

Abstract
Brain-derived neurotrophic factor (BDNF), like other neurotrophins, has long-term effects on neuronal survival and differentiation; furthermore, recent work has shown that BDNF also can induce rapid changes in synaptic efficacy. We have investigated the mechanism(s) of these synaptic effects on cultured embryonic hippocampal neurons. In the presence of the GABAA receptor antagonist, picrotoxin, the application of BDNF (100 ng/ml) for 1–5 min increased the amplitude of evoked synaptic currents by 48 ± 9% in 10 of 15 pairs of neurons and increased the frequency of EPSC bursts to 205 ± 20% of the control levels. There was no detectable effect of BDNF on various measures of electrical excitability, including the resting membrane potential, input resistance, action potential threshold, and action potential amplitude. In addition, BDNF did not change the postsynaptic currents induced by the exogenous application of glutamate. BDNF did increase the frequency of miniature EPSCs (mEPSCs) (268.0 ± 46.8% of control frequency), however, without affecting the mEPSC amplitude. The effect of BDNF on mEPSC frequency was blocked by the tyrosine kinase inhibitor K252a and also by the removal of extracellular calcium ([Ca2+]o). Fura-2 recordings showed that BDNF elicited an increase in intracellular calcium concentration ([Ca2+]c). This effect was dependent on [Ca2+]o; it was blocked by K252a and by thapsigargin, but not by caffeine. The results demonstrate that BDNF enhances glutamatergic synaptic transmission at a presynaptic locus and that this effect is accompanied by a rise in [Ca2+]c that requires the release of Ca2+ from IP3-gated stores.
Keywords: BDNF, calcium, glutamate receptors, hippocampal, neurotrophins, synaptic plasticity, neurotransmitter release
The neurotrophins play important roles in the survival and differentiation of many types of neurons (for review, see Thoenen, 1991; Lindsay et al., 1994). These effects generally occur over the course of hours or days. Recent studies indicate that, within minutes of application, neurotrophins can influence synaptic transmission and plasticity (for review, seeThoenen, 1995; Berninger and Poo, 1996; Schuman, 1997). In developing neuromuscular synapses, the application of NT-3 and BDNF resulted in a marked increase in the frequency, but not the amplitude, of spontaneous synaptic currents (Lohof et al., 1993; Stoop and Poo, 1996), suggesting that NT-3 and BDNF act by increasing neurotransmitter release.
Neurotrophins and their receptors, the trk tyrosine kinases, are found in many areas of the brain (Fryer et al., 1996). Studies subsequent to that of Lohof et al. (1993) have examined neurotrophin effects on cultured cortical neurons (Kim et al., 1994), cultured hippocampal cells (Levine et al., 1995, 1998; Li et al., 1998), hippocampal slices (Kang and Schuman, 1995, 1996; Gottschalk et al., 1998), entorhinal/hippocampal slices (Scharfman, 1997), visual cortex slices (Akaneya et al., 1997; Carmignoto et al., 1997), and hippocampal neurons in vivo (Messaoudi et al., 1998). In these studies somewhat varied results have been reported under a range of conditions.Levine et al. (1995) reported that BDNF enhanced excitatory synaptic transmission in cultured hippocampal neurons. In cultured cortical neurons, Kim et al. (1994) described an NT-3-induced potentiation of neuronal activity that appears to result from an NT-3-induced reduction of GABAergic inhibitory synaptic currents. Inhibition of GABAA synaptic responses by BDNF in rat hippocampal slices was reported by Tanaka et al. (1997). Do neurotrophins influence synaptic transmission by acting presynaptically, postsynaptically, or both? Kang and Schuman (1995) have suggested that there is a presynaptic component of the long-lasting neurotrophin-induced synaptic plasticity observed in acute hippocampal slices. They also have shown that local protein synthesis, likely occurring in the postsynaptic dendrites, was required for this effect (Kang and Schuman, 1996). In other studies, both presynaptic modulation (Lohof et al., 1993; Lebmann et al., 1994; Gottschalk et al., 1998) and postsynaptic modulation (Kim et al., 1994; Levine et al., 1995, 1998) by neurotrophins have been suggested.
In the present study we have investigated further the effects of BDNF on synaptic transmission. We find that, in the presence of a GABAA receptor antagonist, brief (1–5 min) application of BDNF enhances the amplitudes of evoked glutamatergic synaptic currents and increases the frequency, but not the amplitude, of mEPSCs. We also find that BDNF did not affect electrical excitability or glutamate-induced responses. These results demonstrate that BDNF enhances presynaptic neurotransmitter release. Our data also show that these effects are accompanied by a rise in presynaptic [Ca2+]c, which is dependent on the Ca2+ release from IP3-sensitive Ca2+ stores.
MATERIALS AND METHODS
Cell culture. Pregnant Wistar rats were killed by the inhalation of CO2, and embryos (at day 18) were removed immediately by cesarean section. Hippocampi were removed rapidly under stereomicroscopic observation under sterile conditions, cut into 1 mm pieces, and digested with 0.25% trypsin/0.25 mg/ml DNase (Sigma, St. Louis, MO) in HBSS without calcium or magnesium (Life Technologies, Gaithersburg, MD) at 36°C for 15 min. The pieces then were rinsed gently in HBSS, washed twice in plating medium [Neurobasal with B27 supplement (Life Technologies), with 500 μm Glutamax (Life Technologies), 25 μmglutamate, and 5% horse serum], and gently triturated in 1 ml of plating medium with five passes through the 0.78 mm opening of a tip of a P-1000 Pipetman. Suspended cells were decanted, and the remaining pieces were triturated once more. Cell suspensions were gravity-filtered through a 70 μm Nylon mesh (Falcon, Oxnard, CA) to remove large debris and were centrifuged for 2 min at 150 ×g. Pellets were resuspended by gentle trituration as above. Approximately 35,000 cells were plated in an area 15 mm in diameter at the middle of a 35 mm plastic culture dish (Corning, Corning, NY) that had been coated with poly-d-lysine and laminin. Cultures were maintained at 36°C in a 5% CO2 incubator. One-half volume of the medium was changed twice weekly with culture medium (Neurobasal with B27 supplement and with 500 μmGlutamax). Recordings were made after ∼2 weeks in culture.
For the measurement of [Ca2+]c the cells were plated on 15 mm glass coverslips instead of plastic culture dishes. All other conditions were the same as described above.
