

Abstract
Nitrous oxide (N2O; laughing gas) has been a widely used anesthetic/analgesic since the 19th century, although its cellular mechanism of action is not understood. Here we characterize the effects of N2O on excitatory and inhibitory synaptic transmission in microcultures of rat hippocampal neurons, a preparation in which anesthetic effects on monosynaptic communication can be examined in a setting free of polysynaptic network variables. Eighty percent N2O occludes peak NMDA receptor-mediated (NMDAR) excitatory autaptic currents (EACs) with no effect on the NMDAR EAC decay time course. N2O also mildly depresses AMPA receptor-mediated (AMPAR) EACs. We find that N2O inhibits both NMDA and non-NMDA receptor-mediated responses to exogenous agonist. The postsynaptic blockade of NMDA receptors exhibits slight apparent voltage dependence, whereas the blockade of AMPA receptors is not voltage dependent. Although the degree of ketamine and Mg2+ blockade of NMDA-induced responses is dependent on permeant ion concentration, the degree of N2O blockade is not. We also observe a slight and variable prolongation of GABAA receptor-mediated (GABAR) postsynaptic currents likely caused by previously reported effects of N2O on GABAA receptors. Despite the effects of N2O on both NMDA and non-NMDA ionotropic receptors, glial glutamate transporter currents and metabotropic glutamate receptor-mediated synaptic depression are not affected. Paired-pulse depression, the frequency of spontaneous miniature excitatory synaptic currents, and high-voltage–activated calcium currents are not affected by N2O. Our results suggest that the effects of N2O on synaptic transmission are confined to postsynaptic targets.
Keywords: NMDA receptor, glutamate, nitrous oxide, GABA, postsynaptic, presynaptic
Despite much attention, cellular mechanisms underlying general anesthesia remain elusive. Many anesthetics share the ability to potentiate exogenously or synaptically generated GABAA receptor-mediated (GABAR) currents (Franks and Lieb, 1994). Halothane, isofluorane, barbiturates, neurosteroids, and propofol are examples of known anesthetic GABAA modulators. Some anesthetics like halothane inhibit high-voltage–activated calcium currents (Herrington et al., 1991; Miao et al., 1995), suggesting the possibility that presynaptic effects contribute to some of the anesthetic actions of these agents. Inhibitors of NMDA glutamate receptors, like ketamine, phencyclidine, and MK-801, have anesthetic properties, with ketamine enjoying widespread clinical use in pediatric populations.
Nitrous oxide (N2O) has been used as an inhalation anesthetic for over a century and as a recreational drug of abuse since at least the 18th century; yet the mechanism(s) of the effects of nitrous oxide on signaling in the CNS is not understood. N2O is widely used clinically because of its good analgesic properties; however, it is a relatively weak anesthetic, requiring high volume percent and hyperbaric conditions to achieve the minimal alveolar concentration for anesthesia in 50% of subjects (MAC) (Gonsowski and Eger, 1994). Because of its low potency, N2O is often used in combination with other anesthetics.
In an initial study, we showed that N2O possesses several properties of a noncompetitive NMDA receptor antagonist, including the ability to protect brain tissue against excitotoxic damage, the ability to damage neurons in posterior cingulate and retrosplenial cortex, and the ability to block NMDA-gated currents in CNS neurons. In addition, N2O weakly potentiates GABAR currents (Dzoljic and Duiijn, 1998; Jevtovic-Todorovic et al., 1998). A complete understanding of N2O actions requires an analysis of effects on neural communication, in which potential presynaptic and postsynaptic contributions can be assessed. To increase understanding of the mechanisms of N2O actions, we explored in the current study the actions of N2O on neurotransmission in a simple setting of rat hippocampal microcultures (Segal and Furshpan, 1990). Solitary neurons grown in synaptic isolation in microcultures form autaptic (self-synaptic) connections (Bekkers and Stevens, 1991;Segal, 1991), thereby allowing the exploration of N2O synaptic actions in an environment free of many complicating variables, such as feedback inhibition and other network properties associated with more intact preparations.
MATERIALS AND METHODS
Hippocampal cultures. Hippocampal cells were prepared from 1–3 d postnatal Sprague Dawley rats. Slices of hippocampus 500–800 μm thick were digested with 1 mg/ml papain in oxygenated Leibovitz’s L-15 medium. Digested slices were mechanically triturated in modified Eagle’s medium containing 5% horse serum, 5% fetal calf serum, 17 mmd-glucose, 400 μmglutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin. Cells (75/mm2) were plated onto plastic culture dishes coated with a layer of collagen microdots over a layer of dried 0.15% agarose (microcultures) as described previously (Mennerick et al., 1995). Cultures were treated with cytosine arabinoside (10 μm) after 3 d in vitro and were used for experiments 1–16 d after plating.
Electrophysiology. The extracellular bath solution for synaptic physiology contained (in mm): NaCl 140, KCl 4.0, CaCl2 2.0, MgCl2 1.0, and HEPES 10. For experiments examining isolated NMDA receptor-mediated (NMDAR) excitatory autaptic currents (EACs), 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo(F)quinoxaline (NBQX; 1 μm), glycine (10 μm), and bicuculline (25 μm) were added to the bath, and Mg2+was removed from the bath. For examination of AMPA receptor-mediated (AMPAR) EACs, d-2-amino-5-phosphonovalerate (d-APV; 25 μm) and bicuculline (25 μm) were added to the bath. For examination of inhibitory autaptic currents (IACs), NBQX (1 μm) andd-APV (50 μm) were added. For examination of miniature EPSCs (mEPSCs), tetrodotoxin (500 nm),d-APV (25 μm), and bicuculline (25 μm) were included in the bath. In experiments in which currents were induced by exogenous NMDA, the extracellular solution contained no added Mg2+ and contained reduced Ca2+ (0.1–0.5 mm) to diminish calcium-dependent fade of NMDA-induced currents (Mayer and Westbrook, 1985; Zorumski et al., 1989; Legendre et al., 1993). For examination of high-voltage–activated (HVA) calcium currents, 5 mmBaCl2 replaced CaCl2 and MgCl2 and served as the charge carrier. Additionally, 1 μmtetrodotoxin was present in the bath. Calcium currents were leak subtracted using a P/5 (subpulse) protocol. Voltage pulses from −70 to −10 mV delivered every 20 sec were used to elicit HVA calcium currents.
For addition of gas, the extracellular solution was bubbled with air or N2O/O2 mixtures using a bubbling stone. The bubbling container was sealed with Parafilm punctured with a small escape hole. The solution was equilibrated with gas for at least 30 min, at which time gas-equilibrated solutions were drawn into a closed glass syringe. The syringe served as a solution reservoir for a gravity-driven local perfusion system consisting of glass tubes connecting the reservoirs with a multibarrel pipette (List Electronic, Darmstadt, Germany). The common tip of the multibarrel pipette was placed 400 μm from the recorded cell. Solution flow rates were 0.8–1.5 ml/min. The slower flow rates were used for synaptic experiments; faster flow rates (attained with larger-bore glass connecting tubes) were used for exogenous applications. Based on junction-current measurements using solutions of different chloride concentrations, rise times of solution exchanges with the faster flow rates were <30 msec (10–90% rise) at the tip of an open recording pipette. For most experiments 80% N2O/20% O2was used so that bottled air (80% N2/20% O2) could be used as a control.
