

Abstract
The transforming growth factor-β (TGF-β) family consists of three isoforms and is part of a larger family of cytokines regulating differentiation, development, and tissue repair. Previous work from our laboratory has shown that TGF-β1 can increase amyloid-β protein (Aβ) immunoreactive (Aβir) plaque-like deposits in rat brain. The aim of the current study was to evaluate all three isoforms of TGF-β for their ability to affect the deposition and neurotoxicity of Aβ in an organotypic, hippocampal slice culture model of Aβ deposition. Slice cultures were treated with Aβ either with or without one of the TGF-β isoforms. All three isoforms can increase Aβ accumulation (over Aβ treatment alone) within the slice culture, as determined by ELISA. However, there are striking differences in the pattern of Aβir among the three isoforms of TGF-β. Isoforms 1 and 3 produced a cellular pattern of Aβ staining that colocalizes with GS lectin staining (microglia). TGF-β2 produces dramatic Aβ staining of pyramidal neurons in layers CA1–CA2. In addition to cellular Aβ staining, plaque-like deposits are increased by all of the TGF-βs. Although no gross toxicity was observed, morphological neurodegenerative changes were seen in the CA1 region when the slices were treated with Aβ plus TGF-β2. Our results demonstrate important functional differences among the TGF-β isoforms in their ability to alter the cellular distribution and degradation of Aβ. These changes may be relevant to the pathology of Alzheimer’s disease (AD).
Keywords: Alzheimer’s disease, aging, trauma, injury, growth factor, amyloid
Transforming growth factors-β (TGF-βs) are a family of cytokines with three closely related isoforms, TGF-β1–TGF-β3. Within the CNS these isoforms are expressed within neurons, astrocytes, and microglia (Constam et al., 1992; Krieglstein et al., 1995). Some of the functions proposed for the TGF-βs include control of inflammation and immune responses, cell adhesion, proliferation and differentiation, extracellular matrix formation, and wound healing. TGF-βs are expressed in a number of CNS insults, including traumatic injury (Klempt et al., 1992; Logan et al., 1992; Roberts and Sporn, 1993), hypoxic injury (Lindholm et al., 1992), and neurodegenerative diseases such as Parkinson’s (Vawter et al., 1996) and Alzheimer’s (Chao et al., 1994; Flanders et al., 1995; Lippa et al., 1995; Peress and Perillo, 1995; Vawter et al., 1996).
The roles of the TGF-βs in the CNS and the differences among the isoforms are not well understood. TGF-βs may be a key factor regulating inflammatory and tissue-specific wound responses. Although TGF-β generally is known as an anti-inflammatory cytokine, it exerts proinflammatory effects under certain pathological conditions (Yao et al., 1990; Feldmann et al., 1996; Sharma et al., 1996). Transgenic mice that overexpress TGF-β1 are susceptible to the immune-mediated disease experimental autoimmune encephalomyelitis (Wyss-Coray et al., 1997). Other documented actions of TGF-βs include microglial chemotaxis (Yao et al., 1990), induction of apoptosis (Langer et al., 1996; Xiao et al., 1997) and the glial limitans (Logan et al., 1994), impairment of astrocyte function (Chao et al., 1992), increased amyloid precursor protein (APP) expression (Gray and Patel, 1993; Monning et al., 1994), enhanced amyloid-β (Aβ) deposition in an in vivo model of Alzheimer’s disease (AD) (Frautschy et al., 1996), and exacerbation of neurotoxicity after long-term excitotoxicity (Prehn and Krieglstein, 1994; Prehn and Miller, 1996). TGF-β has been reported to be neurotrophic (Chalazonitis et al., 1992; Poulsen et al., 1994) and neuroprotective against Aβ toxicity (Prehn et al., 1996; Ren and Flanders, 1996) and short-term excitotoxicity (Prehn and Krieglstein, 1994; Prehn and Miller, 1996).
The expression of TGF-βs was found to be altered in AD (Flanders et al., 1995; Vawter et al., 1996), and increases in TGF-β have been found in AD CSF and serum (Chao et al., 1994). Detailed studies of TGF-β isoforms in AD brain have revealed increased TGF-β1 labeling of senile plaques (van der Wal et al., 1993) and TGF-β2 labeling of neurofibrillary tangle-bearing neurons, astrocytes (Flanders et al., 1995), and plaque neurites (Peress and Perillo, 1995). TGF-β localization in microglia surrounding senile plaques (van der Wal et al., 1993), and its synthesis in microglia after brain injury (Morgan et al., 1993) suggest that inflammation may play a key role in plaque formation. Microglia, the immune cells in the CNS, are associated with senile plaques and are speculated to participate directly in plaque formation (Mackenzie et al., 1995). Microglia may be the source of increased Aβ production or may respond to Aβ by becoming activated and increasing the production of toxic cytokines, reactive oxygen species, and nitric oxide (El Khoury et al., 1996).
The present study was performed to analyze the inflammation and neurodegeneration of TGF-β-mediated deposition of Aβ in organotypic slice cultures.
MATERIALS AND METHODS
Slice cultures. Hippocampal slice cultures were prepared according to the method of Stoppini et al. (1991) with some modification. Briefly, slice cultures were prepared from 6- to 7-d-old ICR mouse pups (Harlan Laboratories, San Diego, CA). Slices were cut at 400 μm on a Stoelting tissue chopper and transferred to Costar (Cambridge, MA) membrane inserts (0.4 μm). Initially, slice culture media consisted of Minimal Essential Medium plus HEPES (50%; Life Technologies, Grand Island, NY), heat-inactivated horse serum (25%; Sigma, St. Louis, MO), and HBSS (25%; Sigma) containing a total of 6.5 mg/ml glucose and penicillin–streptomycin (50 U/ml and 0.05 mg/ml, respectively). After the first 4 d in culture, the slice medium was replaced gradually with a serum-free medium prepared by replacing the horse serum with the supplement TCM (final concentration 2%; ICN Pharmaceuticals, Costa Mesa, CA). The exchange of serum-containing medium with serum-free medium was as follows: (1) 75% serum medium/25% serum-free medium on day 4 in vitro, (2) 50% serum medium/50% serum-free medium on day 6 in vitro, and (3) 100% serum-free medium on day 7.
Experimental treatment. On day 7 in vitro, treatment of the slices was started. All reagents were added to serum-free medium and equilibrated in a 5% CO2 incubator at 37°C before their addition to the slices. All treatment solutions were prepared from stock solutions of TGF-β (10 ng/μl in ddH2O), Aβ40 (1 mg/ml in ddH2O; US Peptide, Fullerton, CA), or Aβ42 (1 mg/ml in 20% DMSO; C. Glabe, University of California Irvine, Irvine, CA). Final concentrations of the reagents were 10 ng/ml (TGF-βs), 10 μg/ml (Aβ40), and 1 μg/ml (Aβ42). Controls were prepared containing the appropriate vehicles. Medium containing the treatments was allowed to remain with the slices for 4 d (days 1–4 of treatment), at which time the slice medium was replaced with fresh medium (no new treatment added); the medium subsequently was changed three times per week (over days 5–12) until the conclusion of the experiment (day 12). For some experiments (early time course) the slices were fixed for immunohistochemistry at days 2–3 of treatment. Data from these early time course experiments are discussed in Results. All figures are from data collected on day 12 slices unless otherwise stated in the figure.
