

Abstract
Neuronal degeneration and cell death can result from excessive activation of receptors for the excitatory neurotransmitter glutamate; however, the very earliest changes in cytoskeletal organization have not been well documented. We have used an in vitro model system to examine the early consequences of intense glutamate receptor activation on dendritic spine synapses. Cultured hippocampal neurons exposed to NMDA for as little as 5 min exhibited a rapid and extensive loss of dendritic spines. Staining for the presynaptic marker synapsin 1 and the postsynaptic density proteins PSD-95 and the NR1 subunit of NMDA receptors remained intact. The disappearance of spines was accompanied by a selective loss of filamentous actin staining at synapses. The NMDA-induced loss of spine actin was time- and concentration-dependent and blocked by NMDA receptor antagonists. The effect was mimicked by l-glutamate, AMPA, and ionomycin but not by agonists of L-type calcium channels or of metabotropic glutamate receptors. The effect of NMDA on local actin assembly was strongly attenuated by pretreatment with an actin stabilizing compound or by an antagonist of the calcium-dependent protein phosphatase calcineurin. Immunoreactivity for calcineurin was enriched at synapses together with F-actin. These results indicate that the actin-mediated stability of synaptic structure is disrupted by intense glutamate receptor activity and that calcineurin blockers may be useful in preventing such destabilization.
Keywords: dendrite, synapse, dendritic spine, postsynaptic, calcineurin, excitotoxicity, actin, cytoskeleton, glutamate, NMDA, neuronal culture
Dendritic spines are microspecializations of the postsynaptic membrane present on many types of neurons. These specialized structures function as integrative units in synaptic circuitry (Yuste and Denk, 1995). A pronounced decrease in dendritic spine synapses occurs in several neurological conditions, especially ones associated with cognitive impairments. For example, spines are morphologically abnormal in certain forms of mental retardation (Purpura, 1974; Hinton et al., 1991), and decreases in spine number and changes in spine morphology occur during normal aging in rodents and humans (Terry et al., 1987; Geinisman et al., 1992;Masliah et al., 1993). In addition, changes in spines accompany temporal lobe epilepsy (Scheibel et al., 1974), Huntington’s disease (Graveland et al., 1985), and acquired immunodeficiency syndrome-related dementia (Masliah et al., 1997).
Although their precise functions remain somewhat obscure, spines have been proposed to mediate both chemical and structural synaptic plasticity (for review, see Harris and Kater, 1994). Their density and overall morphology vary widely, and this structural diversity likely reflects functional diversity (Harris and Sultan, 1995). In addition, dendritic spines are rapidly modified in response to transmembrane signals, including those associated with behavior, hormonal status, and synaptic activity (Brandon and Coss, 1982; Woolley et al., 1990;Horner, 1993). It is reasonable to hypothesize that spines possess the biochemical machinery to support rapid modifications in cytoskeletal organization in response to glutamate or other physiological stimuli; thus, mechanisms controlling spine stability may be relevant to normal synaptic reorganization.
Actin seems to provide the only structural basis for cytoskeletal organization in dendritic spines, because spines lack microtubules and intermediate filaments. Staining of cultured neurons with rhodamine–phalloidin suggests that F-actin is highly concentrated in spines, appearing as intense punctae along the dendrites (Kaech et al., 1997; Wyszynski et al., 1997). The β and γ isoforms of actin are expressed in brain and specifically targeted to dendritic spines (Kaech et al., 1997). Immunoelectron microscopy studies demonstrated that actin is highly enriched at spines as compared with the surrounding neuropil (Matus et al., 1982; Cohen et al., 1985), and direct observations of actin filaments identified by heavy meromyosin labeling confirm a much greater density of actin filaments in spines compared with that in the adjacent dendritic shaft or presynaptic terminal (Fifkova and Delay, 1982). Electron microscopic studies therefore support the inference in light microscopic studies that the dendritic punctae intensely labeled with rhodamine–phalloidin mainly correspond to dendritic spines.
Reports have suggested that dendritic spines undergo some of the earliest structural changes during excitotoxic injury to neurons (Olney et al., 1979). Excitatory amino acids, including the endogenous neurotransmitter glutamate, activate several subtypes of glutamate receptors in the vertebrate CNS. Excessive stimulation of these receptors results in neuronal cell death 12–24 hr later and occurs during hypoxia and ischemia (stroke), hypoglycemia, and CNS trauma (Choi, 1994; Rothman and Olney, 1995). Animal models have been used to investigate the structural and ultrastructural changes preceding cell death. Such changes include cell body swelling, chromatin clumping, development of dendritic varicosities, and swelling or loss of dendritic spines (Olney et al., 1979; Choi, 1994). Some of these morphological changes have been observed within hours of an excitotoxic event; however, few studies have investigated changes in neuronal structure occurring within the first several minutes. Incubation of cultured neurons with high concentrations of glutamate or NMDA mimics the effects of excitoxicity in vivo (Choi, 1994; Park et al., 1996). The present study used cultured hippocampal neurons to investigate mechanisms underlying glutamate-induced structural changes in dendritic spines.
MATERIALS AND METHODS
Dissociated cultures. Hippocampal neurons were cultured at low density from embryonic day 19 Sprague Dawley rats essentially as described (Goslin and Banker, 1991), except that the feeder layer of cultured primary astrocytes was derived from frozen stocks stored at −80°C for up to 2 months. Cells were plated onto poly-l-lysine–coated glass coverslips at a density of 5400 cells per cm2. Most neurons developed the characteristic morphology of hippocampal pyramidal cells, and these neurons constituted >90% of the cells on the coverslip. Dendritic spines appeared after 16–19 d in vitro and correlated with the appearance of punctate labeling by rhodamine–phalloidin. The density of such actin punctae varied among culture preparations (range, 0.75–1.5 “spine” punctae/μm of dendrite) but was consistent within a given preparation. Cultures were examined with phase-contrast optics before use in experiments to assess the overall health and morphology of the neurons. Coverslips exhibiting abnormal morphologies or significant signs of neuronal degeneration were excluded from experiments. Pharmacological compounds were diluted from concentrated stocks to their indicated final concentrations in conditioned culture medium. Vehicle-only controls were analyzed and found to produce no detectable effects on neuronal morphology or spine density.
