

Abstract
The medium collected from cultured astrocytes transiently exposed to the group-II metabotropic glutamate (mGlu) receptor agonists (2S,1′R,2′R,3′R)-2-(2,3-dicarboxycyclopropyl)glycine (DCG-IV) or (S)-4-carboxy-3-hydroxyphenylglycine (4C3HPG) is neuroprotective when transferred to mixed cortical cultures challenged with NMDA (Bruno et al., 1997). The following data indicate that this particular form of neuroprotection is mediated by transforming growth factor-β (TGFβ). (1) TGFβ1 and -β2 were highly neuroprotective against NMDA toxicity, and their action was less than additive with that produced by the medium collected from astrocytes treated with DCG-IV or 4C3HPG (GM/DCG-IV or GM/4C3HPG); (2) antibodies that specifically neutralized the actions of TGFβ1 or -β2 prevented the neuroprotective activity of DCG-IV or 4C3HPG, as well as the activity of GM/DCG-IV or GM/4C3HPG; and (3) a transient exposure of cultured astrocytes to either DCG-IV or 4C3HPG led to a delayed increase in both intracellular and extracellular levels of TGFβ. We therefore conclude that a transient activation of group-II mGlu receptors (presumably mGlu3 receptors) in astrocytes leads to an increased formation and release of TGFβ, which in turn protects neighbor neurons against excitotoxic death. These results offer a new strategy for increasing the local production of neuroprotective factors in the CNS.
Keywords: metabotropic glutamate receptors, glial cells, transforming growth factor-β, neuroprotection, excitotoxic neuronal death, astrocyte, cortical cultures
Metabotropic glutamate (mGlu) receptors form a family of eight subtypes (mGlu1–8), which have been subdivided into three groups. Group-I includes mGlu1 and -5, which are coupled to polyphosphoinositide (PI) hydrolysis as well as to various classes of K+ channels via a Go or Gq GTP binding protein (for review, see Pin and Duvoisin, 1995). Group-II (mGlu2 and -3) and group-III (mGlu4, -6, -7, -8) receptors are coupled to a Gi-protein, and their activation inhibits adenylyl cyclase activity in heterologous expression systems (Tanabe et al., 1992, 1993). However, native group-II or group-III mGlu receptors in the CNS are coupled to multiple transduction pathways, including inhibition of voltage-sensitive Ca2+channels, stimulation or inhibition of cAMP formation, stimulation of PI hydrolysis, and activation of the mitogen-activated protein (MAP) kinase pathway (Winder and Conn, 1992; Genazzani et al., 1994; for review, see Pin and Duvoisin, 1995).
Recently, mGlu receptors have been considered a potential target for neuroprotective drugs. In particular, activation of group-II or group-III mGlu receptors protects neurons against excitotoxic death or other forms of degeneration (for review, see Nicoletti et al., 1996). However, the mechanism responsible for neuroprotection differs between these two classes of mGlu receptor subtypes. mGlu4, -7, and -8 receptors are exclusively localized in neurons, and their activation inhibits glutamate release (Shigemoto et al., 1997). Hence, group-III mGlu receptor agonists are of potential value in the experimental therapy of epilepsy or neurodegenerative disorders of excitotoxic origin. In contrast, inhibition of glutamate release may contribute to, but is not sufficient to explain, neuroprotection mediated by group-II mGlu receptors. Accordingly, mGlu2 or -3 receptor agonists protect not only against excitotoxic death but also against apoptosis induced by β-amyloid peptide or hypoxia combined with glucose deprivation in the presence of a mixture of ionotropic receptor antagonists, i.e., under conditions in which neurodegeneration develops in the absence of any excitotoxic component (Buisson and Choi, 1995; Copani et al., 1995). We recently showed that neuroprotection mediated by group-II mGlu receptors involves a novel form of glial–neuronal interaction, which is promoted by the activation of mGlu3 receptors present in astrocytes (Bruno et al., 1997). The medium collected from cultured astrocytes 2–20 hr after a brief exposure to mGlu3 receptor agonists is highly neuroprotective against NMDA toxicity (Bruno et al., 1997,1998). Neuroprotection is attenuated after treating the astrocytes with cycloheximide or after heating the medium, suggesting that astrocytes produce and release a proteic neuroprotective factor in response to mGlu3 receptor activation (Bruno et al., 1997). This novel mechanism may offer a new strategy to increase the local production of neurotrophic factors in the CNS. We now demonstrate that neuroprotection by glial mGlu3 receptors is mediated by transforming growth factor-β1 (TGFβ1) and TGFβ2, which are released from astrocytes and exert a potent neuroprotective activity in in vitro and in vivo models of excitotoxic death.
MATERIALS AND METHODS
Mixed cortical cultures. Mixed cortical cultures containing both neurons and astrocytes were prepared from fetal mice at 14–16 d of gestation, as described by Rose et al. (1992). In brief, dissociated cortical cells were plated in 15 mm multiwell vessels (Falcon Primaria, Lincoln Park, NY) on a layer of confluent astrocytes [prepared as described by Rose et al. (1992)], using a plating medium of MEM-Eagle’s salts (supplied glutamine-free) supplemented with 5% heat-inactivated horse serum, 5% fetal bovine serum, glutamine (2 mm), glucose (21 mm), and NaHCO3 (25 mm). After 3–5 d in vitro (DIV), non-neuronal cell division was halted by a 1–3 d exposure to 10 μm cytosine arabinoside, and cultures were shifted to a maintenance medium identical to plating medium but lacking fetal bovine serum. Subsequent partial medium replacement was performed twice a week. Cultures at 13–14 DIV were used.
Glial cultures. Glial cell cultures were prepared from postnatal mice (1–3 d after birth), as described previously (Rose et al., 1992). Dissociated cortical cells were grown in 15 mm multiwell vessels using a plating medium of MEM-Eagle’s salts supplemented with 10% of heat-inactivated horse serum, 10% fetal bovine serum, 2 mm glutamine, sodium bicarbonate (25 mm), and glucose (21 mm). Cultures were kept at 37°C in a humidified CO2 atmosphere until they reached confluency (7–14 DIV). Confluent cultures were then used for the experiments or as a support for mixed cultures.