Histology and image analysis. At the conclusion of the experiment the slices were submersion-fixed in 4% paraformaldehyde for 1 hr, followed by three rinses in Tris-buffered saline (TBS). Slices then were cryopreserved in increasing concentrations of sucrose (10, 15, and 20%), sectioned at 10 μm, and mounted onto gelatin-coated slides. Alternatively, slices were whole-mounted onto poly-l-lysine-coated slides.
Aβ antibodies used in the present study were 10G4 monoclonal to the amino acid 5–13 region of native human Aβ1–40 (Yang et al., 1994); anti-34–40 specific for AβX-40 (Mak et al., 1994); and affinity-purified rabbit anti-42 absorbed on Aβ43, which labels peptides ending at 42, but not at 43 (Saido et al., 1995). For Aβ immunohistochemistry the slices were pretreated with 70% trichloroacetic acid (6 min) and rinsed with TBS. Endogenous peroxidase activity was suppressed by a peroxidase suppressor buffer (Vector Labs, Burlingame, CA) for 30 min at room temperature (RT), and nonspecific binding sites were blocked with Superblock blocking buffer (Pierce, Rockford, IL) for 1 hr at RT. The 10G4 antibody was applied (overnight at 4°C) to slices at a 1:800 dilution in TBS containing 0.1% Tween 20, 3% bovine serum albumin, and 8 mm sodium azide. Then the slices were rinsed and incubated with a biotinylated anti-mouse secondary antibody (1:500 in TBS plus Tween 20; 1 hr at RT). The slices were rinsed and incubated with ABC solution (Elite ABC kit, Vector Labs) for 45 min at RT. Diaminobenzidine (DAB) metal-enhanced chromagen (Pierce) was used to reveal the 10G4 antibody binding. The specificity of Aβ staining was verified by preincubating 10G4 antibody in the presence of Aβ1–40 peptide before the immunostaining. Colocalization of antigens was determined by additional staining [glial fibrillary acidic protein (GFAP) or biotinylated Griffonia Simplicifolia lectin I (GS lectin) binding for microglial analysis; Sigma] with the use of another enzyme system, alkaline phosphatase, and Vector Blue chromagen (Vector Labs).
Preabsorption of the 10G4 antibody was performed by adding 1 μl of antibody to 50 μl of 3% BSA in TBS containing 30 μg of Aβ40 peptide. The solution was incubated overnight at 4°C, brought up to 800 μl with 3% BSA in TBS, and centrifuged at 14 × 103 g for 30 min at 4°C. The resulting supernatant was used for immunocytochemistry.
All histological and immunohistochemical images were acquired from an Olympus Vanox-T (AHBT) microscope with an Optronix Engineering LX-450A CCD video camera system. Then the video signal was routed into a Power Center 120 Macintosh-compatible microcomputer via a Scion Corporation AG-5 averaging frame grabber. Once digitized, the images were analyzed with National Institutes of Health–Image public domain software (developed at National Institutes of Health and available on the internet at http://rsb.info.nih.gov/nih-image/). Custom Pascal macro subroutines were written for Aβ immunoreactive protein (Aβir) to calculate plaque number/mm2 and average plaque diameter. Throughout the image analysis process the sections for all treatments were done with identical microscope light, condenser settings, and density slice threshold settings, which differentiate between stained and unstained regions. Double-blind image analysis was done with respect to treatment.
Terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL). The “Apoptag” kit (Oncor, Gaithersburg, MD) was used for in situ end labeling of DNA fragments as per the manufacturer’s instructions, with 10 min of proteinase K pretreatment (37°C) and DAB (Pierce) as the peroxidase substrate.
Hematoxylin and eosin Y staining. Slices were stained with Harris’s modified hematoxylin (two quick dips or ∼10 sec), followed by rinses with tap water. This was followed by 10 dips in acid alcohol, one water rinse, 20 dips in ammonia water, and one water rinse. Slices then were submerged in eosin Y (two quick dips), followed by three changes of 95% ethanol (10 dips each), three changes of 100% ethanol (10 dips each), and 10 dips in Hemo-De. Slices were coverslipped, and eosin Y fluorescence was visualized with a Nikon Microphot-FX fluorescence microscope and a fluorescein emission (520 nm) filter set or with light microscopy.
Sandwich ELISA for Aβ. Hippocampal slices were solubilized by being vortexed in 30 μl of 70% formic acid. After centrifugation, the formic acid supernatant was diluted 1:20 in 0.25 m Tris base, pH 8.0, containing 30% acetonitrile and neutralized with 5N NaOH. The neutralized sample was diluted 1:2 with ELISA Capture (EC) buffer (TBS, pH 7.4, containing 0.1 mm EDTA, 1% BSA, and 0.05% CHAPS).
The capture antibody (monoclonal 4G8) was loaded at a concentration of 3 μg/ml in 100 μl of 0.1 m carbonate buffer, pH 9.6, onto a 96-well plate (Nunc Maxisorp, Naperville, IL). After incubation at 4°C for 16 hr, the plate was washed three times with TBS. Blocking was completed with 2% BSA in TBS at 25°C for 3 hr. The neutralized samples, diluted with EC buffer, were loaded onto the wells for antigen capture. A standard curve in the range of 0.02–5 ng of Aβ also was prepared and loaded onto the ELISA plate. The detector antibody (biotinylated monoclonal 10G4; 1 mg/ml) was loaded simultaneously in 50 μl of EC buffer at a final dilution of 1:1500. After an overnight incubation at 4°C the plate was washed three times with TBS, followed by the addition of 100 μl of streptavidin–alkaline phosphatase (1:4000 dilution with 1% BSA). After a 2 hr incubation at 25°C the plate was washed six times with ddH2O, and 100 μl of Attophos substrate (JBL, San Luis Opisbo, CA) was added. Fluorescence of the Attophos product was monitored at an excitation wavelength of 450 nm and an emission wavelength of 580 nm in a Cytofluor II plate reader.
Tumor necrosis factor-α ELISA. For the quantitative determination of mouse tumor necrosis factor-α (TNF-α) in slice culture media, the Quantikine M, Murine TNF-α ELISA kit (R & D Systems, Minneapolis, MN) was used per the manufacturer’s instructions.
Lactate dehydrogenase. Lactate dehydrogenase (LDH) in the culture media was measured with the CytoTox 96 nonradioactive cytotoxicity assay (Promega, Madison, WI) per the manufacturer’s instructions.
TGF-β neutralization. Pan-specific anti-TGF-β antibody (AB100NA; R & D Systems), anti-TGF-β2 antibody (AB112NA; R & D Systems), and anti-TGF-β1 antibody (AB101NA; R & D Systems) were used on slice cultures at concentrations of 100 μg/ml (95% neutralization), 10 μg/ml (99% neutralization), and 80 μg/ml (99% neutralization), respectively. The appropriate nonimmune serum was used as a control. Neutralizing antibodies were added to slice culture media 1 d before experimental treatment with Aβ/TGF-βs. Neutralizing antibodies remained in the culture media throughout the entire treatment and survival interval.
RESULTS
We have developed a hippocampal slice culture model to study the formation and progression of Aβir plaque-like deposits. This slice culture model has several advantages. First, slice cultures contain multiple cell types and maintain appropriate cell–cell contacts while in culture. This is crucial to studying the complex cellular interactions involved in Aβ plaque formation, progression, and the resulting neurotoxicity. Second, slice cultures are easy to manipulate, and Aβir plaque-like deposits can be induced within the slice cultures in a matter of days, allowing us to study the initial events involved in plaque formation.
