

Abstract
Mitochondria within cultured rat cerebellar granule cells have a complex influence on cytoplasmic free Ca2+([Ca2+]c) responses to glutamate. A decreased initial [Ca2+]celevation in cells whose mitochondria are depolarized by inhibition of the ATP synthase and respiratory chain (conditions which avoid ATP depletion) was attributed to enhanced Ca2+ extrusion from the cell rather than inhibited Ca2+ entry via the NMDA receptor. Even in the presence of elevated extracellular Ca2+, when [Ca2+]cresponses were restored to control values, such cells showed resistance to acute excitotoxicity, defined as a delayed cytoplasmic Ca2+ deregulation (DCD) during glutamate exposure. DCD was a function of the duration of mitochondrial polarization in the presence of glutamate rather than the total period of glutamate exposure. Once initiated, DCD could not be reversed by NMDA receptor inhibition. In the absence of ATP synthase inhibition, respiratory chain inhibitors produced an immediate Ca2+deregulation (ICD), ascribed to an ATP deficit. In contrast to DCD, ICD could be reversed by subsequent ATP synthase inhibition with or without additional NMDA receptor blockade. DCD could not be ascribed to the failure of an ATP yielding metabolic pathway. It is concluded that mitochondria can control Ca2+ extrusion from glutamate-exposed granule cells by the plasma membrane in three ways: by competing with efflux pathways for Ca2+, by restricting ATP supply, and by inducing a delayed failure of Ca2+ extrusion. Inhibitors of the mitochondrial permeability transition only marginally delayed the onset of DCD.
Keywords: glutamate, excitotoxicity, mitochondria, calcium, granule cell, NMDA
In common with many other cultured neurons, rat cerebellar granule cells die after extended NMDA receptor activation (Garthwaite, 1986; Novelli et al., 1988; Manev et al., 1989). There is increasing evidence that mitochondrial dysfunction plays a primary role in the initiation of both necrotic and apoptotic neuronal cell death (for review, see Beal, 1996; Petit et al., 1996;Richter et al., 1996; Henneberry, 1997; Nicotera and Leist, 1997;Nicholls and Budd, 1998). However, unraveling the multiple roles of the organelle in this context is not trivial, because each mitochondrial function can in turn influence, and be influenced by, the other parameters. In particular, the mitochondrial membrane potential (ΔΨm) or the full proton electrochemical potential (Δp) controls ATP generation by the ATP synthase (Nicholls and Ferguson, 1992), Ca2+ sequestration within the mitochondrial matrix (Nicholls and Åkerman, 1982; Gunter and Gunter, 1994), and superoxide generation by the respiratory chain (Beal et al., 1997; Korshunov et al., 1997; Nègre-Salvayre et al., 1997), and may additionally affect the mitochondrial permeability transition (Halestrap et al., 1997; Scorrano et al., 1997), outer membrane rupture, and cytochrome c release (Liu et al., 1996; Yang et al., 1997; Kroemer et al., 1998; but see Kluck et al., 1997;Bossy-Wetzel et al., 1998).
Glutamate-induced neuronal necrosis is preceded by a rapid uncontrolled increase in cytoplasmic free Ca2+([Ca2+]c) concentration, which occurs stochastically within a field of neurons (Dubinsky et al., 1995) and has been termed delayed Ca2+ deregulation (DCD) (Tymianski et al., 1993). Mitochondrial depolarization after glutamate exposure has been reported to be an early event associated with neuronal Ca2+ loading (Ankarcrona et al., 1996;Isaev et al., 1996; Khodorov et al., 1996b; Schinder et al., 1996; White and Reynolds, 1996; Keelan et al., 1998). However, the distinction is not always drawn between a reversible, physiological response to increased energy demand and a pathological depolarization after mitochondrial damage. Because we (Budd and Nicholls, 1996b) and others (Tan et al., 1997; Stout et al., 1998) have reported that complete depolarization of mitochondria before glutamate exposure is acutely neuroprotective, the implication is that polarized mitochondria, perhaps in response to excessive Ca2+loading, generate a factor or create a condition triggering subsequent necrotic cell death. However, the nature of this factor, which could be a high cytoplasmic Ca2+ per se, the mitochondrial permeability transition, reactive oxygen-induced damage, metabolic inhibition, etc., remains unclear.