Assessment of neuronal death in culture. For induction of excitotoxic death, mixed cultures were exposed to NMDA for 10 min at room temperature in a HEPES-buffered salt solution containing (in mm): 120 NaCl, 5.4 KCl, 0.8 MgCl2, 1.8 CaCl2, 20 HEPES, and 15 glucose. Afterward, the cultures were extensively washed and incubated in MEM-Eagle’s (supplemented with 25 mm NaHCO3 and 21 mm glucose) (MS) at 37°C. In some experiments, glial conditioned medium (GM) was added to the cultures immediately after the NMDA pulse and maintained during the following 24 hr of incubation. GM was prepared as follows. Glial cortical cultures were exposed for 10 min to group-II mGlu receptor agonists, and then drugs were washed out and cultures were kept in MS (which does not contain serum) at 37°C for the following 20 hr. At the end of this incubation, the medium was collected and immediately transferred to mixed cultures.
Neuronal injury was estimated by examining the cultures with phase-contrast microscopy 24 hr after the insult, when the process of cell death was largely complete. Neuronal damage was quantitatively assessed by trypan blue staining. Stained neurons were counted from three random fields per well. Neuronal injury was also assessed by measuring the activity of lactate dehydrogenase (LDH) into the extracellular medium, as described in Koh and Choi (1987).
Western blot analysis. Cultured astrocytes were harvested at 4°C in a 10 mm Tris buffer, pH 7.4, containing 5 mm EDTA, 1 mm PMSF, 25 μg/ml leupeptin, and 0.5% aprotinin. After sonication, samples were centrifuged at 8000 rpm for 10 min, and an aliquot of the supernatants was processed for the assessment of protein concentration by the Bredford method. Samples were diluted in SDS-bromophenol blue buffer and boiled for 5 min before loading. Electrophoresis was performed in 15% SDS-PAGE using 40 μg of total protein per lane. After separation, proteins were transferred onto a nitrocellulose membrane (Hybond ECL) for 35 min using a Bio-Rad transblot system (Bio-Rad, Munchen, Germany). After blocking, membranes were incubated with primary antibodies for 1 hr at room temperature and then repeatedly washed and exposed to horseradish peroxidase-conjugated secondary antibodies for 1 hr at room temperature. Proteins were visualized using the enhancing chemiluminescence detection system (ECL). The following primary antibodies were used: rabbit polyclonal TGFβ2 antibody (Santa Cruz Biotechnology, Tebu, France) (final dilution: 500 ng/ml) and monoclonal anti-actin antibody (Sigma, St. Louis, MO) (1:1000 dilution).
Measurement of TGFβ in the astrocyte medium. The amount of TGFβ released from cultured astrocytes in the medium was measured by using a sensitive bioassay based on the ability of TGFβ to reduce the proliferation rate of mink lung cells. Mink lung cells were purchased from American Type Culture Collection and plated in 15 mm multiwell vessels (Falcon Primaria) using a plating medium of DMEM containing 0.1 mm nonessential amino acids, 1 mm sodium pyruvate, 2 mm glutamine, and 10% fetal bovine serum. Twenty-four hours after plating, cells were exposed to GM in the presence of 0.5 μCi/well of [methyl-3H]thymidine at 37°C for 24 hr. [Methyl-3H]thymidine incorporation has been assessed as described by Ciccarelli et al. (1997). Standard curves were constructed by using concentrations of human recombinant TGFβ1 or -β2 ranging from 0.1 pg/ml to 10 ng/ml.
The amount of TGFβ1 present in the GM was also assessed by the TGFβ1 Emax ELISA System (Promega) which is designed to specifically detect biologically active TGFβ1.
Assessment of in vivo neuronal injury. Male Sprague Dawley rats (250–300 gm, body weight) were anesthetized with pentobarbital (50 mg/kg, i.p.) and infused with NMDA (100 nmol/0.5 μl per 2 min) or NMDA + TGFβ1 or -β2 (0.5 ng/0.5 μl) in the left caudate nucleus, at +2.0 mm anteroposterior (AP), 2.6 mm lateral (L), and 5 mm ventral (V), according to the Pellegrino and Cushman (Pellegrino et al., 1992) atlas. The injection was repeated at a second site (+1 mm AP, 2.6 mm L, and 5 mm V) to obtain a more consistent loss of striatal neurons. Animals were killed by decapitation 7 d later. Neuronal toxicity was evaluated either by histological examination or by measuring striatal glutamate decarboxylase (GAD) activity as a marker for GABAergic neurons. For histological analysis, the brains were removed, frozen rapidly in isopentane at −40°C, and then stored at −80°C. Cryostat sections (20 μm) were Nissl-stained and examined in light microscopy. For measurements of GAD activity, the corpus striatum was dissected bilaterally and homogenized in 5 mm imidazol buffer containing 0.2% Triton X-100 and 10 mm dithiothreitol. An aliquot of the homogenate was incubated in 400 μl of 10 mm phosphate buffer, pH 7.0, containing 10 mm2-mercaptoethanol, 0.02 mm pyridoxalphosphate, and 1 μCi of [3H]-glutamate (Amersham; specific activity 46 Ci/mmol) for 1 hr at 37°C; the reaction was stopped with 15 μl of ice-cold 11.8N HClO4. After centrifugation in a microfuge at maximal speed, 10 μl of the supernatant was diluted with 0.01N HCl and derivatized with O-phthalaldehyde and mercaptoethanol for 1 min at room temperature before injection into HPLC. The HPLC apparatus consisted of a programmable solvent module 126 (Beckman Instruments, Fullerton CA), an analytical C-18 reverse-phase column kept at 30°C (Ultrasphere ODS 5 μm spherical, 80 A pore, 2 mm × 15 cm; Beckman Instruments), and an RF-551 spectrofluorometric detector (Shimadzu). Excitation and emission were set at 360 and 450 nm, respectively. The mobile phase consisted of (1) 50 mm sodium phosphate/10% methanol, pH 7.2, and (2) 50 mm sodium phosphate/70% methanol, pH 7.2. After 8 min of isocratic conditions with 98% (1) and 2% (2), (2) was increased up to 40% in 30 min and then to 98% in 1 min and maintained at 98% for 11 min before returning to the initial conditions. The radioactivity coeluting with GABA was collected and counted by scintillation spectrometry. Protein concentrations in the original samples were determined by using a commercially available kit (Bio-Rad protein assay).
Materials. Ciliary neurotrophic factor (CNTF), glial-derived neurotrophic factor (GDNF), basic fibroblast growth factor (bFGF), tumor necrosis factor-α (TNF-α), and human recombinant TGFβ1 or -β2 were purchased from Sigma. Antibodies specific for TGFβ1 (catalog #sc-146) or TGFβ2 (catalog #sc-90) were purchased from Santa Cruz Biotechnology. Both antibodies are reactive against human, rat, and mouse TGFβ1 or -2. DCG-IV, 4C3HPG,l-2-amino-4-phosphonobutanoic acid (l-AP4), and NMDA were purchased from Tocris Cookson.