Immunolocalization of Aβ within the hippocampal slice
Unlike other studies we have not encountered a problem with the accessibility of treatment reagents to our slice cultures. After 1 weekin vitro we did not find that our slice cultures were covered with an astrocytic layer that limited the access of Aβ peptides to the slice culture. We have avoided the problems of having to inject Aβ peptides directly into the slice (Malouf, 1992) or having to submerge the slices in very high concentrations of Aβ peptides (Allen et al., 1995; Bruce et al., 1996), which could lead to ischemic changes within the slice culture. Aβ was used in the culture media and allowed to diffuse into or be taken up by the slices. Furthermore, we chose to begin our treatments at 7 d in vitro to avoid the robust inflammatory response that occurs immediately after the hippocampi are sliced and placed into culture. Using monoclonal antibody 10G4 (Aβ 5–13), we observed diffuse plaque-like deposit staining in slice cultures treated with Aβ alone (slices fixed at day 12; see Fig. 2A). Preabsorption of the 10G4 antibody with Aβ40 peptide eliminated this staining in the slice cultures (data not shown). Most of these deposits occurred in the stratum radiatum and stratum moleculare regions (Imaging Area 2; see Fig.1 for description of imaging areas). Immunohistochemical analysis of slices fixed at earlier time points (days 2–3 of treatment; data not shown) showed Aβir that was mostly cellular (microglial). By day 7 the slices treated with Aβ began to show diffuse plaque-like deposits, and by day 12 these deposits were well formed. On day 7 the Aβ deposits showed reactive glia (GFAP and GS lectin-positive) associated with these deposits.
Fig. 2.

Demonstration of an image analysis of Aβir deposits in a hippocampal slice. A, 10G4 staining of deposits in a slice treated with Aβ. B, The same region after processing with NIH Image software to analyze Aβ deposits. The deposits are outlined by the program andnumbered for quantitation. Magnification is 10×.
Fig. 1.

Schematic of a hippocampal slice depicting the regions of the hippocampus analyzed by our image analysis system.Imaging Area 1 is composed of the CA1 pyramidal layer and the stratum oriens. Imaging Area 2 is composed of the stratum radiatum and stratum moleculare.
The most dramatic change occurring with the addition of TGF-β isoforms to Aβ treatment was the switch to a cellular distribution of Aβir (Fig. 3). Aβir was increased in small round cells (identified by GS lectin staining as microglia) in area 2 by TGF-β1 and areas 1 and 2 by TGF-β3. Aβ plus TGF-β2 yielded the most striking cellular distribution of Aβir. Heavy Aβir was localized to cells in the pyramidal layer of CA1 and CA2. These cells were identified morphologically as neurons by their pyramidal neuron characteristics. We do not, at this time, know if the Aβir is intra- or extracellular. Cellular Aβir in Aβ plus TGF-β1-treated slices was found in GS lectin-positive microglia (Fig.4B) (Aβ plus TGF-β3 slices were similar; data not shown). Microglia in Aβ plus TGF-β2-treated slices were not Aβir. Instead, large microglia were always found invading and surrounding the Aβir pyramidal neurons of CA1 (Fig. 4C).
Fig. 3.
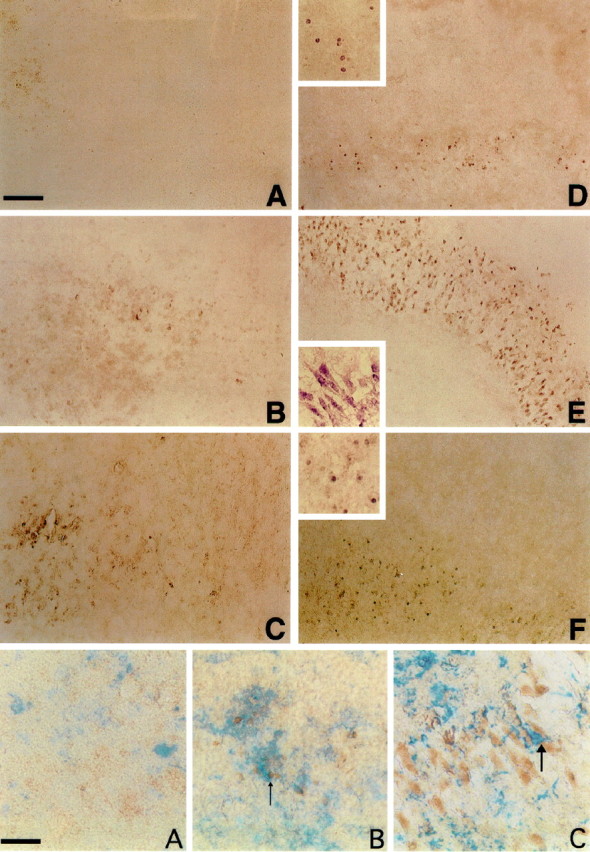
Aβir (detected by using the 10G4 antibody;brown) in the hippocampal slice culture.A, Control. B, Aβ. C, Rev Aβ. D, TGF-β1 plus Aβ. E, TGF-β2 plus Aβ. F, TGF-β3 plus Aβ. Note the pyramidal neuron staining when TGF-β2 is added with Aβ. TGF-β1 and TGF-β3 give a predominantly microglial cellular staining pattern. Large photographs are 10× magnification. Insets are 20× magnification and are taken from a region in the larger photographs. Scale bar, 40 μm.
Fig. 4.
Double labeling of slices for GS lectin and Aβ. Microglia are blue, and Aβ (10G4) isbrown. A, Control slice revealing no Aβir. B, Aβ plus TGF-β1-treated slice demonstrating double labeling of microglia for Aβ and GS lectin (arrows). C, Aβ plus TGF-β2-treated slice demonstrating intense labeling of neuronal structures with large invading microglia (arrow). Scale bar, 20 μm.
Aβ-, Aβ plus TGF-β1-, and Aβ plus TGF-β2-treated slices also were stained with end-specific antibodies to Aβ40 and Aβ42. Diffuse extracellular plaque-like staining occurred with the Aβ40 antibody, and cellular staining occurred with the Aβ42 antibody, whereas 10G4 stained both the diffuse plaque-like Aβ as well as cellular Aβ. Figure 5 shows the Aβ40 (Fig.5A) and Aβ42 (Fig. 5B) staining of Aβ plus TGF-β2-treated slices.
Fig. 5.

Aβ staining that uses end-specific antibodies for Aβ40 and Aβ42. A, Aβ42 staining in the CA1 pyramidal region of the slice culture after treatment with Aβ plus TGF-β2. B, Aβ40 staining in the stratum radiatum region of a slice treated with Aβ plus TGF-β1. Most of the Aβ40 is diffuse with some apparent microglial staining, whereas the Aβ42 staining is predominately cellular. Scale bar, 40 μm.