The whole-cell recording pipette solution for studies of EACs contained (in mm): potassium gluconate 130, NaCl 4.0, CaCl2 0.5, EGTA 5.0, HEPES 10, MgATP2 2.0, and GTP 0.5. For study of IACs, gluconate was replaced with chloride. For studies of responses to exogenous glutamate receptor agonists and for studies of mEPSCs, cesium methanesulfonate replaced potassium gluconate. For study of the voltage dependence of NMDA-receptor blockade, cesium chloride or choline chloride replaced potassium gluconate in the patch-pipette solution. The pH of solutions was adjusted to 7.25.
For evoked synaptic responses, whole-cell, voltage-clamp recordings of autaptic currents were performed from solitary neurons using pipettes with an open-tip resistance of 2–5 MΩ and with series resistance compensated 90–100% using the compensation circuitry of an Axopatch 1-D amplifier (Axon Instruments, Foster City, CA). Neurons were stimulated with a brief (1.5 msec) voltage-command pulse to 0 mV from a holding potential of −70 mV. Successive sweeps were triggered at intervals of ≥20 sec to allow recovery from short-term depression and facilitation. Paired-pulse stimuli were delivered at 100 msec intervals for EACs and 500 msec intervals for IACs to allow conditioning responses to decay before delivery of a test stimulus. Averages of two to five sweeps in each condition were used for display and analysis. For miniature synaptic currents and exogenous currents, experiments were not limited to solitary-neuron microcultures, and series-resistance compensation was usually not used to achieve the lowest background noise levels possible. EAC decays were fit with a single exponential or biexponential function, generated from a Chebychev-transform algorithm (Pclamp 6.0; Axon Instruments). Unless otherwise indicated, results are presented as mean ± SE.
RESULTS
Effect of N2O on NMDAR EPSCs
Because we showed previously that N2O blocks NMDAR currents in hippocampal neurons, we first explored the effect of N2O on NMDAR synaptic currents in hippocampal microcultures. We found that a subanesthetic concentration of 80% (volume percent) N2O applied to isolated microculture neurons inhibited peak NMDAR EACs by 49 ± 6% (n= 6) compared with control responses (−1314 ± 317 pA) examined in extracellular saline bubbled with air. The degree of synaptic blockade was similar to that observed with exogenous applications of NMDA (Jevtovic-Todorovic et al., 1998) (see below).
NMDAR EACs in microcultures decay with a biexponential time course (Clements et al., 1992). In the present experiment, control EACs decayed with time constants of 89.5 ± 4.6 and 491 ± 16 msec, with the fast component representing 53 ± 3% of the initial amplitude of the EAC (Fig.1B). The time course of NMDAR EACs was not significantly affected by the application of 80% N2O (Fig. 1A,B). The lack of N2O effect on NMDAR EAC time course has interesting implications for the mechanism of action of N2O. Ketamine, another anesthetic known to inhibit NMDA receptor function, is a slowly dissociating antagonist that requires channel opening to inhibit NMDA receptors (MacDonald et al., 1987). When we examined the effect of 5–10 μm ketamine, we found a degree of peak NMDAR EAC inhibition similar to that induced by 80% N2O (Fig.1A,C; 45 ± 4% depression with baseline amplitudes of −1545 ± 248 pA; n = 5). However, in contrast to N2O, ketamine also significantly decreased the decay time of the NMDAR EAC. The decrease in decay time in the presence of ketamine was expressed as a decrease in both the fast and slow time constants of decay as well as an increase in the relative amplitude of the fast component of decay (Fig.1D). These effects of ketamine are similar to the effects of MK-801 (Rosenmund et al., 1993) at NMDA receptors and to the actions of slowly dissociating barbiturates acting on neuromuscular endplate currents (Adams, 1976).
Fig. 1.

Effect of N2O and noncompetitive NMDA receptor antagonists on NMDAR EACs. A, Effect of N2O on peak amplitude and on time course of NMDAR EACs is shown. Left, The effect of N2O on peak NMDAR responses. Control responses were obtained in the presence of extracellular solution bubbled with air and exhibit the larger peak current. Right, The EAC in the presence of N2O scaled to the peak NMDAR response in the absence of N2O. No effect of N2O could be detected on the time course of NMDAR EACs. B, Bar graphs summarize the effect of N2O (hashed bars) on the biexponential decay of NMDAR EACs. Open bars represent EACs in the presence of air. No significant difference was detected either in the time constant or in the relative contribution of the two components to the total amplitude (n = 6; pairedt test). C, Effect of 5 μmketamine on NMDAR EACs is shown. Note the speeding of the NMDAR EAC apparent in the scaled traces. D, Ketamine induced speeding of both the fast and slow time constant of decay and an increase in the relative contribution of the fast component (an asterisk indicates p< 0.02; n = 5; paired t test).E, F, Mg2+ (15 μm) has effects on peak and time course similar to those of N2O. No significant effect on time course was detected in six cells. For synaptic current traces in this and subsequent figures, stimulus transients have been partially blanked and truncated for clarity of presentation.
In contrast to the actions of ketamine and of N2O on NMDAR EACs, classical open-channel blockers, like procaine or methyprylone, acting at neuromuscular nicotinic receptors prolong the late phase of endplate currents (Kordas, 1970; Adams, 1976) by increasing the burst duration of channels (Neher and Steinbach, 1978). Our data suggest that N2O acts neither like a classical open-channel blocker nor like a use-dependent, slowly reversible channel blocker. As a representative of another class of NMDA channel blockers, we examined the effects of the noncompetitive antagonist Mg2+. At a concentration of 15 μm, a concentration lower than that at which presynaptic effects of Mg2+ are observed (Tong and Jahr, 1994a), Mg2+ occluded peak NMDAR EACs by an amount similar to that obtained with ketamine and N2O (Fig. 1E). Like N2O, Mg2+ had no detectable effect on the EAC decay (Fig.1F). These effects of Mg2+ are consistent with the rapid block by Mg2+ of NMDA receptor channels, the ability of NMDA channels to close with Mg2+ still bound, and the lack of prolongation of channel burst duration (Nowak et al., 1984). These results suggest that if N2O acts via a channel block mechanism, the mechanism is unlikely that of a classical open-channel blocker or of a slowly dissociating open-channel blocker like ketamine.