The existence of specific mitochondrial inhibitors allows each function of the organelle to be investigated separately. Thus, it is possible to eliminate a failure of oxidative phosphorylation as a cause of DCD in cerebellar granule cells, because the delay and extent of glutamate-induced deregulation are unaffected in cells whose mitochondrial ATP synthase has been inhibited by oligomycin (Budd and Nicholls, 1996b) and thus rely on glycolysis to maintain ATP/ADP ratios (Budd and Nicholls, 1996a). In the present report, we investigate the significance of other mitochondrial parameters in the induction of DCD, as well as the basis of the counter-intuitive finding that cytoplasmic Ca2+ transients in these cells are consistently decreased when mitochondria are depolarized, without depleting ATP (Budd and Nicholls, 1996b), an observation which has recently been challenged (Khodorov et al., 1996c). It is concluded that mitochondrial Ca2+ accumulation influences Ca2+efflux from the granule cells both acutely, accounting for the effect on the initial Ca2+ transients, and progressively, culminating in DCD.
MATERIALS AND METHODS
Materials. Fura-2 acetoxymethyl ester (fura-2 AM) was obtained from Molecular Probes (Leiden, The Netherlands). (5R,10S)-(+)-5-methyl-10,11-dihydro[a,d]cyclohepten-5,10-imine hydrogen maleate (MK-801) was obtained from Research Biochemicals (SEMAT, St. Albans, Hertfordshire, UK). Fetal calf serum and minimal essential medium were obtained from Life Technologies (Paisley, Strathclyde, UK). Bongkrekic acid andN-methylval-4-cyclosporin (SDZ 220–384) were kindly donated by Dr. M. Klingenberg (University of Munich, München, Germany) and Novartis Pharma Inc. (Basel, Switzerland), respectively. Cyclosporin A, ionomycin, ketamine, oligomycin, rotenone, and all other reagents were obtained from Sigma (Poole, Dorset, UK).
Preparation of cerebellar granule cells. Granule cells were prepared as described previously (Courtney et al., 1990) from 7-d-old Wistar rats. Cells were plated on poly-d-lysine-coated glass coverslips (13 mm circular for nonperfusion experiments and 22 mm square for use with the perfusion chamber in Fig.1A–C) at a density of 280,000 cells per coverslip for imaging studies and 750,000 per coverslip for 45Ca2+ determination. Cells were cultured in minimal essential medium containing Earle’s salts (Life Technologies) plus 10% (v/v) fetal calf serum, 25 mm KCl, 30 mm glucose, 2 mmglutamine, 100 μg/ml streptomycin, and 100 U/ml penicillin. After 24 hr, 10 μm cytosine arabinoside was added to inhibit non-neuronal cell proliferation. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2–95% air and were used after 6–8 d in vitro.
Fig. 1.

Mitochondrial depolarization depresses the cytoplasmic free Ca2+ response to glutamate.A–C, Granule cells were perfused with incubation medium at 25°C and exposed to three sequential 60 sec pulses of 100 μm glutamate–10 μm glycine (glut). [Ca2+]cwas monitored in a minimum of 10 randomly selected somata.A, Individual somata show highly reproducible [Ca2+]c transients in response to three pulses. B, Oligomycin (2.5 μg/ml) (oligo) did not affect the second [Ca2+]c transient, but the addition of 2 μm rotenone (rot) depressed the third transient. C, Antimycin A (0.5 μm) (AA) caused a similar depression of the response in the presence of oligomycin. Under energy-deficient conditions, when in the presence of rotenone minus oligomycin (D) or in the absence of glucose (E), glutamate–glycine produces a large transient, followed by immediate deregulation. Data are representative of at least four independent experiments.
Incubation conditions. The incubation medium contained 120 mm NaCl, 3.1 mm KCl, 0.4 mmKH2PO4, and 20 mmN-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid, pH adjusted to 7.4 at the temperature of the subsequent experiment (37°C, except for Fig. 1), with NaOH, 5 mmNaHCO3, 1.2 mmNa2SO4, and 1.3 mmCaCl2. Unless otherwise stated, all experiments were performed at 37°C in Mg2+-free incubation medium in the presence of 15 mm glucose. Experiments shown in Figure 1A–C were designed to correspond to the conditions used by Khodorov et al. (1996c) and were performed at 25°C in the presence of 5 mm glucose.
Imaging of [Ca2+]c in single cells. Single cell imaging was performed in a MiraCal Imaging facility (Life Science Resources, Cambridge, UK) using a Nikon DIAPHOT-TMD inverted epifluorescence microscope equipped with a 40× oil immersion objective and Sutter filter wheel (excitation, 340 and 380 nm; emission >505 nm). Cells were loaded with 3 μm fura-2 AM for 25 min in incubation medium containing an additional 30 μg/ml bovine serum albumin, 15 mmglucose, and 1.2 mm MgCl2. In all experiments, the objective was focussed on cell somata rather than neurites. All experiments, apart from those in Figure 1, were performed in a nonperfusing thermostatted chamber with a volume of 300 μl.