Fluorescence histochemistry. Cultures were fixed at 37°C for 20 min in PBS containing 4% paraformaldehyde and 4% sucrose. Cells were permeabilized in 0.25% Triton X-100 and incubated with 10% bovine serum albumin to block nonspecific binding. Filamentous actin was detected by incubating coverslips for 2 hr at room temperature with rhodamine- or Oregon Green 514-conjugated phalloidins (1:10,000 and 1:100 dilutions, respectively; Molecular Probes, Eugene, OR). Primary antibodies were incubated at 4°C overnight at the following dilutions: calcineurin antiserum (gift of Dr. E. Villafranca, Agouron Pharmaceuticals) at 1:50,000; monoclonal antibody to synapsin-1 (Chemicon, Temecula, CA) at 1:100; monoclonal antibody to PSD-95 (Affinity Bioreagents) at 1:100; and monoclonal antibody to the NR1 subunit of NMDA receptors (PharMingen, San Diego, CA) at 1:100. Immunolabeling studies for PSD-95 and NR1 were conducted on neurons fixed for 5 min in −20°C methanol and blocked as above. Anti-mouse and anti-rabbit IgGs coupled to Oregon Green 514 or Cascade Blue (Molecular Probes) were used to detect the immunoreactive signal. 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI; Molecular Probes) or 3,3′-dioctadecyloxaarbocyanine perchlorate (DiO) were diluted in a small volume of cod liver oil and applied dropwise via a micropipet to the cell body of individual neurons after cultures were fixed as above; the dye was allowed to incorporate for ≥1 hr before image collection. Before labeling with DiI or DiO, neurons were double-labeled for F-actin as above, except that the Triton permeabilization step was omitted and coverslips were incubated overnight at 4°C with appropriate fluorescent-phalloidins (Molecular Probes) diluted 1:100.
Light microscopy and spine quantification. Fluorescently labeled neurons were imaged using a 60× oil immersion objective (numerical aperture, 1.4) affixed to an Olympus IX-70 inverted microscope. Digital images were collected using a Photometrix PXL cooled CCD camera equipped with a KAF-1400 silicon chip (6.8 μm pixels) and were analyzed using ISee software (Inovision, Chapel Hill, NC). High quality excitation/emission filter sets (Chroma Technologies) were used to select appropriate wavelengths for visualizing fluorescent probes. For double-labeling experiments, the relative intensities of the two probes were balanced, and neutral density filters were used to prevent bleed-through between channels. For quantification of spine density, neurons were selected randomly, and images of rhodamine fluorescence were digitized. Spines were defined operationally as 0.5–1.5 μm actin-enriched puncta along dendrites (having an average pixel intensity at least 50% above that in the adjacent dendritic region) and were manually counted. Many such F-actin punctae might be associated with nonspinous (“shaft”) synapses. However, because it is difficult, using light microscopy, to distinguish reliably a shaft synapse from a stubby spine or from a spine projecting into the z dimension, we quantified all F-actin punctae detectable along dendrites in the image. For convenience we have used the term “spine” to designate F-actin punctae in bar graphs in several figures (see Figs. 5, 6, 8). Regions of primary, secondary, and tertiary branches were selected at random and were traced as far as possible from their point of origin at the cell body or branch point; their length was determined against microscope calibration standards, and the number of spines per unit length was calculated. A minimum of five dendrites per neuron and 6–15 neurons were included for each experimental group. Approximately 350 μm of total dendritic length was analyzed for each cell. Statistical differences among groups were assessed via one-way ANOVA and appropriate post hoc tests using Prism software (Graph Pad, San Diego, CA). Digital images were transferred to Adobe Photoshop for display purposes.
Fig. 5.
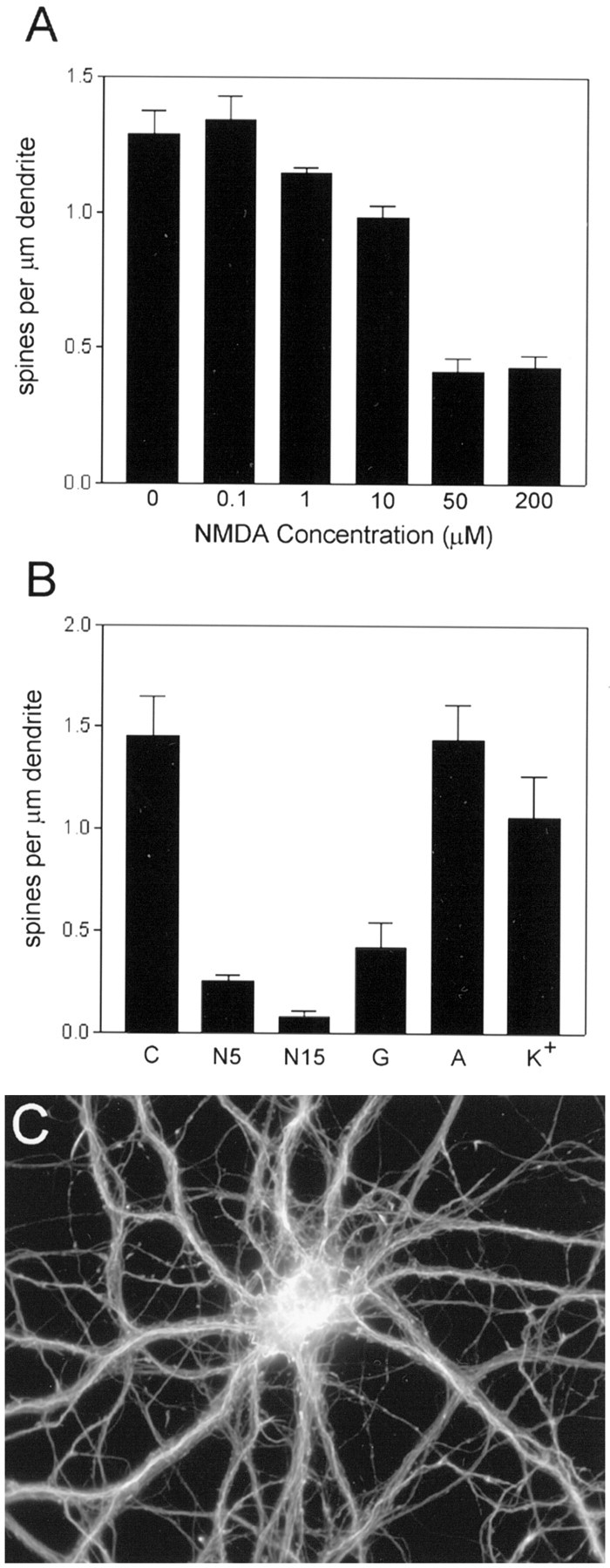
Pharmacological properties of the regulation of F-actin punctae. A, NMDA dose–response relationship. Cultures were incubated for 5 min with the indicated concentrations of NMDA before fixation and staining for F-actin. Concentrations of NMDA ≥ 10 μm caused a statistically significant decrease in the density of F-actin punctae (spines) along dendrites (p < 0.05 for 10, 50, and 200 μm NMDA compared with control, Dunnett’s post hoc test). Vertical bars indicate means; error bars indicate SEM (n = 7 neurons per group).B, Effects of various compounds on F-actin hot spots.Vertical bars indicate spine density (mean ± SEM) observed for neurons incubated under the following conditions: (1) control (C); (2) 50 μm NMDA for 5 min (N5); (3) 50 μm NMDA for 15 min (N15); (4) 50 μml-glutamate for 5 min (G); (5) 200 μm ACPD for 5 min (A); and (6) 40 mm KCl for 5 min (K+). The effects of ACPD and KCl were not significantly different from that of control, but densities of F-actin punctae for l-glutamate and for the two NMDA conditions were significantly reduced compared with control (p < 0.05, Dunnett’s post hoc test). C, Effect of a calcium ionophore on dendritic F-actin punctae. Cultured hippocampal neurons were incubated in the presence of 10 μm ionomycin for 5 min before labeling with rhodamine–phalloidin. Ionomycin had an effect similar to NMDA in selectively reducing the density of F-actin punctae along dendrites, without apparent change in F-actin staining intensity in cell bodies and neurites.