Effect of N2O on responses to exogenous NMDA
To explore further the effects of N2O at NMDA receptors and to compare these effects with the other clinically used NMDA receptor antagonist/anesthetic, we examined the effects of ketamine and N2O on NMDA-induced currents. We found that ketamine blockade developed more slowly than did N2O blockade when the antagonist was rapidly coapplied with NMDA (Fig.2). This slow blockade resulted primarily from the use dependence of ketamine actions rather than from slow binding of ketamine, because ketamine preapplied for 2 sec before the addition of NMDA resulted in peak NMDAR currents nearly equal in amplitude to those responses obtained with simultaneous coapplications of agonist and antagonist (Fig. 2A). In contrast to ketamine blockade, N2O blockade was nearly immediate when the application of agonist and antagonist was simultaneous (Fig.2B). This indicates that the N2O effect develops more rapidly than does that of ketamine. In combination with the lack of effect of N2O on NMDAR EAC time course, this result suggests that N2O blockade is not dependent on channel opening.
Fig. 2.

N2O exhibits rapid block of NMDAR currents. A, Responses to 20 μm NMDA (0.5 mm Ca2+ and nominally 0 mmMg2+ present in the extracellular recording solution) in the presence or absence of 3 μm ketamine. The horizontal bars over the tracesindicate the timing of NMDA and ketamine applications.B, The same application protocol performed on another cell using N2O as the antagonist. Note the rapid development of block with coapplication of agonist and antagonist.
We also compared the voltage dependence of N2O and ketamine blockade. For these experiments antagonists were coapplied with agonists during the steady-state phase of agonist-induced responses (Fig. 3). When the degree of blockade of antagonists was compared at −60 and +60 mV, blockade with both drugs exhibited significant voltage dependence. However, N2O exhibited much weaker voltage dependence than did ketamine (Fig.3D) at a concentration that yielded an equivalent amount of block when effects were collapsed across voltages (Fig. 3D, legend). When the complete current–voltage relationships for ketamine and N2O were examined, currents both in the presence and in the absence of antagonist showed reversal potentials near 0 mV (data not shown). We also examined the voltage dependence of Mg2+ blockade, which, as expected, exhibited strong voltage dependence, similar to that of ketamine (55 ± 1% block relative to baseline amplitude of −195 ± 37 pA at −60 mV and 2 ± 1% block relative to baseline of +222 ± 40 pA at +60 mV; data not shown).
Fig. 3.
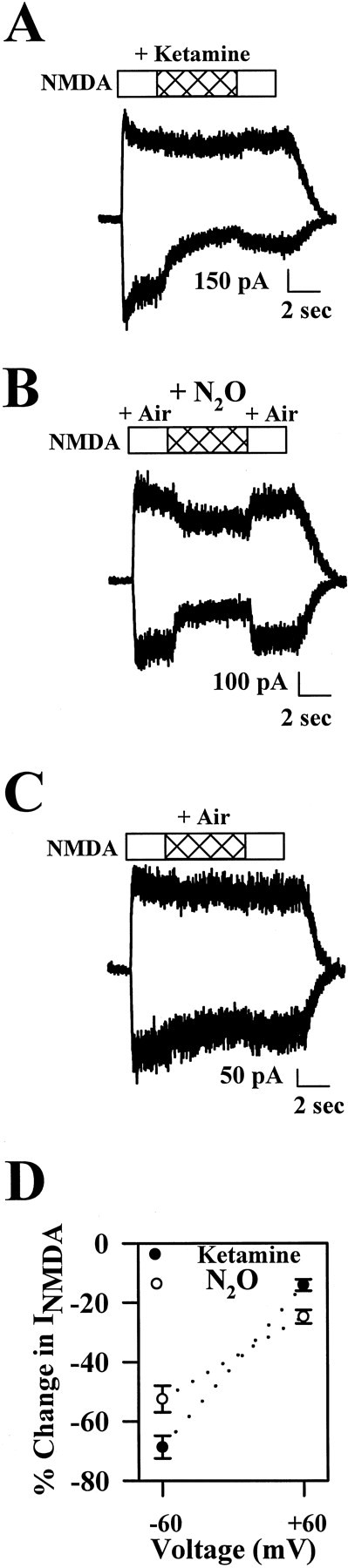
Voltage dependence of N2O and ketamine block of NMDA receptors. A–C, Raw tracesat holding potentials of −60 mV (inward currents) and +60 mV (outward currents) exemplifying the effects of ketamine (A), N2O (B), and air (C) on NMDAR currents. Baseline holding currents have been subtracted. The horizontal bars overthe traces indicate the timing of drug applications.D, Summary data showing fractional inhibition of steady-state NMDAR currents by ketamine (5 μm;closed circles) or N2O (80%; open circles) at −60 and +60 mV. Collapsed across voltages, there was no significant difference between antagonists (at the concentrations used) in the amount of block [repeated measures ANOVA with drug as a between-cell variable and voltage as a within-cell variable, F(1,17) = 0.66;p = 0.42; n = 9 ketamine and 10 N2O cells]. There was significant voltage dependence to the degree of inhibition [F(1,17) = 156.4;p < 0.001]. Post hoc comparisons revealed that both drugs exhibited significant voltage dependence (p < 0.001 for both drugs, pairedt test). However, there was also a significant interaction between antagonist and voltage [F(1,17) = 16.7;p = 0.001], indicating that ketamine exhibits significantly stronger voltage dependence than N2O exhibits.
The voltage dependence of N2O blockade of NMDA receptors, although weak, is puzzling because N2O is an uncharged molecule. Some ion channel blockers derive some of their apparent voltage dependence from direction of current flow rather than from voltage per se. For instance, derivatives of tetraethylammonium exhibit less block of potassium channels when the membrane potential is maintained but when the driving force on potassium is decreased by elevating extracellular potassium concentration (Armstrong, 1971). In addition, the degree of potassium channel block by both Ba2+ and charybdotoxin can be influenced by permeant ion concentration (Armstrong et al., 1982; MacKinnon and Miller, 1988).
To investigate the possibility that this mechanism might underlie the apparent voltage dependence of N2O, we used a pipette solution of either cesium chloride or choline chloride and examined the effect of N2O on NMDA responses. As expected, when choline was used as the main intracellular cation, reversal potentials for NMDA currents were shifted to approximately +50 mV from the typical 0 mV (data not shown). When examined at −30 mV, N2O block of inward NMDA currents was not significantly different between cesium- and choline-loaded cells (Fig.4A–C). However, when 5 μm ketamine was used as an antagonist, blockade of NMDA currents at −30 mV was significantly enhanced in choline-loaded cells versus cesium-loaded cells (Fig. 4D–F). The blockade of NMDA currents by Mg2+ (15 μm) was also greater in choline-loaded cells than in cesium-loaded cells examined at the same membrane potential (Fig.4G–I). These results suggest that enhancing the inward driving force on ions through NMDA receptors does not affect the degree of block by N2O and make it unlikely that the apparent voltage dependence of N2O blockade can be explained by an effect of current flow through the NMDA channel. On the other hand, current flow through the NMDA channel significantly interacts with ketamine and Mg2+ blockade, suggesting this mechanism plays a role in the apparent voltage dependence of ion channel block.
Fig. 4.