Electrophysiological recording. Before recording, a dish was removed from the incubator, and the culture medium was replaced with recording saline (in mm): NaCl 145, KCl 5.4, CaCl2 1.8, MgCl2 0.8, HEPES 10,d-glucose 10, and picrotoxin 0.1, titrated to pH 7.4 with NaOH; the osmolarity was adjusted to 300 mOsm with sucrose. In zero Ca2+ saline, 5 mm EGTA was added instead of CaCl2. Voltage-gated Ca2+ currents were measured by adding 1 μm TTX and replacing CaCl2 and MgCl2 with 5 mmBaCl2. The inward Ca2+(Ba2+) currents were triggered by depolarization to different membrane potentials from a holding potential of −70 mV. Dual or single whole-cell recordings (Hamill et al., 1981) were made at room temperature with pipettes pulled in four stages from 1.5 mm outer diameter glass capillary tubes (WPI, Sarasota, FL) with a P-80/PC micropipette puller (Sutter Instruments, Novato, CA). Patch pipettes were filled with a solution containing (in mm): K-gluconate 140, CaCl2 0.1, EGTA 1.1, MgCl2 5, HEPES 20, Mg-ATP 3, Na2-phosphocreatine 5, and Na-GTP 0.3, pH 7.2, adjusted to 290 mOsm. To measure voltage-gated calcium currents, we replaced the K-gluconate by CsCl to block the potassium currents. Ionic currents or voltages were measured with patch-clamp amplifiers (Axopatch 200 and Axopatch-1D, Axon Instruments, Foster City, CA), filtered at 2 kHz, digitized at 10 kHz, recorded on a computer, and monitored on both a storage oscilloscope and a chart recorder. Synaptic activities simultaneously were recorded digitally at 9 kHz on pulse code-modulated magnetic tape (Instrutech VR-100, Great Neck, NY) for off-line analysis. The series resistance was monitored by measuring the instantaneous current in response to a 5 mV voltage step command. Results were discarded if the series resistance changed by >10% during the course of an experiment.
In the presence of the GABAA receptor antagonist picrotoxin, EPSCs occurred in bursts (see Fig. 1A). To quantify the bursts, we considered the detection of more than three peaks (>30 pA) of EPSCs within 100 msec as the start of one burst; we considered the detection of a latency between EPSCs longer than 1 sec as the end of the burst. The frequency of bursts was defined as the number of bursts detected in 1 min. The synaptic charge was measured by integrating all inward synaptic currents over successive 1 min intervals.
Fig. 1.

Bursts of synaptic activity in whole-cell recordings of E18 hippocampal neurons cultured after 2 weeks. The inward current deflections are produced by postsynaptic currents.A, Shown is an example of recordings. B, Shown is the time course of the BDNF effect. The effect of BDNF (100 ng/ml) on the burst frequency appeared ∼30 sec after application. The increased frequency and the increased magnitude of the integrated current per minute (denoted synaptic charge) recovered to baseline ∼5 min after washout of BDNF. C, D, Average values of bursting frequency and of synaptic charge in and after BDNF application.
In the presence of 100 nm tetrodotoxin (TTX), mEPSCs were recorded. The mEPSCs were detected and measured by FETCHEX in the pClamp 6 series (Axon Instruments). Threshold detection levels were between 3 and 10 pA [with lower series resistance the size of both the noise and signal was larger, thus requiring a higher detection threshold (∼10 pA)]. The selected threshold level remained constant for the duration of the experiment. All events larger than this level were included if they had (1) rise times <3 msec and (2) a waveform similar to that of the evoked EPSC. Events that began during the decay phase of a previous event were included in the frequency measurements, but not in the amplitude measurements. The frequency of mEPSCs was measured over a 30 sec interval. The distributions of mEPSC amplitudes were normalized by the median control value for each experiment.
For dual whole-cell recording, both presynaptic and postsynaptic cells were voltage-clamped at a holding potential of −70 mV. When the presynaptic neuron was stimulated with an 8 msec depolarizing voltage step (to −20 or −10 mV) every 15 sec, a large transient inward current was activated in the presynaptic cell, and an EPSC was elicited in the postsynaptic neuron (see Fig. 2B). Only monosynaptic EPSCs were studied. Two criteria for recognizing monosynaptic EPSCs were used: (1) short and constant latency (<5 msec) and (2) the absence of multiple peaks (Arancio et al., 1995).
Fig. 2.
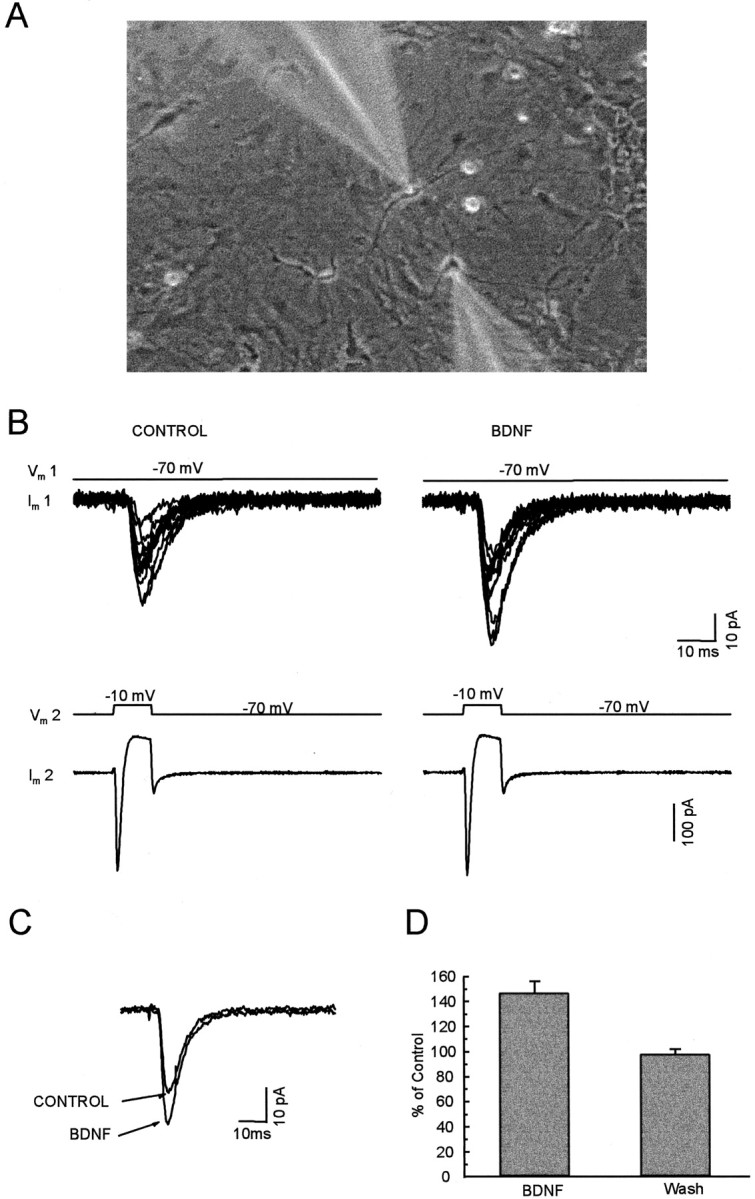
The effect of BDNF on evoked synaptic currents between paired neurons. A, Micrograph taken during dual whole-cell recordings was made from a pair of nearby cultured hippocampal neurons; note the images of the micropipette electrodes.B, The bottom panel shows the current in the presynaptic cell during a voltage step to −10 mV from a holding potential of −70 mV. The top panel superimposes 12 continuous EPSCs recorded simultaneously from the postsynaptic cell before and during BDNF application. C, The average of EPSCs recorded during 5 min before (CONTROL) and 2–5 min after the beginning of BDNF application. D, The average of the amplitude of evoked EPSCs during and after the application of BDNF.