Fig. 6.

Prevention of NMDA-induced spine loss by jasplakinolide. A, B, F-actin labeling in cultures treated with 50 μm NMDA after preincubation in the absence (A) or presence (B) of the actin-stabilizing compound jasplakinolide (2 μm; 2 hr). This compound competes with phalloidin for binding to F-actin. To obtain similar pixel intensities, we collected the image in B using a fivefold longer shutter open time than that used to collect the image inA. This 80% decrease in phalloidin-staining intensity confirms the presence of jasplakinolide bound to intracellular actin filaments. C, Quantification of the protective effect of jasplakinolide (Jasp) on NMDA-induced loss of F-actin punctae (spines). Vertical bars indicate means; error bars indicate SEM (n = 10). NMDA caused a statistically significant loss of F-actin punctae that was fully prevented by preincubation with Jasp (2 μm; 2 hr). The density of F-actin punctae was significantly different in all three experimental groups compared with control (p < 0.05, Dunnett’s post hoc test). D–F. Dendritic spines preserved in the presence of NMDA, in parallel with the preservation of F-actin punctae. DiI-labeled neurons from cultures treated with 50 μm NMDA after preincubation in the absence (D) or presence (E) of jasplakinolide (2 μm; 2 hr), illustrating that the drug indeed prevents spine loss in addition to F-actin loss.F, Enlargement of boxed region inE.
Fig. 8.

Protective effect of the calcineurin antagonist ascomycin. Cultures were preincubated with 20 μm ascomycin (Asc) for 3 hr before treatment in the absence or presence of NMDA (50 μm; 5 min). The Ascalone group was not significantly different from control; theAsc+NMDA group was different from both control and NMDA-treated groups (p < 0.05, Tukey’s multiple comparisons test). Vertical bars indicate means; error bars indicate SEM (n = 7 neurons per group). Similar effects of ascomycin were observed in two additional independent experiments.
RESULTS
NMDA induces rapid loss of F-actin at dendritic spines
To investigate the mechanisms underlying glutamate-induced structural changes, we maintained hippocampal neurons in low-density culture for 18–23 d in vitro before pharmacological manipulation and examination by light microscopy. At this stage most neurons have attained a mature morphology, with highly branched dendrites bearing numerous dendritic spines, which were visualized using the lipophilic fluorescent dyes DiI or DiO to label the plasma membrane (Fig.1A,C).
Fig. 1.
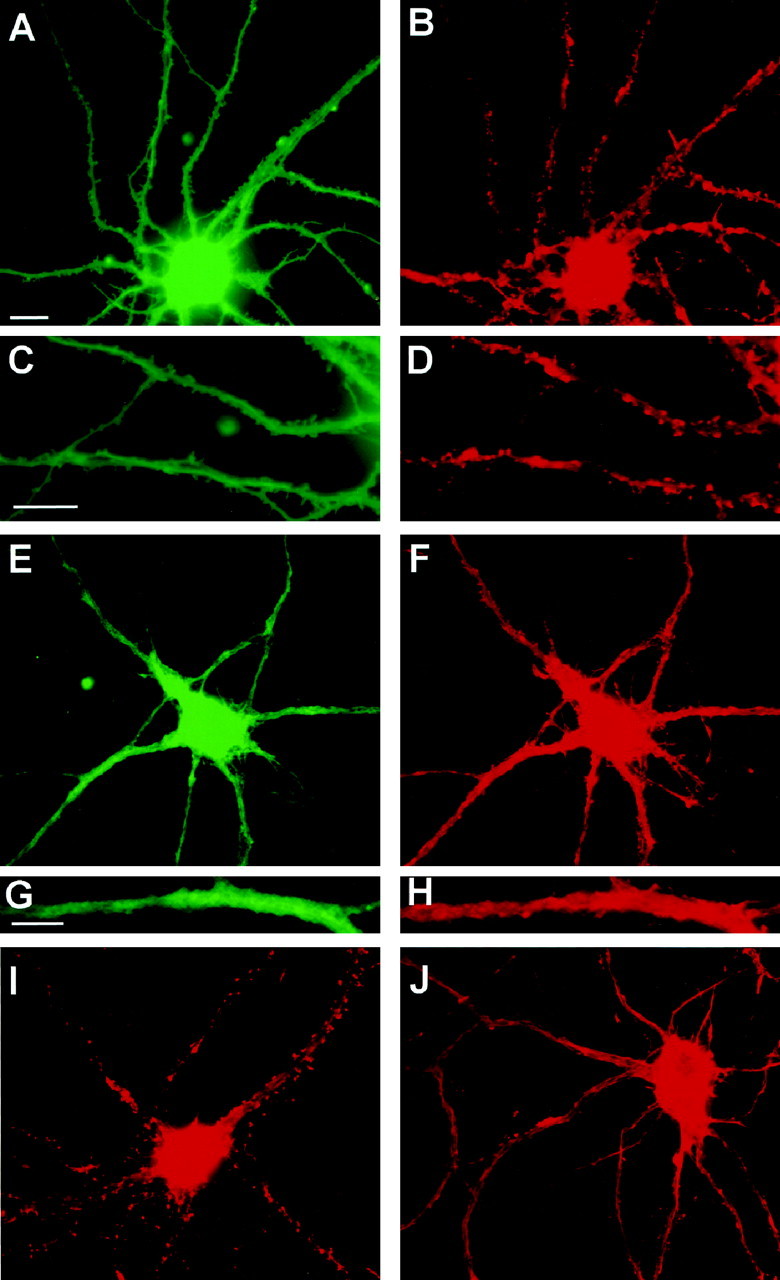
Parallel reduction in spines and F-actin punctae after glutamate receptor stimulation. A–H, Cultures were incubated in the absence (A–D) or presence (E–H) of 50 μm NMDA for 5 min before double-labeling with DiI to stain plasma membrane (A, C, E, G) or with Oregon Green phalloidin to stain F-actin (B, D, F, H). F-actin punctae colocalize with spine-like protrusions of the plasma membrane in control neurons. DiI labeling in NMDA-treated neurons was typically less uniform than that in control neurons. C, D,G, and H are enlargements of dendritic regions in A, B, E, andF, respectively. Note the close correspondence of spine-like profiles with F-actin punctae in the control neuron but not in the NMDA-treated neuron. I, J, Cultures were incubated for 5 min in the absence (I) or presence (J) of 50 μm NMDA before fixation and staining with rhodamine–phalloidin alone. The contrast in J was increased to illustrate clearly the relatively nonpunctate nature of F-actin staining after NMDA stimulation; however, pixel values over the cell body and dendrite shaft were similar in the original digital images for Iand J, which were obtained using identical image collection times and neutral density filters. Scale bars:A, B, E, F,I, J, 10 μm; C,D, 10 μm; G, H, 5 μm.