Effect of changing the reversal potential of the NMDAR current on blockade of NMDAR responses. A,B, Raw traces from two different cells illustrate the N2O block with cesium loading (A) and choline loading (B). The horizontal bars over thetraces indicate the timing of drug applications.C, At a holding potential of −30 mV, the degree of N2O block in cesium-loaded cells was not significantly different from the degree of block in cells loaded with choline (p > 0.3, unpaired t test;n = 14 cesium- and 11 choline-loaded cells). Choline loading changed the reversal potential of currents from ∼0 to approximately +50 mV. D, E, Rawtraces from two different cells illustrate the ketamine block in cesium- versus choline-filled cells. F, At a holding potential of −30 mV, the degree of ketamine (5 μm) block in choline-loaded cells was significantly greater than the block in cesium-loaded cells (p < 0.001, unpairedt test; n = 7 cesium- and 6 choline-loaded cells). G–I, A protocol similar to that shown in A–C and D–F was used except that Mg2+ was used as the antagonist and the holding potential was −60 mV. Raw traces in allpanels have been filtered at 10 Hz for display.
Effect of N2O on AMPAR EACs and tests of presynaptic N2O effects
We next examined the effect of N2O on AMPAR EACs in the presence of 25–50 μmd-APV and 1 mm Mg2+ to block the NMDAR component of EACs. AMPAR EACs were also inhibited by N2O, but less than were NMDAR EACs (Fig. 5). The inhibition by N2O of both NMDAR and AMPAR EACs compared with air controls may suggest a presynaptic effect of N2O because AMPAR and NMDAR EACs are similarly affected by presynaptic manipulations (Tong and Jahr, 1994a). To determine whether the effects on AMPAR EACs were caused by presynaptic or postsynaptic effects of N2O, we first examined paired-pulse modulation, a form of plasticity that is susceptible to presynaptic modulation both in situ and in microcultures (McNaughton, 1980; Mennerick and Zorumski, 1995). Previously, we found that many different presynaptic modulators alter the degree of paired-pulse depression of microculture AMPAR EACs (Mennerick and Zorumski, 1995). However, N2O failed to significantly change the degree of paired-pulse depression observed in response to paired stimulation delivered 100 msec apart (Fig. 5C,D).
Fig. 5.

Effect of N2O on AMPAR EACs.A, Averaged waveforms of interleaved AMPAR EACs in the presence of air or N2O are shown. B, Average depression induced by 80% N2O on peak AMPAR EACs (n = 8) is shown. Depression was calculated as: (Pn/Pa) − 1, where Pn is the peak response in the presence of N2O, and Pa is the peak response in the presence of air. C,D, Paired-pulse depression of AMPAR EACs was unaffected by N2O. C, Example currents are from the same cell shown in A. Paired-pulse interval was 100 msec. D, The degree of paired-pulse modulation was calculated by subtracting 1 from the test peak (P2)/conditioning peak (P1) ratio (P2/P1). Therefore, depression yields negative values, and facilitation yields positive values.
As another test for presynaptic effects of N2O, we examined the frequency of AMPAR mEPSCs (recorded with 500 nmtetrodotoxin in the extracellular recording solution) in the presence of air and N2O. In six neurons we could detect no change in mEPSC frequency with exchange of N2O for air in the extracellular bath (1.1 ± 0.7 Hz in air; 1.1 ± 0.6 Hz in N2O; data not shown). However, we were also unable to detect a significant change in mEPSC amplitude in the presence of N2O in this experiment. For instance, in the cell with the highest frequency of mEPSCs, mEPSC peak amplitude was −33.7 ± 2.1 pA in air (n = 118 events) and −36.1 ± 1.9 pA in N2O (n = 146 events;p > 0.4). Although this could represent a lack of postsynaptic effect, the negative result is more likely attributable to the small effect of N2O on peak synaptic responses coupled with the large variability in mEPSC amplitudes (Bekkers et al., 1990).
As a third experiment to determine whether N2O might have presynaptic effects, we examined effects on voltage-gated calcium currents in hippocampal neurons. Because a common target of many modulators of presynaptic function is HVA calcium channels (Miller, 1990; Wu and Saggau, 1997), we first examined the effect of N2O on HVA calcium current in hippocampal neurons. We found no reliable effect of 80% N2O on soma HVA currents in these cells compared with currents in the presence of air (98.7 ± 1.5% of control; control peak amplitude, −327 ± 74 pA;n = 5; Fig. 6). Because of the difficulty of spatially voltage-clamping cultured neurons, we also examined HVA calcium currents in acutely dissociated rat dorsal root ganglia neurons, which do not have extensive processes. These results also showed no effect of N2O on HVA currents (data not shown).
Fig. 6.

Effect of N2O on HVA calcium currents in hippocampal neurons. A, Superimposed leak-subtractedtraces from a hippocampal neuron in the presence of air and 80% N2O. Voltage pulses from −70 to −10 mV were used to elicit the currents; 5 mm BaCl2 was used as the charge carrier as detailed in Materials and Methods.B, Time course plot showing the lack of effect of N2O on the amplitude of leak-subtracted barium current in another neuron.
Effect of N2O on exogenously activated AMPAR currents
The above experiments suggest that presynaptic depression does not contribute to the depression of excitatory synaptic transmission. Therefore, we designed experiments to assess more directly the possibility of postsynaptic modulation of AMPA glutamate receptors by N2O. We examined the effect of N2O on currents elicited by kainic acid, a weakly desensitizing agonist at AMPA receptors. Steady-state responses to 50 μm kainate averaged −568 ± 146 pA in the presence of air and were inhibited by 32 ± 2% at −60 mV (n = 10). With a higher concentration of agonist (1 mm KA), the degree of blockade was similar (−29 ± 2% change; n = 11; data not shown). When blockade at −60 and +60 mV was compared, a similar degree of antagonism was observed at both potentials (p> 0.15, paired t test; Fig.7). These results suggest that like blockade of NMDA receptors, N2O blockade of AMPA receptors appears noncompetitive. However, unlike the blockade of NMDA receptors, there is no detectable voltage dependence to the block. The effects observed are unlikely to be caused by blockade of high-affinity kainate receptors, because these receptors are rapidly desensitized with the drug application protocols used (Wilding and Huettner, 1997). Also, at the high concentration of kainate, mostly AMPA receptors are activated in hippocampal cells because of the greater numbers of these receptors. Therefore, the similar degree of block at the two concentrations suggests that blockade is primarily of postsynaptic AMPA receptors.
Fig. 7.
