

Abstract
Idiopathic Parkinson’s disease involves the loss of midbrain dopaminergic neurons, resulting in the presynaptic breakdown of dopaminergic transmission in the striatum. Huntington’s disease and some neurodegenerative diseases with Parkinsonian features have postsynaptic defects caused by striatal cell death. Mice were generated in which an attenuated form of the diphtheria toxin gene (tox-176) was expressed exclusively in D1 dopamine receptor (D1R)-positive cells with the aim of determining the effect of this mutation on development of the basal ganglia and on the locomotor phenotype. Transgenic mice expressing Cre, a site-specific DNA recombinase, were crossed with a second line in which a transcriptionally silenced tox-176 gene was inserted into the D1R gene locus by homologous recombination. Young doubly transgenic mutant mice expressing the tox-176 gene displayed bradykinesia, dystonia, and had falls caused by myoclonic jerks. The mutant brain had evidence of apoptosis and reactive gliosis and, consistent with the D1R expression pattern, the striatum was reduced in volume, and the Islands of Calleja were absent. In contrast, the cortex was of normal thickness. D1Rs were not detectable in mutants by in situ hybridization or ligand autoradiography, whereas D2 dopamine receptor (D2R) mRNA and protein was present in the striatum. In addition, substance P and dynorphin, neuropeptides known to be expressed in D1R-positive striatonigral projection neurons were not detectable. Enkephalin, a marker found in D2-positive striatopallidal projection neurons was expressed in the mutant brain. The mutant represents a novel neurodegenerative disease model with a dramatic extrapyramidal phenotype.
Keywords: D1 dopamine receptor, basal ganglia, Crerecombinase, gene targeting, striatum, Parkinson’s disease
The dopaminergic system of the brain is prone to several degenerative conditions that disrupt normal basal ganglia function resulting in progressive motor impairment. Idiopathic Parkinson’s disease is defined by loss of nigrostriatal dopaminergic neurons (Forno, 1982) and is characterized by the presence of tremor, bradykinesia, rigidity, and postural instability. Many CNS diseases with similar clinical features also show loss of striatal neurons. Recent evidence suggests that striatal degeneration may not be confined to rare Parkinsonian syndromes (Steele et al., 1964; Lees, 1987; Quinn, 1994; Watts et al., 1994) but may be more common than generally appreciated because of misdiagnosis of cases as idiopathic Parkinson’s disease (Rajput et al., 1991; Hughes et al., 1992). In addition, hyperkinetic movement disorders such as Huntington’s disease (Ferrante et al., 1985; Albin et al., 1989) and dystonia (Rothwell and Obeso, 1987; Waters et al., 1993) are also associated with striatal degeneration.
Of the five cloned dopamine receptors (Sibley and Monsma, 1992), D1 and D2 dopamine receptors are highly expressed in the adult striatum (Gerfen, 1992). The degree of coexpression of D1 and D2 dopamine receptors on striatal projection neurons is the subject of considerable debate (Surmeier et al., 1993). A strategy of targeted expression of a diphtheria toxin gene was used to investigate the effect of loss of D1R-positive neurons on the formation of the basal ganglia and the control of body movement. The specific hypothesis examined is that D1R-positive cells are essential for locomotor control and that the lack of D1R-positive cells results in an extrapyramidal phenotype. A single copy of the attenuated diphtheria toxin A-chain gene (tox-176) (Maxwell et al., 1987), located downstream of a LoxP flanked cassette consisting of a neomycin phosphotransferase gene (NEO) and DNA sequence known to inhibit downstream translation (STOP) (Lakso et al., 1992), was inserted into the D1R gene locus by homologous recombination. The B-chain of the diphtheria toxin gene was not encoded in the transgene ensuring that the protein synthesis-inhibiting effects of tox-176 would be restricted to cells that normally express the D1R (Maxwell et al., 1987). Transgenic mice heterozygous (HZ) for this event were phenotypically normal as expression of tox-176 was blocked by NEO/STOP. The HZ mice were crossed with transgenic mice homozygous for adenovirus EIIa promoter driven Cre recombinase (EIIa/Cre), an enzyme that recognizes LoxP motifs and excises intervening DNA (Sauer and Henderson, 1988a). Transactivation of the EIIapromoter, an event that is thought to occur at the fertilized oocyte stage of development (Lakso et al., 1996), results in the removal of NEO/STOP and the tox-176 gene being placed under the control of D1R regulatory elements. This double transgenic paradigm was chosen because it made possible the establishment of a healthy HZ line in which the tox-176 gene is functionally silenced, allowing it to be activated at will by crossing with a Cre-producing line. Activated doubly transgenic mice were bradykinetic and displayed dystonic posturing and myoclonic jerks, suggesting that D1R neurons are important regulators of body movement.
MATERIALS AND METHODS
All procedures involving the use of live animals conformed to the Australian National Health and Medical Research Council code of practice.
Cloning. Figure1A represents a genomic map of the D1R gene (Drago et al., 1994). A targeting construct (pF) was made (Fig. 1) using flanking sequence derived from D1R genomic clones (Drago et al., 1994). A modified pSTOP.2 vector, called pSTOP.2.M.S, was used as a base for construction of pF. AnEcoRI/NotI fragment containing the thymidine kinase gene and plasmid pUC backbone (subcloned from pPNT) (Tybulewicz et al., 1991) was ligated with an EcoRI/NotI fragment containing the LoxP-NEO/STOP-LoxP fragment to generate pSTOP.2. The LoxP-NEO/STOP-LoxP cassette was constructed by cloning the NEO gene (also from pPNT) into the XhoI/BamHI sites within the STOP cassette of pBS302 (Sauer, 1993). The pSTOP.2.M.S vector was completed by introducing a NotI/SalI cloning site (into the NotI site) 5′ of the first LoxP site and a Meganuclease (I-Sce I) site (into the EcoRI site) downstream of the second LoxP site of pSTOP.2. The initiation codon of the D1R gene was identified by sequencing and mutated using a PCR-based mutagenesis strategy. A 3.0 kb stretch of homologous sequence spanning from the BamHI site to a destroyed ATG initiation codon (located 20 bp upstream of the NcoI restriction site; Fig. 1) provided the 5′ homologous flank and was subcloned into the NotI/SalI sites in pSTOP.2.M.S. The tox-176 gene (Harrison et al., 1991) (a gift from Dr. I. Maxwell, University of Colorado) with a polyadenylation sequence (SalI/BamHI fragment from pMSG vector; Pharmacia, Dorval, Québec, Canada; containing the SV40 early splice region and the SV40 polyadenylation sequence) was subcloned immediately downstream of the LoxP-NEO/STOP-LoxP cassette and 5′ of a 3.3 kb (XbaI/BamHI) D1R genomic flank. The tox-176 cassette and theXbaI/BamHI genomic flank was first subcloned into an intermediate subcloning plasmid pDOUG. The pDOUG plasmid contained a polylinker flanked by two I-Sce I restriction sites. The 5′ flank was excised from pDOUG using I-Sce I and then cloned into the I-Sce I site of pSTOP.2.M.S vector. The integrity of the LoxP sites in pF, as evidenced by recombination, was confirmed before electroporation by transforming the plasmid into anEscherichia coli strain that constitutively expresses theCre gene (Sauer and Henderson, 1988b).
Fig. 1.

Construction of targeting vector and Southern blot analysis of ES clones and mutant mice. Representation of the genomic map of the D1R gene (A), the targeting vector pF (B), the expected allelic disruption after homologous recombination (C), and the final chromosomal organization after Cre-mediated excision of the Lox P-bracketed DNA (D). The restriction sites shown are abbreviated as follows: N,NotI; H, HindIII;E, EcoRI;B, BamHI;Nc, NcoI; Xb,XbaI. PK represents the phosphoglycerate kinase-1 promoter, TK the thymidine kinase gene,pA a polyadenylation signal, and Lindicates LoxP sites. The origin of the probes used for homologous recombination screening are also shown. Probe A is anEcoRI-BamHI fragment used for detection of recombination at the 5′ end, and probe B is aBamHI-HindIII fragment for detection of 3′ recombination. E, Southern blot ofEcoRI-digested ES cell genomic DNA (left panel) showing correctly targeted ES cell clones (C35, C62, C85, and C160). C159 is a randomly selected nontargeted clone, and J1 is DNA from normal ES cells. Wild-type allele is 4.2 kb, and the recombinant allele is 12.6 kb. Southern blot ofHindIII-digested DNA (right panel) probed with probe B, the wild-type allele is 5.0 kb, and the mutant allele is 4.3 kb.