Fluorescently tagged phalloidin was used to label filamentous actin in the cultures. Phalloidin labels F-actin in cell bodies and axons and throughout the dendrites; however, in control neurons, “hot spots” of F-actin are seen at dendritic spine synapses (Fig.1B,D,I). The majority of neurons in the culture bore a morphology resembling pyramidal type neurons and had a spiny appearance after several days in culture; however, a small percentage (∼5–7%) were of a nonspiny type, having long and relatively unbranched dendrites. Such neurons were specifically labeled by antibodies to glutamic acid decarboxylase, identifying them as GABAergic neurons, known to be present in such cultures (Benson et al., 1994). Neurons of this morphological type were excluded from the analysis.
Cultured hippocampal pyramidal neurons possess both spinous and nonspinous synapses (Bartlett and Banker, 1984), the latter often representing GABAergic synapses that are present together with glutamatergic synapses along the dendrites (Craig et al., 1994). An antibody against the synaptic vesicle protein synapsin-1 was used to label presynaptic nerve terminals in the cultures. Phalloidin hot spots were detected at all synapsin-1–positive punctae, suggesting that both spinous and nonspinous synapses are enriched for F-actin. However, the relative size and intensity of F-actin punctae were variable, perhaps reflecting differences in the amount of filamentous actin present at different classes of synapses. For the purposes of this study, all F-actin punctae along pyramidal cell dendrites (but not those on cell bodies) were quantified regardless of synapse type.
Stimulation of cultures for 5 min with 50 μm NMDA resulted in characteristic changes in dendritic morphology. In many neurons, labeling for the lipophilic membrane dyes became nonuniform as compared with the generally homogeneous appearance of dye incorporation in control cells, perhaps reflecting NMDA-induced membrane damage. Spine-like profiles were rare in NMDA-treated neurons (Fig.1E,G). A selective loss of phalloidin-positive punctae occurred in parallel with this loss of spines, with no apparent change in phalloidin-staining intensity in cell bodies or along the dendrites themselves (Fig.1F,H,J). Together these observations suggest that F-actin at hippocampal synapses becomes destabilized after intense NMDA receptor activation, whereas F-actin within dendritic shafts and axons is comparatively resistant. Thirty minutes or more after a 5 min exposure to 50 μm NMDA, many neurons showed characteristic signs of degeneration, including formation of dendritic varicosities (“beading”), cell body swelling, and disruption of axonal and dendritic morphology; extensive neuronal cell death was observed 24 hr later (data not shown). However, such changes were almost never observed 5 min after NMDA. Cell body diameters assessed in phalloidin-stained neurons measured 22.9 ± 3.6 μm for control cultures and 22.5 ± 3.0 μm for cultures incubated for 5 min with 50 μm NMDA (mean ± SD; n = 15). A previous study reported a reduction in rhodamine–phalloidin staining in cell bodies of cultured cerebellar neurons after brief exposures to NMDA (Shorte, 1997). In our studies, no consistent change in the intensity of staining for F-actin in hippocampal somata was detected, although it was difficult to assess this quantitatively because the cell nucleus occupies variable proportions of somal area in two-dimensional images.
Presynaptic and postsynaptic density markers are stable during early NMDA-induced loss of spines
In contrast to the change in spine actin, the presynaptic compartment of cultured hippocampal neurons appeared relatively resistant to early changes, in agreement with findings from animal models of excitotoxicity (Olney et al., 1979). The punctate pattern of immunoreactivity for the synaptic vesicle protein synapsin-1 was unchanged in cultures incubated for 5–15 min with 50 μmNMDA (Fig. 2). Furthermore, two transmembrane proteins tightly associated with the postsynaptic density (PSD) (Kennedy, 1997), the NR1 subunit of NMDA receptors and PSD-95, still displayed a punctate pattern of staining and were not reduced in numbers after 5–15 min of incubation in the presence of NMDA (Fig. 3). These observations suggest that intense NMDA receptor activity leads to local reorganization of postsynaptic F-actin that is not immediately accompanied by complete breakdown of synaptic structure.
Fig. 2.

Preservation of punctate staining for presynaptic terminals. Cultured hippocampal neurons were incubated in the absence (A, B) or presence (C,D) of 50 μm NMDA for 15 min before double-labeling for F-actin (A, C) or for the synaptic vesicle marker synapsin-1 (B,D). Even while F-actin hot spots were lost, punctate staining for nerve terminals was preserved. Scale bar, 10 μm.
Fig. 3.
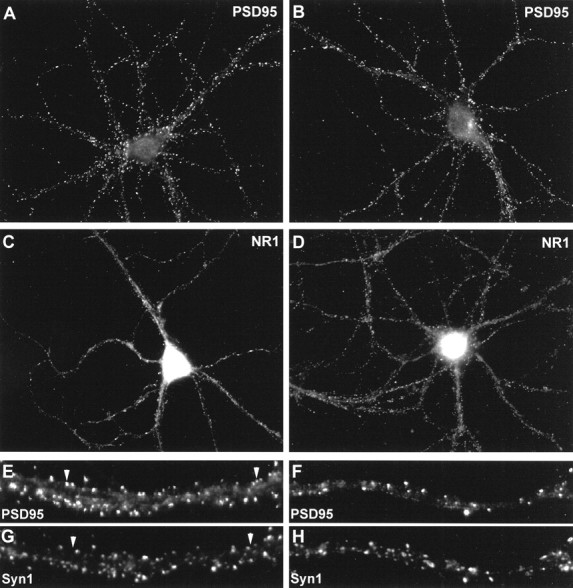
Preservation of punctate staining for proteins of the postsynaptic density. Cultured hippocampal neurons were incubated in the absence (A, C, E,G) or presence (B, D,F, H) of 50 μm NMDA for 5 min before double-labeling for PSD-95 (A,B) or for the NR1 subunit of NMDA receptors (C, D). In other experiments cells were double-labeled for PSD-95 (E, F) and the presynaptic marker synapsin-1 (G,H) to identify synaptic versus extrasynaptic clusters of PSD-95. Extrasynaptic clusters of PSD-95 (arrowheads in E, G) were rare in control cultures. Similarly, cultures incubated with NMDA for 5 min exhibited mainly synaptic clusters of PSD-95 (F,H).