BDNF was applied by a local perfusion system (except in the experiments that involved glutamate application) in which up to eight solutions connected to one port. The port fed a delivery tube (250 μm in internal diameter). The tube was mounted ∼500 μm away from the recorded cells. The flow rate was ∼0.5 ml/min. Changes among the eight solutions occurred within 2 sec. Before and after BDNF application the saline was perfused constantly. Heat-inactivated BDNF was used as a control in every set of experiments. Faster application of glutamate was performed with the DAD-12 superfusion system (ALA Scientific Instruments, Westbury, NY), which produced concentration rise times ≤15 msec. In these experiments BDNF also was applied by the DAD-12 system and induced the same degree of synaptic potentiation.
Fura-2 imaging. Intracellular calcium ([Ca2+]c) levels were measured by using fura-2 AM and ratiometric imaging techniques (Grynkiewicz et al., 1985). Cells were loaded with fura-2 AM (2 μm, Molecular Probes, Eugene, OR) for 40 min at 37°C. One microliter of 25% (w/w) Pluronic F-127 (Molecular Probes) was mixed per 1 ml of fura-2 AM loading solution to help solubilize the ester in aqueous medium. After the cells were washed with control saline, they were incubated for 40 min at 37°C while fura-2 AM was deesterified by intracellular esterases. All recordings then were performed at room temperature. The saline solution used was the same as that for electrophysiological recordings (with 100 nm TTX).
Digital video imaging fluorescence microscopy was used for measuring the fluorescence of identified neurons (at 40× magnification, with a Zeiss inverted microscope); images captured at excitation wavelengths of 340 and 380 nm were stored every 5–15 sec. The ratio of fluorescence at the two exciting wavelengths was calculated for each pixel within a cell boundary. The calcium level was calculated by the equation:
where KD is the fura–Ca2+ binding constant (∼220 nm), and R is a ratio of fluorescence at two wavelengths (Tsien and Poenie, 1986). Calibration of Rmin (the limiting value that the ratio can have at zero [Ca2+]), Rmax (the limiting value that the ratio can have at saturating [Ca2+]), andF0/F∞ (the ratio of fluorescence at 380 nm with zero Ca2+ and saturating [Ca2+]) was performed by using standard Ca2+ buffers with 0 and 39.8 μm[Ca2+] (Molecular Probes) in custom-made micro glass chambers that were 150 μm deep and 3 mm2 in area.
In the experiments examining the mechanisms involved in the BDNF-induced elevation of [Ca2+]c, we first tested the BDNF-induced responses. After the level of [Ca2+]c recovered to baseline, solutions were applied containing zero Ca2+, 200 nm K252a, 2 μm thapsigargin, or 10 mm caffeine. The response to a second application of BDNF was tested 10–15 min later.
All data are shown as mean ± SEM.
BDNF was kindly provided by Dr. Andrew Welcher of Amgen (Thousand Oaks, CA). BDNF was inactivated by boiling for 15 min. Thapsigargin (RBI, Natick, MA) was dissolved in DMSO as a 2 mm stock solution.
RESULTS
Potentiation of spontaneous bursting by BDNF
The hippocampal cultures contain networks of interacting neurons. Whole-cell recordings (VH = −70 mV) in one cell of the network in the presence of the GABAA receptor antagonist (picrotoxin, 100 μm) revealed bursts of synaptic activity in the vast majority of neurons after ∼2 weeks in culture (Fig. 1). Similar bursting behavior has been documented by Bijak et al. (1991) and Rohrbacher et al. (1997). These currents represent action potential-dependent spontaneous EPSCs mediated by non-NMDA receptors, because they were abolished by 20 μm CNQX, a non-NMDA receptor antagonist (data not shown).
Bath application of BDNF at 100 ng/ml increased the frequency of the bursts. This effect appeared ∼30 sec after the addition of BDNF; gradual recovery to baseline levels occurred within 5–10 min after BDNF washout (Fig. 1B). On average, the burst rates increased to 205 ± 20% of control levels (n = 9). In some cases the frequency of bursts increased, but their duration decreased. The effect of BDNF was characterized further by measuring the integrated inward current over a specific interval; this is denoted as synaptic charge in Figure 1D. The synaptic charge was increased to 158 ± 13% of control level (n = 9) by BDNF application. As a control for BDNF exposure, treatment with heat-inactivated BDNF had no effect on either the frequency of bursts (110 ± 9% of control; n = 5) or the synaptic charge (105 ± 3% of control; n = 5).
Potentiation of evoked synaptic currents by BDNF
Synaptic transmission was evoked in pairs of nearby cultured hippocampal neurons during dual whole-cell recording. A typical pair of neurons under study is shown in Figure2A. An inward current lasting ∼2 msec was produced in the presynaptic cell (cell 2) by depolarization to −10 mV from a holding potential of −70 mV. A resulting EPSC was recorded in the postsynaptic cell (cell 1). The amplitudes of the evoked EPSCs were increased (by >115% of control levels) by BDNF application in 10 of 15 pairs of neurons; there was no change in the remaining five pairs. The average increase for the 10 pairs was 48 ± 9%, as shown in Figure 2, C andD. The mean increase was 32 ± 8% for all 15 tested pairs. Heat-inactivated BDNF did not induce any change in all seven control experiments (101 ± 1% of control).
BDNF does not affect parameters determining electrical excitability
In principle, BDNF-induced enhancement of neuronal electrical excitability could increase firing rate and increase synaptic transmission in the network of bursting neurons. To address possible effects on electrical excitability, we simultaneously blocked (1) non-NMDA receptor-mediated EPSPs by the addition of CNQX (20 μm), (2) NMDA receptor-mediated EPSPs by APV (50 μm), (3) GABAA receptor-mediated IPSPs by picrotoxin (100 μm), and (4) GABABreceptor-mediated IPSPs by 2-hydroxy-saclofen (100 μm). In the presence of this antagonist cocktail no synaptically evoked response remained. Action potentials were triggered by the injection of inward currents under current clamp. As shown in Table1, there were no detectable effects of BDNF on the resting membrane potential, input resistance, action potential threshold, action potential amplitude, or the current–frequency relationship (Fig. 3). We conclude that BDNF does not affect the voltage-dependent currents that underlie excitability under the present experimental conditions.