Embryonic stem cell culture and molecular analysis of transgenic mice. Linearized targeting construct (25 μg) was electroporated into the J1 line (a gift from Dr. R. Jaenisch, MIT) of ES cells using standard techniques (Drago et al., 1994). A total of 150 clones were isolated from a single electroporation experiment after a strategy of simultaneous positive selection with G418 (400 μg/ml, total powder; Life Technologies, Gaithersburg, MD) and negative selection with Gancyclovir (2 μm) (a gift from Syntex Corporation). A Southern blot of an EcoRI digest of ES cell clone derived genomic DNA probed with probe A (Fig. 1) was used to detect recombination at the 5′ end (recombinant allele 12.6 kb band, wild-type 4.2 Kb). A Southern blot of a HindIII digest probed with probe B detected 3′ recombination (recombinant 4.3 kb, wild-type 5.0 kb). A single incorporation event was confirmed for each of the clones by probing EcoRI-digested DNA with a NEO gene cDNA probe (data not shown). Four recombinants were identified by Southern blotting (Fig. 1), one of which (C35) was injected into Balb/C blastocysts, and germline transmitting chimeras were generated. Heterozygous (HZ) progeny, derived by mating male chimeras with either Balb/C or CD1 females, were identified by Southern blotting. Doubly transgenic mice as defined by Southern blotting were generated by mating HZ mice with EIIa/Cre (CD1 background) homozygous transgenic mice. Activated heterozygous (HZac) mice were detected by probing BamHI digested DNA with a NEO cDNA (Fig. 3F). In vivo Cre-mediated recombination was also verified by PCR amplification across the sole remaining regenerated LoxP site (Fig. 3C). Normal mice were homozygous wild-type with respect to the targeted allele but heterozygous for the EIIa/Cre transgene. Only one HZ line was generated because HZ mice per se have a normal phenotype, it is only after backcrossing to EIIa/Cre mice that progeny with a mutant phenotype are generated, making random events during production of targeted ES cells an unlikely explanation for any aspects of the phenotype. From the breeding protocol described above it can be seen that HZac mice and their littermates had a mixed genetic background (i.e., 129/Sv, Balb/C, and CD1), a factor that may have contributed to the variability seen in the length of survival (see Results).
Fig. 3.

Molecular analysis of transgenic mice.A, Southern blot of HindIII-digested whole-brain-derived DNA probed with probe B. B, Southern blot of BamHI-digested whole-brain-derived DNA probed with a NEO cDNA showing failure of Cre-mediated recombination (asterisk) in some mice. C, PCR with oligonucleotide probes that span the residual Lox P site afterCre-mediated recombination (marker is 1 kb ladder). There is PCR evidence of recombination in phenotypically normal HZnon-ac pups despite a prominentBamHI fragment that hybridizes with the NEO cDNA.
Genomic DNA derived from whole mouse brain was PCR-amplified with primers Tox F (5′-TCTAAGCCACCGAAGTGCTTT-3′) and Tox R (5′-TCTACAGAGTATCCCGTAGTG-3′), situated 50 nucleotides upstream from the NcoI site in the 5′UTR of the D1R gene and in the attenuated diphtheria toxin A-chain gene 185 nucleotides downstream from the initiation codon, respectively. Each reaction mixture contained 2.5 μg of DNA in a 50 μl reaction (in mm: 50 KCl, 15 Tris, pH 8.4, 1 MgCl2, and 0.1 dNTP with 2.5 U Taq polymerase). Amplification was performed in a Perkin-Elmer Gene Amp PCR system 2400 under the following conditions: an initial denaturation step of 5′ at 94°C was followed by 40 cycles comprising 30 sec at 94°C, 30 sec at 55°C, and 1 min at 72°C with a final extension step of 7 min at 72°C. PCR products were analyzed by electrophoresis in a 1.5% agarose gel containing 1× Tris–borate–EDTA.
Locomotor studies. Mice aged 6–10 d were placed in an open field (18 × 18 cm) with a grid pattern made up of 0.5 cm spaced lines drawn on the floor and videotaped for 5 min. Locomotion was scored when the tip of the snout completely crossed a line. Animals were scored for falls during this same period of observation.
Receptor autoradiography. Postnatal day 3 (P3) and P4 pups [HZac (n = 12), doubly transgenic mice in which Cre failed to cut (see Results) called HZnon-ac (n = 4), and normal (n = 5)] were decapitated, and brains were removed and snap frozen by immersion in super-cooled isopentane. Twenty micrometer sections were cut in a cryostat and mounted onto gelatin and chrome–alum-coated slides (0.1% BSA was added for D2 receptor autoradiography). D1 autoradiography was as described in Drago et al. (1994) except that 10 μm SCH-23390 was used to define nonspecific binding. D2R autoradiography was as described in Lawrence et al. (1995). Slides were apposed overnight at room temperature to XAR5 film (Eastman Kodak, Rochester, NY) in the presence of standard microscales, developed, and autoradiograms were quantified on an MCID M4 image analysis system (Imaging Research, Brock University, St. Catherine’s, Ontario, Canada) under constant illumination.
In situ hybridization. Pups (P3–P4) were killed by decapitation [HZac (n = 6), HZnon-ac (n = 7), and normal (n = 3)], and the brains were snap frozen in cold isopentane and stored at −70°C before use. Twenty micrometer frozen coronal sections were cut and thaw-mounted onto 3-aminopropyltriethoxysilane-coated (Sigma, St. Louis, MO) slides. The antisense mouse D2R oligonucleotide sequence comprised the 48 bases located between nucleotides 556 and 600 from the sequence reported byMontmayeur et al. (1991). The mouse antisense D1R oligonucleotides were as described (Drago et al., 1994). Both D1.1 and D1.3 oligonucleotides were used to identify D1R mRNA. The 48-base oligonucleotide to detect substance P corresponded to nucleotides 224–271 of the sequence published by Kako et al. (1993). The oligonucleotides used to detect enkephalin and dynorphin corresponded to nucleotides 304–351 and nucleotides 862–909, respectively, based on the sequences published byZurawski et al. (1986) and Civelli et al. (1985). The oligonucleotide probes were then 5′-end labeled using a standard kinase protocol (Wong et al., 1997) with [γ-33P]ATP (NEN Life Science Products, Boston, MA) and T4 polynucleotide kinase (New England Biolabs, Beverly, MA). In situ hybridization was performed according to the protocols as reported in Wong et al. (1997). Specificity of all probes used in this study was determined by using a 100-fold excess of unlabeled antisense oligonucleotide that was added to the in situ hybridization reactions to competitively inhibit probe hybridization. Slides were apposed (together with laboratory-prepared 33P standards) to Hyperfilm (Amersham International, Amersham, UK), and the density of mRNA expression was subsequently quantified using an MCID M4 image analysis system. Standardization was achieved by comparing autoradiographic images with standards exposed with each film. All values are expressed as counts per minute per square millimeter for mRNA expression (mean ± SEM).
TUNEL processing. Apoptotic cells were detected using a variation of the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) protocol as described byGavrieli et al. (1992). In brief, 8 μm sections of paraffin-embedded tissue fixed in vivo by perfusing mice with freshly prepared 4% formaldehyde solution were cut, and the paraffin was removed with xylene. Sections were incubated with proteinase K (1 μg/ml) (Boehringer Mannheim, Castle Hill, New South Wales, Australia) for 5 min at 37°C and then incubated for 60 min at 37°C with the labeling mixture, containing digoxigenin-labeled UTP in the presence of terminal transferase (Boehringer Mannheim). Sections were washed in Tris-buffered saline, and digoxigenin was detected using an alkaline phosphatase-conjugated sheep anti-digoxigenin antibody (Boehringer Mannheim). Alkaline phosphatase was detected using Fast Red TR/Napthol AS-MX (Sigma Fast; Sigma-Aldrich, Castle Hill, New South Wales, Australia). Sections were counterstained with 1% Alcian blue (BDH Chemicals, Poole, UK) in 3% acetic acid and coverslipped under Aquamount (BDH Chemicals). Negative control sections were subjected to an identical protocol, but the terminal transferase was omitted. Immersion-fixed interdigital web tissue from embryonic day 13 mice was used as a positive control.