To approximate the conditions used by Khodorov et al. (1996c), the experiments shown in Figure 1 used square coverslips mounted in a Warner RC-21B closed bath imaging chamber on a Warner PH-2 heater platform. Cells were superfused at 2 ml/min with incubation medium (25°C) lacking Mg2+ and containing 5 mm glucose. Cells were exposed to three 60 sec pulses of 100 μm glutamate–10 μm glycine separated by 10 min intervals by introducing concentrated glutamate–glycine into a mixing chamber immediately upstream of the chamber. Where indicated, incubation medium was supplemented with 2.5 μg/ml oligomycin, 1 μm rotenone, and 0.5 μm antimycin A. Calibration was performed by the sequential addition of 10 μm ionomycin and 10 mm Na-EGTA. The time course for [Ca2+]c was plotted from a minimum of ten randomly selected cell somata.
Manganese quenching of cytoplasmic fura-2 was monitored by alternate excitation at 340 and 380 nm and emission at >505 nm. Emission intensity corresponding to the isobestic point was calculated by interpolation using the following algorithm:Fisobestic = {(2 ×F340nm) +F380nm}/3. Cells were incubated under nonperfusing conditions at 37°C in the presence of 2.5 μg/ml oligomycin and 1 μm nifedipine, with the further additions of 2 μm rotenone or 2 μmketamine, where indicated. To each experiment, 100 μmMnCl2 was added, followed by 100 μmglutamate–10 μm glycine.
45Ca2+ content of cells.Coverslip-attached granule cells (7–9 d in vitro) were removed from culture medium, transferred to 24-well plates, and preincubated for 30 min at 37°C in 350 μl of incubation medium lacking Mg2+. Oligomycin (8 μg/ml) and rotenone (5 μm) were added to one series of experiments 2 min before addition of 100 μm NMDA plus 10 μm glycine and 1 μm nifedipine. 45CaCl2(6000 Bq/well) was added 20 sec before addition of the agonist. At specified times, individual coverslips were removed from their wells and rapidly washed three times with ice-cold incubation medium containing 1.2 mm MgCl2, 1 mm EGTA, and 1.3 mm unlabeled CaCl2. 45Ca2+ associated with the cells was counted by liquid scintillation counting. Results reported are the mean ± SEM of four independent experiments.
Statistics. Each set of single cell responses shown is representative of at least three independent experiments from different cell preparations. Significance was assessed by unpaired-variance Student’s t test (see Fig. 3).
Fig. 3.
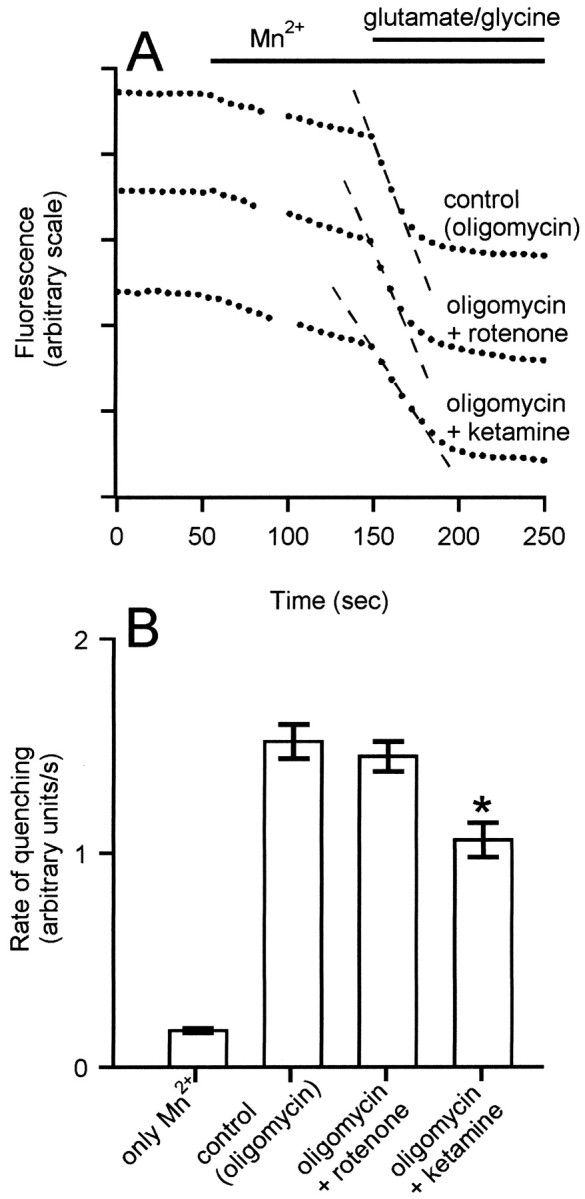
Mitochondrial depolarization does not inhibit the rate of NMDA receptor-induced manganese quenching of fura-2. Cells were incubated in the presence of 5 μg/ml oligomycin (control) or with the further addition of 2 μm rotenone or 2 μm ketamine, as indicated. Where indicated, 100 μm Mn2+ and 100 μm glutamate–10 μm glycine were added. *p < 0.05, statistically significant compared with the same condition in the absence of ketamine; n = 5.