Table 1.
Neuronal excitability before and during BDNF application
| Vm (mV) | Rin(GΩ) | AA (mV) | AT (mV) | |
|---|---|---|---|---|
| Control | −60.2 ± 1.2 | 0.51 ± 0.08 | 103.2 ± 3.9 | −43.1 ± 0.9 |
| BDNF | −61.6 ± 1.4 | 0.52 ± 0.08 | 105.9 ± 4.4 | −42.8 ± 1.0 |
Resting membrane potential, Vm; input resistance, Rin; action potential amplitude, AA; action potential threshold, AT. Data are averaged from seven cells. There were no significant differences between the parameters before and during BDNF application (p > 0.05).
Fig. 3.
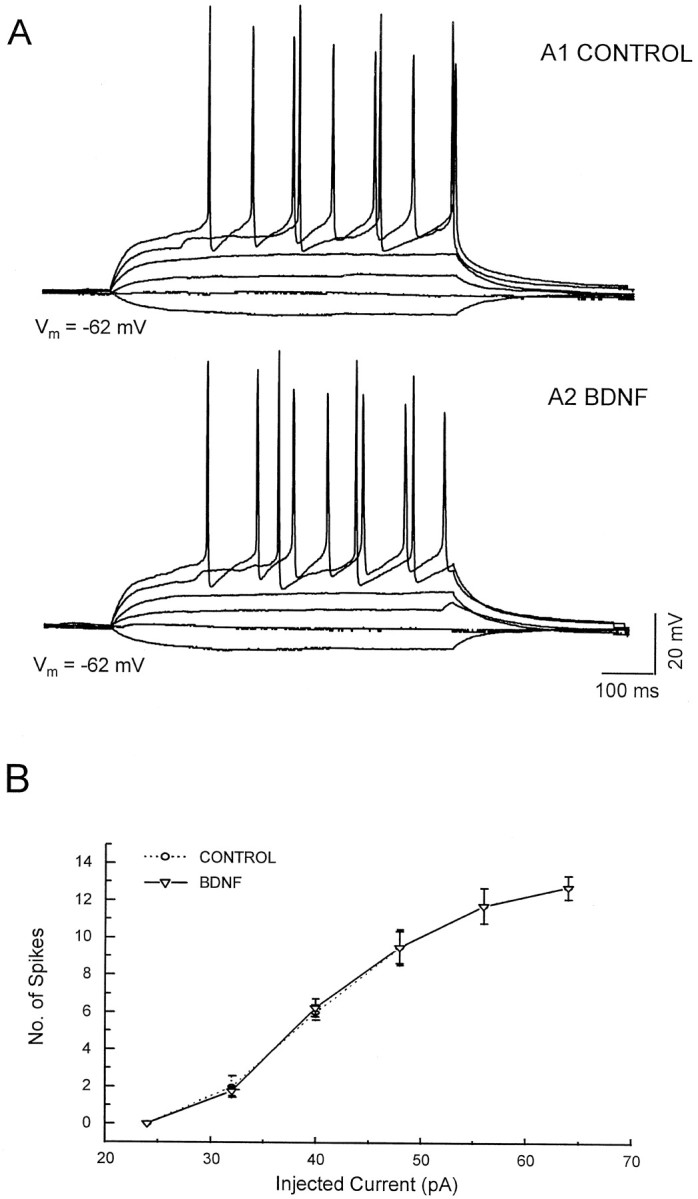
BDNF did not affect the electrical excitability of neurons. APV, CNQX, and picrotoxin were used to block the NMDA-, AMPA-, and GABA-induced responses from the input of other cells. Recordings were made in current-clamp mode. A, Shown are the I–V responses recorded from cells both before (A1) and during (A2) BDNF application. B, Shown is the relation between the amplitude of the injected current and the number of action potentials during the 600 msec current pulses.
BDNF increases the frequency, but not the amplitude, of mEPSCs
To investigate the synaptic locus of the action of BDNF, we measured mEPSCs under conditions in which there was no action potential-dependent release. Specifically, mEPSCs were recorded in the presence of 100 nm TTX and 100 μm picrotoxin. Typical records are shown in Figure4A. The application of BDNF increased the frequency of mEPSCs to 268 ± 47% of the control level (n = 9). The increased frequency became noticeable ∼30 sec after the beginning of BDNF application. Just as for the spontaneous and evoked synaptic currents, the effect returned to control levels within several minutes after the washout of BDNF (Fig. 4B,C).
Fig. 4.

BDNF increased the frequency, but not the amplitude, distribution of mEPSCs. A, Shown are examples of recordings. TTX (100 nm) was present to block the release of transmitter caused by spontaneous action potentials.B, Shown is the time course of the BDNF effect. At ∼30 sec after BDNF application the frequency of mEPSCs was increased. The increased frequency recovered to the baseline value 5 min after the washout of BDNF. C, The average values of frequency during and after BDNF application. D, Averages of cumulative amplitude distributions of mEPSCs obtained before and during BDNF application (n = 9). There is no obvious difference (p > 0.05) between the distributions of amplitudes before and during BDNF application.
In another set of experiments, treatment with heat-inactivated BDNF had no effect on the frequency of mEPSCs (109 ± 3% of control;n = 5; Fig. 5). Bath application of 200 nm K252a, an inhibitor of tyrosine kinases (Berg et al., 1992), completely blocked the effect of BDNF (111 ± 4% of control; n = 6; Fig. 5). The removal of external Ca2+ caused a slight decrease in the basal mEPSC frequency. The effect of BDNF on mEPSC frequency was decreased significantly (128 ± 5% of control; n= 5; Fig. 5) by removing [Ca2+]o. Kang and Schuman (1996) showed a requirement for protein synthesis in neurotrophin-induced long-lasting enhancement of synaptic transmission in adult hippocampal slices. However, pretreatment for 30 min with one of two protein synthesis inhibitors, either anisomycin (40 μm) or cycloheximide (40 μm), did not block the BDNF-induced enhancement of mEPSC frequency on the cultured embryonic hippocampal neurons. The application of BDNF increased the frequency of mEPSCs to 275 ± 33% (n = 5) and 238 ± 27% (n = 5) of control levels on anisomycin-treated and cycloheximide-treated neurons, specifically.
Fig. 5.

The increased frequency of mEPSCs caused by BDNF was blocked by K252a and was dependent on the extracellular calcium.