A previous report (Allison et al., 1998) showed that incubation of cultures for 24 hr with the actin-destabilizing drug latrunculin A caused a reduction in the proportion of synaptic NMDA receptor clusters (as compared with extrasynaptic clusters). However, they reported that PSD-95 remained associated with both synaptic and nonsynaptic NMDA receptor clusters. We therefore tested whether NMDA (50 μm; 5 min) induced a decrease in the proportion of synaptic PSD-95 clusters in our cultures by double-labeling for PSD-95 and synapsin-1. As shown in Figure 3E–H, in both control and NMDA-treated cultures, PSD-95 punctae were mainly located at synaptic sites. A small number of punctae appeared to be extrasynaptic, having no apposing synapsin-1–labeled terminal (Fig.3E,G, arrowheads). However, we detected no change in the fraction of PSD-95 clusters associated with presynaptic terminals after a 5 min incubation with 50 μm NMDA. Coverslips treated in parallel and stained for rhodamine–phalloidin showed the characteristic reduction in F-actin punctae described above. Therefore, it would seem that any NMDA-induced loss of PSD-95 and NMDA receptors at synapses probably occurs more slowly than the F-actin reorganization that occurs within the first few minutes.
Pharmacological properties of the stimulus-induced decrease in F-actin punctae
Preincubation of cultures with the specific NMDA receptor antagonists MK-801 (data not shown) ord-amino-5-phosphonopentanoic acid (AP-5; 10 μm; Fig. 4) fully blocked the effect of NMDA in reducing F-actin punctae. Loss of spine F-actin was a function of both NMDA concentration and incubation time. Concentrations ≥10 μm NMDA, when added to the culture medium for 5 min, reliably induced a significant decrease in the number of dendritic F-actin punctae, with an approximate EC50 of 20 μm (Fig. 5A). The time dependence of synaptic F-actin disappearance was determined using 50 μm NMDA. More extensive loss was observed after a 15 min incubation with NMDA than was observed after a 5 min incubation (Fig. 5B), suggesting that synapses may exhibit variable sensitivity to intense glutamate receptor activation. The magnitude of the decrease was variable across experiments, ranging from 30 to 90% loss of dendritic actin punctae; however, statistically significant reductions in the density of punctae were observed in every experiment. Synapses located proximal to the cell body appeared to be more resistant to the effects of NMDA. In the presence of 10 μm AP-5, AMPA (10 μm; 5 min) also induced a decrease in F-actin punctae (data not shown). l-Glutamate (50 μm; 5 min) was nearly as effective as NMDA in inducing loss of F-actin punctae (Fig. 5B), an effect that was not fully inhibited by AP-5. Glutamate induces many intracellular effects by evoking a large increase in intracellular calcium. The NMDA-induced reduction in F-actin punctae was mimicked by brief exposures to the calcium ionophore ionomycin (Fig. 5C). Therefore, multiple routes of calcium entry might contribute to destabilization of spine actin.
Fig. 4.

Blockade of NMDA-induced loss of F-actin punctae by an NMDA antagonist. Cultured hippocampal neurons were exposed to 50 μm NMDA for 5 min after preincubation in the absence (A) or presence (B) of the NMDA receptor antagonist AP-5 (10 μm; 10 min). Scale bars, 10 μm.
However, not all stimuli that raise intracellular calcium were able to mimic the effect of NMDA. The metabotropic glutamate receptor agonist 1S,3R-1-aminocyclopentane-1,3-dicarboxylic acid (ACPD; 200 μm; 5–15 min) was ineffective in reducing spine actin (Fig. 5B). The L-type calcium channel agonist BAYK-50861 (10 μm; 5–10 min) had little or no effect on spine actin, although it appeared to induce qualitative changes in the phalloidin-staining pattern in many neuronal cell bodies (A. Hipolito and S. Halpain, unpublished observations). Depolarization of cultures with 40 mm KCl had no significant effect on the density of F-actin punctae (Fig. 5B), suggesting either that release of endogenous glutamate failed to sufficiently activate glutamate receptors in the culture, that calcium levels failed to rise sufficiently, or that factors coreleased along with glutamate inhibit its action on F-actin.
Prevention of spine loss by prestabilization of actin filaments
The rapid and selective effect of NMDA in disrupting F-actin in spines, compared with that on F-actin along dendritic shafts, suggests that biochemical mechanisms closely associated with the subsynaptic compartment act to destabilize actin filaments locally in response to glutamate. The actin-stabilizing compound jasplakinolide was used to examine whether stabilization of F-actin confers NMDA resistance on dendritic spines. Jasplakinolide is more membrane permeable than phalloidin but competes with phalloidin for the same binding site on actin filaments. It is also more effective at stabilizing actin filaments in vitro (Bubb et al., 1994). The NMDA-induced loss of F-actin punctae was completely prevented when cultures were preincubated for 2 hr with 2 μm jasplakinolide (Fig.6A–C). In parallel, the NMDA-induced loss of spines visualized by DiI labeling was also prevented by jasplakinolide (Fig. 6D–F).
Involvement of calcineurin in regulating spine actin
One consequence of the activation of NMDA receptors is stimulation of the calcium- and calmodulin-dependent protein phosphatase calcineurin (Halpain et al., 1990; Quinlan and Halpain, 1996). We therefore investigated the subcellular localization of calcineurin and its potential role in regulating spine actin. Calcineurin immunoreactivity was present throughout the neuron, but intense spots of immunoreactivity were colocalized with synaptic F-actin punctae along dendrites (Fig.7A,B). Such calcineurin punctae were disrupted in parallel with F-actin punctae in response to NMDA (Fig.7C,D), suggesting that calcineurin is enriched in the postsynaptic compartment together with F-actin. A recent electron microscopic study confirms that calcineurin immunoreactivity is present in dendritic spines of hippocampal neurons (Sik et al., 1998).
Fig. 7.

Colocalization of calcineurin and F-actin at spine-like punctae. Cultured hippocampal neurons were incubated for 5 min in the absence (A, B) or presence (C, D) of 50 μm NMDA before double-labeling for F-actin (A, C) or calcineurin (B, D).Arrowheads indicate examples of spine-like punctae double-labeled for both probes.