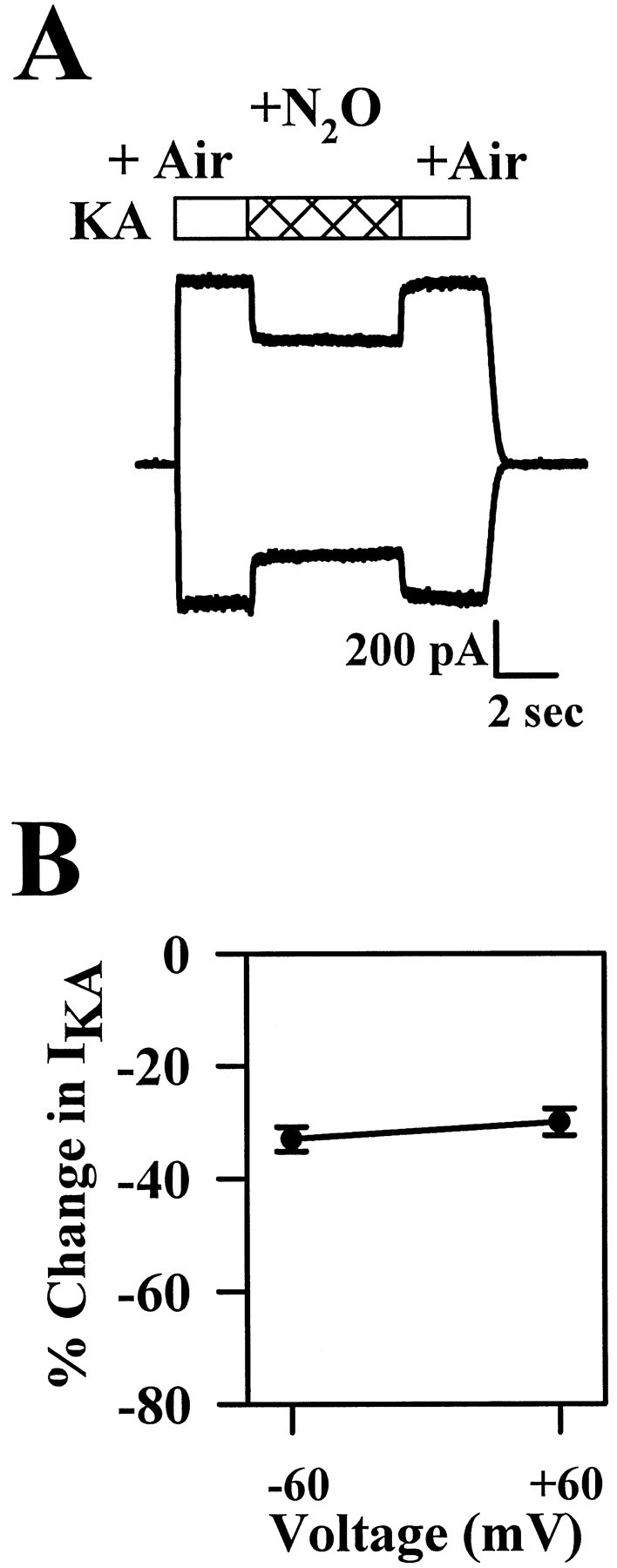
Effect of N2O on kainic acid (KA)-induced currents. A, Exemplartraces obtained from one neuron at −60 mV (inward current trace) and at +60 mV (outward currenttrace). Drug applications were made during the periods indicated by the horizontal bars. B, Summary of the effect of N2O on the responses to 50 μmKA at −60 and at +60 mV (n = 10).
Tests of N2O interaction with presynaptic metabotropic receptors and with glutamate transporters
Because the present results show that N2O interacts with both NMDA and non-NMDA subtypes of ionotropic glutamate receptors, we also assessed whether N2O targets presynaptic metabotropic glutamate receptors. We showed previously that activation of metabotropic receptors causes presynaptic depression at microculture autaptic synapses (Mennerick and Zorumski, 1995). Similarly, in the present study we found that the combination of 100 μm1S,3R-1-aminocyclopentane-1,3-dicarboxylic acid (1S,3R-ACPD) plus 100 μml-AP-4 to activate groups I, II, and III metabotropic receptors (Conn and Pin, 1997) depressed AMPAR EACs by 36 ± 9% from a baseline amplitude of −3379 ± 859 pA (all responses recorded in the presence of air; n = 7). The effect of N2O on peak AMPAR EAC was similar in the absence or presence of metabotropic agonists (−13 ± 2% N2O-induced change relative to responses in the absence of metabotropic agonists; −11 ± 2% change relative to responses in the presence of metabotropic agonists; n = 8; Fig.8). From this experiment, we conclude that N2O does not interact with the metabotropic receptors involved in mediating depression of synaptic responses. We cannot exclude an effect of N2O on other physiological effects of metabotropic receptors, such as phosphoinositide hydrolysis.
Fig. 8.

N2O does not interact with metabotropic glutamate receptor-induced synaptic depression.Left, The effect of N2O on the AMPAR EAC obtained in the absence of metabotropic agonists (air vs N2O). Right, The effects of N2O after the addition of 100 μm1S,3R-ACPD plus 100 μml-AP-4, which depressed transmission.
Glutamate transporters are another class of plasma membrane glutamate-binding proteins that might be a target for N2O actions. Glutamate transporters in situ and in culture rapidly bind glutamate after synaptic release (Mennerick and Zorumski, 1994; Tong and Jahr, 1994b). Under certain conditions, transport inhibitors can influence the peak glutamate concentration obtained (Tong and Jahr, 1994b) or increase the effective lifetime of glutamate acting at postsynaptic receptors (Mennerick and Zorumski, 1994). Additionally, glutamate transporters may be the target of other volatile anesthetics (Hirofumi et al., 1997). For these reasons, we directly tested the possibility that glutamate transporters are a target of N2O actions by exploiting the electrogenicity of glutamate transporters (Brew and Attwell, 1987). We examined the effect of N2O on d-aspartate currents elicited from microculture glia filled with a potassium gluconate solution. Under the conditions of this experiment, the current generated byd-aspartate application should result from the electrogenic transport current produced by the net influx of positive charge on each transporter cycle but not from the recently described anion conductance also gated by glutamate transporter activation (Wadiche et al., 1995). In four glial cells, d-aspartate currents averaged −45.5 ± 21 pA. We observed little change in the d-aspartate current with coapplication of 80% N2O (+8 ± 2% change compared with the current withd-aspartate in the presence of air). In two additional cells there was an apparent enhancement of the glutamate transporter current when d-aspartate and N2O were coapplied (the trace shown in Fig. 9 is an example of a modest enhancement). However, this apparent change was unrelated to an effect on glutamate transporter currents, because the current was elicited by N2O alone in the absence ofd-aspartate (data not shown) and was present in one cell despite an undetectable d-aspartate current. Furthermore, the response to N2O alone was not observed in all glial cells in which transporter currents were detected. Finally, transporter currents under the present conditions reverse direction at potentials more positive than +50 mV (Brew and Attwell, 1987), whereas the N2O-induced glial current reversed direction near −70 mV (data not shown). Because of its small amplitude and variability, the direct effect of N2O on glial membrane conductance was not studied further in the present work.
Fig. 9.

Effect of N2O on glial glutamate transporter currents induced by 100 μmd-aspartate. The trace shows the response of a microculture glial cell to the application of 100 μmd-asp in the presence of air and N2O. Thehorizontal bars denote application times.