RESULTS
TGFβ protects cultured neurons against excitotoxic death and mediates neuroprotection by glial group-II mGlu receptors
A 10 min exposure of mixed cortical cultures to 100 μm NMDA produced the delayed degeneration of ∼80% of the neuronal population (Table 1). NMDA toxicity was attenuated by the medium collected from pure cultures of astrocytes (GM) 2 or 20 hr after a 10 min exposure to 1 μm DCG-IV (GM/DCG-IV) or 100 μm 4C3HPG (GM/4C3HPG) [Table 1; see Bruno et al. (1997) for a more detailed characterization]. We started the search for the neuroprotective agent present in the GM/DCG-IV or GM/4C3HPG by screening a number of trophic factor for their neuroprotective activity against NMDA toxicity. Factors were either combined with NMDA for 10 min or applied immediately after the NMDA pulse and then maintained in the medium for the following 20 hr. Under both conditions, TGFβ1 and -β2 displayed the highest neuroprotective activity, followed by that of bFGF. GDNF, CNTF, and TNF-α produced little, if any, neuroprotection (Fig.1). We have not examined the effect of nerve growth factor or other neurotrophins, because they are reported not to affect or even amplify NMDA toxicity in mixed cortical cultures (Koh et al., 1995).
Table 1.
Neuroprotection by the medium collected from glial cultures 2 or 20 hr after a transient exposure to group-II mGlu receptor agonists
| NMDA toxicity | ||
|---|---|---|
| Number of dead cells | LDH release (mO.D./min) | |
| Basal | 7 ± 0.7 | 15 ± 7 |
| NMDA, 100 μm | 155 ± 9 | 119 ± 8 |
| + GM/control, 2 hr | 168 ± 11 | 116 ± 22 |
| + GM/control, 20 hr | 183 ± 24 | 104 ± 17 |
| + GM/DCG-IV, 2 hr | 67 ± 7* | 59 ± 9* |
| + GM/DCG-IV, 20 hr | 74 ± 15* | 57 ± 6* |
| + GM/4C3HPG, 2 hr | 71 ± 9* | 63 ± 11* |
| + GM/4C3HPG, 20 hr | 56 ± 5* | 60 ± 8* |
Values are means ± SEM of 12–16 determinations. *p < 0.01 (one-way ANOVA + Fisher PLSD) versus NMDA alone. GM/control, Glial medium (GM) collected 2 or 20 hr after a 10 min exposure of pure cultures of astrocytes to the buffer alone; GM/DCG-IV and GM/4C3HPG, medium collected after a 10 min exposure of cultured astrocytes to 1 μm DCG-IV or 100 μm 4C3HPG. Collected medium was transferred to mixed cultures immediately after the toxic pulse with NMDA and maintained for the following 20 hr. The astrocyte medium did not affect neuronal viability in the absence of NMDA (data not shown). The number of dead cells was determined by counting neurons stained with trypan blue in three random microscopic fields. See Bruno et al. (1997) for a more detailed characterization. mO.D., Milli-Optical density.
Fig. 1.

Effect of trophic factors on NMDA toxicity in mixed cultures of cortical cells. Factors were either combined with NMDA (coadded) or applied to the cultures immediately after the NMDA pulse and maintained in the medium during the following 20 hr (post). Values are means ± SEM of four to six determinations from two different multiplates and were calculated from the counts of neurons stained with trypan blue. Results were virtually identical when calculated from the extracellular LDH activity, which was always measured in parallel. In each multiplate, the mean of values obtained from individual dishes treated with NMDA alone, after subtracting the basal values, was considered as 100% or NMDA toxicity. Each individual determination was expressed as percentage of NMDA toxicity, always after subtracting the respective basal values. The SD calculated from the original counts of neurons stained with trypan blue was always <10% of the mean value for each experimental group. *p < 0.01 (one-way ANOVA + Fisher PLSD), if compared with values obtained with NMDA alone.
TGFβ1 and -β2 were equipotent as neuroprotectants against NMDA toxicity, and their efficacy was essentially similar (Fig.2). TGFβ1 or -β2 did not produce any further neuroprotection when combined with DCG-IV or 4C3HPG (Fig.3A) or with GM/DCG-IV (Fig.3B). In contrast, the neuroprotective activity ofl-AP4 (100 μm) was additive with that produced by GM/DCG-IV (Fig. 3B).
Fig. 2.

Concentration-dependent neuroprotection by TGFβ1 or -β2 against NMDA toxicity in mixed cultures of cortical cells. TGFβ1 or -β2 were applied immediately after the NMDA pulse and maintained in the medium during the following 20 hr. Values are means ± SEM of four individual determinations and were calculated from the counts of neurons stained with trypan blue, as described in Figure 1.
Fig. 3.