Ascomycin (a more water-soluble analog of FK-506, also known in the literature as FK-520 and L-683,590) is an immunosuppressant compound that specifically inhibits calcineurin activity after binding to the intracellular immunophilin FKBP-12 (Dumont et al., 1992). The NMDA-induced reduction in F-actin punctae in cultured neurons was significantly attenuated by preincubation for 3 hr with 20 μm ascomycin (Fig. 8), consistent with a role for calcineurin in regulating F-actin stability at synapses.
DISCUSSION
It has long been presumed that F-actin plays a central role in regulating spine shape. The present data provide direct experimental confirmation of this idea with the demonstration that loss of spine integrity is prevented by a compound that stabilizes the actin network. In addition, the results show that an early consequence of NMDA receptor activation is local actin filament reorganization at synapses. Furthermore, the data indicate that, at least at initial stages, NMDA does not simply induce a generalized breakdown of synapse structure but rather that specific mechanisms target the actin cytoskeleton for reorganization, resulting in spine contraction or collapse.
Actin plays multiple roles in cells but is often associated with motile functions, such as growth, contractility, and migration. In such cases the length of individual actin filaments is dynamic and responsive to transmembrane signals (for review, see Cooper, 1991; Carlier and Pantaloni, 1997). Cellular motility is arrested within seconds to minutes by agents that cap the fast-growing ends of actin filaments, such as the cytochalasins (Cooper, 1987) or those that sequester actin monomers, such as the latrunculins (Spector et al., 1989). However, there are well established exceptions to the notion of “dynamic” actin. For example, actin is involved in the maintenance of specialized cell shape in striated muscle and in the submembrane cytoskeleton of erythrocytes. In these structures actin filament length is tightly regulated and stable over lengthy periods, and the filaments are resistant to actin-depolymerizing drugs (Fowler, 1996; Littlefield and Fowler, 1998).
Until recently it has been unclear whether the actin network of dendritic spines serves to maintain spine shape rigidly or whether it is dynamic, thereby supporting rapid morphological changes. It is likely that spines contain a heterogeneous population of actin filaments having differential susceptibilities to depolymerization. Net actin disassembly at synapses appears to be slow under basal conditions, because synaptic actin punctae persist for many hours in the presence of cytochalasin D (Allison et al., 1998). This result correlates with the observed persistence of individual dendritic spines over similar time periods (Hosokawa et al., 1992; Dailey and Smith, 1996). However, individual spines undergo shape changes on the order of seconds to minutes (Hosokawa et al., 1992; Dailey and Smith, 1996), and brief NMDA application induces shortening of spine necks within 1–2 hr (Segal, 1995a). A recent study demonstrated that, in cultured hippocampal neurons, actin distribution in spines changes on a time scale of seconds to minutes in a manner that is perturbed by latrunculin A (Fischer et al., 1998). It is reasonable to speculate that this rapid, actin-based motility is regulated by endogenous levels of synaptic glutamatergic activity in the cultures. The glutamate receptor-dependent changes reported here might target either dynamic filaments or a distinct, stable pool of F-actin in spines. Although further studies will be required to determine whether physiological glutamate receptor activity alters spine shape and F-actin organization, the present study clearly suggests a functional tie between glutamate receptor activation and the actin-mediated shape of dendritic spines.
Both calcineurin and F-actin are involved in controlling the efficacy of NMDA receptors (Rosenmund and Westbrook, 1993; Lieberman and Mody, 1994; Tong et al., 1995). The present results provide evidence that, in turn, activation of NMDA receptors and calcineurin can rapidly modify the local actin organization at synapses. Together these data suggest a close reciprocal relationship among NMDA receptors, calcineurin, and actin filaments in regulating postsynaptic structure and function. Actin filaments participate in membrane targeting of NMDA receptor clusters (Allison et al., 1998), an effect that is regulated by activity at NMDA receptors themselves (Rao and Craig, 1997). Recent findings indicate that NMDA receptors are linked to the actin cytoskeleton via the actin-binding protein α-actinin (Wyszynski et al., 1997). Other interactions may additionally contribute to linking proteins of the postsynaptic junction to F-actin (Kennedy, 1997). Calcineurin is therefore in a position to modify such linkages.
The colocalization of calcineurin immunoreactivity with F-actin at synapses and its parallel regulation by NMDA indicate that a significant fraction of calcineurin may be tethered directly or indirectly to the actin cytoskeleton. In developing cerebellar (Ferreira et al., 1993) and hippocampal (Halpain et al., 1997) neurons, calcineurin is highly concentrated in growth cones, structures that, like spines, contain a dense network of F-actin. Actin filaments dynamically regulate growth cone motility during axon elongation (Forscher et al., 1992), and the calcineurin antagonist cyclosporin A was shown to inhibit neurite outgrowth in cultured cerebellar neurons (Ferreira et al., 1993). Our data suggest that calcineurin may have additional roles in the development or structural plasticity of dendritic spines. Calcineurin might be generally involved in regulating neuronal structures having highly dynamic populations of actin filaments.
The link demonstrated here between actin filament integrity and the maintenance of spines is consistent with the idea that local membrane tension in spines is dependent on actin filaments. One consequence of disrupting such membrane tension would be alteration of the mechanosensitive properties of NMDA receptors (Paoletti and Ascher, 1994). Loss of local mechanisms regulating NMDA receptor function could, in turn, contribute to the cascade of events leading ultimately to cell death. Indeed, dendritic spines have been postulated to serve a neuroprotective role (Segal, 1995b); thus, neurons in which spines have collapsed as part of an early response to intense NMDA receptor activity might become more vulnerable to subsequent excitotoxic stimuli. Alternatively, perhaps the ability of the spine to alter its local cytoskeletal arrangements represents a neuroprotective mechanism. Disruption of F-actin in cultured neurons has been shown to attenuate excitotoxic cell death (Furukawa et al., 1995).
The excessive activation of glutamate synapses underlying excitotoxicity clearly has acute effects on synapse stability in models of cerebral ischemia. Furthermore, it is widely believed that excitotoxic mechanisms can exacerbate the loss of neurons and synapses occurring in more chronic neurodegenerative diseases, including amyotrophic lateral sclerosis, Huntington’s disease, and Alzheimer’s disease. Our data suggest that calcineurin plays an important role in regulating actin filament stability at synapses and that calcineurin antagonists may attenuate some of the earliest structural changes accompanying excitotoxic injury to the nervous system.
Footnotes
This work was supported by a grant from the National Institute of Mental Health to S.H. We thank Dr. Ernest Villafranca, Agouron Pharmaceuticals, for the gift of calcineurin antiserum, Mr. Phong Nguyen for assistance with data analysis, and Dr. Velia Fowler and members of the Halpain laboratory for helpful discussions.