Using our imaging system and the National Institutes of Health imaging software, we quantitated deposit numbers in two areas of the hippocampal slices (see Fig. 2B, which demonstrates the use of the imaging system). Treatment of slice cultures with Aβ plus TGF-β1, TGF-β2, or TGF-β3 altered the distribution of deposits within the slice culture (Fig.6). Aβ plus TGF-β1 increased deposit numbers mostly in area 2, with some increase in area 1 (pyramidal cell layer and stratum oriens of CA1). Aβ plus TGF-β3 shifted deposit staining from area 2 to area 1 similarly to Aβ plus TGF-β1, but it did not increase the number of deposits in area 2 as TGF-β1 did. Aβ plus TGF-β2 shifted deposit staining from area 2 to area 1. TGF-βs 2 and 3 decreased the total number of deposits, whereas TGF-β1 increased the total number of plaque-like deposits. In addition, all three TGF-β isoforms showed a trend toward decreasing the diameter and increasing the 10G4 staining density of deposits occurring in any region of the slice culture (data not shown).
Fig. 6.

Number of Aβir deposits (detected by using the 10G4 antibody) in areas 1 and 2 of hippocampal slice cultures. In addition to increasing cellular Aβir, TGF-βs also increase the plaque-like deposition of Aβ. TGF-β1 strongly increases Aβ deposition into the stratum radiatum region (Area 2), whereas TGF-β2 increases deposition in the pyramidal and stratum oriens region (Area 1). *p < 0.05 versus control (CON); **p < 0.05 versus Aβ and TGF-β1; n = 4–6 from three independent experiments.
Quantitation of Aβ within the slices
The treatment of slices with Aβ, alone or in combination with TGF-β, increased Aβ within the slice as quantitated by an Aβ ELISA developed in our laboratory (Fig.7). Treatment with Aβ alone resulted in 97 ± 43 ng of Aβ per milligram of protein (p < 0.05 vs untreated control). The addition of TGF-β isoforms with Aβ increased the detectable Aβ approximately twofold to threefold (Aβ plus TGF-β1, TGF-β2, or TGF-β3 was significantly different from Aβ alone; p< 0.05).
Fig. 7.

Total Aβ quantitated via ELISA assay. All TGF-β isoforms increase Aβ within the hippocampal slice. *p < 0.05 versus Aβ plus TGF-β1, Aβ plus TGF-β2, and Aβ plus TGF-β3. Results are the average of triplicate measurements from six slices in two independent experiments.
TNF-α ELISA
TGF-βs are known to suppress TNF-α levels. We monitored the activity of TGF-β added to the slice culture. Using an ELISA assay specific for murine TNF-α, we quantitated TNF-α levels in slice culture media (data not shown). We assayed two time points during the treatment period, days 1–4 and days 8–12. During days 1–4 the TNF-α levels were increased by treatment of the slices with Aβ alone (p < 0.05 vs control). TGF-βs in combination with Aβ suppressed TNF-α levels (Aβ plus TGF-β1, TGF-β2, or TGF-β3 vs Aβ alone; p < 0.05). Although the medium was changed at day 4 of the treatment period and no additional treatment was added to the slices, we wanted to know if the changes in TNF-α levels induced by Aβ and/or TGF-β treatment would persist to the end of the experimental period (day 12). Analysis of the media from days 8–12 showed that those TNF-α levels had returned to control levels.
Degenerative changes
No detectable release of LDH into the slice culture media was found in any treatment group at any time point examined (days 1–4 or 8–12), indicating a lack of gross toxicity within the slice cultures (data not shown). To examine more subtle degenerative changes, we used the hematoxylin and eosin Y staining procedure. This procedure uses eosin Y, a tetrabrominated derivative of fluorescein. Eosin Y staining has been shown to be a good histochemical marker for damaged neurons (Stinchcombe et al., 1995; Chen and Liu, 1996). Eosin Y staining can be visualized via fluorescence or light microscopy (damaged cells are dark pink/red). Figure 8 demonstrates some of the degenerative changes occurring in cells of Aβ plus TGF-β2-treated slices. Cellular eosin Y staining occurred in the CA1 of slices treated with Aβ plus TGF-β2 (Fig. 8A). Double staining of these same slices for eosin Y and 10G4 revealed double labeling of cells only in the Aβ plus TGF-β2-treated slices. Often, the double-labeled cells were outlined heavily in 10G4 immunoreactivity (“halo”; Fig. 8B). No eosin Y staining was seen in any control, Aβ alone, Aβ plus TGF-β3, reverse Aβ40, or TGF-β1-, TGF-β2-, or TGF-β3-only treated slices. Some eosin Y staining was found in Aβ plus TGF-β1-treated slices, although this appeared to stain plaque-like deposits as opposed to the cellular staining seen in the Aβ plus TGF-β2-treated slices. Note the very dark eosin Y labeling of cells in the CA1 layer (Fig. 8,arrows), some of which have a pyramidal morphology.
Fig. 8.

Neurodegenerative changes in hippocampal slice cultures treated with Aβ plus TGF-β2. A, Hematoxylin and eosin Y-stained slice. B, Same field stained with 10G4 to identify Aβ. Many of the same cells that are eosin Y-positive are also Aβir (arrows). Scale bar, 20 μm.
The slices also were stained for TUNEL to examine DNA damage. There was no significant difference among any of the treatment groups.
TGF-β neutralization
All three neutralizing antibodies were toxic to the slices irrespective of Aβ/TGF-β treatment. Pan anti-TGF-β antibody was the most toxic, followed by anti-TGF-β1 antibody and anti-TGF-β2 antibody. No slices survived the Pan TGF-β treatment, which caused the slices to lose their morphology and dissociate. Treatment with any of the three antibodies caused an increase in the number of microglia and astrocytes displaying a reactive phenotype.
DISCUSSION
This is the first study to demonstrate that TGF-β isoforms not only increase Aβ but also influence the cellular and extracellular deposition of Aβ differentially within hippocampal slice cultures. This study also confirms the in vivo finding of increased Aβ deposition in the rat brain by the TGF-β1 isoform (Frautschy et al., 1996). The role of Aβ in AD and the toxicity of the Aβ protein remain controversial. There are abundant studies citing both toxic and trophic effects of Aβ. This is to be expected, given the diversity of culture conditions under which Aβ has been studied and considering that AD is a disease that takes many years to develop, a factor difficult to replicate in vitro. It is advantageous to find a culture model that will allow for conditions most similar to thein vivo situation and that also will allow for a longer time course for the development of neurotoxicity. The results of this study demonstrate that Aβ (no exogenous TGF-β) added to the culture media can result in increased Aβir within the hippocampal slice culture in as few as 2 d. Most of this early Aβir is cellular (microglial), supporting a role for microglia in the formation of Aβ deposits. This cellular staining progresses over another week into what appear to be diffuse (extracellular) deposits with increased microglial staining surrounding the deposits. The addition of any of the TGF-β isoforms along with Aβ increased the amount of Aβ within the slice culture and the number of plaque-like deposits and prolonged the time course of cellular Aβ staining.
The role of microglia in plaque/deposit formation is unclear. There are many descriptive papers demonstrating increased microglia surrounding amyloid plaques as a key feature of AD. Aβ can be toxic to microglia (Korotzer et al., 1993) and can lead to increased production of reactive nitrogen and oxygen intermediates and neuron toxicity (Meda et al., 1995; El Khoury et al., 1996). Microglia also appear to have a limited ability to degrade phagocytosed Aβ (Frackowiak et al., 1992;Ard et al., 1996), which may result in the death of the microglia and the extracellular deposition of aggregated Aβ. In vivostudies in our laboratory have shown that phagocytic cells can internalize exogenous amyloid and attempt to clear it from the brain (Frautschy et al., 1992, 1994). In our Aβ-only-treated slice cultures, Aβ staining progressed from intracellular in very small round microglia to diffuse deposits surrounded by microglia that are large and round with short thick processes. In addition, Aβ plus either TGF-β1 or TGF-β3 increased Aβir within small round microglia. These data support a primary role for microglia in the formation and progression of Aβir deposits.