In contrast to the large changes in mEPSCs frequency, the distribution of mEPSCs amplitudes was not changed by BDNF (see Fig.4D). Additionally, the amplitudes and dose–response curve of currents evoked by the direct application of 2–200 μm glutamate to individual neurons (blocked by CNQX; data not shown) were unchanged by BDNF (Fig.6). Fast application of glutamate (>50 μm) induced a rapidly desensitizing (within 200 msec) response, followed by a sustained response (Patneau and Mayer, 1990). The peak amplitudes of currents induced by 200 μmglutamate are 64 ± 10 pA/pF before and 65 ± 10 pA/pF after BDNF application (n = 5; paired Student’s ttest; p ≥ 0.05). The amplitudes of sustained currents were measured 900 msec after glutamate application as the mean current over an interval of 100 msec. The maximal sustained currents induced by 200 μm glutamate are 23.1 ± 5.5 pA/pF before BDNF and 22.6 ± 4.7 pA/pF after BDNF (n = 5;p > 0.05). The EC50 and Hill coefficient are 14.3 μm and 1.8 before and 13.8 μm and 1.7 after BDNF application.
Fig. 6.

BDNF did not affect the sensitivity of glutamate receptors. A, Shown are inward currents induced by glutamate (2–200 μm) before and during BDNF application.B, Shown is the dose–response relation for the glutamate-induced current, measured during the plateau (900 msec after the start of application; n = 5).
These data indicate that BDNF probably induces an increase in presynaptic efficacy rather than an increase in postsynaptic receptor sensitivity.
Effect of BDNF on [Ca2+]c
How does BDNF enhance presynaptic efficacy? One possibility is via the activation of phospholipase C-γ (PLCγ), which causes the release of intracellular Ca2+ (Bothwell, 1995;Zirrgiebel et al., 1995; Segal and Greenberg, 1996). PLCγ is abundant in hippocampal neurons (Yamada et al., 1991). In addition, elevations in [Ca2+]c induced by BDNF and NT-3 in hippocampal neurons have been reported by Berninger et al. (1993). It is well known that the frequency of spontaneous synaptic currents is affected directly by changes in [Ca2+]c levels in the nerve terminal (Miledi, 1973). Thus, an increase in [Ca2+]c might explain a higher frequency of mEPSCs after BDNF exposure. We therefore used ion-sensitive optical recording to examine the level of [Ca2+]c in cultured hippocampal neurons. Because the signals in neuronal processes are too small for systematic measurements, our measurements were averaged over the cell body. After ∼30 sec of BDNF application (100 ng/ml) the neurons showed an elevation in [Ca2+]c (to 305 ± 9 nm from 98 ± 7 nm before BDNF; n = 73); this elevation was reversed with the washout of BDNF and could be elicited repetitively (Fig.7A,D). Just as for the effect of BDNF on the mini frequency, this increase was dependent on extracellular Ca2+([Ca2+]o). Depletion of [Ca2+]o for 10 min slightly reduced the basal level of [Ca2+]c, and the subsequent addition of BDNF produced no further change in [Ca2+]c. BDNF application increased the [Ca2+]c by 204 ± 24 nm in normal saline and by 26 ± 3 nmunder Ca-free conditions (n = 22) (Fig. 7A). After pretreatment of the neurons for 10 min with K252a (200 nm), an inhibitor of tyrosine kinases, the response also was decreased (221 ± 20 nm before K252a and 31 ± 4 nm after K252a; n = 21) (Fig.7B).
Fig. 7.
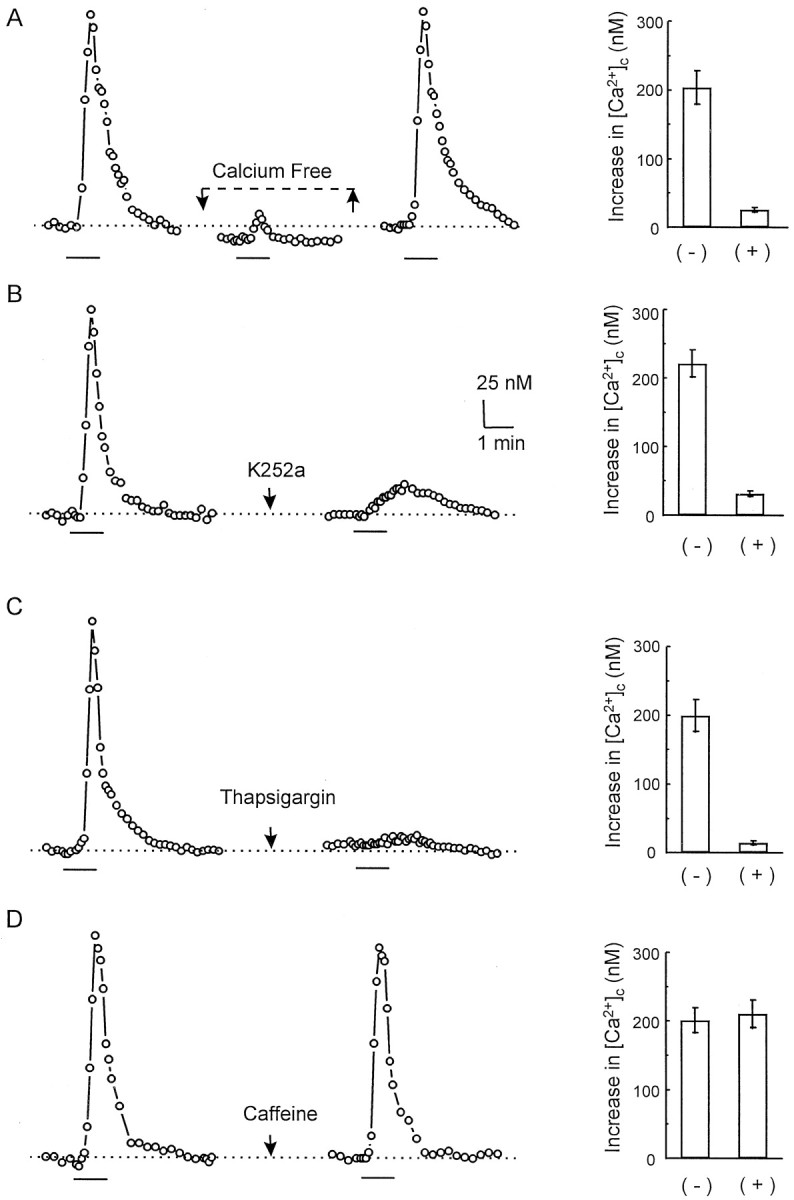
BDNF induced an increase in intracellular Ca2+. [Ca2+]c was measured by image ratio fluorescence microscopy with fura-2.A, A hippocampal cell responded to BDNF with an increase in [Ca2+]c. The BDNF-induced increase in [Ca2+]c disappeared in Ca2+-free solution and was restored after a switch to normal saline. B, K252a blocked the BDNF-induced increase in [Ca2+]c. C, Exposure to 2 μm thapsigargin blocked the [Ca2+]c response. D, The [Ca2+]c response was present during exposure to caffeine-treated cells. (+) and (−) in the graphs indicate the BDNF-induced responses in the presence and absence of the drug, respectively. The bar in each panel indicates the period of BDNF application.