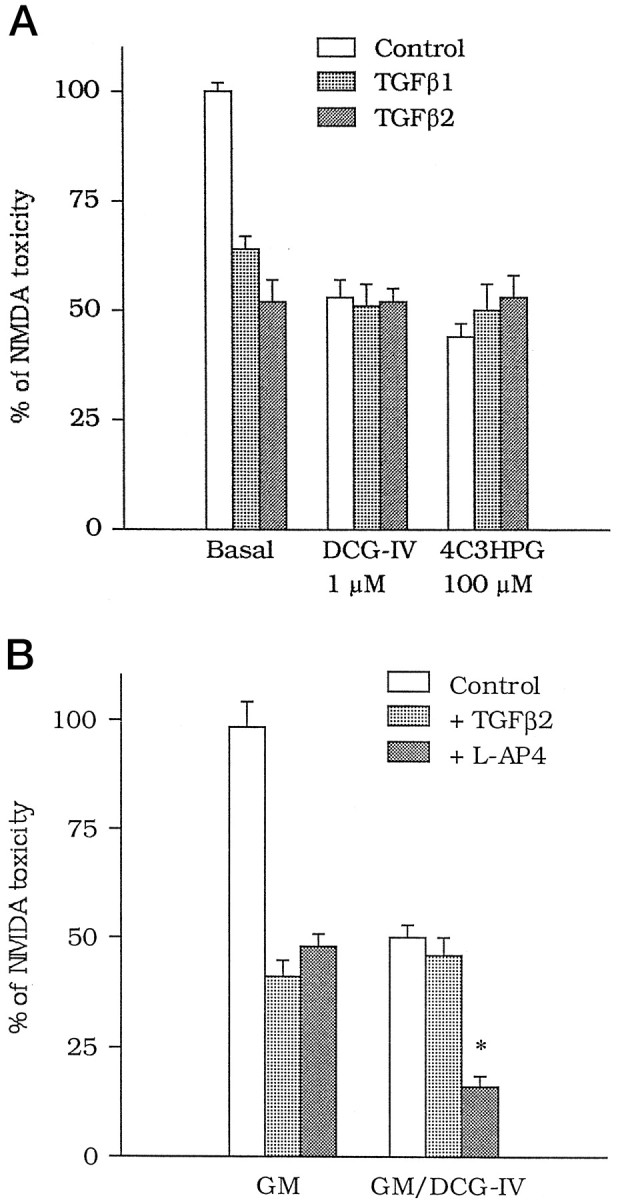
A, Lack of additive effects between TGFβ1 or -β2 and group-II mGlu receptor agonists on NMDA toxicity in mixed cortical cultures. DCG-IV (1 μm) or 4C3HPG (100 μm) were applied in combination with NMDA, whereas TGFβ1 or -β2 (both at 1 ng/ml) were applied after the NMDA pulse. Values are means ± SEM of four determinations. B, The neuroprotective activity of TGFβ2 (1 ng/ml) is obliterated when the factor is combined with the glial medium collected 20 hr after a 10 min exposure to 1 μm DCG-IV (GM/DCG-IV). Note that the protective activity of l-AP4 (100 μm) is instead additive to that produced by the medium of DCG-IV-treated astrocytes. GM, Glial medium. Values are means ± SEM of four determinations. *p < 0.01 (one-way ANOVA + Fisher PLSD) if compared with the respective controls. InA and B, values were calculated from the counts of neurons stained with trypan blue, as described in Figure1.
We used antibodies specific for TGFβ1 or -β2 (TGFβ1Ab or TGFβ2Ab), which were able to neutralize the neuroprotective activity of exogenously applied TGFβ1 or -β2 (Fig.4A) but not that of bFGF (see legend of Fig. 4A). TGFβ1Ab or TGFβ2Ab added to the GM/DCG-IV or GM/4C3HPG abolished neuroprotection, whereas a neutralizing antibody against GDNF was inactive (Fig.4B). Interestingly, TGFβ1Ab and TGFβ2Ab also prevented the neuroprotective activity of group-II mGlu receptor agonists applied in combination with NMDA (Fig. 4C).
Fig. 4.
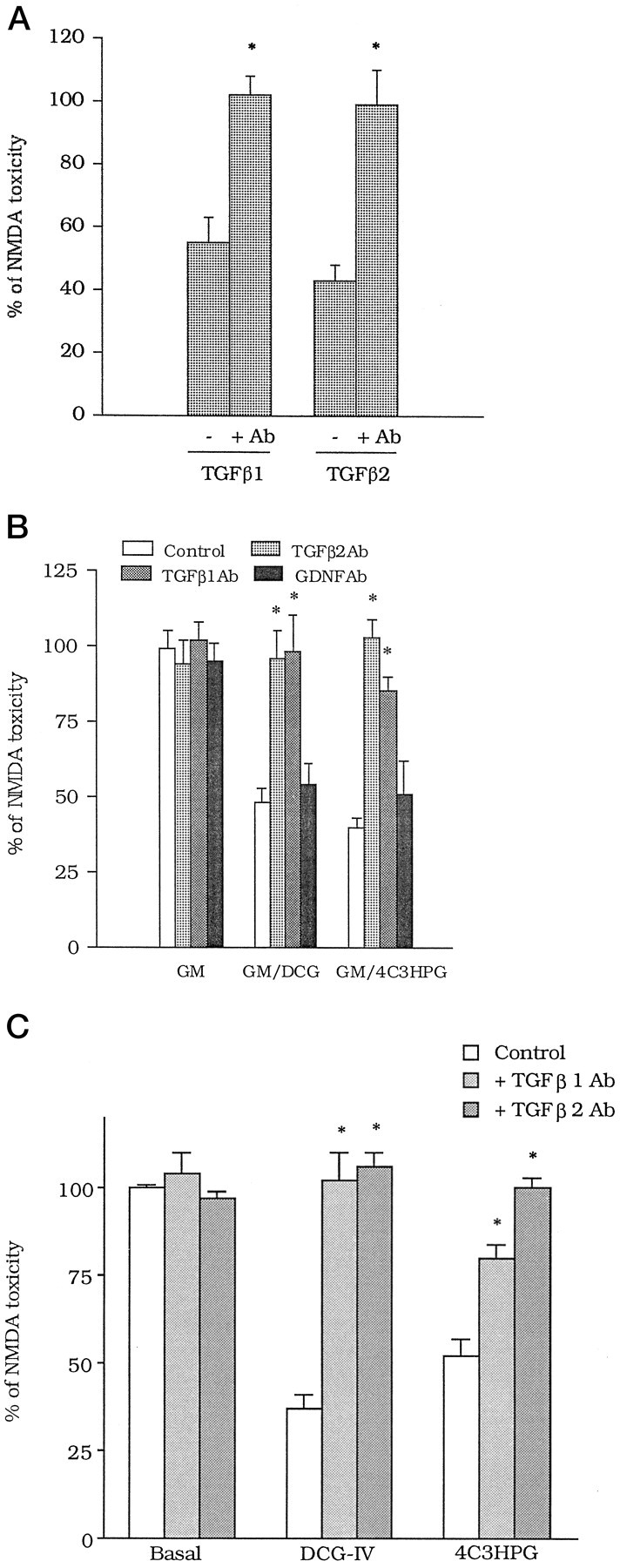
A, Antibodies (Ab) specific for TGFβ1 or -β2 (both at 100 ng/ml) abolish the neuroprotective activity of TGFβ1 and -β2 (both at 10 ng/ml) in mixed cortical cultures. Each of the factors and the respective antibody were added to the cultures immediately after the NMDA pulse and maintained in the medium during the following 20 hr. The concentration of antibodies was 100 ng/ml. Values are means ± SEM of eight determinations from two different experiments. *p < 0.01 (Student’s t test), if compared with the respective values obtained in the absence of the antibody. TGFβ1Ab at least did not prevent neuroprotection induced by bFGF applied after the NMDA pulse [bFGF, 10 ng/ml = 70 ± 3.5; bFGF + TGFβ1Ab (100 ng/ml) = 66 ± 4.2; TGFβ1Ab alone = 92 ± 6.3; n = 4, expressed as percentage of NMDA toxicity]. B, TGFβ1Ab and TGFβ2 Ab (both at 100 ng/ml) prevent the neuroprotective activity of the glial medium collected 20 hr after treating cultured astrocytes with 1 μm DCG-IV (GM/DCG-IV) or 100 μm 4C3HPG (GM/4C3HPG). Note that antibodies against GDNF (GDNFAb, 100 ng/ml) are inactive. All antibodies were applied to the glial medium before it was transferred to mixed cortical cultures. Values are means ± SEM of eight determinations from two different experiments; *p < 0.01 (one-way ANOVA + Fisher PLSD) if compared with the respective controls.C, TGFβ1Ab and TGFβ2Ab (all at 100 ng/ml) reduce the neuroprotective activity of group-II mGlu receptor agonists in mixed cortical cultures. DCG-IV (1 μm) or 4C3HPG (100 μm) were applied to the cultures during the 10 min pulse with NMDA. Antibodies were applied immediately after the pulse and maintained in the medium during the following 20 hr. Values are means ± SEM of four determinations. *p < 0.01 (one-way ANOVA + Fisher PLSD), if compared with the respective controls. In A–C, values were calculated from the counts of neurons stained with trypan blue, as described in Figure1.