Indeed, AD has been hypothesized to be an inflammatory disease. TGF-βs have been localized to plaques and activated glial cells around plaques, suggesting that increased expression of this cytokine functions to counteract the inflammation. Although TGF-βs have been shown to have positive effects on neuron survival under certain conditions, it has been suggested that the enrichment of TGF-β in lesions of AD actually may dampen the inflammatory processes necessary to clear toxic Aβ, leading to a progressive accumulation of lesions (Peress and Perillo, 1995). Two other features of the TGF-β response to injury may be important in this regard: induction of increased deposition of extracellular matrix (ECM) and autoinduction of TGF-β production (Sporn et al., 1983; Roberts et al., 1986; Massague, 1987;Kim et al., 1989). Positive feedback and an inability to shut off TGF-β production can lead to an accelerated, unregulated ECM production (Border and Ruosiahti, 1992). Several ECM proteoglycans are known to accumulate in senile plaques (Snow et al., 1990, 1992; Su et al., 1992; De Witt et al., 1993) and may bind Aβ and increase its resistance to enzymatic proteolysis (Brunden et al., 1993; Gupta-Bansal et al., 1993). Perlecan, a proteoglycan component of diffuse and fibrillar plaques in AD, can enhance fibril formation and stability (Castillo et al., 1997) and enhance fibrillar Aβ deposition and persistence in brain (Snow et al., 1994). A balance of TGF-β is obviously important, because too much or too little can have devastating consequences. TGF-β1 null mice develop a multifocal inflammatory disease (Kulkarni and Karlsson, 1993; Christ et al., 1994;Kulkarni et al., 1995), and loss of TGF-β responsiveness may promote tumorigenesis directly (Bottinger et al., 1997). Overexpressing TGF-β1 leads to experimental autoimmune encephalomyelitis (Wyss-Coray et al., 1997). This may explain why our TGF-β neutralization experiments were not successful. Toxicity as a result of neutralization of TGF-β may be the result of the inhibition of ECM production or aberrant immune function in the slices.
Components of the ECM can affect microglial migration, target recognition, and binding (Chamak and Mallat, 1991). Microglia may play a key role in the initial events of plaque formation by initiating the accumulation of amyloidogenic APP fragments in response to an altered ECM (Monning et al., 1995). In an immortalized microglial cell line, TGF-β increased the accumulation of cellular mature APP, the putative precursor of Aβ (Monning et al., 1994). Preliminary immunocytochemical findings indicate that APP is upregulated by Aβ plus TGF-β treatment in slice cultures (our unpublished observations). In our slices, TGF-β alone does not increase Aβir. Aβir was dependent on the addition of Aβ to the culture medium, and it is likely that Aβ, either taken up from the culture media or produced endogenously (in response to Aβ/TGF-β treatment), is not being degraded or eliminated. Induction of neurodegeneration has been shown to increase intracellular Aβ accumulation (Leblanc, 1995;Frautschy et al., 1998). Last, in preliminary studies Aβir as detected by the 10G4 antibody and APPir did not colocalize (data not shown), suggesting that the 10G4 antibody was not detecting APP in either the diffuse deposits or cells. The results of the end-specific antibodies to Aβ40 and Aβ42 provide further support for the 10G4 labeling being Aβ. The regulation of APP metabolism and Aβ degradation by TGF-β currently is being studied in our slice culture model.
The increase in Aβ deposition and altered cellular distribution of Aβ by the three TGF-β isoforms in our slice model may be relevant to the progression of AD. Differential expression of the three TGF-β isoforms in AD has been documented (Flanders et al., 1995; Peress and Perillo, 1995), and TGF-β1 immunoreactivity was found in neuritic profiles of senile plaques. Interestingly, TGF-β2, which produced striking neuronal Aβ staining in our slice cultures, was abundant in neurofibrillary tangle-bearing neurons of AD brains (Peress and Perillo, 1995). TGF-β3 immunoreactivity was highly localized to Hirano bodies in AD brains. Although there is some evidence for different mechanisms of action of the three TGF-β isoforms, it is not yet possible to explain the altered distribution and cellular targeting of Aβ in our slice culture model. The three isoforms display remarkable structural and sequence homology and are highly conserved across species, suggesting important specific functions for each isoform. TGF-β knock-out experiments are demonstrating that there are no phenotypic overlaps between TGF-β1 or TGF-β3 null mice (Sanford et al., 1997), suggesting noncompensated functions among the three isoforms. Peptide sequence analysis of the immunomodulatory properties of the TGF-βs suggests that the isoforms should exert different influences on the immune response (Wieczorek et al., 1995). TGF-β1 should possess both immunosuppressive and stimulative properties, whereas TGF-β2 and TGF-β3 should be immunosuppressive and immunostimulative, respectively. Several receptors and binding proteins for TGF-β have been identified. Endoglin, a binding protein thought to present TGF-β isoforms to the type II signaling receptor, has been shown to bind only isoforms β1 and β3 (Lastres et al., 1996). TGF-β1 and TGF-β2 have been shown to bind to receptors in different ways: β1 binds directly to the type II receptor, whereas type II binding of β2 requires coexpression of the type I or II receptors (Rodriguez et al., 1995). In addition, TGF-β1 and TGF-β3, but not TGF-β2, inhibit the growth of some endothelial cells (Jennings et al., 1988; Merwin et al., 1991) and TGF-β1, but not TGF-β2, inhibits the growth of the LS513 colorectal cancer cell line (Suardet et al., 1992).
In conclusion, the TGF-β isoforms cause a differential cellular distribution of Aβir in the hippocampal slice culture and a generalized inhibition of Aβ degradation. The accumulation of Aβir by Aβ plus TGF-β2 on neurons and the resulting degenerative changes in regions of the hippocampus known to be vulnerable in AD suggest a possible relationship of TGF-β2 to neurotoxicity. TGF-β2 increases glutamate-induced neuron death (Kane et al., 1996). A stable complex between TGF-β2 and APP has been observed (Bodmer et al., 1990), suggesting an interaction that is important to the function of APP and TGF-β2 in vivo and in Aβ deposition in AD. Given that both TGF-β2 and APP exhibit growth-promoting properties, the regulation of these proteins may be important to inflammatory and injury repair processes. The presence of TGF-β2 in glial cells in other brain diseases suggests a more generalized function for TGF-β2 in brain injury, a known risk factor for AD. Although it is not yet known whether TGF-β2 itself is a risk factor in AD, it is interesting to note that the gene for TGF-β2 shares chromosome site 1q41 with the gene for presenilin-2.
Footnotes
This work was supported by a French Foundation Fellowship, a Los Angeles Alzheimer’s Association Turken Fellowship, and a University of California Los Angeles Alzheimer’s Disease Center Pilot Grant (to M.E.H.W.). This work was also supported by AG10685 and a Veterans Affairs Merit Award (to S.A.F.). We thank Greg Cole for valuable advice and commentary.
Correspondence should be addressed to Dr. Marni E. Harris-White, University of California Los Angeles and Veterans Affairs Medical Center Sepulveda, 16111 Plummer Street (151), Sepulveda, CA 91343.