Histology, immunohistochemistry, and volumetric analyses.Pups were anesthetized with sodium pentobarbitone (100 mg/kg, i.p.) and either perfused intracardially or decapitated and brains immersed in fixative consisting of 4% paraformaldehyde and 0.2% (v/v) of saturated picric acid solution in 0.1 m phosphate buffer, pH 7.4. The former mice were first perfused with PBS (10–50 ml at 37°C) followed by chilled fixative solution (20 ml). The brains were then collected into fixative with 10% sucrose (w/v) and stored overnight in the dark at 4°C. The cryoprotected brains were quick-frozen onto a microtome chuck with compressed CO2. Serial sections were cut coronally at 50 μm and collected into ordered wells containing PBS. Every fourth section was counterstained to generate a reference map, and appropriate serial sections were selected and processed for immunocytochemistry. The sections were stained with rabbit polyclonal antisera directed against either substance P (a gift from Prof. John Furness, University of Melbourne) or enkephalin (Incstar, Stillwater, MN). Briefly, immunolabeling was performed on floating sections by preblocking with 10% normal goat serum followed by incubation in primary antisera for 24–48 hr at 4°C with gentle agitation. Enkephalin primary antisera was used at dilutions of 1:1500, substance P antisera at 1:5000, glial fibrillary acidic protein (GFAP) primary antisera (Dako, Glostrup, Denmark) was used at 1:4000. Sections were rinsed in PBS (3 × 5 min) and then incubated in biotinylated anti-rabbit secondary antibody (1:400 for 1 hr) (Vector Laboratories, Burlingame, CA). Sections were then rinsed in PBS (3 × 5 min) and labeled with avidin peroxidase (Sigma) (1:5000) for 1 hr, a standard cobalt nickel diaminobenzidine reaction was performed, and sections were mounted onto glass slides in a 0.5% gelatin solution, dehydrated, and coverslipped.
Volumetric analysis was performed on immersion-fixed brains (48 hr in fixative and an additional 24 hr in fixative and 10% sucrose). The volume of the striatum was calculated by measuring the area of the striatum in each 50 μm section. The cortical thickness was assessed in the barrel field area of the somatosensory cortex as depicted in the mouse brain atlas of Franklin and Paxinos (1997). Morphometric analysis was performed using a computerized image analysis system (Jandel Scientific, Corte Madera, CA) on images obtained using a 100× oil immersion lens and a drawing tube.
Electron microscopy. The mice (wild-type, n= 4; HZnon-ac, n = 1; HZac, n = 5) were deeply anesthetized with sodium pentobarbitone (100 mg/kg, i.p.) and perfused with fixative consisting of 3% paraformaldehyde and 3% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, via the left ventricle. The brain was removed and left in fixative solution overnight at 4°C. Serial 50 μm coronal sections were cut on a vibratome. The sections were post-fixed in 0.4% osmium tetroxide (60 min), dehydrated in ethanol, washed in epoxy propane, and flat embedded in Epon Araldite. Semithin sections (0.5 μm) of the whole coronal section were cut to identify the striatum. Ultrathin (silver gray) sections of the central striatum, at the level of the anterior commissure, were cut on a Reichert-Jung Ultracut, mounted on grids, stained with 2% aqueous uranyl acetate and 2% lead citrate, and examined with a Jeol 100S electron microscope.
Statistics. All data presented in the text are mean ± SEM. Statistical comparisons were performed using Sigmastat (Jandel Scientific). Student’s t tests were used for parametric data (locomotor studies, volume of striatum).
RESULTS
Behavioral phenotype of mutant mice
Mutant mice (HZac), produced by mating HZ mice with homozygous Cre transgenic mice, were readily identified by P2 by a small or absent milk spot, periodic breathing, and frequent falls (Fig. 2). Most pups died in the first postnatal week, but some survived to P19. HZacpups (P6–P10) were markedly bradykinetic [normal (n = 11), 288 ± 40 crossings/5 min; HZac(n = 8), 88 ± 36 crossings/5 min (p < 0.003)], demonstrated twisting movements of the limbs characteristic of peripheral dystonia, maintained abnormal postures at rest, and failed to right after spontaneous falls. The limb dystonia contributed to some falls; however, most falls were erratic, violent, intrusive, and associated with attempted movement or tactile stimulation. These falls were more typical of myoclonic jerks than ataxia. Although infrequent falls caused by myoclonic jerks were sometimes seen in normal pups (2 of 11) (usually <P6) all mobile HZac pups examined (8 of 8) had myoclonic jerks, suggesting that the jerks were locomotor-activated. At later stages (P10) myoclonic jerks were only seen in HZac mice (n = 8). In addition, periodic breathing was transient and not seen in older pups. Overall, the phenotype was suggestive of basal ganglia disease (Marsden, 1982) and shown not to be caused by hypocalcemia or renal failure (J. Drago and P. Padungchaichot, unpublished observations).
Fig. 2.
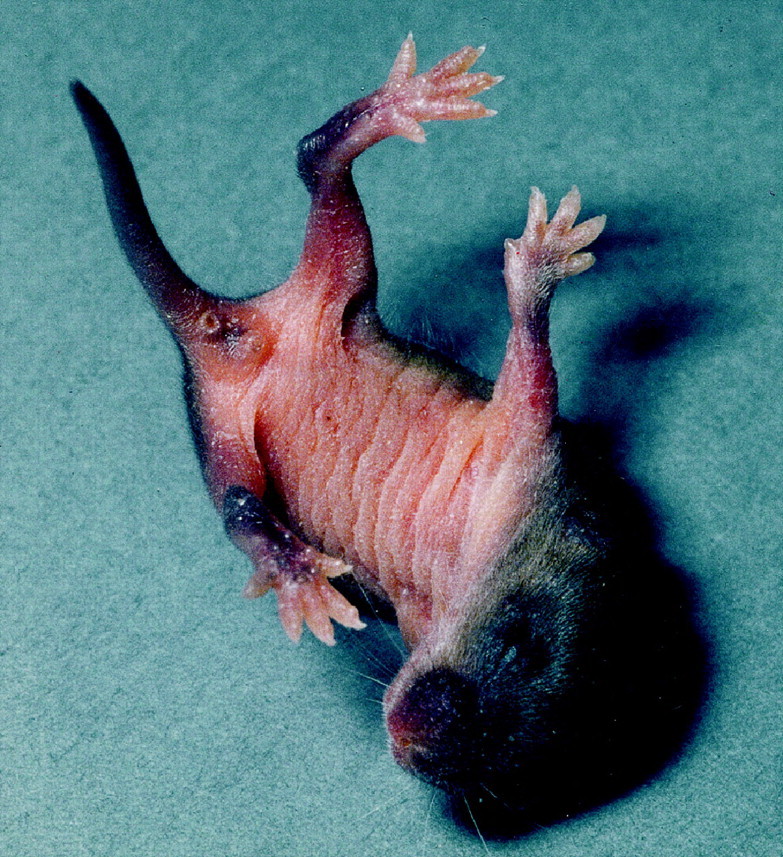
A P8 HZac mouse after a myoclonic jerk. The hindlimbs show dystonic posturing.
Failure of Cre-mediated recombination in some mice
Given that the EIIa/Cre transgenic activator mice were homozygous for the transgene, the frequency of the mutant phenotype was expected to approach 50% of all live births, however, analysis demonstrated that only 45% (24 of 53 HZ progeny) had the phenotype. The divergence from the expected frequency reflected failure ofCre-mediated recombination in 23% of doubly transgenic mice (HZnon-ac) (7 of 31) (Fig.3). Brain-derived genomic DNA was examined by PCR to verify Cre-mediated in vivorecombination. Oligonucleotide primers were designed to generate a small PCR product that spans the single LoxP site generated after recombination. As expected, all HZac pups examined (Fig. 3) were shown to have evidence of Cre-mediated recombination. A PCR product was also evident in HZnon-ac mice, although the biological significance of this finding was unclear, particularly given the nonquantitative and highly sensitive nature of PCR. In addition, PCR gives no information regarding the origin of the tissue contributing the DNA. Recombination may have occurred in tissues such as circulating blood elements, endothelial cells, or glial cells co-purified from the brain. Such tissues do not express the D1R and would, therefore, not maketox-176. Furthermore, myoclonic jerks, dystonia, or apparent locomotor retardation was not seen in a large number (n= 52) of HZnon-ac mice. Finally, HZnon-ac mice were fertile and survived to a normal age, casting further doubt on the biological significance of PCR evidence for recombination in HZnon-ac pups.
Neuroanatomical studies
The brains of HZac mice were smaller and lighter than those of normal or HZnon-ac mice [normal phenotype mice (n = 28 at P4) 0.147 ± 0.003 gm, HZac (n = 5 at P4) 0.099 ± 0.003 gm (p < 0.02)], and gross inspection suggested some distortion in the normal relationship of the forebrain and hippocampus. HZnon-ac mice were not significantly different to genetically normal mice with respect to brain morphology or total brain weight [normal mice (n= 16) 0.144 ± 0.004 gm, HZnon-ac mice (n = 12) 0.151 ± 0.005 gm]. The forebrain of HZac mice appeared smaller, and the hippocampus was displaced anteriorly. Volumetric analysis confirmed reduction in the size of the striatum by ∼40% [normal phenotype pups (n = 8, seven were genetically wild-type, and one was HZnon-ac), 2.3 ± 0.2 mm3; HZac (n = 7), 1.3 ± 0.2 mm3; p < 0.001] with compensatory enlargement of the lateral ventricles but no change in the cortical thickness (Fig. 4). The Islands of Calleja, which, like the striatum, are known to express D1R at high levels were not identified in HZac mice but were identified in all phenotypically normal mice examined (Fig. 4).