N2O and GABAR IACs
Approximately 50% of the neurons in postnatally derived hippocampal microcultures are GABAergic (Bekkers and Stevens, 1991;Segal, 1991). We studied the effect of N2O on IACs by replacing gluconate in the whole-cell pipette with chloride to cause IACs to appear as inward currents at −70 mV. Consistent with weak effects of N2O on GABAA receptor-mediated currents in response to exogenous GABA (Dzoljic and Duiijn, 1998;Jevtovic-Todorovic et al., 1998), we observed small effects of N2O on GABAR IACs. Peak GABAR IACs were not affected by 80% N2O (2 ± 3% potentiation compared with air;n = 21). Measured using the 10–90% decay time to quantify the IAC time course, IAC decays were variably affected, ranging from no effect to 50% prolongation (n = 21; Fig. 10). Overall, N2O resulted in a 15 ± 5% increase in the IAC charge transfer (n = 21). As a comparison we used pentobarbital, an anesthetic known to potentiate GABAergic transmission (Franks and Lieb, 1994). Because 80% N2O is approximately one-half the MAC required to produce anesthesia in rats (Jevtovic-Todorovic et al., 1998), we used 25 μm pentobarbital, which should represent an anesthetic concentration equivalent to 80% N2O (Franks and Lieb, 1994), as a comparison. Slowing of IAC decays was much more pronounced with pentobarbital compared with N2O; pentobarbital treatment increased the IAC charge transfer by 86 ± 13%, with little effect on IAC peak amplitude (8 ± 7% change; n = 4; Fig.10A,B). Paired-pulse depression of IACs was not affected or was slightly reduced by N2O (−36 ± 4% paired-pulse change in air vs −27 ± 7% change in N2O; n = 14; p > 0.08, paired t test; Fig. 10C), suggesting that a presynaptic potentiation of IACs is unlikely. As another anesthetic comparison, we examined the effect of ketamine on IPSCs. At 100 μm, a concentration approximately two orders of magnitude higher than anesthetic concentrations (Franks and Lieb, 1994), ketamine had no effect on peak IACs or on total IAC charge transfer (−4 ± 8% change in peak; 6 ± 9% change in charge; n = 4).
Fig. 10.
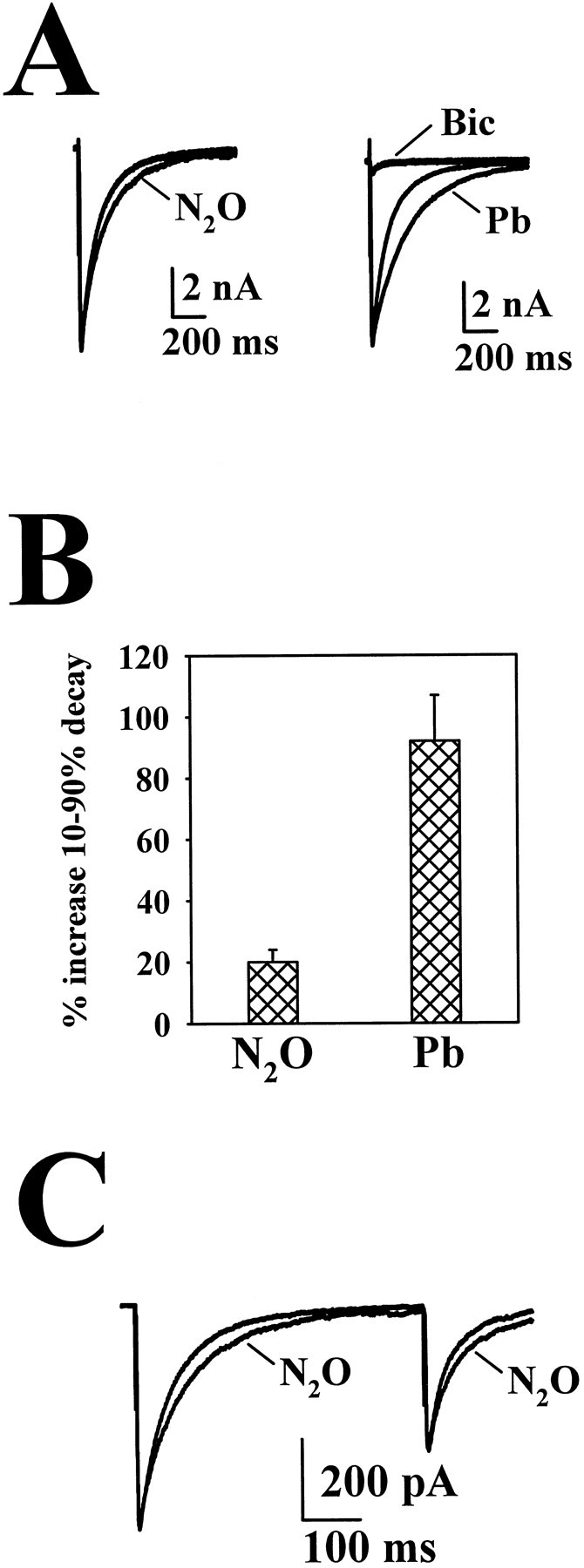
Effect of N2O on GABAergic IACs.A, Left, The effect of N2O on the IAC in baseline conditions. Right, The effect of 25 μm pentobarbital (Pb) on IACs from the same neuron. After the experiment, 25 μm bicuculline (Bic) was added to confirm that the response was a GABAergic IAC. Average peak amplitude of 21 baseline IACs was −4029 ± 797 pA. B, Summary data showing the effect of 80% N2O (n = 21) and 25 μmPb (n = 4) on the 10–90% decay time of GABAR IACs. The average 10–90% decay time for 21 baseline IACs was 101.4 ± 18.1 msec. C, Paired-pulse stimulation of the GABAergic neuron depicted inA demonstrating that there is no change in paired-pulse depression in cells that show a prolonged IAC. Responses are superimposed IACs in the presence of air and N2O.
DISCUSSION
Our results show that synaptic targets of the widely used inhalation anesthetic N2O are confined to postsynaptic sites in hippocampal microcultures. Although a lack of effect of N2O on GABAA receptors has been reported previously (Little and Thomas, 1986), a recent paper using isolated hippocampal neurons and a local perfusion system similar to that used here found potentiation of responses to exogenously applied muscimol (Dzoljic and Duiijn, 1998). Similarly, a study of invertebrate glutamate receptors reported blockade by N2O (Macdonald and Ramsey, 1995). Our present results combined with our previous results (Jevtovic-Todorovic et al., 1998) suggest that several postsynaptic effects may contribute to the anesthetic/analgesic effects of N2O.
As part of our effort to examine possible presynaptic targets of N2O, we examined HVA calcium currents, the general class of calcium current thought to underlie fast neurotransmitter release at glutamate and GABA synapses. We conclude that presynaptic effects via calcium channels are unlikely to underlie the modulation of synaptic transmission observed. Although we cannot eliminate the possibility that the presynaptic terminal may possess a different complement of calcium channels than those examined in the somata, previous studies suggest that effects on soma calcium currents are predictive of synaptic effects (Wu and Saggau, 1997).