RESULTS
Mitochondrial depolarization and glutamate-induced cytoplasmic Ca2+ transients
In the absence of selective cell-permeant inhibitors of the mitochondrial Ca2+ uniporter, the most common approach to establishing the functional role of mitochondrial Ca2+ accumulation in neuronal Ca2+ homeostasis has been to collapse ΔΨm (Thayer and Miller, 1990; Duchen and Biscoe, 1992;Friel and Tsien, 1994; White and Reynolds, 1995; Budd and Nicholls, 1996a,b; Park et al., 1996). Because ΔΨm is the major component of the Δp driving ATP synthesis, simple protonophore addition results in reversal of the ATP synthase and rapid depletion of cytoplasmic ATP (Duchen and Biscoe, 1992a; Budd and Nicholls, 1996a;Leyssens et al., 1996). We have reported previously that the increased [Ca2+]c transients observed in the presence of protonophores were a consequence of inhibited ATP-dependent Ca2+ efflux from the cell rather than inhibited mitochondrial Ca2+ sequestration (Budd and Nicholls, 1996a). Indeed, when granule cells maintained by glycolytic ATP, i.e., in the presence of the ATP synthase inhibitor oligomycin, are depolarized by the further addition of a respiratory chain inhibitor, acute cytoplasmic Ca2+ responses to KCl (Budd and Nicholls, 1996a) and glutamate (Budd and Nicholls, 1996b) are instead decreased.
The inference from these studies, that mitochondrial control of plasma membrane Ca2+ fluxes is more important than mitochondrial Ca2+ sequestration itself in regulating cytoplasmic Ca2+ responses, has been questioned by a report (Khodorov et al., 1996c) in which granule cells were superfused under somewhat different conditions, and addition of oligomycin plus the complex III inhibitor antimycin A resulted in an initial [Ca2+]c transient in response to glutamate that was essentially unchanged but was followed by a secondary increase in [Ca2+]c, similar to that seen under conditions of ATP depletion (Budd and Nicholls, 1996a). Because fundamentally opposite conclusions were reached in these studies concerning the effect of mitochondria on neuronal Ca2+ homeostasis, it is important to resolve this discrepancy.
In Figure 1, [Ca2+]c responses are reported for granule cells superfused under the conditions used byKhodorov et al. (1996c). Eighty percent of coverslips showed highly reproducible [Ca2+]c responses in individual somata to three 60 sec pulses of 100 μmglutamate–10 μm glycine with full subsequent recovery (Fig. 1A); these were used for the following experiments to avoid complications caused by acute glutamate excitotoxicity that was seen in the remaining fraction of coverslips. In subsequent experiments, the first pulse acted as a control, oligomycin was present during the second pulse, and oligomycin plus rotenone (Fig. 1B) or antimycin A (Fig.1C) was present for the third pulse. Although oligomycin did not affect the response of individual somata, oligomycin plus rotenone (Fig. 1B) or oligomycin plus antimycin A (Fig.1C) substantially reduced the response to the subsequent glutamate–glycine pulse. The only conditions in which we have been able to reproduce the rapid secondary rise in [Ca2+]c after glutamate addition reported by Khodorov et al. (1996c) has been when the respiratory chain is inhibited in the absence of oligomycin (Fig. 1D) or when substrate is limiting (Fig. 1E), both of which will result in ATP depletion.
To account for the counter-intuitive decreased cytoplasmic Ca2+ response in cells whose major Ca2+ sink is inhibited, we have investigated whether this is attributable to inhibited uptake of Ca2+ or to accelerated efflux across the plasma membrane. We have observed previously that the total 45Ca2+accumulation of granule cells exposed to glutamate for 20 min is decreased in cells with depolarized mitochondria. Figure2 shows that this is caused by a rapid inhibition of net accumulation within 5 min of NMDA receptor activation.
Fig. 2.

Net accumulation of45Ca2+ into NMDA-exposed cells is decreased by mitochondrial depolarization. Cells were incubated in the presence (•) or absence (○) of 8 μg/ml oligomycin plus 5 μm rotenone for 2 min before the addition of 100 μm NMDA, 10 μm glycine, and 1 μm nifedipine (t = 0).45Ca2+ was added 20 sec before the agonist. Results are the mean ± SEM of four independent experiments.