Fig. 4.
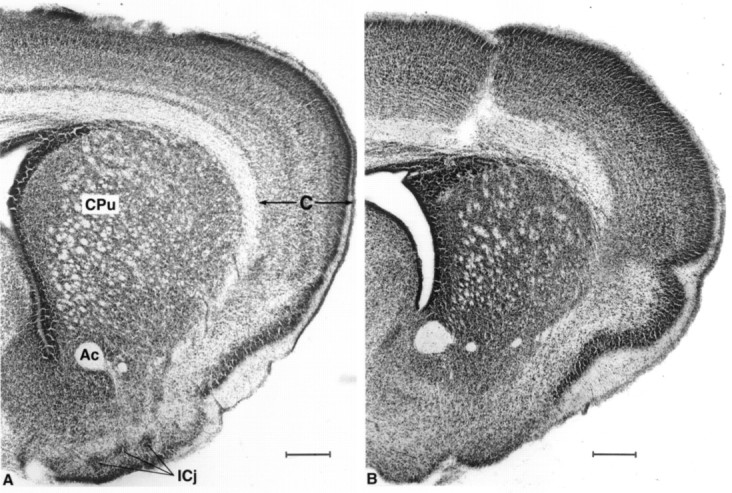
Neutral red-stained coronal sections (50 μm) at the level of the striatum of normal (A) and HZac (B) pups showing reduction in the size of striatum and absence of Islands of Calleja in HZac mice. CPu, Striatum (also called caudate putamen); ICj, Islands of Calleja;Ac, anterior commissure; C, cortex. Scale bar, 250 μm.
In situ hybridization
In situ hybridization for D1R mRNA performed on a large number of pups (wild-type, n = 5; HZnon-ac, n = 2; and HZac, n = 6) showed a typical distribution of mRNA in normal and HZnon-ac pups with signal in the striatum, nucleus accumbens, and olfactory tubercle but without specific hybridization evident in HZacmice (Fig. 5, compare B,C). Substance P (wild-type, n = 3; HZnon-ac, n = 3; HZac, n = 6) and dynorphin (wild-type, n = 4; HZnon-ac,n = 3; HZac, n = 5)in situ hybridization confirmed the absence of both neuropeptides in the mutant brain (Fig.6). D1R, dynorphin, and substance P mRNA studies show a homogenous loss of striatonigral pathway markers, suggesting that at least within the resolution of this assay, there is no mosaicism with respect to failure of Cre-mediated recombination within the brain. Enkephalin mRNA (wild-type,n = 4; HZnon-ac, n = 1; HZac, n = 5), a neuropeptide normally expressed in striatopallidal projection neurons was readily detectable in the mutant striatum (Fig. 6) and appeared to be upregulated (J. Y. F. Wong, unpublished observations). Although there was an expected decrease in D1R mRNA levels in HZnon-ac mice, they were normal with respect to D2R, dynorphin, substance P, enkephalin mRNA levels, and mazindol binding (J. Y. F. Wong, J. Drago, and P. Padungchaichot, unpublished observations). Quantitative in situ hybridization for D2R mRNA (wild-type, n = 3; HZnon-ac,n = 3; HZac, n = 6) showed upregulated expression by 22% in the striatum of HZac mice (Fig.7E).
Fig. 5.

In situ hybridization for D1R mRNA in normal (A) and HZac(B) P4 mice. Strong hybridization is seen in the striatum, nucleus accumbens, and olfactory tubercle of normal pup with only low level signal seen in the cortex. C, Nonspecific signal obtained when excess unlabeled oligonucleotide was used to competitively inhibit labeled probe hybridization to a normal P4 mouse brain. Specific hybridization was not seen in the HZac mouse brain.
Fig. 6.
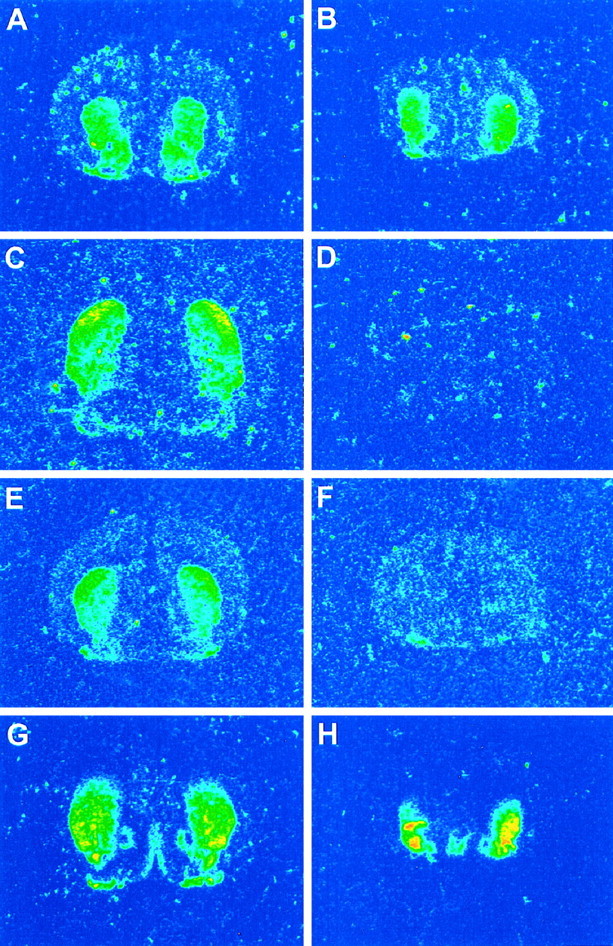
In situ hybridization for D2R (A, B), dynorphin (C, D), substance P (E,F), and enkephalin (G,H) in P3/P4 normal (A,C, E, G) and mutant (B, D, F,H) pups.
Fig. 7.
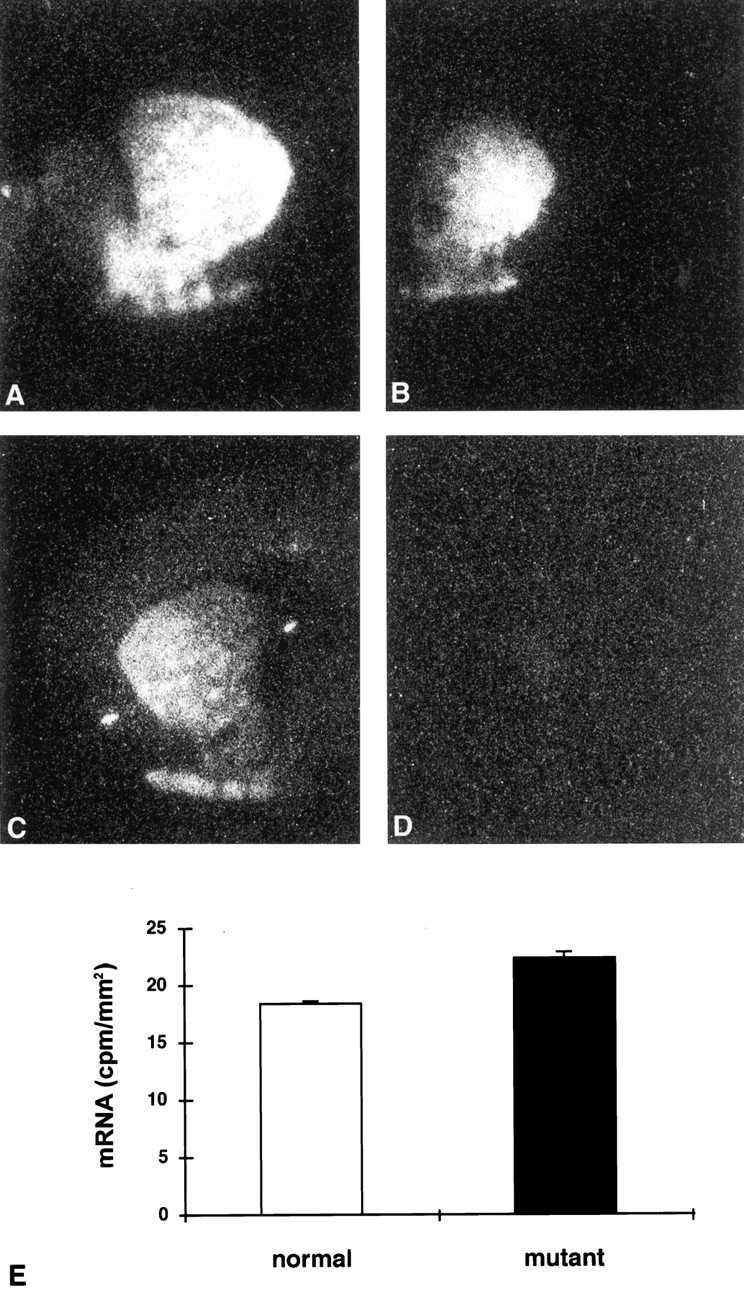
Receptor autoradiography and quantitativein situ hybridization for D2R mRNA in the striatum. Each panel (A–D) shows one half of a coronal section. D2 receptor autoradiography ([125I] NCQ-298) on normal (A) and mutant (B) pups. D1R autoradiography ([125I] SCH-23982) on normal (C) and mutant (D) pups showing complete lack of signal in HZac mice.E, The graph demonstrates the increase of D2R mRNA expression in the mutant striatum. Bar graph represents mean ± SEM.