REFERENCES
- 1.Allen YS, Devanathan PH, Owen GP. Neurotoxicity of β-amyloid protein: cytochemical changes and apoptotic cell death investigated in organotypic cultures. Clin Exp Pharmacol Physiol. 1995;22:370–371. doi: 10.1111/j.1440-1681.1995.tb02021.x. [DOI] [PubMed] [Google Scholar]
- 2.Ard MD, Cole GM, Wei J, Mehrle AP, Fratkin JD. Scavenging of Alzheimer’s β-protein by microglia in culture. J Neurosci Res. 1996;43:190–202. doi: 10.1002/(SICI)1097-4547(19960115)43:2<190::AID-JNR7>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 3.Bodmer S, Podlisny MB, Selkoe DJ, Heid I, Fontana A. Transforming growth factor-β bound to soluble derivatives of the β-amyloid precursor protein of Alzheimer’s disease. Biochem Biophys Res Commun. 1990;171:890–897. doi: 10.1016/0006-291x(90)91229-l. [DOI] [PubMed] [Google Scholar]
- 4.Border WA, Ruosiahti E. Transforming growth factor-β in disease: the dark side of tissue repair. J Clin Invest. 1992;90:1–7. doi: 10.1172/JCI115821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bottinger EP, Letterio JJ, Roberts AB. Biology of TGF-β in knock-out and transgenic mouse models. Kidney Int. 1997;51:1355–1360. doi: 10.1038/ki.1997.185. [DOI] [PubMed] [Google Scholar]
- 6.Bruce AJ, Malfroy B, Baudry M. β-Amyloid toxicity in organotypic hippocampal cultures: protection by EUK-8, a synthetic catalytic free radical scavenger. Proc Natl Acad Sci USA. 1996;93:2312–2316. doi: 10.1073/pnas.93.6.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunden KR, Richter-Cook NJ, Chaturvedi N, Frederickson RCA. pH-Dependent binding of synthetic β-amyloid peptides to glycosaminoglycans. J Neurochem. 1993;61:2147–2154. doi: 10.1111/j.1471-4159.1993.tb07453.x. [DOI] [PubMed] [Google Scholar]
- 8.Castillo GM, Ngo C, Cummings J, Wight TN, Snow AD. Perlecan binds to the β-amyloid (Aβ) of Alzheimer’s disease, accelerates Aβ fibril formation, and maintains Aβ fibril stability. J Neurochem. 1997;69:2452–2465. doi: 10.1046/j.1471-4159.1997.69062452.x. [DOI] [PubMed] [Google Scholar]
- 9.Chalazonitis A, Kalberg J, Twardzik DR, Morrison RS, Kessler JA. Transforming growth factor-β has neurotrophic actions on sensory neurons in vitro and is synergistic with nerve growth factor. Dev Biol. 1992;152:121–132. doi: 10.1016/0012-1606(92)90162-a. [DOI] [PubMed] [Google Scholar]
- 10.Chamak B, Mallat M. Fibronectin and laminin regulate the in vitro differentiation of microglial cells. Neuroscience. 1991;45:513–527. doi: 10.1016/0306-4522(91)90267-r. [DOI] [PubMed] [Google Scholar]
- 11.Chao CC, Hu S, Tsang M, Weatherbee J, Molitor TW, Anderson WR, Peterson PK. Effects of transforming growth factor-β on murine astrocyte glutamine synthetase activity. Implications in neuronal injury. J Clin Invest. 1992;90:1786–1793. doi: 10.1172/JCI116053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chao CC, Hu S, Frey WH, Ala TA, Tourtellotte WW, Peterson PK. Transforming growth factor-β in Alzheimer’s disease. Clin Diagn Lab Immunol. 1994;1:109–110. doi: 10.1128/cdli.1.1.109-110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen HH, Liu HM. A new fluorescent histological marker for ischemic neurons, EA 50: correlated with Fos and Jun/AP-1 immunoreactivity. Histochem Cell Biol. 1996;105:375–382. doi: 10.1007/BF01463658. [DOI] [PubMed] [Google Scholar]
- 14.Christ M, McCartney-Francis NL, Kulkarni AB, Ward JM, Mizel DE, Mackall CL, Gress RE, Hines KL, Tian H, Karlsson S, Wahl SM. Immune dysregulation in TGF-β1-deficient mice. J Immunol. 1994;153:1936–1946. [PubMed] [Google Scholar]
- 15.Constam DB, Philipp J, Malipiero UV, ten Dijke P, Schachner M, Fontana A. Differential expression of transforming growth factor-β1, -β2, and -β3 by glioblastoma cells, astrocytes, and microglia. J Immunol. 1992;148:1404–1410. [PubMed] [Google Scholar]
- 16.DeWitt DA, Silver J, Canning DR, Perry G. Chondroitin sulfate proteoglycans are associated with the lesions of Alzheimer’s disease. Exp Neurol. 1993;121:149–152. doi: 10.1006/exnr.1993.1081. [DOI] [PubMed] [Google Scholar]
- 17.El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature. 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- 18.Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- 19.Flanders KC, Lippa CF, Smith TW, Pollen DA, Sporn MB. Altered expression of transforming growth factor-β in Alzheimer’s disease. Neurology. 1995;45:1561–1569. doi: 10.1212/wnl.45.8.1561. [DOI] [PubMed] [Google Scholar]
- 20.Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce β-amyloid fibrils. Acta Neuropathol (Berl) 1992;84:225–233. doi: 10.1007/BF00227813. [DOI] [PubMed] [Google Scholar]
- 21.Frautschy SA, Cole GM, Baird A. Phagocytosis and deposition of vascular β-amyloid in rat brains injected with Alzheimer β-amyloid. Am J Pathol. 1992;140:1389–1399. [PMC free article] [PubMed] [Google Scholar]
- 22.Frautschy SA, Albright T, Cole GM. Ultrastructural effects of β-amyloid protein (Aβ) and injury in the rat hippocampus. Soc Neurosci Abstr. 1994;20:25219. [Google Scholar]
- 23.Frautschy SA, Calderon L, Yang F, Cole GM. Rodent models of Alzheimer’s disease: a comparison of transgenic APP mouse and rat Aβ infusion approaches. Neurobiol Aging. 1996;17:311–321. doi: 10.1016/0197-4580(95)02073-x. [DOI] [PubMed] [Google Scholar]
- 24.Frautschy SA, Horn D, Sigel JJ, Harris-White ME, Yang F, Saido TC, Cole GM. Protease inhibitor co-infusion with amyloid-β protein results in enhanced deposition and toxicity in rat brain. J Neurosci. 1998;18:8311–8321. doi: 10.1523/JNEUROSCI.18-20-08311.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray CW, Patel AJ. Regulation of β-amyloid precursor protein isoform mRNAs by transforming growth factor-β1 and interleukin-1β in astrocytes. Mol Brain Res. 1993;19:251–256. doi: 10.1016/0169-328x(93)90037-p. [DOI] [PubMed] [Google Scholar]
- 26.Gupta-Bansal R, Ziehler W, Wujek JR, Brunden KR. Potential role of proteoglycans in the accumulation and persistence of β-amyloid (Aβ) in Alzheimer’s disease (AD) senile plaques. Soc Neurosci Abstr. 1993;19:1471. [Google Scholar]
