10.1016/0896-6273(92)90183-e. [DOI] [PubMed] [Google Scholar]
- 67.Schwaber JS, Chronwall BM, Lewis ME. In situ hybridization histochemistry combined with markers of neuronal connectivity. Methods Enzymol. 1989;168:778–791. doi: 10.1016/0076-6879(89)68058-5. [DOI] [PubMed] [Google Scholar]
- 68.Sebert ME, Shooter EM. Expression of mRNA for neurotrophic factors and their receptors in the rat dorsal root ganglion and sciatic nerve following nerve injury. J Neurosci Res. 1993;36:357–367. doi: 10.1002/jnr.490360402. [DOI] [PubMed] [Google Scholar]
- 69.Segal RA, Greenberg ME. Intracellular signalling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 70.Sendtner M, Holtmann B, Kolbeck R, Thoenen H, Barde YA. Brain-derived neurotrophic factor prevents the death of motoneurons in newborn rats after nerve section. Nature. 1992;360:757–759. doi: 10.1038/360757a0. [DOI] [PubMed] [Google Scholar]
- 71.Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 72.Sofroniew MV, Galletly NP, Isacson O, Svendsen CN. Survival of adult basal forebrain cholinergic neurons after loss of target neurons. Science. 1990;247:338–342. doi: 10.1126/science.1688664. [DOI] [PubMed] [Google Scholar]
- 73.Springer JE, Mu X, Bergmann LW, Trojanowski JQ. Expression of GDNF mRNA in rat and human nervous tissue. Exp Neurol. 1994;127:167–170. doi: 10.1006/exnr.1994.1091. [DOI] [PubMed] [Google Scholar]
- 74.Tetzlaff W, Kobayashi NR, Giehl KM, Tsui BJ, Cassar SL, Bedard AM. Response of rubrospinal and corticospinal neurons to injury and neurotrophins. Prog Brain Res. 1994;103:271–286. doi: 10.1016/s0079-6123(08)61142-5. [DOI] [PubMed] [Google Scholar]
- 75.Treanor JJS, Goodman L, de Sauvage F, Stone DM, Poulsen KT, Beck CD, Gray C, Armanini MP, Pollock RA, Hefti F, Phillips HS, Goddard A, Moore MW, Buj-Bello A, Davies AM, Asai N, Takahashi M, Vandlen R, Henderson CE, Rosenthal A. Characterization of a multicomponent receptor for GDNF. Nature. 1996;382:80–83. doi: 10.1038/382080a0. [DOI] [PubMed] [Google Scholar]
- 76.Wang CY, Ni J, Jiang H, Hsu TA, Dufich-Djordjevic M, Feng L, Zhang M, Mei L, Gentz R, Lu B. Cloning and characterization of glial cell line-derived neurotrophic factor receptor-B: a novel receptor for members of glial cell line-derived neurotrophic factor family of neurotrophic factors. Neuroscience. 1998;83:7–14. doi: 10.1016/s0306-4522(97)00462-4. [DOI] [PubMed] [Google Scholar]
- 77.Yan Q, Elliott J, Snider WD. Brain-derived neurotrophic factor rescues spinal motor neurons from axotomy-induced cell death. Nature. 1992;360:753–755. doi: 10.1038/360753a0. [DOI] [PubMed] [Google Scholar]
- 78.Yan Q, Matheson C, Lopez DT. In vivo neurotrophic effects of GDNF on neonatal and adult facial motor neurons. Nature. 1995;373:341–344. doi: 10.1038/373341a0. [DOI] [PubMed] [Google Scholar]
- 79.Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennet L, Welcher AA. Expression of brain-derived neurotrophic factor (BDNF) in the adult rat central nervous system. Neuroscience. 1997a;78:431–448. doi: 10.1016/s0306-4522(96)00613-6. [DOI] [PubMed] [Google Scholar]
- 80.Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of trkB in the central nervous system of the adult rat. J Comp Neurol. 1997b;378:135–157. [PubMed] [Google Scholar]