N2O has somewhat nonselective but differential effects on excitatory and inhibitory synaptic currents. It remains to be determined which of the three postsynaptic effects described here might be most important in determining the anesthetic/analgesic properties of N2O. Of the three effects of N2O detected in the present study, blockade of NMDAR EACs was quantitatively the largest effect of N2O. Although GABAA receptors are a common target of many anesthetics, including volatile anesthetics and barbiturates, the effects of N2O on IACs were significantly weaker than were the effects of an anesthetically equivalent concentration of pentobarbital (Franks and Lieb, 1994). An anesthetic with the property of an AMPA receptor antagonist has not been described. Because blockade of glutamate receptors and potentiation of GABAA receptors would both tend to dampen CNS excitability, it is possible that all three effects work in concert to produce clinical anesthesia. It is of note that the MAC necessary for N2O to produce anesthesia in 50% of rats is 150% (v/v) (Gonsowski and Eger, 1994), a concentration unachievable except under hyperbaric conditions. We assume that the effects observed in the present work at subanesthetic concentrations of N2O can be extrapolated to anesthetic doses (and may be more pronounced at these doses). However, the extent to which the postsynaptic effects described here underlie the anesthetic/analgesic properties of N2O remains to be determined.
Our results do not address the mechanism by which N2O blocks ionotropic glutamate receptors and potentiates GABA receptors. Because of the larger magnitude of the effect on NMDA receptors and because of the novelty of this effect, we examined NMDA receptor blockade in most detail. Via these analyses we can make some qualified statements regarding the mechanism of N2O block of NMDA receptors. Our previous work showed that N2O exhibits a noncompetitive inhibition profile in dose–response relationships (Jevtovic-Todorovic et al., 1998), as does the other clinically used NMDAR antagonist anesthetic ketamine. However, the present work shows that N2O blockade of NMDA receptors is much faster and more easily reversible than the block exhibited by ketamine. In addition, the lack of effect on the NMDAR EAC time course suggests that N2O is unlikely to possess a use dependence like that of classical local anesthetic agents such as procaine acting at neuromuscular nicotinic acetylcholine receptors (Neher and Steinbach, 1978). The term “uncompetitive inhibition” has been used to describe antagonists with a requirement for agonist binding (Pennefather and Quastel, 1992). Our data show no evidence of an uncompetitive mechanism for N2O.
A difference in the blockade of AMPA versus NMDA glutamate receptors is the voltage dependence exhibited by N2O block of NMDA but not of AMPA receptors. Because N2O is not charged, it seems unlikely that this voltage dependence signifies that N2O senses the transmembrane electrical field and binds within the channel pore. Our results suggest that the direction of current flow is unlikely to impart apparent voltage dependence to the N2O blockade via a knock-on/knock-off type mechanism. Rather, because NMDA receptor gating has been shown to exhibit inherent voltage dependence aside from physiological blockade by Mg2+ (Nowak and Wright, 1992), it is likely that the apparent voltage dependence of N2O is caused by increased N2O binding to channel conformations adopted preferentially at negative potentials. In contrast, changing the reversal potential of NMDAR currents clearly influences the degree of ketamine and Mg2+ blockade, suggesting that a knock-on/knock-off mechanism likely explains at least part of the apparent voltage dependence of blockade of these two agents (MacDonald and Nowak, 1990).
Ketamine and Mg2+ represent two different classes of molecules that interact with the NMDAR ion channel. Although ketamine exhibits use dependence, Mg2+ apparently is capable of interacting with the closed NMDAR (Nowak et al., 1984; MacDonald et al., 1987; MacDonald and Nowak, 1990). Despite these differences in mechanism, the degree of block by both agents shares strong voltage dependence and at least partial dependence on permeant ion concentration (Fig. 4). N2O shares neither of these features. Although our data do not definitively suggest a mechanism of N2O action, this dissimilarity between N2O and two known channel blockers makes it less likely that N2O interacts directly with the NMDAR ion channel.
Although our results suggest that N2O targets multiple postsynaptic sites, we failed to detect effects on responses generated by several other classes of transmembrane proteins. We detected no effect on currents mediated by HVA calcium channels, no effect on electrogenic glutamate transporter currents, and no interaction with presynaptic metabotropic glutamate receptors. The lack of N2O effect on these responses suggests some specificity of action toward ligand-gated ion channels. The extent to which N2O may interact with other classes of ligand-gated ion channels awaits further study.
Footnotes
This work was supported by a Lucille P. Markey Postdoctoral Fellowship (S.M.); by a Foundation for Anesthesiology Education and Research/Abbott New Investigator Award (V.J.-T.); by National Institutes of Health Grants AG11355 (J.W.O), DA05072 (J.W.O), MH45493 (C.F.Z), GM47969 (C.F.Z.), and MH00964 (C.F.Z.); and by a grant from the Bantly Foundation (C.F.Z.). We thank Drs. Jim Huettner, Joe Henry Steinbach, and Chris Lingle for advice on the NMDA receptor experiments and Ann Benz for assistance with the primary cultures.
Correspondence should be addressed to Dr. Steven Mennerick, Department of Psychiatry, Washington University School of Medicine, 4940 Children’s Place, St. Louis, MO 63110.