- 27.Jennings JC, Mohan S, Linkhart TA, Widstrom R, Baylink DJ. Comparison of the biological effects of TGF-β1 and TGF-β2: differential activity in endothelial cells. J Cell Physiol. 1988;137:167–172. doi: 10.1002/jcp.1041370120. [DOI] [PubMed] [Google Scholar]
- 28.Kane JM, Brown GJ, Phelan KD. Transforming growth factor-β2 increases NMDA receptor-mediated excitotoxicity in rat cerebral cortical neurons independently of glia. Neurosci Lett. 1996;204:93–96. doi: 10.1016/0304-3940(96)12332-6. [DOI] [PubMed] [Google Scholar]
- 29.Kim S-J, Jeang K-T, Glick AB, Sporn MB, Roberts AB. Promoter sequences of the human transforming growth factor-β. J Biol Chem. 1989;264:7041–7045. [PubMed] [Google Scholar]
- 30.Klempt ND, Sirimanne E, Gunn AJ, Klempt M, Singh K, Williams C, Gluckman PD. Hypoxia–ischemia induces transforming growth factor-β1 mRNA in the infant rat brain. Brain Res. 1992;13:93–101. doi: 10.1016/0169-328x(92)90048-g. [DOI] [PubMed] [Google Scholar]
- 31.Korotzer AR, Pike CJ, Cotman CW. β-Amyloid peptides induce degeneration of cultured rat microglia. Brain Res. 1993;624:121–125. doi: 10.1016/0006-8993(93)90068-x. [DOI] [PubMed] [Google Scholar]
- 32.Krieglstein K, Rufer M, Suter-Crazzolara C, Unsicker K. Neural functions of the transforming growth factors-β. Int J Dev Neurosci. 1995;13:301–315. doi: 10.1016/0736-5748(94)00062-8. [DOI] [PubMed] [Google Scholar]
- 33.Kulkarni AB, Karlsson S. Transforming growth factor-β1 knock-out mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am J Pathol. 1993;143:3–9. [PMC free article] [PubMed] [Google Scholar]
- 34.Kulkarni AB, Ward JM, Yaswen L, Mackall CL, Bauer SR, Huh C-G, Gress RE, Karlsson S. Transforming growth factor-β1 null mice. An animal model for inflammatory disorders. Am J Pathol. 1995;146:264–275. [PMC free article] [PubMed] [Google Scholar]
- 35.Langer C, Jurgensmeier JM, Bauer G. Reactive oxygen species act at both TGF-β-dependent and -independent steps during induction of apoptosis of transformed cells by normal cells. Exp Cell Res. 1996;222:117–124. doi: 10.1006/excr.1996.0015. [DOI] [PubMed] [Google Scholar]
- 36.Lastres P, Letamendia A, Zhang H, Rius C, Almendro N, Raab U, Lopez LA, Langa C, Fabra A, Letarte M, Bernabeu C. Endoglin modulates cellular responses to TGF-β1. J Cell Biol. 1996;133:1109–1121. doi: 10.1083/jcb.133.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.LeBlanc A. Increased production of 4 kDa amyloid-β peptide in serum-deprived human primary neuron cultures: possible involvement of apoptosis. J Neurosci. 1995;15:7837–7846. doi: 10.1523/JNEUROSCI.15-12-07837.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindholm D, Castren E, Kiefer E, Zafra F, Thoenen H. Transforming growth factor-β1 in the rat brain: increase after injury and inhibition of astrocyte proliferation. J Cell Biol. 1992;117:395–400. doi: 10.1083/jcb.117.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lippa CF, Smith TW, Flanders KC. Transforming growth factor-β: neuronal and glial expression in CNS degenerative diseases. Neurodegeneration. 1995;4:425–432. doi: 10.1006/neur.1995.0051. [DOI] [PubMed] [Google Scholar]
- 40.Logan A, Frautschy SA, Gonzalez A-M, Sporn MB, Baird A. Enhanced expression of transforming growth factor-β1 in the rat brain after a localized cerebral injury. Brain Res. 1992;587:216–225. doi: 10.1016/0006-8993(92)91000-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Logan A, Berry M, Gonzalez AM, Frautschy SA, Sporn MB, Baird A. Effects of transforming growth factor-β1 on scar production in the injured central nervous system of the rat. Eur J Neurosci. 1994;6:355–363. doi: 10.1111/j.1460-9568.1994.tb00278.x. [DOI] [PubMed] [Google Scholar]
- 42.Mackenzie IRA, Hao C, Munoz DG. Role of microglia in senile plaque formation. Neurobiol Aging. 1995;16:797–804. doi: 10.1016/0197-4580(95)00092-s. [DOI] [PubMed] [Google Scholar]
- 43.Mak K, Yang F, Vinters HV, Frautschy SA, Cole GM. Polyclonals to β-amyloid(1–42) identify most plaque and vascular deposits in Alzheimer cortex, but not striatum. Brain Res. 1994;667:138–142. doi: 10.1016/0006-8993(94)91725-6. [DOI] [PubMed] [Google Scholar]
- 44.Malouf AT. Effect of β-amyloid peptides on neurons in hippocampal slice cultures. Neurobiol Aging. 1992;13:543–551. doi: 10.1016/0197-4580(92)90054-2. [DOI] [PubMed] [Google Scholar]
- 45.Massague J. The TGF-β family of growth and differentiation factors. Cell. 1987;49:437–438. doi: 10.1016/0092-8674(87)90443-0. [DOI] [PubMed] [Google Scholar]
- 46.Meda L, Cassatella MA, Szendrei GI, Otvos L, Jr, Baron P, Villalba M, Ferrari D, Rossi F. Activation of microglial cells by β-amyloid protein and interferon-γ. Nature. 1995;374:647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 47.Merwin JR, Newman W, Beall LD, Tucker A, Madri J. Vascular cells respond differentially to transforming growth factors β1 and β2 in vitro. Am J Pathol. 1991;138:37–51. [PMC free article] [PubMed] [Google Scholar]
- 48.Monning U, Sandbrink R, Banati RB, Masters C, Beyreuther K. Transforming growth factor-β mediates increase of mature transmembrane amyloid precursor protein in microglial cells. FEBS Lett. 1994;342:267–272. doi: 10.1016/0014-5793(94)80514-8. [DOI] [PubMed] [Google Scholar]
- 49.Monning U, Sandbrink R, Weidemann A, Banati RB, Masters CL, Beyreuther K. Extracellular matrix influences the biogenesis of amyloid precursor protein in microglial cells. J Biol Chem. 1995;270:7104–7110. doi: 10.1074/jbc.270.13.7104. [DOI] [PubMed] [Google Scholar]
- 50.Morgan TE, Nichols NR, Pasinetti GM, Finch CE. TGF-β1 mRNA increases in macrophage/microglial cells of the hippocampus in response to deafferentation and kainic acid-induced neurodegeneration. Exp Neurol. 1993;120:291–301. doi: 10.1006/exnr.1993.1063. [DOI] [PubMed] [Google Scholar]
- 51.Peress NS, Perillo E. Differential expression of TGF-β1, 2, and 3 isotypes in Alzheimer’s disease: a comparative immunohistochemical study with cerebral infarction, aged human, and mouse control brains. J Neuropathol Exp Neurol. 1995;54:802–811. doi: 10.1097/00005072-199511000-00007. [DOI] [PubMed] [Google Scholar]
- 52.Poulsen KT, Armanini MP, Klein RD, Hynes MA, Phillips HS, Rosenthal A. TGF-β2 and TGF-β3 are potent survival factors for midbrain dopaminergic neurons. Neuron. 1994;13:1245–1252. doi: 10.1016/0896-6273(94)90062-0. [DOI] [PubMed] [Google Scholar]