Correspondence should be addressed to Dr. Shelley Halpain, Department of Cell Biology, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037.
Dr. Saffer’s present address: Department of Internal Medicine, University of Virginia Health Sciences Center, Charlottesville, VA 22908.
REFERENCES
- 1.Allison DW, Gelfand VI, Spector I, Craig AM. Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci. 1998;18:2423–2436. doi: 10.1523/JNEUROSCI.18-07-02423.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartlett WP, Banker GA. An electron microscopic study of the development of axons and dendrites by hippocampal neurons in culture. II. Synaptic relationships. J Neurosci. 1984;4:1954–1965. doi: 10.1523/JNEUROSCI.04-08-01954.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benson DL, Watkins FH, Steward O, Banker G. Characterization of GABAergic neurons in hippocampal cell cultures. J Neurocytol. 1994;23:279–295. doi: 10.1007/BF01188497. [DOI] [PubMed] [Google Scholar]
- 4.Brandon JG, Coss RG. Rapid dendritic spine stem shortening during one-trial learning: the honeybee’s first orientation flight. Brain Res. 1982;252:51–61. doi: 10.1016/0006-8993(82)90977-5. [DOI] [PubMed] [Google Scholar]
- 5.Bubb MR, Senderowicz AMJ, Sausville EA, Duncan KLK, Korn ED. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J Biol Chem. 1994;269:14869–14871. [PubMed] [Google Scholar]
- 6.Carlier M, Pantaloni D. Control of actin dynamics in cell motility. J Mol Biol. 1997;269:459–467. doi: 10.1006/jmbi.1997.1062. [DOI] [PubMed] [Google Scholar]
- 7.Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- 8.Cohen RS, Chung SK, Pfaff DW. Immunocytochemical localization of actin in dendritic spines of the cerebral cortex using colloidal gold as a probe. Cell Mol Neurobiol. 1985;5:271–284. doi: 10.1007/BF00711012. [DOI] [PubMed] [Google Scholar]
- 9.Cooper JA. Effects of cytochalasin and phalloidin on actin. J Cell Biol. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooper JA. The role of actin polymerization in cell motility. Annu Rev Physiol. 1991;53:585–605. doi: 10.1146/annurev.ph.53.030191.003101. [DOI] [PubMed] [Google Scholar]
- 11.Craig AM, Blackstone CD, Huganir RL, Banker G. Selective clustering of glutamate and gamma-aminobutyric acid receptors opposite terminals releasing the corresponding neurotransmitters. Proc Natl Acad Sci USA. 1994;91:12373–12377. doi: 10.1073/pnas.91.26.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dailey ME, Smith SJ. The dynamics of dendritic structure in developing hippocampal slices. J Neurosci. 1996;16:2983–2994. doi: 10.1523/JNEUROSCI.16-09-02983.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dumont FJ, Staruch MJ, Koprak SL, Siekierka JJ, Lin CS, Harrison R, Sewell T, Kindt VM, Beattie TR, Wyvratt M, Sigal NH. The immunosuppressive and toxic effects of FK-506 are mechanistically related: pharmacology of a novel antagonist of FK-506 and rapamycin. J Exp Med. 1992;176:751–760. doi: 10.1084/jem.176.3.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferreira A, Kincaid R, Kosik KS. Calcineurin is associated with the cytoskeleton of cultured neurons and has a role in the acquisition of polarity. Mol Biol Cell. 1993;4:1225–1238. doi: 10.1091/mbc.4.12.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fifkova E, Delay RJ. Cytoplasmic actin in neuronal processes as a possible mediator of synaptic plasticity. J Cell Biol. 1982;95:345–350. doi: 10.1083/jcb.95.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer M, Kaech S, Knutti D, Matus A. Rapid actin-based plasticity in dendritic spines. Neuron. 1998;20:847–854. doi: 10.1016/s0896-6273(00)80467-5. [DOI] [PubMed] [Google Scholar]
- 17.Forscher P, Lin CH, Thompson C. Novel form of growth cone motility involving site-directed actin filament assembly. Nature. 1992;357:515–518. doi: 10.1038/357515a0. [DOI] [PubMed] [Google Scholar]
- 18.Fowler VM. Regulation of actin filament length in erythrocytes and striated muscle. Curr Opin Cell Biol. 1996;8:86–96. doi: 10.1016/s0955-0674(96)80052-4. [DOI] [PubMed] [Google Scholar]
- 19.Furukawa K, Smith-Swintosky VL, Mattson MP. Evidence that actin depolymerization protects hippocampal neurons against excitotoxicity by stabilizing [Ca2+]. Exp Neurol. 1995;133:153–163. doi: 10.1006/exnr.1995.1018. [DOI] [PubMed] [Google Scholar]
- 20.Geinisman Y, de Toledo-Morrell L, Morrell F, Persina IS, Rossi M. Age-related loss of axospinous synapses formed by two afferent systems in the rat dentate gyrus as revealed by the unbiased stereological dissector technique. Hippocampus. 1992;2:437–444. doi: 10.1002/hipo.450020411. [DOI] [PubMed] [Google Scholar]
- 21.Goslin K, Banker G. Rat hippocampal neurons in low density culture. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT; Cambridge, MA: 1991. pp. 251–281. [Google Scholar]
- 22.Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington’s disease. Science. 1985;227:770–773. doi: 10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- 23.Halpain S, Girault J-A, Greengard P. Activation of NMDA receptors induces dephosphorylation of DARPP-32 in rat striatal slices. Nature. 1990;343:369–372. doi: 10.1038/343369a0. [DOI] [PubMed] [Google Scholar]
- 24.Halpain S, Hipolito A, Batinica A, duPont N, Saffer L. Co-localization of calcineurin and F-actin in dendritic spines. Soc Neurosci Abstr. 1997;23:1677. [Google Scholar]
- 25.Harris KM, Kater SB. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341–371. doi: 10.1146/annurev.ne.17.030194.002013. [DOI] [PubMed] [Google Scholar]
- 26.Harris KM, Sultan P. Variation in the number, location and size of synaptic vesicles provides an anatomical basis for the nonuniform probability of release at hippocampal CA1 synapses. Neuropharmacology. 1995;34:1387–1395. doi: 10.1016/0028-3908(95)00142-s. [DOI] [PubMed] [Google Scholar]