REFERENCES
- 1.Adams PR. Drug blockade of open end-plate channels. J Physiol (Lond) 1976;260:531–552. doi: 10.1113/jphysiol.1976.sp011530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong CM. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J Gen Physiol. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armstrong CM, Swenson RP, Jr, Taylor SR. Block of squid axon K channels by internally and externally applied barium ions. J Gen Physiol. 1982;80:663–682. doi: 10.1085/jgp.80.5.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci USA. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bekkers JM, Richerson GB, Stevens CF. Origin of variability in quantal size in cultured hippocampal neurons and hippocampal slices. Proc Natl Acad Sci USA. 1990;87:5359–5362. doi: 10.1073/pnas.87.14.5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brew H, Attwell D. Electrogenic glutamate uptake is a major current carrier in the membrane of axolotl retinal glial cells. Nature. 1987;327:707–709. doi: 10.1038/327707a0. [DOI] [PubMed] [Google Scholar]
- 7.Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- 8.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 9.Dzoljic M, Duiijn BV. Nitrous oxide-induced enhancement of γ-aminobutyric acidA-mediated chloride currents in acutely dissociated hippocampal neurons. Anesthesiology. 1998;88:473–480. doi: 10.1097/00000542-199802000-00026. [DOI] [PubMed] [Google Scholar]
- 10.Franks NP, Lieb WR. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- 11.Gonsowski CT, Eger EI., II Nitrous oxide minimum alveolar anesthetic concentration in rats is greater than previously reported. Anesth Analg. 1994;79:710–712. doi: 10.1213/00000539-199410000-00016. [DOI] [PubMed] [Google Scholar]
- 12.Herrington J, Stern RC, Evers AS, Lingle CJ. Halothane inhibits two components of calcium current in clonal (GH3) pituitary cells. J Neurosci. 1991;11:2226–2240. doi: 10.1523/JNEUROSCI.11-07-02226.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hirofumi M, Nakamura Y, Arai T, Kiyoshi K. Increase of glutamate uptake in astrocytes: a possible mechanism of action of volatile anesthetics. Anesthesiology. 1997;86:1359–1366. doi: 10.1097/00000542-199706000-00018. [DOI] [PubMed] [Google Scholar]
- 14.Jevtovic-Todorovic V, Todorovic SM, Mennerick S, Powell S, Dikranian K, Benshoff N, Zorumski CF, Olney JW. Nitrous oxide (laughing gas) is an NMDA antagonist, neuroprotectant and neurotoxin. Nat Med. 1998;4:460–464. doi: 10.1038/nm0498-460. [DOI] [PubMed] [Google Scholar]
- 15.Kordas M. The effect of procaine on neuromuscular transmission. J Physiol (Lond) 1970;209:689–699. doi: 10.1113/jphysiol.1970.sp009186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Legendre P, Rosenmund C, Westbrook GL. Inactivation of NMDA channels in cultured hippocampal neurons by intracellular calcium. J Neurosci. 1993;13:674–684. doi: 10.1523/JNEUROSCI.13-02-00674.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Little HJ, Thomas DL. The effects of anesthetics and high pressure on the responses of the rat superior cervical ganglion in vitro. J Physiol (Lond) 1986;374:387–399. doi: 10.1113/jphysiol.1986.sp016086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macdonald AG, Ramsey RL. The effects of nitrous oxide on a glutamate-gated ion channel and their reversal by high pressure; a single channel analysis. Biochim Biophys Acta. 1995;1236:135–141. doi: 10.1016/0005-2736(95)00029-3. [DOI] [PubMed] [Google Scholar]
- 19.MacDonald JF, Nowak LM. Mechanisms of blockade of excitatory amino acid receptor channels. Trends Pharmacol Sci. 1990;11:167–172. doi: 10.1016/0165-6147(90)90070-O. [DOI] [PubMed] [Google Scholar]
- 20.MacDonald JF, Miljkovic Z, Pennefather P. Use-dependent block of excitatory amino acid currents in cultured neurons by ketamine. J Neurophysiol. 1987;58:251–266. doi: 10.1152/jn.1987.58.2.251. [DOI] [PubMed] [Google Scholar]
- 21.MacKinnon R, Miller C. Mechanism of charybdotoxin block of the high-conductance, Ca2+-activated K+ channel. J Gen Physiol. 1988;91:335–349. doi: 10.1085/jgp.91.3.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayer ML, Westbrook GL. The action of N-methyl-d-aspartic acid on mouse spinal neurones in culture. J Physiol (Lond) 1985;361:65–90. doi: 10.1113/jphysiol.1985.sp015633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNaughton BL. Evidence for two physiologically distinct perforant pathways to the fascia dentata. Brain Res. 1980;199:1–19. doi: 10.1016/0006-8993(80)90226-7. [DOI] [PubMed] [Google Scholar]
- 24.Mennerick S, Zorumski CF. Glial contributions to excitatory neurotransmission in cultured hippocampal cells. Nature. 1994;368:59–62. doi: 10.1038/368059a0. [DOI] [PubMed] [Google Scholar]
- 25.Mennerick S, Zorumski CF. Paired-pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. J Physiol (Lond) 1995;488:85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mennerick S, Que J, Benz A, Zorumski CF. Passive and synaptic properties of neurons grown in microcultures and in mass cultures. J Neurophysiol. 1995;73:320–332. doi: 10.1152/jn.1995.73.1.320. [DOI] [PubMed] [Google Scholar]
- 27.Miao N, Frazer MJ, Lynch C., III Volatile anesthetics depress Ca2+ transients and glutamate release in isolated cerebral synaptosomes. Anesthesiology. 1995;83:593–603. doi: 10.1097/00000542-199509000-00019. [DOI] [PubMed] [Google Scholar]
- 28.Miller RJ. Receptor-mediated regulation of calcium channels and neurotransmitter release. FASEB J. 1990;4:3291–3299. [PubMed] [Google Scholar]
- 29.Neher E, Steinbach JH. Local anesthetics transiently block currents through single acetylcholine-receptor channels. J Physiol (Lond) 1978;277:153–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nowak L, Bregestoversuski P, Ascher P, Herbert A, Prochiantz Z. Magnesium gates glutamate activated channels in mouse central neurons. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 31.Nowak LM, Wright JM. Slow voltage-dependent changes in channel open-state probability underlie hysteresis of NMDA responses in Mg2+-free solutions. Neuron. 1992;8:181–187. doi: 10.1016/0896-6273(92)90119-x. [DOI] [PubMed] [Google Scholar]
- 32.Pennefather P, Quastel DMJ. Modification of dose–response curves by effector blockade and uncompetitive antagonism. Mol Pharmacol. 1992;22:369–380. [PubMed] [Google Scholar]
- 33.Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- 34.Segal MM. Epileptiform activity in microcultures containing one excitatory hippocampal neuron. J Neurophysiol. 1991;65:761–770. doi: 10.1152/jn.1991.65.4.761. [DOI] [PubMed] [Google Scholar]
- 35.Segal MM, Furshpan EJ. Epileptiform activity in microcultures containing small numbers of hippocampal neurons. J Neurophysiol. 1990;64:1390–1399. doi: 10.1152/jn.1990.64.5.1390. [DOI] [PubMed] [Google Scholar]
- 36.Tong G, Jahr CE. Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron. 1994a;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 37.Tong G, Jahr CE. Block of glutamate transporters potentiates postsynaptic excitation. Neuron. 1994b;13:1195–1203. doi: 10.1016/0896-6273(94)90057-4. [DOI] [PubMed] [Google Scholar]
- 38.Wadiche JI, Amara SG, Kavanaugh MP. Ion fluxes associated with excitatory amino acid transport. Neuron. 1995;15:721–728. doi: 10.1016/0896-6273(95)90159-0. [DOI] [PubMed] [Google Scholar]
- 39.Wilding TJ, Huettner JE. Activation and desensitization of hippocampal kainate receptors. J Neurosci. 1997;17:2713–2721. doi: 10.1523/JNEUROSCI.17-08-02713.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu LG, Saggau P. Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 1997;20:204–212. doi: 10.1016/s0166-2236(96)01015-6. [DOI] [PubMed] [Google Scholar]
- 41.Zorumski CF, Yang J, Fischbach GD. Calcium-dependent, slow desensitization distinguishes different types of glutamate receptors. Cell Mol Neurobiol. 1989;9:95–104. doi: 10.1007/BF00711446. [DOI] [PMC free article] [PubMed] [Google Scholar]