To determine whether the reduction in net45Ca2+ accumulation in the presence of the mitochondrial inhibitors was attributable to an inhibition of the NMDA receptor, the rate at which cytoplasmic fura-2 fluorescence is quenched by exogenous manganous ions (Miyata et al., 1991) was determined in the presence of oligomycin plus or minus rotenone. This technique has been used extensively to monitor NMDA receptor activity in granule cells (Simpson et al., 1995; Khodorov et al., 1996a;Savidge and Bristow, 1997). Figure 3shows that addition of Mn2+ to the extracellular medium initiated a slow quenching of fura-2 fluorescence in oligomycin-treated cells. This rate was not inhibited by MK-801 (data not shown). Addition of glutamate–glycine increased the rate of quenching ninefold. No significant decrease in the rate of glutamate-induced quenching was seen in cells whose mitochondria were depolarized in the presence of both rotenone and oligomycin (Fig.3).
The Mn2+ quench technique can detect a partial inhibition of the NMDA receptor. Thus, the competitive antagonist ketamine (2 μm), which reduced the initial [Ca2+]c transient to the same extent as that seen in the presence of oligomycin plus rotenone (data not shown), produced a 30% inhibition in the rate of Mn2+ quenching. Thus, if mitochondrial depolarization were producing its effects on [Ca2+]c by inhibiting NMDA receptor activity, this would have been clearly detectable. Importantly, the residual fluorescence of cells after exposure to Mn2+ and glutamate was not punctate; thus, there was no contribution to the cytoplasmic [Ca2+]c response by fluorescence from any fura-2 accumulated within the mitochondria. As a consequence, the influence of mitochondrial depolarization on the cellular fluorescence cannot be ascribed to a decrease in a mitochondrial signal.
Mitochondrial polarization and glutamate-induced delayed [Ca2+]c deregulation
When granule cells are exposed continuously to glutamate, a high proportion of cells show a delayed failure of Ca2+homeostasis. This DCD precedes and accurately predicts subsequent, predominantly necrotic cell death (Tymianski et al., 1993). As reported previously (Budd and Nicholls, 1996b), glutamate-induced DCD also occurs in oligomycin-treated cells and thus cannot be ascribed to a failure of mitochondrial ATP synthesis, whereas mitochondrial depolarization by the further addition of rotenone before glutamate greatly decreases DCD. Representative traces are shown in Figure 4, A and B. The decreased initial transient in the presence of rotenone–oligomycin is again apparent, after which the cells maintain a stable plateau with only occasional cells showing a rather sudden deregulation (Fig.4B).
Fig. 4.
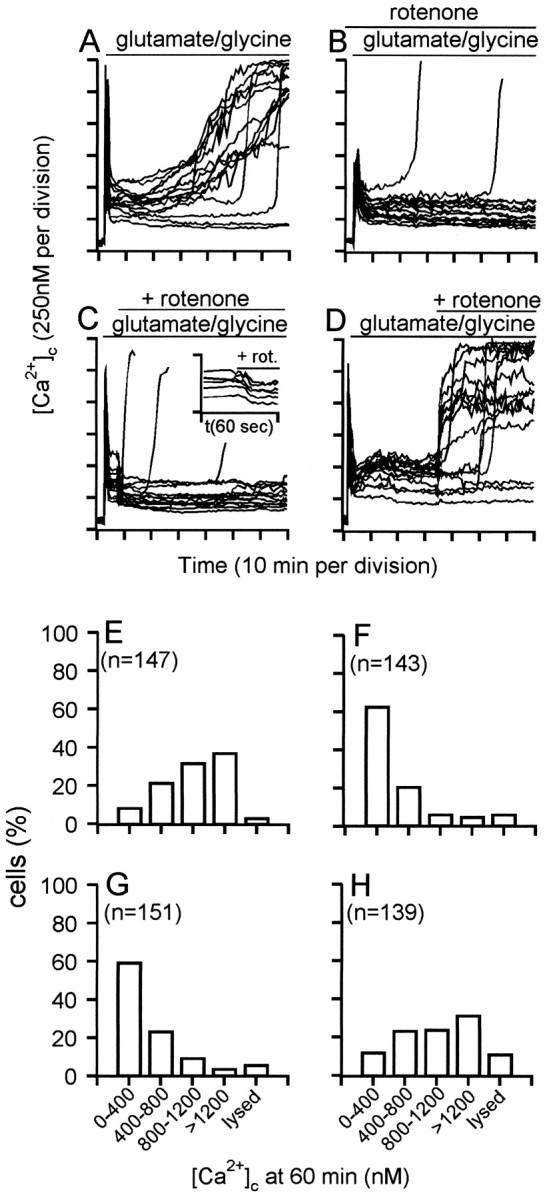
DCD is controlled by the duration of mitochondrial polarization. Granule cells were incubated in the presence of 5 μg/ml oligomycin. Two micromolar rotenone was added 2 min before (B), 5 min after (C), or 35 min after (D) addition of 100 μmglutamate–10 μm glycine. Inset inC shows the change in [Ca2+]c produced by rotenone addition 5 min after the amino acids. E–H, The frequency with which the indicated range of [Ca2+]cis observed in individual somata 60 min after glutamate–glycine addition, corresponding to experiments shown in A–D, respectively. Each histogram contains the data from at least 130 somata and four independent experiments. Lysed cells were characterized by a loss of fluorescence and the disappearance of phase-bright somata.