To examine the requirement of intracellular Ca2+stores in the BDNF-induced responses, we compared the effects of (1) thapsigargin, which depletes all intracellular Ca2+stores by inhibiting endosomal Ca2+-ATPase activity (Thastrup et al., 1990), and (2) caffeine, which selectively depletes just the Ca2+-activated pool (Garaschuk et al., 1997). We assessed the effects of these agents on the BDNF-induced Ca2+ responses. Neither thapsigargin nor caffeine changed the baseline levels of [Ca2+]cunder our experimental conditions (also see Garaschuk et al., 1997). Figure 7C shows, however, that when BDNF was applied during exposure to 2 μm thapsigargin, there was only a small increase in [Ca2+]c (200 ± 23 nm without thapsigargin and 15 ± 3 nmwith thapsigargin; n = 18). On the other hand, Figure7D shows that applications of BDNF induced similar amplitudes of Ca2+ responses in 10 mmcaffeine (201 ± 18 nm without caffeine and 211 ± 20 nm with caffeine; n = 12). These data suggest that the increase in [Ca2+]cis attributable to a Ca2+ release from intracellular stores, perhaps from the inositol trisphosphate (IP3)-sensitive stores. Thapsigargin itself dramatically increased the basal level of mEPSC frequency (more than fivefold; data not shown), thus preventing direct studies of additional effects of BDNF.
The effects of BDNF on voltage-gated calcium currents also were investigated. The peak calcium current densities were 32.1 ± 2.1 pA/pF and 31.3 ± 1.9 pA/pF (n = 5) before and during BDNF treatment. Thus, BDNF had no obvious effect on the macroscopic Ca2+ current. The activation and inactivation curves of the calcium current also were not affected by BDNF treatment (data not shown).
DISCUSSION
Several recent studies have shown that BDNF enhances synaptic efficacy in neurons of the CNS (Kim et al., 1994; Lebmann et al., 1994; Kang and Schuman, 1995, 1996; Levine et al., 1995; Akaneya et al., 1997; Carmignoto et al., 1997; Scharfman, 1997; Tanaka et al., 1997; Messaoudi et al., 1998). To gain insight into the mechanisms of BDNF action, we have examined its effects on cultured E18 hippocampal neurons. Our results demonstrate that the brief application of BDNF enhances glutamatergic synaptic activity and increases the amplitude of evoked synaptic currents. These effects were induced independently of GABAA receptor-mediated responses. Specifically, our experiments indicate that BDNF likely acts by increasing the probability of transmitter release at the presynaptic terminals, although we cannot rule out completely the possibility of unsilencing postsynaptically silent synapses or an increase in the number of presynaptic release sites (Liao et al., 1995). Under our conditions, however, BDNF did not change the postsynaptic response to exogenously applied glutamate. We also observed that BDNF does not affect the parameters that influence electrical excitability (membrane potential, input resistance, action potential magnitude, and/or threshold). The BDNF effects were blocked by K252a and by thapsigargin, but not by caffeine. The results demonstrate that BDNF enhances glutamatergic synaptic transmission at a presynaptic locus and that this effect is accompanied by a rise in [Ca2+]c that requires the release of Ca2+ from IP3-gated stores.
Neurotrophins have been shown to inhibit GABAA-mediated currents and thus may affect synaptic transmission (Kim et al., 1994;Rutherford et al., 1997; Tanaka et al., 1997). These effects were not examined in our study because the GABAA inhibitor picrotoxin was present in the bath solution.
BDNF enhanced both EPSC amplitudes and mEPSC frequencies within a few minutes of application. The increases in the EPSC amplitudes and mEPSC frequencies lasted, without decrease, for the entire time that BDNF was present, and these parameters returned to baseline values within 5–15 min of BDNF washout. Reversible actions of BDNF on the 5–15 min time scale also have been reported in other studies of cultured neurons (Lohof et al., 1993; Kim et al., 1994; Levine et al., 1995). In contrast, studies in slices and in vivo have shown that BDNF can produce more slowly developing, but also more stable, changes in synaptic efficacy (Kang and Schuman, 1995, 1996; Akaneya et al., 1997;Carmignoto et al., 1997; Scharfman, 1997; Messaoudi et al., 1998). The effects on slices (Kang and Schuman, 1995, 1996) occur over a 20 min period on the application of BDNF and do not reverse on washout. There are clearly many differences between embryonic neurons in culture and the adult acute slice preparation. These differences include neuron density, number of glia, and, perhaps most importantly, developmental maturity. Kang and Schuman (1996) showed a requirement for protein synthesis in neurotrophin-induced synaptic enhancement in hippocampal slices. In the present study, however, we found that the protein synthesis inhibitors anisomycin and cycloheximide did not block the reversible actions in the cultured embryonic hippocampal neurons.
In agreement with earlier observations (Berninger et al., 1993), we have observed an increase in intracellular Ca2+ in hippocampal neurons when BDNF was administered under conditions in which action potentials were inhibited by the addition of TTX. Because thapsigargin, but not caffeine, blocked the increase in [Ca2+]c induced by BDNF, we conclude that the release of Ca2+ from IP3receptor-gated pools is critical. This result is consistent with activation of the BDNF–trkB signal transduction pathway leading to the activation of PLCγ/IP3. Although our measurements were obtained from the cell body, it is known that endoplasmic reticulum-based calcium stores are present at synaptic terminals (Sharp et al., 1993; Reyes and Stanton, 1996). In addition, Stoop and Poo (1996) also have reported that BDNF increases the calcium level in presynaptic nerve terminals. Interestingly, the BDNF-induced increase in [Ca2+]c that we and others have observed also depends on extracellular Ca2+ (Stoop and Poo, 1996) (Fig. 7). However, BDNF did not change the voltage-gated calcium currents. In many non-neuronal cell types intracellular Ca2+ stores have a limited capacity, and the sustained increases in cytoplasmic Ca2+ arise via capacitative Ca2+ entry from the external solution (Mikoshiba, 1997). Recent data suggest that neurons also express capacitative Ca2+ entry, so that the dependence on extracellular Ca2+ may arise in our experiments because the Ca2+ influx is induced by Ca2+ released from intracellular stores (Mathes and Thompson, 1994; Grudt et al., 1996; Garaschuk et al., 1997).
Although capacitative Ca2+ entry to the endoplasmic reticulum in some non-neural cells is manifested by a small inward current (Hoth and Penner, 1993), such currents typically are not reported in CNS neurons (Garaschuk et al., 1997). In the cultured hippocampal neurons we also detected no inward current during the BDNF-induced increase in [Ca2+]c. It is possible that the capacitative-like Ca2+ entry is not detected directly at the plasmalemma in hippocampal neurons because the endoplasmic reticulum directly contacts the plasmalemma (Putney, 1986; Tsien and Tsien, 1990).