We therefore examined whether astrocytes treated with group-II mGlu receptor agonists release TGFβ into the medium by using a highly sensitive and specific bioassay, based on the ability of TGFβ to reduce the proliferation rate of mink lung epithelial cells. The control GM (i.e., the medium collected from astrocytes 20 hr after simple addition of the buffer) showed an antiproliferative activity corresponding to that produced by 5 pg/ml of authentic human TGFβ1 or -β2. This activity increased several-fold 20 hr after a 10 min exposure to DCG-IV or 4C3HPG (Table 2). These results were confirmed by measuring the extracellular levels of TGFβ1 by ELISA (medium from control astrocytes = 6.4 ± 0.32 pg/ml; medium from astrocytes treated with 4C3HPG = 38 ± 0.45 pg/ml).
Table 2.
A transient activation of group-II mGlu receptors increases the extracellular levels of TGFβ in cultured astrocytes
| TGFβ1-like activity (arbitrary units) | TGFβ2-like activity (arbitrary units) | |
|---|---|---|
| Control astrocytes | 1.0 ± 0.31 | 1.0 ± 0.18 |
| DCG-IV/astrocytes | 7.1 ± 0.42* | 4.5 ± 0.25* |
| 4C3HPG/astrocytes | 6.9 ± 0.51* | 5.9 ± 0.69* |
Values are means ± SEM from 12 individual determinations. *p < 0.01 (one-way ANOVA + Fisher PLSD), if compared with control astrocytes. TGFβ1- or TGFβ2-like activity was determined by measuring the ability of the medium to reduce [3H]thymidine incorporation in mink lung epithelial cells. One arbitrary unit corresponds to the antiproliferative activity produced by 5 pg/ml of authentic human TGFβ1 or -β2. Values were calculated from reference curves constructed with authentic human TGFβ1 and -β2 (from 0.1 to 10,000 pg/ml). Maximal concentrations of TGFβ1 or -β2 (300–1000 pg/ml in different assays) reduced [3H]thymidine incorporation by 70–80%. The different numbers between TGFβ1- and TGFβ2-like activities reflect the slightly different potency we have found between authentic TGFβ1 and -β2 in reducing [3H]thymidine incorporation. bFGF failed to reduce [3H]thymidine incorporation in mink lung epithelial cells [[3H]thymidine incorporation (cpm/well): basal = 28840 ± 1737; TGFβ1, 300 pg/ml = 6900 ± 2349; bFGF, 3 ng/ml = 34560 ± 4510;n = 4].
Finally, we measured the intracellular levels of TGFβ2 in response to group-II mGlu receptor activation in astrocytes. Immunoblots of cultured astrocytes showed a single band of 24–25 kDa, corresponding to the dimeric form of TGFβ (Fig. 5). In control astrocytes, the intensity of this band decreased from 2 to 10 hr after a 10 min treatment with buffer followed by incubation in serum-free medium. The intracellular levels of dimeric TGFβ2 were substantially higher 2 or 10 hr after treating the cultures with DCG-IV (Fig. 6). A similar increase in TGFβ2 was induced by the A1 adenosine receptor agonist 2-chloro-N6-cyclopentyladenosine (CCPA) (Fig. 5).
Fig. 5.

Western blot analysis of TGFβ2 in protein extracts from cultured astrocytes transiently exposed to 1 μm DCG-IV or to 100 nm CCPA.CTRL, Control astrocyte cultures treated with buffer alone for 10 min, and then incubated for 2 or 10 hr in serum-free medium (see Materials and Methods); DCG-IV, cultures treated for 10 min with 1 μm DCG-IV and then incubated for 2 or 10 hr in serum-free medium; CCPA, cultures treated for 10 min with CCPA and then incubated for 2 or 10 hr in serum-free medium. Expression of β-actin is shown in the same protein extracts. Authentic monomeric human TGFβ2 is shown in thefirst lane.
Fig. 6.
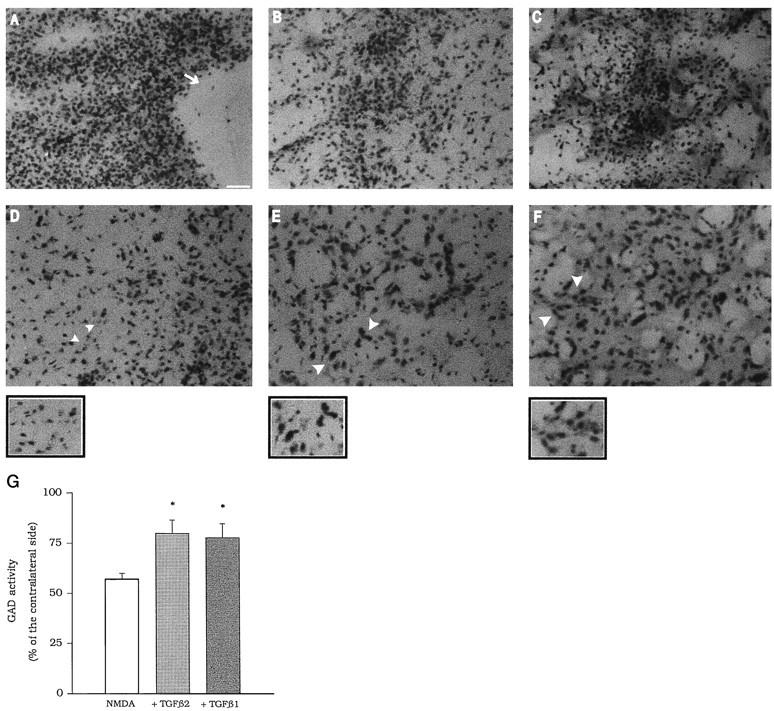
A–F, Local infusion of TGFβ1 or -β2 protects against NMDA toxicity in the rat caudate nucleus. The core of the lesion in animals injected with NMDA, NMDA + TGFβ1, and NMDA + TGFβ2 is shown in A, B, andC, respectively. The white arrow points to a large area of necrosis, which is absent in B andC. Neuronal degeneration present at the periphery of the lesion in animals injected with NMDA is shown in D. The corresponding regions in animals injected with NMDA + TGFβ1 and NMDA + TGFβ2 are shown in E and F. The small areas indicated by the white arrowheads are also shown at higher magnification. Note the greater number of spared neurons in animals treated with TGFβ1 or -β2. G, Striatal GAD activity in animal locally infused with NMDA or with NMDA + TGFβ1 or NMDA + TGFβ2. Results are expressed as percentage of the respective contralateral unlesioned site for each individual determination. GAD activity was calculated as counts per minute of authentic [3H]GABA/μg of protein. *p< 0.05 (one-way ANOVA + Fisher PLSD) versus NMDA alone.