The net accumulation of 45Ca2+ by glutamate-exposed cells with polarized mitochondria is virtually complete within the first 5 min of glutamate exposure (Fig. 2, ○). By then, [Ca2+]c has peaked and declined to an initially stable plateau (Fig. 4A). Thus, because neither free cytoplasmic Ca2+ nor total cellular Ca2+ are changing at 5 min, this suggests that mitochondrial Ca2+ accumulation has reached a steady state by this time. To establish whether the initial period of mitochondrial Ca2+ loading during the first 5 min of glutamate exposure initiates the sequence of events leading to deregulation or whether chronic maintenance of the mitochondria in the Ca2+-loaded state is necessary, the mitochondria were subjected to delayed depolarization by the further addition of rotenone after either 5 (Fig. 4C) or 35 min (Fig.4D) of glutamate exposure. Previous experiments have shown that rotenone plus oligomycin causes ΔΨm to decay within 60 sec (Budd and Nicholls, 1996a), while protons leak back into the matrix.
In contrast to the transient spike of [Ca2+]c that is seen when the mitochondria within Ca2+-loaded cells suddenly release their Ca2+ to the cytoplasm (Budd and Nicholls, 1996a), after rotenone addition, the [Ca2+]c of individual cells falls to a lower plateau level with no detectable spike but with a time course consistent with the relatively slow depolarization of the mitochondrial membrane (Fig. 4C, inset). Because the Mn2+ quench rate is unaffected by mitochondrial depolarization (Fig. 3), it can be concluded that the activity of the plasma membrane Ca2+ efflux pathways is enhanced such that the steady-state between NMDA receptor-mediated uptake and efflux from the cell is achieved at a lower bulk cytoplasmic free Ca2+.
The proportion of cells treated with rotenone 5 min after glutamate addition (Fig. 4C) that maintain a low [Ca2+]c over the next 60 min is not significantly different from those in which rotenone is added before glutamate (Fig. 4B). In contrast, cells whose mitochondria have remained Ca2+-loaded for 35 min before rotenone addition show extensive deregulation, either at the time of rotenone addition or shortly afterward (Fig.4D). In Figure 4, a good correlation is seen between the behavior of individual cells before and after the delayed rotenone addition; those with a low, stable [Ca2+]c do not undergo deregulation after rotenone, those with a rapidly rising [Ca2+]c deregulate immediately, and intermediate cells deregulate after a short delay. The sudden deregulation of Ca2+ after rotenone at 35 min may be attributable to the release of Ca2+ from the mitochondria, overwhelming an already compromised Ca2+ extrusion mechanism. Thus, it appears that it is the duration for which the mitochondria maintain an accumulation of Ca2+, rather than the initial extent of Ca2+ loading (or the duration of glutamate exposure), that defines the sensitivity of the cells to subsequent DCD. Figure 4, E–H, shows the frequency histograms of [Ca2+]c observed at 60 min in randomly selected cells on four to six coverslips for each condition.
DCD is not controlled by [Ca2+]c
Because the effect of mitochondrial depolarization is to lower the initial peak and subsequent plateau of cytoplasmic free Ca2+ (Fig. 4B) and to decrease the extent of 45Ca2+ accumulation by the cells (Fig. 2), it could be argued that the improved survival of the cells was a consequence of a decreased cytoplasmic Ca2+ rather than a specific depletion of Ca2+ from the mitochondrial matrix. To distinguish these possibilities, the survival of cells was compared in the presence of normal (1.3 mm) and elevated (2.6 mm) extracellular Ca2+. In the presence of oligomycin and 2.6 mm Ca2+, glutamate induced rapid and complete deregulation (Fig.5A). In contrast, nearly all the cells survived elevated external Ca2+ for at least 60 min of glutamate exposure in the presence of rotenone plus oligomycin (Fig. 5B), despite a substantial elevation in the [Ca2+]c plateau to levels (470 ± 23.9 nm; n = 15) in excess of the initial plateau seen 10 min after glutamate addition to oligomycin-treated cells in the presence of the normal 1.3 mmCa2+ (401 ± 23.2 nm;n = 15) (Fig. 4A). Thus, deregulation is not a function of the cytoplasmic Ca2+elevation.
Fig. 5.