Calcium-dependent potassium channels can be activated in rat cortical neurons by NT3 and NGF (Holm et al., 1997). These channels help to set the resting membrane potential and input resistance and, therefore, firing frequencies; they may modulate transmitter release in some neurons (Robitaille et al., 1993). However, we did not observe any change in parameters determining electrical excitability, including the resting membrane and input resistance, K+ current waveforms, and Na+ current waveforms. These results show that electrical excitability changes cannot account for the effect of BDNF on synaptic transmission. That electrical excitability remains unchanged argues against a direct role for modulated channels in the increased frequency of spontaneous EPSC bursts produced by BDNF. We therefore suggest that the most likely source of this increase is a BDNF-induced increase in the probability of neurotransmitter release.
It is well known that synaptic transmission is affected by changes in presynaptic Ca2+ level. Although our observations concerned neuronal somata, similar results have been reported for axon terminals (Stoop and Poo, 1996). The effects of BDNF on synaptic transmission and on intracellular calcium concentration were compared under several conditions. K252a, a protein kinase inhibitor, decreased the [Ca2+]c response induced by BDNF. Pretreatment of cells with K252a for 30 min, which by itself did not affect basal mEPSC activity, completely blocked the effect of BDNF on mEPSC frequency (see Fig. 5). The BDNF-induced [Ca2+]c response is dependent on extracellular Ca2+, as is the synaptic enhancement (see Figs. 5, 7). Because the effects of BDNF on synaptic transmission and on [Ca2+]c are similar, the increase in [Ca2+]c induced by BDNF may account for the increase in synaptic transmission. However, other mechanisms, such as neurotrophin-induced phosphorylation of synaptic related proteins (Jovanovic et al., 1996), may be involved.
Because BDNF increased the frequency, but not the amplitude, of mEPSCs and because the currents induced by exogenous glutamate application were not affected by BDNF, our results strongly suggest that BDNF ultimately exerts its effects mainly on presynaptic function under our conditions. Levine et al. (1995) also have reported that BDNF enhances synaptic transmission in cultured E18 hippocampal neurons. On the basis of the observation that postsynaptic injection of either K252a or the general serine/threonine phosphatase inhibitor okadaic acid reduced or potentiated the BDNF-induced potentiation, respectively, Levine et al. (1995) concluded that the actions of BDNF were mainly postsynaptic. However, we suggest that the membrane-permeant agents used in their experiments could have acted by diffusing to presynaptic compartments adjacent to the injected postsynaptic cells. There were also differences in culture conditions: namely, Levine et al. (1995) used cultures that contain virtually pure neurons, whereas substantial numbers of glial cells also were present in our cultures. Although our experiments indicate that the ultimate site of enhancement is the presynaptic terminal, they do not address directly the site of initial signal transduction. It is therefore possible that the BDNF signal initially was transduced postsynaptically, resulting in the generation of a retrograde signal that ultimately enhanced presynaptic function. Indeed, experiments in hippocampal slices have suggested both pre- and postsynaptic actions of BDNF (Kang and Schuman, 1995, 1996). Future experiments that block BDNF signal transduction selectively in pre- or postsynaptic neurons will address this question directly.
Regardless of the synaptic locus of the actions of BDNF, our studies clearly document fast (<5 min), reversible, and direct actions of BDNF on synaptic transmission. These results indicate that one immediate consequence of endogenous BDNF release in the hippocampus will be the enhancement of synaptic strength at glutamatergic synapses near the release site. It will be of interest to determine whether electrical or pharmacological manipulations can convert the enhancement studied here to a less reversible form. Moreover, the accessibility of the cell culture system lends itself particularly well to studies of BDNF signaling mechanisms and the coordination of the local actions of BDNF at the synapse as well as potential influences on translation and transcription (Segal and Greenberg, 1996).
Footnotes
This work was supported by the Silvio Conte Center for Neuroscience Research at Caltech sponsored by the National Institute of Mental Health and by Amgen, Thousand Oaks, CA.
Correspondence should be addressed to Dr. Erin M. Schuman, Division of Biology 216-76, California Institute of Technology, Pasadena, CA 91125.
REFERENCES
- 1.Akaneya Y, Tsumoto T, Kinoshita S, Hatanaka H. Brain-derived neurotrophic factor enhances long-term potentiation in rat visual cortex. J Neurosci. 1997;17:6707–6716. doi: 10.1523/JNEUROSCI.17-17-06707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arancio O, Kandel ER, Hawkins RD. Activity-dependent long-term enhancement of transmitter release by presynaptic 3′,5′-cyclic GMP in cultured hippocampal neurons. Nature. 1995;376:74–80. doi: 10.1038/376074a0. [DOI] [PubMed] [Google Scholar]
- 3.Berg MM, Sternberg DW, Parada LF, Chao MV. K252a inhibits nerve growth factor-induced trk proto-oncogene tyrosine phosphorylation and kinase activity. J Biol Chem. 1992;267:13–16. [PubMed] [Google Scholar]
- 4.Berninger B, Poo M-M. Fast actions of neurotrophic factors. Curr Opin Neurobiol. 1996;6:324–330. doi: 10.1016/s0959-4388(96)80115-2. [DOI] [PubMed] [Google Scholar]
- 5.Berninger B, Garcia DE, Inagaki N, Hahnel C, Lindholm D. BDNF and NT-3 induced intracellular Ca2+ elevation in hippocampal neurons. NeuroReport. 1993;4:1303–1306. doi: 10.1097/00001756-199309150-00004. [DOI] [PubMed] [Google Scholar]
- 6.Bijak M, Jarolimek W, Misgeld U. Effects of antagonists on quisqualate and nicotinic receptor-mediated currents of midbrain neurons in culture. Br J Pharmacol. 1991;102:699–705. doi: 10.1111/j.1476-5381.1991.tb12236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci. 1995;18:223–253. doi: 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- 8.Carmignoto G, Pizzorusso T, Tia S, Vicini S. Brain-derived neurotrophic factor and nerve growth factor potentiate excitatory synaptic transmission in the rat visual cortex. J Physiol (Lond) 1997;498:153–164. doi: 10.1113/jphysiol.1997.sp021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fryer RH, Kaplan DR, Feinstein SC, Radeke MJ, Grayson DR, Kromer LF. Developmental and mature expression of full-length and truncated trkB receptors in the rat forebrain. J Comp Neurol. 1996;374:21–40. doi: 10.1002/(SICI)1096-9861(19961007)374:1<21::AID-CNE2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 10.Garaschuk O, Yaari Y, Konnerth A. Release and sequestration of calcium by ryanodine-sensitive stores in the rat hippocampal neurons. J Physiol (Lond) 1997;502:13–30. doi: 10.1111/j.1469-7793.1997.013bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grudt TJ, Usowicz MM, Henderson G. Ca2+ entry following store depletion in SH-SY5Y neuroblastoma cells. Brain Res Mol Brain Res. 1996;36:93–100. doi: 10.1016/0169-328x(95)00248-q. [DOI] [PubMed] [Google Scholar]