Receptor autoradiography for D1 and D2 class binding
Receptor autoradiography on HZac, normal, and HZnon-ac mice confirmed the complete lack of D1-like binding in HZac mice (wild-type, n = 2; HZnon-ac, n = 1; HZac, n = 4) (Fig.7C,D) with a significant reduction evident in striatal D2-like binding (wild-type, n = 5; HZnon-ac, n = 4; HZac, n = 12) (Fig.7A,B) in HZacmice after quantitative analysis (33% reduction compared with normal pups: 3388 ± 100 dpm/mm2 for HZac, n = 12 and 5063 ± 173 dpm/mm2 for normal pups, n = 9;p < 0.001). HZnon-ac mice had reduced D1-ligand binding consistent with the gene dosage (Drago et al., 1994).
Evidence of apoptosis and reactive gliosis in mutant mice
Hematoxylin and eosin-stained mutant brain sections (wild-type,n = 8; HZnon-ac, n = 2; HZac, n = 9) showed a hypercellular striatum with frequent condensed nuclear bodies suggestive of apoptosis (Fig.8A,B). Subsequent analysis using TUNEL staining to detect nuclear DNA fragmentation confirmed the presence of apoptosis in the mutant striatum (wild-type, n = 1; HZac,n = 1) (70 TUNEL-positive cells per square millimeter compared with six cells per square millimeter in the normal pup), nucleus accumbens (60 cells per square millimeter in the mutant brain compared with two cells per square millimeter in normal brain) (Fig.9) and to a lesser degree in the cortex (25 cells in mutant cortex compared with one cell per square millimeter in the normal cortex), although the septal area that is known to be D1R-negative was essentially free of TUNEL-positive cells. The presence of apoptosis was also verified by electron microscopy (Kerr et al., 1972) with cells containing condensed nuclear chromatin and displaying nuclear budding with preservation of cellular organelles identified consistently in the striatum of HZac mice (Fig.8C,D). Immunohistochemical staining for the glial cell marker GFAP (wild-type,n = 6; HZac, n = 6) showed reactive gliosis in the lateral striatum and along the corpus callosum of all HZac mice examined (Fig. 10).
Fig. 8.
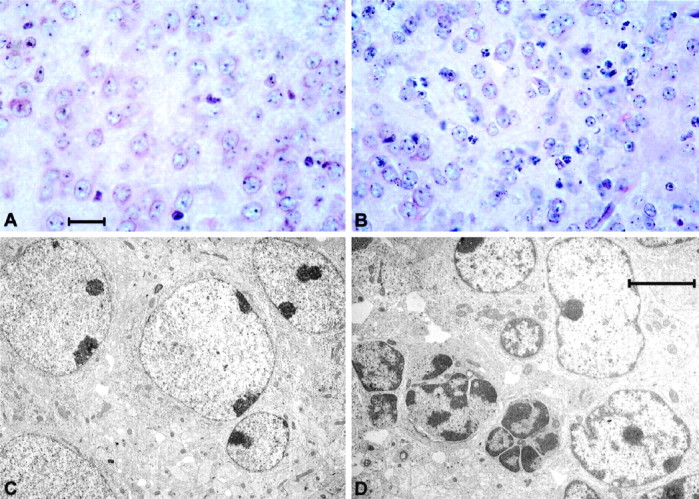
Histology and electron microscopy. High-power photomicrographs of hematoxylin- and eosin-stained sections through the normal (A) and mutant (B) brain at the level of the striatum showing frequent condensed nuclear bodies (B, white arrow). Low-power electron photomicrograph through the normal (C) and mutant brain (D). Cell showing nuclear chromatin condensation and nuclear budding is indicated by thewhite arrow in D. Scale bars:A, 20 μm; D, 5 μm.
Fig. 9.
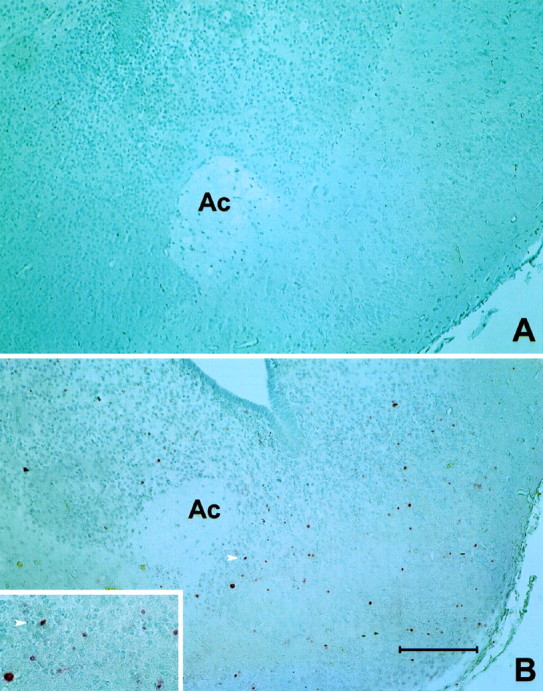
TUNEL processing to identify apoptotic cells.A, Normal mouse P4 brain. B, P4 HZac mutant brain with coronal sections taken at the level of the anterior commissure (Ac). Frequent apoptotic cells can be identified by pink nuclei in HZac brain. The arrow indicates a positive nucleus and can be used to orient within the high magnification insert. Scale bar: B, 100 μm.
Fig. 10.
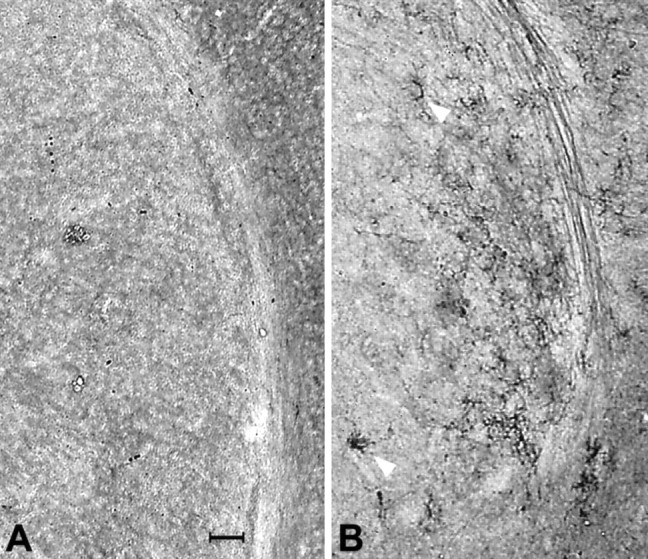
GFAP immunohistochemistry in normal (A) and HZac mouse brain (B) at the level of the striatum.Arrows indicate GFAP-positive cells in the mutant striatum. Scale bar: A, 100 μm.
Neuropeptide immunohistochemistry in mutant brain
Immunohistochemistry for neuropeptides (substance P: wild-type,n = 4; HZnon-ac, n = 1; HZac, n = 4; enkephalin: wild-type, n = 2; HZac,n = 2) used by striatal projection neurons showed consistent presence of substance P in the axons and terminals within the substantia nigra pars reticulata (Fig.11A) and enkephalin immunoreactivity (Fig. 11C) in the globus pallidus of normal pups. In contrast, HZac mice were devoid of substance P in the midbrain but exhibited normal levels of enkephalin staining in the globus pallidus (Fig.11B,D). In addition, scattered enkephalin-immunoreactive fibers were also identified in the caudate putamen of the mutant brain (Fig. 11D). A smaller number of such fibers was also seen in the normal brain at the same postnatal age. The prominence of the fibers in the mutant brain may be secondary to changes in the structure of the striatum or represent trans-synaptic effects.