- 53.Prehn JH, Krieglstein J. Opposing effects of transforming growth factor-β1 on glutamate neurotoxicity. Neuroscience. 1994;60:7–10. doi: 10.1016/0306-4522(94)90198-8. [DOI] [PubMed] [Google Scholar]
- 54.Prehn JH, Miller RJ. Opposite effects of TGF-β1 on rapidly and slowly triggered excitotoxic injury. Neuropharmacology. 1996;35:249–256. doi: 10.1016/0028-3908(96)00001-9. [DOI] [PubMed] [Google Scholar]
- 55.Prehn JHM, Bindokas VP, Jordan J, Galindo MF, Ghadge GD, Roos RP, Boise LH, Thompson CB, Krajewski S, Reed JC, Miller RJ. Protective effect of transforming growth factor-β1 on β-amyloid neurotoxicity in rat hippocampal neurons. Mol Pharmacol. 1996;49:319–328. [PubMed] [Google Scholar]
- 56.Ren RF, Flanders KF. Transforming growth factors-β protect primary rat hippocampal neuronal cultures from degeneration induced by β-amyloid peptide. Brain Res. 1996;732:16–24. doi: 10.1016/0006-8993(96)00458-1. [DOI] [PubMed] [Google Scholar]
- 57.Roberts AB, Sporn MB. Physiological actions and clinical applications of transforming growth factor-β. Growth Factors. 1993;8:1–9. doi: 10.3109/08977199309029129. [DOI] [PubMed] [Google Scholar]
- 58.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, Fauci AS. Transforming growth factor-β: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci USA. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodriguez C, Chen F, Weinberg RA, Lodish HF. Cooperative binding of transforming growth factor (TGF)-β2 to the types I and II TGF-β receptors. J Biol Chem. 1995;270:15919–15922. doi: 10.1074/jbc.270.27.15919. [DOI] [PubMed] [Google Scholar]
- 60.Saido TC, Iwatsubo T, Mann DM, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct β-amyloid peptide species, AβN3(PE), in senile plaques. Neuron. 1995;14:457–466. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- 61.Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGF-β2 knock-out mice have multiple developmental defects that are nonoverlapping with other TGF-β knock-out phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharma K, Jin Y, Guo J, Ziyadeh FN. Neutralization of TGF-β by anti-TGF-β antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes. 1996;45:522–530. doi: 10.2337/diab.45.4.522. [DOI] [PubMed] [Google Scholar]
- 63.Snow AD, Mar H, Nochlin D, Sekiguchi RT, Kimata K, Koika Y, Wight TN. Early accumulation of heparan sulfate in neurons and in the β-amyloid protein-containing lesions of Alzheimer’s disease and Down’s syndrome. Am J Pathol. 1990;137:1253–1270. [PMC free article] [PubMed] [Google Scholar]
- 64.Snow AD, Mar H, Nochlin D, Kresse H, Wight TN. Peripheral distribution of dermatan sulfate proteoglycans (decorin) in amyloid-containing plaques and their presence in neurofibrillary tangles of Alzheimer’s disease. J Histochem Cytochem. 1992;40:105–113. doi: 10.1177/40.1.1370306. [DOI] [PubMed] [Google Scholar]
- 65.Snow AD, Sekiguchi R, Nochlin D, Fraser P, Kimata K, Mitzutani A, Arai M, Schreier WA, Morgan DA. An important role of heparan sulfate proteoglycan (perlecan) in a model system for the deposition and persistence of fibrillar Aβ amyloid in rat brain. Neuron. 1994;12:219–234. doi: 10.1016/0896-6273(94)90165-1. [DOI] [PubMed] [Google Scholar]
- 66.Sporn MB, Roberts AB, Shull JH, Smith JM, Ward JM. Polypeptide transforming growth factors isolated from bovine sources and used for wound healing in vivo. Science. 1983;219:1329–1330. doi: 10.1126/science.6572416. [DOI] [PubMed] [Google Scholar]
- 67.Stinchcombe S, Buchmann A, Bock KW, Schwarz M. Inhibition of apoptosis during 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated tumour promotion in rat liver. Carcinogenesis. 1995;16:1271–1275. doi: 10.1093/carcin/16.6.1271. [DOI] [PubMed] [Google Scholar]
- 68.Stoppini L, Buchs P-A, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 69.Su JH, Cummings BJ, Cotman CW. Location of heparan sulfate glycosaminoglycan and proteoglycan core protein in aged brain and Alzheimer’s disease. Neuroscience. 1992;51:801–814. doi: 10.1016/0306-4522(92)90521-3. [DOI] [PubMed] [Google Scholar]
- 70.Suardet L, Gaide AC, Calmes JM, Sordat B, Givel JC, Eliason JF, Odartchenko N. Responsiveness of the three newly established human colorectal cancer cell lines to transforming growth factors β1 and β2. Cancer Res. 1992;52:3705–3712. [PubMed] [Google Scholar]
- 71.van der Wal EA, Gomez-Pinilla F, Cotman CW. Transforming growth factor-β1 is in plaques in Alzheimer and Down pathologies. NeuroReport. 1993;4:69–72. doi: 10.1097/00001756-199301000-00018. [DOI] [PubMed] [Google Scholar]
- 72.Vawter MP, Dillon-Carter O, Tourtellotte WW, Carvey P, Freed WJ. TGF-β1 and TGF-β2 concentrations are elevated in Parkinson’s disease in ventricular cerebrospinal fluid. Exp Neurol. 1996;142:313–322. doi: 10.1006/exnr.1996.0200. [DOI] [PubMed] [Google Scholar]
- 73.Wieczorek Z, Slon J, Kluczyk A, Zbozien R, Stafanowicz P, Siemion IZ. The immunomodulatory diversity of the proteins of the transforming growth factor-β (TGF-β) family. Int J Pept Protein Res. 1995;46:113–118. doi: 10.1111/j.1399-3011.1995.tb01326.x. [DOI] [PubMed] [Google Scholar]
- 74.Wyss-Coray T, Borrow P, Brooker MJ, Mucke L. Astroglial overproduction of TGF-β1 enhances inflammatory central nervous system disease in transgenic mice. J Neuroimmunol. 1997;77:45–50. doi: 10.1016/s0165-5728(97)00049-0. [DOI] [PubMed] [Google Scholar]
- 75.Xiao BG, Bai XF, Zhang GX, Link H. Transforming growth factor-β1 induces apoptosis of rat microglia without relation to bcl-2 oncoprotein expression. Neurosci Lett. 1997;226:71–74. doi: 10.1016/s0304-3940(97)00234-6. [DOI] [PubMed] [Google Scholar]
- 76.Yang F, Mak K, Vinters HV, Frautschy SA, Cole GM. Monoclonal antibody to the C-terminus of β-amyloid. NeuroReport. 1994;5:2117–2120. doi: 10.1097/00001756-199410270-00032. [DOI] [PubMed] [Google Scholar]
- 77.Yao J, Harvath L, Gilbert DL, Colton CA. Chemotaxis by a CNS macrophage, the microglia. J Neurosci Res. 1990;27:36–42. doi: 10.1002/jnr.490270106. [DOI] [PubMed] [Google Scholar]