- 13.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 14.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patch. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 15.Holm N, Christophersen P, Olesen SP, Gammeltoft S. Activation of calcium-dependent potassium channels in rat brain neurons by neurotrophin-3 and nerve growth factor. Proc Natl Acad Sci USA. 1997;94:1002–1006. doi: 10.1073/pnas.94.3.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol (Lond) 1993;65:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jovanovic JN, Benfenati F, Siow YL, Sihra TS, Sanghera JS, Pelech SL, Greengard P, Czernik AJ. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. Proc Natl Acad Sci USA. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;264:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 19.Kang H, Schuman EM. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- 20.Kim HG, Wang T, Olafsson P, Lu B. Neurotrophin 3 potentiates neuronal activity and inhibits γ-aminobutyratergic synaptic transmission in cortical neurons. Proc Natl Acad Sci USA. 1994;91:12341–12345. doi: 10.1073/pnas.91.25.12341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lebmann V, Gottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurons. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- 22.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levine ES, Crozier RA, Black IB, Plummer MR. Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing N-methyl-d-aspartic acid receptor activity. Proc Natl Acad Sci USA. 1998;95:10235–10239. doi: 10.1073/pnas.95.17.10235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li YX, Xu Y, Ju D, Lester HA, Davidson N, Schuman EM. Expression of a dominant negative TrkB receptor, T1, reveals a requirement for presynaptic signaling in BDNF-induced synaptic potentiation in cultured hippocampal neurons. Proc Natl Acad Sci USA. 1998;95:10884–10889. doi: 10.1073/pnas.95.18.10884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- 26.Lindsay RM, Wiegand SJ, Altar CA, Sistefano PS. Neurotrophic factors: from molecule to man. Trends Neurosci. 1994;17:182–190. doi: 10.1016/0166-2236(94)90099-x. [DOI] [PubMed] [Google Scholar]
- 27.Lohof AM, Ip NY, Poo M-M. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 28.Mathes C, Thompson SH. Calcium current activated by muscarinic receptors and thapsigargin in neuronal cells. J Gen Physiol. 1994;104:107–121. doi: 10.1085/jgp.104.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Messaoudi E, Bardsen K, Srebro B, Bramham CR. Acute intrahippocampal infusion of brain-derived neurotrophic factor induces lasting potentiation of synaptic transmission in the rat dentate gyrus. J Neurophysiol. 1998;79:496–499. doi: 10.1152/jn.1998.79.1.496. [DOI] [PubMed] [Google Scholar]
- 30.Mikoshiba K. The insP3 receptor and intracellular Ca2+ signaling. Curr Opin Neurobiol. 1997;7:339–345. doi: 10.1016/s0959-4388(97)80061-x. [DOI] [PubMed] [Google Scholar]
- 31.Miledi R. Transmitter release induced by injection of calcium ions into nerve terminals. Proc R Soc Lond [Biol] 1973;183:421–425. doi: 10.1098/rspb.1973.0026. [DOI] [PubMed] [Google Scholar]
- 32.Patneau DK, Mayer ML. Structure–activity relationships for amino acid transmitter candidates acting at NMDA and quisqualate receptors. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Putney JW. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 34.Reyes M, Stanton PK. Induction of hippocampal long-term depression requires release of Ca2+ from separate presynaptic and postsynaptic intracellular stores. J Neurosci. 1996;16:5951–5960. doi: 10.1523/JNEUROSCI.16-19-05951.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- 36.Rohrbacher J, Jarolimek W, Lewen A, Misgeld U. GABAB receptor-mediated inhibition of spontaneous inhibitory synaptic currents in rat midbrain culture. J Physiol (Lond) 1997;500:739–749. doi: 10.1113/jphysiol.1997.sp022055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rutherford LC, Dewan A, Lauer HM, Turrigiano GG. Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci. 1997;17:4527–4535. doi: 10.1523/JNEUROSCI.17-12-04527.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scharfman HE. Hyperexcitability in combined entorhinal/hippocampal slices of adult rat after exposure to brain-derived neurotrophic factor. J Neurophysiol. 1997;78:1082–1095. doi: 10.1152/jn.1997.78.2.1082. [DOI] [PubMed] [Google Scholar]
- 39.Schuman E. Growth factors sculpt the synapse. Science. 1997;275:1277–1278. doi: 10.1126/science.275.5304.1277. [DOI] [PubMed] [Google Scholar]
- 40.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 41.Sharp AH, Dawson TM, Ross CA, Fotuhi M, Mourey RJ, Snyder SH. Inositol 1,4,5-trisphosphate receptors: immunohistochemical localization to discrete areas of rat central nervous system. Neuroscience. 1993;53:927–942. doi: 10.1016/0306-4522(93)90478-x. [DOI] [PubMed] [Google Scholar]
- 42.Stoop R, Poo M-M. Synaptic modulation by neurotrophic factors: differential and synergistic effects of brain-derived neurotrophic factor and ciliary neurotrophic factor. J Neurosci. 1996;6:3256–3264. doi: 10.1523/JNEUROSCI.16-10-03256.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thoenen H. The changing scene of neurotrophic factors. Trends Neurosci. 1991;14:165–170. doi: 10.1016/0166-2236(91)90097-e. [DOI] [PubMed] [Google Scholar]
- 46.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 47.Tsien RY, Poenie M. Fluorescence ratio imaging: a new window into intracellular ionic signaling. Trends Biochem Sci. 1986;11:450–455. [Google Scholar]
- 48.Tsien RW, Tsien RY. Calcium channel, stores, and oscillations. Annu Rev Cell Biol. 1990;6:715–760. doi: 10.1146/annurev.cb.06.110190.003435. [DOI] [PubMed] [Google Scholar]
- 49.Yamada M, Mizuguchi M, Rhee SG, Kim SU. Developmental changes of three phosphoinositide-specific phospholipase C isozymes in the rat nervous system. Dev Brain Res. 1991;59:7–16. doi: 10.1016/0165-3806(91)90023-c. [DOI] [PubMed] [Google Scholar]
- 50.Zirrgiebel U, Ohga Y, Carter B, Berninger B, Inagaki N, Thoenen H, Lindholm D. Characterization of TrkB receptor-mediated signaling pathways in rat cerebellar granule neurons: involvement of protein kinase C in neuronal survival. J Neurochem. 1995;65:2241–2250. doi: 10.1046/j.1471-4159.1995.65052241.x. [DOI] [PubMed] [Google Scholar]