Mitochondrial depolarization is still neuroprotective in the presence of elevated cytoplasmic Ca2+. Cells were incubated in the presence of 5 μg/ml oligomycin and 2.6 mm external Ca2+ and exposed to 100 μmglutamate–10 μm glycine for the indicated period in the absence (A) or presence (B) of 2 μm rotenone. Where indicated, 1 μmMK-801 was added. C, D, Frequency histograms of [Ca2+]c observed at 60 min in individual somata from a minimum of four independent experiments under the same conditions in the absence (C) or presence (D) of 2 μmrotenone.
The retained viability of the cells in Figure 5B is indicated by the ability of the NMDA receptor antagonist MK-801 to restore the basal [Ca2+]c in the surviving cells (Fig. 5, compare A, B). We can thus conclude that a glutamate-induced elevation of cytoplasmic Ca2+ is not acutely excitotoxic as long as the mitochondria are depolarized.
The reversibility of ICD
In the absence of oligomycin, protonophore addition (Fig.6A) or respiratory chain inhibition by antimycin A (Fig. 6B) or rotenone (Fig. 6C) results in an acute failure of cytoplasmic Ca2+ homeostasis. Under these conditions, the glycolytic capacity of granule cells is insufficient to generate sufficient ATP to fuel the plasma membrane Na+/K+-ATPase and Ca2+-ATPase and also to drive the reversed mitochondrial ATP synthase (Budd and Nicholls, 1996b). Because both acute and delayed Ca2+ deregulation are reported as increases in [Ca2+]c, it is important to establish their mechanisms.
Fig. 6.
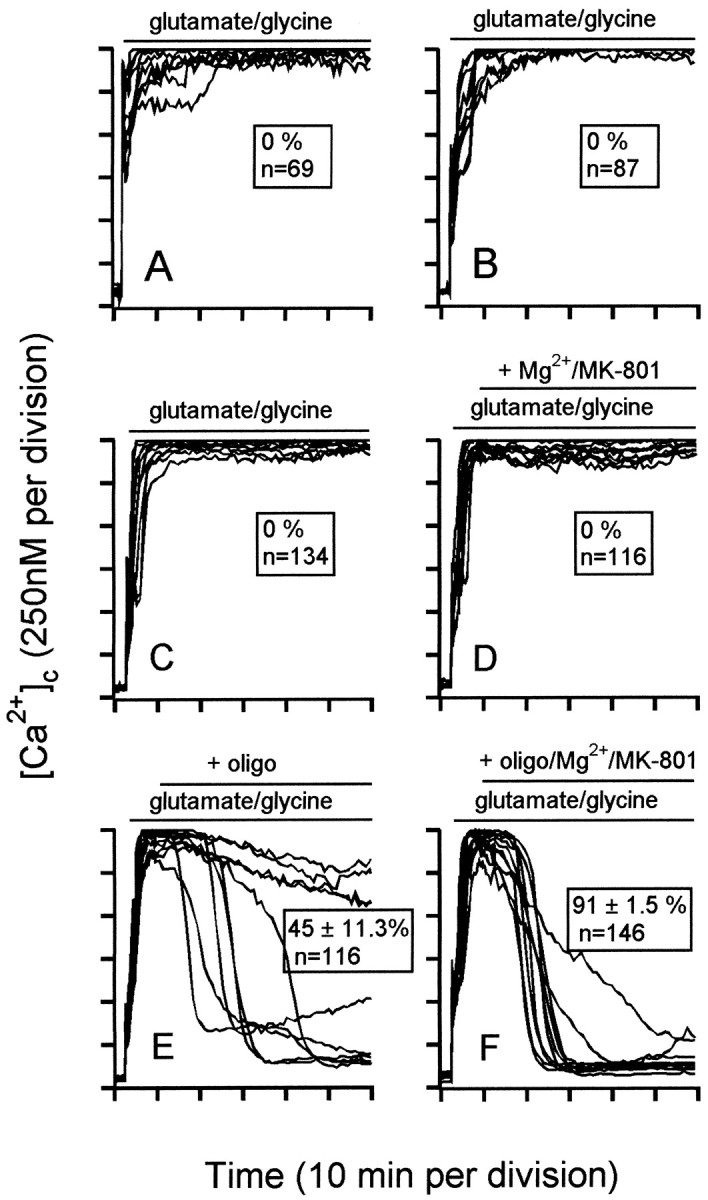
ICD under conditions of energy limitation: conditions for reversal. Granule cells were incubated fromt = 0 in the presence of 1 μmcarbonyl cyanide p-trifluoromethoxyphenyl hydrazone (A), 1 μm antimycin A (B), or 2 μm rotenone (C–F). At 2 min, 100 μmglutamate–10 μm glycine were added. Where indicated, 5 μg/ml oligomycin (oligo) and/or 1 μmMK-801 and 2 mm Mg2+ were added. Note that NMDA receptor inhibition per se does not restore Ca2+ homeostasis. Representative experiments from a minimum of three independent experiments are shown. Values in theinsets represent the proportion of cells (mean ± SEM) showing recovery of [Ca2+]c to <500 nm after 60 min of amino acid exposure.