Fig. 11.
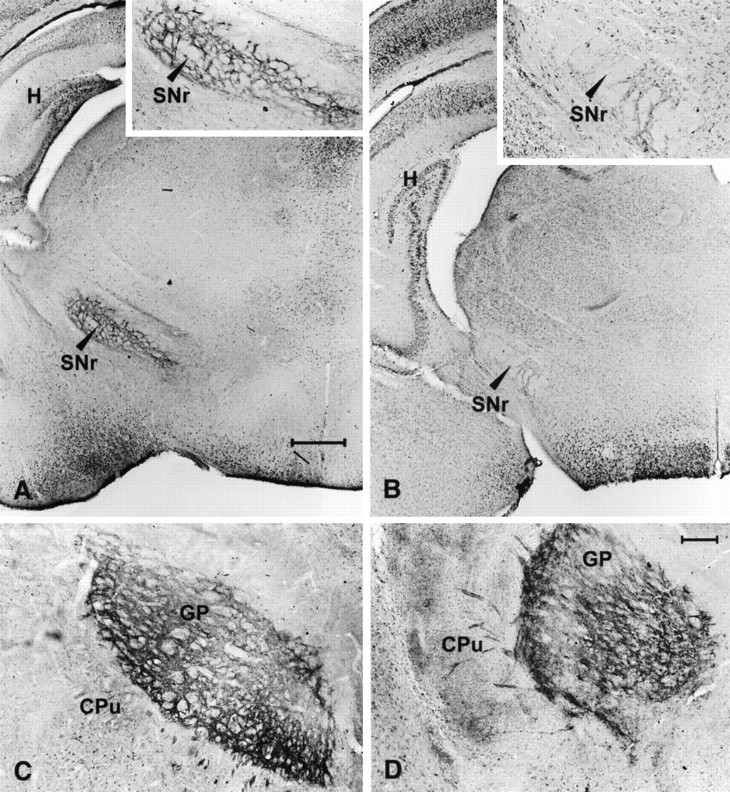
Immunohistochemistry for substance P and enkephalin. A, Substance P immunoreactivity seen at the level of the substantia nigra pars reticulata of normal pups.B, Lack of substance P immunoreactivity shown in mutant brainstem (insert). C, Section through the globus pallidus of a normal pup showing normal distribution of enkephalin immunoreactivity in terminals of the striatopallidal pathway. D, Normal enkephalin immunoreactivity is also seen in mutant pups. Scale bar, 50 μm. SNr, Substantia nigra pars reticulata; CPu, caudate putamen;GP, globus pallidus; H, hippocampus.
DISCUSSION
Striatal cell degeneration is seen in a number of rare Parkinsonian syndromes such as the striatonigral degeneration variant of multiple system atrophy (Davie et al., 1993; Quinn, 1994), progressive supranuclear palsy (Steele et al., 1964; Lees, 1987), and corticobasal ganglionic degeneration (Watts et al., 1994). Given the high diagnostic error rate in the clinical diagnosis of idiopathic Parkinson’s disease (Rajput et al., 1991; Hughes et al., 1992), striatal degeneration may be relatively common particularly in tremor-negative, l-dopa-nonresponsive Parkinsonism (Quinn, 1997). In addition, Huntington’s disease, an autosomal dominant neurodegenerative disease that causes gross involuntary movements is characterized pathologically by early loss of projection neurons within the caudate nucleus of the striatum (Ferrante et al., 1985; Albin et al., 1989). Furthermore, both acquired and genetically determined (Waters et al., 1993) striatal lesions have been reported in association with contralateral dystonia (Rothwell and Obeso, 1987), thus adding to the number of movement disorders associated with striatal cell loss. The relationship between the neurotransmitter phenotype of lost striatal neurons and the clinical manifestation in distinct movement disorders is largely unexplored. Not surprisingly, because of small sample size, clinical diversity and the nonspecific nature of the ligands used, autoradiographic and positron emission tomography studies examining dopamine receptor levels in Parkinsonian syndromes have generated conflicting results (Palacios et al., 1993). An experimental paradigm that involves the selective elimination of a genetically defined neuronal population might help elucidate both the direct effects of cell loss on movement phenotype and the downstream effects on other cell populations.
The striatum is a complex structure made up of projection neurons, interneurons, and afferent input from the brainstem and cerebral cortex (Graybiel, 1990; Gerfen, 1992). Medium-sized spiny projection neurons comprise ∼90% of striatal neurons, with interneurons making up the remainder (Gerfen, 1992). Of the five cloned dopamine receptors (Sibley and Monsma, 1992), D1 and D2 dopamine receptors are expressed at high levels in the adult striatum (Gerfen, 1992). D1R gene expression is detectable in the rat striatum from embryonic day 15 of development, with D1R mRNA being found at increasing levels in the caudate putamen, accumbens nucleus, Islands of Calleja, and the olfactory tubercle during maturation (Caille et al., 1995), whereas D1R mRNA is not detectable in the substantia nigra (Guennoun and Bloch, 1992; Sibley and Monsma, 1992). In situ hybridization studies suggested that D1Rs are preferentially expressed on substance P and dynorphin-positive striatal neurons that project directly to the substantia nigra pars reticulata–entopeduncular complex (the direct pathway), whereas enkephalin-positive D2R neurons project to the same nuclear complex via the external segment of the globus pallidus and subthalamic nucleus (the indirect pathway) (Gerfen et al., 1990). The validity of this dual pathway model has been challenged by recent studies reporting a substantial degree of D1 and D2 receptor colocalization on striatal projection neurons (Surmeier et al., 1992,1993, 1996; Surmeier and Kitai, 1994). The data reported in this study supports the dual pathway model of basal ganglia circuitry and the D1 and D2 dopamine receptor segregation hypothesis.
The movement disorder displayed by mutant mice is dramatic and consists of bradykinesia, dystonia, myoclonus, and postural instability. Bradykinesia, dystonia, and postural instability are seen in untreated idiopathic Parkinson’s disease and Parkinsonian syndromes, but myoclonus is more typical of the spectrum of hyperkinetic involuntary movements seen in Huntington’s disease (Penney and Young, 1993) and only rarely seen in neurodegenerative Parkinsonian syndromes (Marsden et al., 1982). Huntington’s disease, initially characterized as having selective loss of D2R/enkephalin-positive striatopallidal projection neurons has been found more recently to have more extensive dopamine receptor changes involving the D1 system (Ginovart et al., 1997;Stewart et al., 1998) with downregulated D1 binding identified in both presymptomatic and symptomatic Huntington’s disease gene-positive individuals (Andrews et al., 1998; Stewart et al., 1998).
The death of HZac mice was attributed to a combination of hypoxia and malnutrition. The eating disorder was consistent with the role played by the D1R in reward pathways (Drago et al., 1994; El-Ghundi et al., 1998) and the respiratory abnormality suggestive of a major role of dopamine in central respiratory control. Although brainstem D2Rs are thought to be of prime importance in the control of respiration (Mueller et al., 1982), the periodic breathing seen in mutant mice is not surprising given the expression of D1R in the brainstem during embryogenesis (Schambra et al., 1994). Interestingly, the respiratory abnormality was transient and not seen in older mutants that survived past day 9, variability that may reflect the background genetic heterogeneity of the mutant pups. The neuropathological changes of apoptosis and reactive gliosis described in this study, although characteristic of human neurodegenerative CNS disease (Bredesen, 1995; Mochizuki et al., 1996), are unique in a transgenic model. Furthermore, it is unlikely that the transient apneic periods result in symptomatic striatal hypoxia sufficient to explain the apoptosis, reactive gliosis, or the pathophysiology of the locomotor phenotype given the upregulated striatal expression of D2R mRNA, enkephalin mRNA, and vesicular acetylcholine transporter immunoreactivity (J. Drago, D. I. Finkelstein, P. Padungchaichot, and J. Y. F. Wong, unpublished observations) seen in mutant mice.
Brain dopamine receptor and neuropeptide expression pattern is complex in the CNS. Based on the dual pathway model of basal ganglia circuitry (Albin et al., 1989; Kawaguchi et al., 1990), which proposes D1 and D2 receptor segregation (Gerfen et al., 1990), HZacmice were predicted to lack D1R-positive striatonigral projection neurons but have an intact D2R-positive striatopallidal pathway. In addition, the neuropeptides substance P and dynorphin should be absent with preservation of enkephalin expression. As expected, brain D1R expression was undetectable by both ligand autoradiography and in situ hybridization; substance P immunoreactivity and mRNA as well as dynorphin mRNA were absent, whereas enkephalin expression was preserved in the mutant brain. Removal of the D1R-positive cells would be expected to result in increased packing density of remaining D2R-positive cells and a corresponding increase in D2R expression on quantitative in situ hybridization as seen (Fig.7E), however, the reduction of D2R expression as assessed by ligand autoradiography was surprising. The downregulated D2-like binding in the presence of upregulated D2R mRNA levels may be caused by many different effects, including compensatory changes in translation of D2R mRNA into protein in D2R-positive projection neurons, a reduction of dendritic arbors in D2R neurons, changes to baseline striatal expression of D3 and D4 dopamine receptors, or trans-synaptic effects on nigrostriatal or corticostriatal pathways reducing the contribution of D2 autoreceptors present on afferent terminals to total striatal D2-class binding. The methods used in this study do not provide a sensitive analysis at the cellular level to explore these issues.