Neuroprotective activity of TGFβ against invivo excitotoxicity
In animals infused with NMDA alone (100 nmol/0.5 μl per 2 min; double injection) in the left caudate nucleus, histological analysis showed an extensive necrotic region at the injection sites and neuronal loss, reactive gliosis, and edema in the surrounding tissue. In animals coinfused with NMDA and 0.5 ng of TGFβ1 or -β2, the extension of the necrotic area was smaller, and the neuronal loss in the surrounding tissue was substantially reduced (Fig. 6A–F). We have quantified the protective activity of TGFβ1 or -β2 by measuring striatal GAD activity, which reflects the survival of GABAergic projection neurons and interneurons. Infusion of TGFβ1 or -β2 prevented the reduction in striatal GAD activity induced by NMDA (Fig. 6G). Intrastriatal infusion of TGFβ1 or -β2 alone did not produce significant changes in GAD activity after 7 d (data not shown).
DISCUSSION
The novel selective group-II mGlu receptor agonist LY354740 protects pure neuronal cultures against excitotoxic death, but only at concentrations higher than those sufficient to interact with mGlu2 or -3 receptors (Kingston et al., 1977). This casts doubt on the role of group-II mGlu receptors in neuroprotection, although several reports show a clear-cut protective activity of mGluR2/3 receptor agonists (for review, see Nicoletti et al., 1996). We suggest that it is the presence of astrocytes that enables neuroprotection by group-II mGlu receptor agonists. Accordingly, a brief exposure of glial cultures to the mGlu2/3 agonists DCG-IV, 4C3HPG, or (2S,1′S,2′S)-2-(carboxycyclopropyl)glycine, or to the selective mGlu3 receptor agonist α-N-acetylaspartylglutamate renders the medium neuroprotective against excitotoxic death (Bruno et al., 1997, 1998). This protective activity is abolished after heating the medium or after treating the astrocytes with the protein synthesis inhibitor cycloheximide (Bruno et al., 1997). Hence, we have hypothesized that astrocytes treated with mGlu3 receptor agonists produce and release a proteic neuroprotective factor. Present results identify this factor with TGFβ, although they cannot exclude the possibility that other factors are produced, the activity of which depends on TGFβ.
TGFβ1 and -β2 showed a substantial neuroprotective activityin vitro and in vivo, which, unexpectedly, was greater than that exhibited by established neurotrophic factors such as GDNF or CNTF. The protective activity of TGFβ is in agreement with several recent reports (Prehn et al., 1994, 1996; Prehn, 1996; Ren and Flanders, 1996; Buisson et al., 1997) [but see also Kane et al. (1996)]. Protection by TGFβ1 or -β2 was obscured by the medium collected from astrocytes treated with DCG-IV, and not because the response was saturated: the group-III mGlu receptor agonistl-AP4 could in fact further enhance the protective activity of the glial medium.
Untreated astrocytes released a low amount of TGFβ into the medium (∼5 pg/ml), which was below the threshold for neuroprotection. However, astrocytes transiently exposed to DCG-IV or 4C3HPG released a five- to sevenfold greater amount of TGFβ (TGFβ1, -β2, or both), which may account for the protective activity of the glial medium. Finally, we proved that TGFβ was necessary for the neuroprotective activity of the glial medium by using antibodies against TGFβ1 or -β2. Each of these antibodies abolished neuroprotection when applied to the medium collected from astrocytes treated with DCG-IV or 4C3HPG. Because the two antibodies are specific for their respective TGFβ isoforms, we conclude that the combination of TGFβ1 and -β2 released in the glial medium reaches the threshold for neuroprotection or (more likely) that TGFβ is released as a β1/β2 heterodimer.
The glial medium acquires its neuroprotective activity at least 2 hr after a transient activation of mGlu3 receptors, and neuroprotection vanishes after astrocytes are treated with the protein synthesis inhibitor cycloheximide (Bruno et al., 1997). Hence, we have considered the possibility that activation of glial mGlu3 receptors enhances thede novo synthesis of TGFβ. TGFβ is synthesized as a dimer from the cleavage of a high molecular weight precursor (Massaguè et al., 1994: Flanders et al., 1998). A dimeric form of TGFβ containing TGFβ2 was detectable by Western blot analysis in cultured astrocytes, and its levels decreased with time after the cultures were incubated in the absence of serum (as we generally did in all experiments in which we collected the glial medium). Intracellular dimeric TGFβ increased substantially after the cultures were treated with DCG-IV, suggesting that activation of mGlu3 receptors enhances thede novo synthesis of TGFβ. We cannot exclude the possibility, however, that mGlu3 receptor activation inhibits the degradation rate of TGFβ and that neuroprotection is cycloheximide-sensitive because it requires the synthesis of a different protein that is necessary for the accumulation and release of TGFβ. It is noteworthy that an increase in intracellular TGFβ levels was also induced after transient activation of A1 adenosine receptors, which share with group-II mGlu receptors the coupling with the Gi type of GTP binding proteins (for review, see Collis and Hourani, 1994). The βγ subunits released in large amounts from trimeric Gi-proteins exert pleiotropic effects, including activation of adenylyl cyclase types II or IV, phospholipase C, or the MAP kinase pathway (for review, see Sternweis, 1994; Morris and Scarlata, 1997). Whether any of these intracellular pathways contributes to the production and release of TGFβ in response to mGlu3 receptor activation will be the subject of future investigation.