- 27.Hinton VJ, Brown WT, Wisniewski K, Rudelli RD. Analysis of neocortex in three males with the fragile X syndrome. Am J Med Genet. 1991;41:289–294. doi: 10.1002/ajmg.1320410306. [DOI] [PubMed] [Google Scholar]
- 28.Horner CH. Plasticity of the dendritic spine. Prog Neurobiol. 1993;41:281–321. doi: 10.1016/0301-0082(93)90002-a. [DOI] [PubMed] [Google Scholar]
- 29.Hosokawa T, Bliss TV, Fine A. Persistence of individual dendritic spines in living brain slices. NeuroReport. 1992;3:477–480. doi: 10.1097/00001756-199206000-00005. [DOI] [PubMed] [Google Scholar]
- 30.Kaech S, Fischer M, Doll T, Matus A. Isoform specificity in the relationship of actin to dendritic spines. J Neurosci. 1997;17:9565–9572. doi: 10.1523/JNEUROSCI.17-24-09565.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kennedy MB. The postsynaptic density at glutamatergic synapses. Trends Neurosci. 1997;20:264–268. doi: 10.1016/s0166-2236(96)01033-8. [DOI] [PubMed] [Google Scholar]
- 32.Lieberman DN, Mody I. Regulation of NMDA channel function by endogenous Ca2+-dependent phosphatase. Nature. 1994;369:235–239. doi: 10.1038/369235a0. [DOI] [PubMed] [Google Scholar]
- 33.Littlefield R, Fowler VM. Defining actin filament length in striated muscle: rulers and caps or dynamic stability? Annu Rev Cell Dev Biol. 1998;14:487–525. doi: 10.1146/annurev.cellbio.14.1.487. [DOI] [PubMed] [Google Scholar]
- 34.Masliah E, Mallory M, Hansen I, DeTeresa R, Terry RD. Quantitative synaptic alterations in the human neocortex during normal aging. Neurology. 1993;43:192–197. doi: 10.1212/wnl.43.1_part_1.192. [DOI] [PubMed] [Google Scholar]
- 35.Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I, HNRC Group Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. Ann Neurol. 1997;42:963–972. doi: 10.1002/ana.410420618. [DOI] [PubMed] [Google Scholar]
- 36.Matus A, Ackermann M, Pehling G, Byers HR, Fujiwara K. High actin concentrations in brain dendritic spines and postsynaptic densities. Proc Natl Acad Sci USA. 1982;79:7590–7594. doi: 10.1073/pnas.79.23.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olney JW, Fuller T, De Gubareff T. Acute dendrotoxic changes in the hippocampus of kainate treated rats. Brain Res. 1979;176:91–100. doi: 10.1016/0006-8993(79)90872-2. [DOI] [PubMed] [Google Scholar]
- 38.Paoletti P, Ascher P. Mechanosensitivity of NMDA receptors in cultured mouse central neurons. Neuron. 1994;13:645–655. doi: 10.1016/0896-6273(94)90032-9. [DOI] [PubMed] [Google Scholar]
- 39.Park JS, Bateman MC, Goldberg MP. Rapid alterations in dendrite morphology during sublethal hypoxia or glutamate receptor activation. Neurobiol Dis. 1996;3:215–227. doi: 10.1006/nbdi.1996.0022. [DOI] [PubMed] [Google Scholar]
- 40.Purpura DP. Dendritic spine “dysgenesis” and mental retardation. Science. 1974;186:1126–1128. doi: 10.1126/science.186.4169.1126. [DOI] [PubMed] [Google Scholar]
- 41.Quinlan EM, Halpain S. Postsynaptic mechanisms for bidirectional control of MAP2 phosphorylation by glutamate receptors. Neuron. 1996;16:357–368. doi: 10.1016/s0896-6273(00)80053-7. [DOI] [PubMed] [Google Scholar]
- 42.Rao A, Craig AM. Activity regulates the synaptic localization of the NMDA receptor in hippocampal neurons. Neuron. 1997;19:801–812. doi: 10.1016/s0896-6273(00)80962-9. [DOI] [PubMed] [Google Scholar]
- 43.Rosenmund C, Westbrook GL. Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron. 1993;10:805–814. doi: 10.1016/0896-6273(93)90197-y. [DOI] [PubMed] [Google Scholar]
- 44.Rothman SM, Olney JW. Excitotoxicity and the NMDA receptor–still lethal after eight years. Trends Neurosci. 1995;18:57–58. doi: 10.1016/0166-2236(95)93869-y. [DOI] [PubMed] [Google Scholar]
- 45.Scheibel ME, Crandall PH, Scheibel AB. The hippocampal-dentate complex in temporal lobe epilepsy. Epilepsia. 1974;15:55–88. doi: 10.1111/j.1528-1157.1974.tb03997.x. [DOI] [PubMed] [Google Scholar]
- 46.Segal M. Morphological alterations in dendritic spines of rat hippocampal neurons exposed to N-methyl-d-aspartate. Neurosci Lett. 1995a;193:73–76. doi: 10.1016/0304-3940(95)11665-j. [DOI] [PubMed] [Google Scholar]
- 47.Segal M. Dendritic spines for neuroprotection: a hypothesis. Trends Neurosci. 1995b;18:468–471. doi: 10.1016/0166-2236(95)92765-i. [DOI] [PubMed] [Google Scholar]
- 48.Shorte SL. N-Methyl-d-aspartate evokes rapid net depolymerization of filamentous actin in cultured rat cerebellar granule cells. J Neurophysiol. 1997;78:1135–1143. doi: 10.1152/jn.1997.78.2.1135. [DOI] [PubMed] [Google Scholar]
- 49.Sik A, Hajos N, Gulacsi A, Mody I, Freund TF. The absence of a major Ca2+ signaling pathway in GABAergic neurons of the hippocampus. Proc Natl Acad Sci USA. 1998;95:3245–3250. doi: 10.1073/pnas.95.6.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spector I, Shocet NR, Blasberger D, Kashman Y. Latrunculins–novel marine macrolides that disrupt microfilament organization and affect cell growth: I. Comparison with cytochalasin D. Cell Motil Cytoskeleton. 1989;13:127–144. doi: 10.1002/cm.970130302. [DOI] [PubMed] [Google Scholar]
- 51.Terry RD, DeTeresa R, Hansen IA. Neocortical cell counts in normal human adult aging. Ann Neurol. 1987;21:530–539. doi: 10.1002/ana.410210603. [DOI] [PubMed] [Google Scholar]
- 52.Tong G, Shepherd D, Jahr CE. Synaptic desensitization of NMDA receptors by calcineurin. Science. 1995;267:1510–1512. doi: 10.1126/science.7878472. [DOI] [PubMed] [Google Scholar]
- 53.Woolley CS, Gould E, Frankfurt M, McEwen BS. Naturally occurring fluctuation in dendritic spine density on adult hippocampal pyramidal neurons. J Neurosci. 1990;10:4035–4039. doi: 10.1523/JNEUROSCI.10-12-04035.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of α-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- 55.Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]