D1R knock-out mice have been described by two independent groups (Drago et al., 1994; Xu et al., 1994). Although changes in exploratory behavior (Drago et al., 1994; Smith et al., 1998) and baseline locomotor activity are described, the dramatic movement abnormalities and early postnatal death seen in HZac mice are not seen. This suggests that loss of systems other than those mediated through D1Rs are necessary to generate this repertoire of locomotor abnormalities. Furthermore, a null mutation in the D2R resulted in bradykinesia without dystonia, myoclonus, or premature death (Baik et al., 1995).
D1R-positive neurons exist in sites other than the striatum, and the experimental paradigm used in this study may result in a phenotype contributed to by the death of all brain D1R-positive cells. D1R mRNA is first identified at E16 in the frontal and insular cortex of the developing rat brain and persists to involve more extensive regions of the cortex in late embryogenesis and in the postnatal brain (Schambra et al., 1994). Low level cortical D1R mRNA was also identified in the present study in P4 normal pups (Fig. 5). Notwithstanding the preservation of cortical thickness in mutant mice, a small number of apoptotic cells were identified with TUNEL staining. Furthermore, although myoclonus is certainly seen with primary basal ganglia diseases, it has no anatomical localizing value and may indeed reflect underlying cortical pathology (Marsden et al., 1982). In addition, developmental and postnatal compensatory effects, seen commonly in knock-out mutants (Drago et al., 1998), may also contribute to the phenotype. Future studies including controlled delivery ofCre recombinase expressing vectors directly into the striatum or mating HZ mice with striatal specific Cretransgenic strains will determine whether the effect is indeed caused by the loss of striatal D1R neurons rather than other D1R-expressing neurons elsewhere in the brain. Furthermore, there was PCR evidence forCre-mediated recombination in pups that had a normal phenotype making low level mosaicism involving D1R-positive neurons a possibility. This potential for mosaicism may need to be considered in future studies with this model system.
Footnotes
This work is supported in part by the National Health and Medical Research Council of Australia and the Australian Commonwealth Department of Veterans Affairs. J.D. is a Logan Research Fellow at Monash University. P.P. is a recipient of a Royal Thai Government Postgraduate Scholarship. J.F.M. is supported by the National Health and Medical Research Council of Australia Network for Brain Research into Mental Disorders. We thank Prof. Bevyn Jarrott for the gift of iodinated NCQ 298 radioligand, Minnie Cai and Jim Massalas for technical help, and John Secombe and Department of Biochemistry, Monash Medical Centre for performing blood urea and calcium assays.
Correspondence should be addressed to Dr. John Drago, Neurosciences Group, Department of Anatomy, Monash University, Wellington Road, Clayton, Victoria, 3168, Australia.
REFERENCES
- 1.Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- 2.Andrews TC, Weeks RA, Turjanski N, Brooks DJ. Monitoring disease progression in presymptomatic and early Huntington’s disease: D1 and D2 ligand PET and the unified Huntington’s disease rating scale. American Academy of Neurology; Minneapolis: 1998. [Google Scholar]
- 3.Baik JH, Picetti R, Saiardi A, Thiriet G, Dierich A, Depaulis A, Le Meur M, Borrelli E. Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature. 1995;377:424–428. doi: 10.1038/377424a0. [DOI] [PubMed] [Google Scholar]
- 4.Bredesen DE. Neural apoptosis. Ann Neurol. 1995;38:839–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- 5.Caille I, Dumartin B, Le Moine C, Begueret J, Bloch B. Ontogeny of the D1 dopamine receptor in the rat striatonigral system: an immunohistochemical study. Eur J Neurosci. 1995;7:714–722. doi: 10.1111/j.1460-9568.1995.tb00675.x. [DOI] [PubMed] [Google Scholar]
- 6.Civelli O, Douglass J, Goldstein A, Herbert E. Sequence and expression of the rat prodynorphin gene. Proc Natl Acad Sci USA. 1985;82:4291–4295. doi: 10.1073/pnas.82.12.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davie CA, Wenning GK, Barker GJ, Brennan A, Quinn N, Miller DH. MRS to differentiate multiple system atrophy from idiopathic Parkinson’s disease. Lancet. 1993;342:681–682. doi: 10.1016/0140-6736(93)91790-s. [DOI] [PubMed] [Google Scholar]
- 8.Drago J, Gerfen CR, Lachowicz JE, Steiner H, Hollon TR, Love PE, Ooi GT, Grinberg A, Lee EJ, Huang SP, Bartlett PF, Jose PA, Sibley DR, Westphal H. Altered striatal function in a mutant mouse lacking D1A dopamine receptors. Proc Natl Acad Sci USA. 1994;91:12564–12568. doi: 10.1073/pnas.91.26.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drago J, Padungchaichot P, Accili D, Fuchs S. Dopamine receptors and dopamine transporter in brain function and addictive behaviors: insights from targeted mouse mutants. Dev Neurosci. 1998;20:188–203. doi: 10.1159/000017313. [DOI] [PubMed] [Google Scholar]
- 10.El-Ghundi M, George SR, Drago J, Fan T, Fletcher PJ, Fan T, Nguyen T, Liu C, Sibley DR, Westphal H, O’Dowd BF. Disruption of dopamine D1 receptor gene expression attenuates alcohol seeking behavior. Eur J Pharmacol. 1998;353:149–158. doi: 10.1016/s0014-2999(98)00414-2. [DOI] [PubMed] [Google Scholar]
- 11.Ferrante RJ, Kowall NW, Beal MF, Richardson EP, Jr, Bird ED, Martin JB. Selective sparing of a class of striatal neurons in Huntington’s disease. Science. 1985;230:561–563. doi: 10.1126/science.2931802. [DOI] [PubMed] [Google Scholar]
- 12.Forno LS. Pathology of Parkinson’s disease. In: Marsden CD, Fahn S, editors. Movement disorders. Butterworth; London: 1982. pp. 25–30. [Google Scholar]
- 13.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; San Diego: 1997. [Google Scholar]
- 14.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-c. [DOI] [PubMed] [Google Scholar]
- 16.Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- 17.Ginovart N, Lundin A, Farde L, Halldin C, Backman L, Swahn CG, Pauli S, Sedvall G. PET study of the pre- and post-synaptic dopaminergic markers for the neurodegenerative process in Huntington’s disease. Brain. 1997;120:503–514. doi: 10.1093/brain/120.3.503. [DOI] [PubMed] [Google Scholar]
- 18.Graybiel AM. Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci. 1990;13:244–254. doi: 10.1016/0166-2236(90)90104-i. [DOI] [PubMed] [Google Scholar]
- 19.Guennoun R, Bloch B. Ontogeny of D1 and DARPP-32 gene expression in the rat striatum: an in situ hybridization study. Mol Brain Res. 1992;12:131–139. doi: 10.1016/0169-328x(92)90076-n. [DOI] [PubMed] [Google Scholar]
- 20.Harrison GS, Maxwell F, Long CJ, Rosen CA, Glode M, Maxwell IH. Activation of a diphtheria toxin A gene by expression of Human Immunodeficiency Virus-1 Tat and Rev proteins in transfected cells. Hum Gene Ther. 1991;2:53–60. doi: 10.1089/hum.1991.2.1-53. [DOI] [PubMed] [Google Scholar]
- 21.Hughes AJ, Daniel SE, Kilford L, Lees AJ. The accuracy of the clinical diagnosis of idiopathic Parkinson’s disease: a clinic-pathological study of 100 cases. J Neurol Neurosurg Psych. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kako K, Munekata E, Hosaka M, Murakami K, Nakayama K. Cloning and sequence analysis of mouse cDNAs encoding preprotachykinin A and B. Biomed Res. 1993;14:253–259. [Google Scholar]