The protective activity of TGFβ1 or -β2 against NMDA toxicity may provide new insights into the mechanism of neuronal degeneration. Both TGFβ and group-II mGlu receptor agonists protect not only against excitotoxic death but also against other forms of neurodegeneration, including neuronal apoptosis induced by β-amyloid peptide (Copani et al., 1995; Prehn et al., 1996; Ren et al., 1997). One can therefore speculate that TGFβ prevents the execution of a pathway that is common to various forms of neuronal degeneration. TGFβ1 or -β2 interacts with membrane receptors endowed with intrinsic serine/threonine kinase activity. Activation of these receptors leads to the phosphorylation of the latent transcription factor Smad2, which after complexing with Smad4 migrates to the nucleus where it activates gene expression (Massaguè et al., 1994, 1997). Established target genes of TGFβ are the “check points” p27 and p21, which produce growth arrest by inhibiting the activity of cyclin-dependent kinases (Datto et al., 1995; Ravitz, 1996). This mechanism may be relevant for the neuroprotective activity of TGFβ, because the induction of an abortive mitotic cycle has been causally related to the development of neuronal degeneration (Freeman et al., 1995; Herrup and Busser, 1995; Park et al., 1997a,b). Alternatively, TGFβ may act to inhibit the expression of cyclooxygenase-2 (Minghetti et al., 1998; Pruzanski et al., 1998), an enzyme that is upregulated in response to synaptic excitation or β-amyloid peptide and is implicated in the pathophysiology of neuronal degeneration (Yamagata et al., 1993; Adams et al., 1996; Pasinetti, 1997).
In conclusion, activation of glial mGlu3 receptors may provide a mechanism for increasing the local production of TGFβ in the brain, thereby protecting neurons against various toxic insults. Through this particular mechanism, selective mGlu3 receptor agonists are expected to exert a wide-range neuroprotective effect without causing the side effects associated with use of NMDA or AMPA receptor antagonists, such as sedation, impairment of synaptic plasticity, ataxia, or psychotomimetic effects.
Footnotes
Correspondence should be addressed to Dr. Ferdinando Nicoletti, Institute of Pharmacology, School of Pharmacy, University of Catania, Viale AA Doria 6, 95125 Catania, Italy.
REFERENCES
- 1.Adams J, Collaco-Moraes Y, de Belleroche J. Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. J Neurochem. 1996;66:6–13. doi: 10.1046/j.1471-4159.1996.66010006.x. [DOI] [PubMed] [Google Scholar]
- 2.Bruno V, Sureda FX, Storto M, Casabona G, Caruso A, Knopfel T, Kuhn R, Nicoletti F. The neuroprotective activity of group-II metabotropic glutamate receptors requires new protein synthesis and involves a glial-neuronal interaction. J Neurosci. 1997;17:1891–1897. doi: 10.1523/JNEUROSCI.17-06-01891.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bruno V, Wroblewska B, Wroblewska JT, Fiore L, Nicoletti F. Neuroprotective activity of N-acetylaspartylglutamate in cultured cortical cells. Neuroscience. 1998;3:751–757. doi: 10.1016/s0306-4522(97)00531-9. [DOI] [PubMed] [Google Scholar]
- 4.Buisson A, Choi DW. The inhibitory mGluR agonist, S-4-carboxy-3-hydroxyphenylglycine, selectively attenuates NMDA neurotoxicity and oxygen-glucose deprivation-induced neuronal death. Neuropharmacology. 1995;34:1081–1087. doi: 10.1016/0028-3908(95)00073-f. [DOI] [PubMed] [Google Scholar]
- 5.Buisson A, Nicole O, Nouvelot A, MacKenzie ET, Vivien D. Reduction of NMDA-induced toxicity by transforming growth factor-β1. Soc Neurosci Abstr. 1997;23:897. [Google Scholar]
- 6.Ciccarelli R, Sureda FX, Casabona G, Di Iorio P, Caruso A, Spinella F, Condorelli DF, Nicoletti F, Caciagli F. Opposite influence of the metabotropic glutamate receptors subtypes mGlu3 and -5 on astrocyte proliferation in culture. Glia. 1997;21:390–398. doi: 10.1002/(sici)1098-1136(199712)21:4<390::aid-glia6>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 7.Collis MG, Hourani SMO. Adenosine receptor subtypes. Trends Pharmacol Sci. 1993;14:360–366. doi: 10.1016/0165-6147(93)90094-z. [DOI] [PubMed] [Google Scholar]
- 8.Copani A, Bruno V, Battaglia G, Leanza G, Pellitteri R, Russo A, Stanzani S, Nicoletti F. Activation of metabotropic glutamate receptors protects against apoptosis induced by β-amyloid peptide. Mol Pharmacol. 1995;47:890–897. [PubMed] [Google Scholar]
- 9.Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci USA. 1995;92:5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flanders KC, Ren RF, Lippa CF. Transforming growth factor-betas in neurodegenerative disease. Prog Neurobiol. 1998;54:71–85. doi: 10.1016/s0301-0082(97)00066-x. [DOI] [PubMed] [Google Scholar]
- 11.Freeman RF, Estus S, Johnson EM., Jr Analysis of cell-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 12.Genazzani AA, L’Episcopo MR, Casabona G, Shinozaki H, Nicoletti F. (2S,1′R,2′R,3′R)-2-(2,3-dicarboxycyclopropyl)glycine positively modulates metabotropic glutamate receptors coupled to polyphosphoinositide hydrolysis in rat hippocampal slices. Brain Res. 1994;659:10–16. doi: 10.1016/0006-8993(94)90857-5. [DOI] [PubMed] [Google Scholar]
- 13.Herrup K, Busser JC. The induction of multiple cell cycle events precedes target-related neuronal death. Development. 1995;121:2385–2395. doi: 10.1242/dev.121.8.2385. [DOI] [PubMed] [Google Scholar]
- 14.Kane CJ, Brown GJ, Phelan KD. Transforming growth factor-β2 increases NMDA receptor-mediated excitotoxicity in rat cerebral cortical neurons independently of glia. Neurosci Lett. 1996;204:93–96. doi: 10.1016/0304-3940(96)12332-6. [DOI] [PubMed] [Google Scholar]