- 23.Kawaguchi Y, Wilson CJ, Emson PC. Projection subtypes of rat neostriatal matrix cells revealed by intracellular injection of biocytin. J Neurosci. 1990;10:3421–3438. doi: 10.1523/JNEUROSCI.10-10-03421.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lakso M, Sauer B, Mosinger BJ, Lee EJ, Manning RW, Yu S-H, Mulder KL, Westphal H. Targeted oncogene activation by site specific recombination in transgenic mice. Proc Natl Acad Sci USA. 1992;89:6232–6236. doi: 10.1073/pnas.89.14.6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lakso M, Pichel JG, Gorman J, Sauer B, Okamoto Y, Lee E, Alt F, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci USA. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawrence AJ, Krstew E, Jarrott B. Functional dopamine D2 receptors on rat vagal afferent neurones. Br J Pharmacol. 1995;114:1329–1334. doi: 10.1111/j.1476-5381.1995.tb13352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lees AJ. The Steele-Richardson-Olszewski syndrome (progressive supranuclear palsy). In: Marsden CD, Fahn S, editors. Movement disorders 2. Butterworth; Cornwell: 1987. pp. 272–287. [Google Scholar]
- 29.Marsden CD. Basal ganglia disease. Lancet. 1982;2:1141–1147. doi: 10.1016/s0140-6736(82)92797-0. [DOI] [PubMed] [Google Scholar]
- 30.Marsden CD, Hallett M, Fahn S. The nosology and pathophysiology of myoclonus. In: Marsden CD, Fahn S, editors. Movement disorders. Butterworth; London: 1982. pp. 196–248. [Google Scholar]
- 31.Maxwell F, Maxwell IH, Glode LM. Cloning, sequence determination, and expression in transfected cells of the coding sequence for the tox-176 attenuated diphtheria toxin A chain. Mol Cell Biol. 1987;7:1576–1579. doi: 10.1128/mcb.7.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mochizuki H, Goto K, Mori H, Mizuno Y. Histochemical detection of apoptosis in Parkinson’s disease. J Neurol Sci. 1996;137:120–123. doi: 10.1016/0022-510x(95)00336-z. [DOI] [PubMed] [Google Scholar]
- 33.Montmayeur JP, Bausero P, Amlaiky N, Maroteaux L, Hen R, Borrelli E. Differential expression of the mouse D2 dopamine receptor isoforms. FEBS Lett. 1991;278:239–243. doi: 10.1016/0014-5793(91)80125-m. [DOI] [PubMed] [Google Scholar]
- 34.Mueller RA, Lundberg DB, Breese GR, Hedner J, Hedner T, Jonason J. The neuropharmacology of respiratory control. Pharmacol Rev. 1982;34:255–285. [PubMed] [Google Scholar]
- 35.Palacios JM, Landwehrmeyer B, Mengold G. Brain dopamine receptors: characterization, distribution, and alteration in disease. In: Jankovic J, Tolosa E, editors. Parkinson’s disease and movement disorders. Williams and Wilkins; Baltimore: 1993. pp. 35–54. [Google Scholar]
- 36.Penney JB, Young AB. Huntington’s disease. In: Jankovic J, Tolosa E, editors. Parkinson’s disease and movement disorders. Williams and Wilkins; Baltimore: 1993. pp. 205–216. [Google Scholar]
- 37.Quinn N. Multiple system atrophy. In: Marsden CD, Fahn S, editors. Movement disorders 3. Butterworth-Heinemann; Oxford: 1994. pp. 262–281. [Google Scholar]
- 38.Quinn N. Parkinson’s disease: clinical features. Bailliere’s Clin Neurol. 1997;6:1–13. [PubMed] [Google Scholar]
- 39.Rajput AH, Rozdilsky B, Rajput A. Accuracy of clinical diagnosis in parkinsonism: a prospective study. Can J Neurol Sci. 1991;18:275–278. doi: 10.1017/s0317167100031814. [DOI] [PubMed] [Google Scholar]
- 40.Rothwell JC, Obeso JA. The anatomical and physiological basis of torsion dystonia. In: Marsden CD, Fahn S, editors. Movement disorders 2. Butterworth; Cornwell: 1987. pp. 313–331. [Google Scholar]
- 41.Sauer B. Manipulation of transgenes by site-specific recombination; use of Cre recombinase. In: Wassarman PM, DePamphilis ML, editors. Guide to techniques in mouse development. Methods in enzymology. Academic; San Diego: 1993. pp. 890–900. [DOI] [PubMed] [Google Scholar]
- 42.Sauer B, Henderson N. Site specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc Natl Acad Sci USA. 1988a;85:5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sauer B, Henderson N. The cyclization of linear DNA in Escherichia coli by site-specific recombination. Gene. 1988b;70:331–341. doi: 10.1016/0378-1119(88)90205-3. [DOI] [PubMed] [Google Scholar]
- 44.Schambra UB, Ducan GE, Breese GR, Fornaretto MG, Caron MG, Fremeau RTJ. Ontogeny of D1A and D2 dopamine receptor subtypes in rat brain using in situ hybridization and receptor binding. Neuroscience. 1994;62:65–85. doi: 10.1016/0306-4522(94)90315-8. [DOI] [PubMed] [Google Scholar]
- 45.Sibley DR, Monsma FJ., Jr Molecular biology of dopamine receptors. Trends Pharmacol Sci. 1992;13:61–69. doi: 10.1016/0165-6147(92)90025-2. [DOI] [PubMed] [Google Scholar]
- 46.Smith DR, Striplin CD, Geller AM, Mailman RB, Drago J, Lawler CP, Gallager M (1998) Behavioral assessment of mice lacking D1A dopamine receptors. Neuroscience, in press. [DOI] [PubMed]
- 47.Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. Arch Neurol. 1964;10:333–359. doi: 10.1001/archneur.1964.00460160003001. [DOI] [PubMed] [Google Scholar]
- 48.Stewart JD, Hussey D, Jones C, Houle S, Thomson J, Guttman M. PET studies in presymptomatic and symptomatic Huntington’s disease patients comparing [18F] Fluorodeoxyglucose, [11C] Raclopride, and [11C] SCH-23390. American Academy of Neurology; Minneapolis: 1998. [Google Scholar]
- 49.Surmeier DJ, Kitai ST. Dopaminergic regulation of striatal efferent pathways. Curr Opin Neurobiol. 1994;4:915–919. doi: 10.1016/0959-4388(94)90142-2. [DOI] [PubMed] [Google Scholar]
- 50.Surmeier DJ, Eberwine J, Wilson CJ, Cao Y, Stefani A, Kitai ST. Dopamine receptor subtypes colocalize in rat striatonigral neurons. Proc Natl Acad Sci USA. 1992;89:10178–10182. doi: 10.1073/pnas.89.21.10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Surmeier DJ, Reiner A, Levine MS, Ariano MA. Are neostriatal dopamine receptors co-localized? Trends Neurosci. 1993;16:299–305. doi: 10.1016/0166-2236(93)90103-s. [DOI] [PubMed] [Google Scholar]
- 52.Surmeier DJ, Song W-J, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci. 1996;16:6579–6591. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tybulewicz VL, Crawford CE, Jackson PK, Bronson RT, Mulligan RC. Neonatal lethality and lymphopenia in mice with a homozygous disruption of the c-abl proto-oncogene. Cell. 1991;65:1153–1163. doi: 10.1016/0092-8674(91)90011-m. [DOI] [PubMed] [Google Scholar]
- 54.Waters CH, Faust PL, Powers J, Vinters H, Moskowitz C, Nygaard T, Hunt AL, Fahn S. Neuropathology of lubag (X-linked dystonia Parkinsonism). Mov Disord. 1993;8:387–390. doi: 10.1002/mds.870080328. [DOI] [PubMed] [Google Scholar]
- 55.Watts RL, Mirra SS, Richardson EP. Corticobasal ganglionic degeneration. In: Marsden CD, Fahr S, editors. Movement disorders 3. Butterworth-Heinemann; Oxford: 1994. pp. 282–299. [Google Scholar]
- 56.Wong JYF, Liberatore GT, Donnan GA, Howells DW. Expression of brain-derived neurotrophic factor and TrkB neurotrophin receptors after striatal injury in the mouse. Exp Neurol. 1997;148:83–91. doi: 10.1006/exnr.1997.6670. [DOI] [PubMed] [Google Scholar]
- 57.Xu M, Moratalla R, Gold LH, Hiroi N, Koob GF, Graybiel AM, Tonegawa S. Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses. Cell. 1994;79:729–742. doi: 10.1016/0092-8674(94)90557-6. [DOI] [PubMed] [Google Scholar]
- 58.Zurawski G, Benedik M, Kamb BJ, Abrams JS, Zurawski SM, Lee FD. Activation of mouse T-helper cells induces abundant preproenkephalin mRNA synthesis. Science. 1986;232:772–775. doi: 10.1126/science.2938259. [DOI] [PubMed] [Google Scholar]