- 15.Kingston AE, Bales KR, Monn JA, Paul SM, Pullar IA, Schoepp DD. Comparison of the neuroprotective effects of mGluR agonists: (RS)-3,5-dihydroxyphenylglycine, LY354740 and l-amino-4-phosphonobutyric acid on ionotropic glutamate receptor-induced excitotoxicity in rat cortical neurons. Soc Neurosci Abstr. 1997;23:899. [Google Scholar]
- 16.Koh JY, Choi DW. Quantitative determination of glutamate-mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 17.Koh JY, Gwag BJ, Lobner D, Choi DW. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science. 1995;268:573–575. doi: 10.1126/science.7725105. [DOI] [PubMed] [Google Scholar]
- 18.Massaguè J, Attisano L, Wraba JL. The TGF-β family and its composite receptors. Trends Cell Biol. 1994;4:172–178. doi: 10.1016/0962-8924(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 19.Massaguè J, Hata A, Liu F. TGF-β signalling through the Smad pathway. Trends Cell Biol. 1997;7:187–192. doi: 10.1016/S0962-8924(97)01036-2. [DOI] [PubMed] [Google Scholar]
- 20.Minghetti L, Polazzi E, Nicolini A, Levi G. Opposite regulation of prostaglandin E2 synthesis by transforming growth factor-β1 and interleukin 10 in activated microglial cultures. J Neuroimmunol. 1998;82:31–39. doi: 10.1016/S0165-5728(97)00185-9. [DOI] [PubMed] [Google Scholar]
- 21.Morris AJ, Scarlata S. Regulation of effectors by G protein alpha-, and beta gamma-subunits. Recent insights from studies of the phospholipase C-beta isoenzymes. Biochem Pharmacol. 1997;54:429–435. doi: 10.1016/s0006-2952(97)00032-4. [DOI] [PubMed] [Google Scholar]
- 22.Nicoletti F, Bruno V, Copani A, Casabona G, Knoepfel T. Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders? Trends Neurosci. 1996;19:267–271. doi: 10.1016/S0166-2236(96)20019-0. [DOI] [PubMed] [Google Scholar]
- 23.Park DS, Levine B, Ferrari G, Greene LA. Cyclin-dependent kinase inhibitors and dominant negative cyclin-dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 1997a;17:8975–8983. doi: 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park DS, Morris EJ, Greene LA, Geller HM. G1/S Cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J Neurosci. 1997b;17:1256–1270. doi: 10.1523/JNEUROSCI.17-04-01256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pasinetti GM (1997) Cyclooxygenases and inflammation in Alzheimer’s disease: experimental approaches and therapeutical implications. Abstract of the 36th Meeting of the American College of Neuropsychopharmacology (ACNP), Waikoloa, Hawaii, December.
- 26.Pellegrino JL, Pellegrino SA, Cushman JA. A stereotaxic atlas of the rat brain. Plenum; New York: 1992. [Google Scholar]
- 27.Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 28.Prehn JH. Marked diversity in the action of growth factors on N-methyl-d-aspartate-induced neuronal degeneration. Eur J Pharmacol. 1996;306:81–88. doi: 10.1016/0014-2999(96)00225-7. [DOI] [PubMed] [Google Scholar]
- 29.Prehn JH, Bindokas VP, Marcuccilli CJ, Krajewski S, Reed JC, Miller RJ. Regulation of neuronal Bcl-2 protein expression and calcium homeostasis by transforming growth factor type beta confers wide ranging protection on rat hippocampal neurons. Proc Natl Acad Sci USA. 1994;91:12599–12603. doi: 10.1073/pnas.91.26.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prehn JHM, Bindokas VP, Jordan J, Galindo MF, Ghadge GD, Roos RP, Boise LH, Thomson CB, Krajewski SW, Reed JC, Miller RJ. Protective effect of transforming growth factor-β1 on β-amyloid neurotoxicity in rat hippocampal neurons. Mol Pharmacol. 1996;49:319–328. [PubMed] [Google Scholar]
- 31.Pruzanski W, Stefanski E, Vadas P, Kennedy BP, van den Bosch H. Regulation of the cellular expression of secretory and cytosolic phospholipase A2, and cyclooxygenase-2 by peptide growth factors. Biochim Biophys Acta. 1998;1403:47–56. doi: 10.1016/s0167-4889(98)00029-9. [DOI] [PubMed] [Google Scholar]
- 32.Ravitz MJ. Differential regulation of p27 and cyclin D1 by TGFβ2 and EGF in C3H10T1/2 mouse fibroblasts. J Cell Physiol. 1996;168:510–520. doi: 10.1002/(SICI)1097-4652(199609)168:3<510::AID-JCP3>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 33.Ren RF, Flanders KC. Transforming growth factors-β protect primary rat hippocampal neuronal cultures from degeneration induced by β-amyloid. Brain Res. 1996;732:16–24. doi: 10.1016/0006-8993(96)00458-1. [DOI] [PubMed] [Google Scholar]
- 34.Ren RF, Hawver DB, Kim RS, Flanders KC. Transforming growth factor-β protects human hNT cells from degeneration induced by β-amyloid peptide: involvement of the TGF-β type II receptor. Brain Res Mol Brain Res. 1997;48:315–322. doi: 10.1016/s0169-328x(97)00108-3. [DOI] [PubMed] [Google Scholar]
- 35.Rose K, Goldberg MP, Choi DW. Cytotoxicity in murine neocortical cell culture. In: Tyson CA, Frazier JM, editors. Methods in toxicology, Vol 1, in vitro biological systems. Academic; San Diego: 1992. pp. 46–60. [Google Scholar]
- 36.Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sternweis PC. The active role of beta gamma in signal transduction. Curr Opin Cell Biol. 1994;6:198–203. doi: 10.1016/0955-0674(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 38.Tanabe Y, Masu M, Ishii I, Shigemoto R, Mizuno N, Nakanishi S. A family of metabotropic receptors. Neuron. 1992;8:169–172. doi: 10.1016/0896-6273(92)90118-w. [DOI] [PubMed] [Google Scholar]
- 39.Tanabe Y, Nomura A, Masu M, Shigemoto R, Mizuno N, Nakanishi S. Signal transduction, pharmacological properties, and expression patterns of two rat metabotropic glutamate receptors, mGluR3 and mGluR4. J Neurosci. 1993;13:1372–1378. doi: 10.1523/JNEUROSCI.13-04-01372.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winder DC, Conn PJ. Activation of metabotropic glutamate receptors increases cAMP accumulation in hippocampus by potentiating responses to endogenous adenosine. J Neurosci. 1993;13:38–44. doi: 10.1523/JNEUROSCI.13-01-00038.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-t. [DOI] [PubMed] [Google Scholar]