Acute deregulation of rotenone-treated cells is virtually immediate after glutamate addition, well before the initial Ca2+ “spike” decays to a stable plateau (Fig.6C). No recovery (at least to levels detectable by fura-2, below ∼2 μm) occurs in the subsequent hour. The acute Ca2+ deregulation in the presence of rotenone is glutamate-dependent, because rotenone-treated cells maintain Ca2+ homeostasis for at least 2 hr in the presence of MK-801 (data not shown). However, although MK-801 reverses the acute glutamate-evoked [Ca2+]c elevation in granule cells incubated in the absence of respiratory chain inhibitors (Fig. 5B), MK-801 fails to reverse the acute Ca2+ deregulation in rotenone-treated cells when added 5 min subsequent to glutamate (Fig. 6D). Possible explanations for this failure are that the cells have suffered irreversible damage or that the energetic collapse triggered by NMDA receptor activation is self-sustaining, even after the receptor is inhibited. Addition of oligomycin 5 min after glutamate allows a slow partial recovery of Ca2+ homeostasis in a subset of cells (Fig. 6E). Within the next hour, 45% of cells recovered (defined as a [Ca2+]c <500 nm). Inhibition of both the ATP synthase and the NMDA receptor 5 min after glutamate resulted in an almost total restoration of Ca2+ homeostasis (Fig. 6F). Two conclusions can be drawn. First, acute Ca2+deregulation can be readily reversed. Second, inhibition of the ATP synthase facilitates such reversal. The latter suggests that the reversed ATP synthase continues to drain cytoplasmic ATP, even after NMDA receptor inhibition.
The irreversibility of DCD
DCD occurs with the same facility after glutamate addition to both control and oligomycin-treated cells (Budd and Nicholls, 1996b) and is thus not a consequence of a failure of the oligomycin-sensitive ATP synthase. The rise in [Ca2+]c has been proposed to be primarily a consequence of failed Ca2+ efflux rather than accelerated Ca2+ uptake (Khodorov et al., 1993). Consistent with this, when Ca2+ channel inhibitors, including MK-801, are added sequentially to a coverslip whose individual neurons are at different stages of deregulation (Fig.7), it is found that although cells maintain a stable plateau, they generally show a decreased [Ca2+]c on addition of the inhibitors (Fig. 7, trace c). In contrast, cells that are apparently just about to deregulate (Fig. 7, trace b) and cells that have already deregulated (Fig. 7, trace a) are not rescued.
Fig. 7.

Delayed calcium deregulation is not caused by Ca2+ entry through defined calcium channels. Granule cells were exposed to 100 μm glutamate–10 μm glycine at t = 2 min. After 50 min, a population of cells had deregulated. Ca2+channel antagonists were added sequentially, where indicated by thedotted lines: (1) 1 μm nifedipine; (2) 5 μmConus magnus toxin-MVIIC; (3) 1 μm6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione; and (4) 1 μm MK-801. The [Ca2+]cresponses of three representative cells are shown: cella had deregulated before addition of the inhibitors, cell b initiated deregulation, and cell crecovered to a lower [Ca2+]c.
Population ATP/ADP ratios decline in granule cells exposed to glutamate in the presence or absence of oligomycin (Budd and Nicholls, 1996b). Because ATP is required for two steps in glycolysis, a decline in ATP below a critical threshold can lead to an inhibition of glycolysis that could rapidly feed forward to produce a sudden collapse in ATP production. Granule cells can be maintained by glucose in the presence of oligomycin [in which case, ATP generation is purely glycolytic (Fig. 4)] or in the absence of oligomycin and glucose by the addition of lactate (Fig. 8A) or pyruvate (Fig. 8B), when ATP generation is purely by oxidative phosphorylation. In the absence of any added substrate, the cells deregulated within 5–10 min of glutamate addition (Fig.1E). The response of cells in the presence of these nonglycolytic substrates (Fig.8A,B) is remarkably similar to that in the presence of glucose, including a similar time course for DCD. Thus, DCD is not a phenomenon that is seen only during glycolysis. Furthermore, DCD in the presence of glucose cannot be reversed by the subsequent addition of lactate plus pyruvate (Fig. 8C). Because pyruvate, added directly or generated from lactate, is an effective substrate for the in situ mitochondria (Schurr et al., 1997) and does not require ATP for its metabolism, its addition (in the absence of oligomycin) might have reversed a delayed deregulation that was a consequence of failed glycolysis.
Fig. 8.
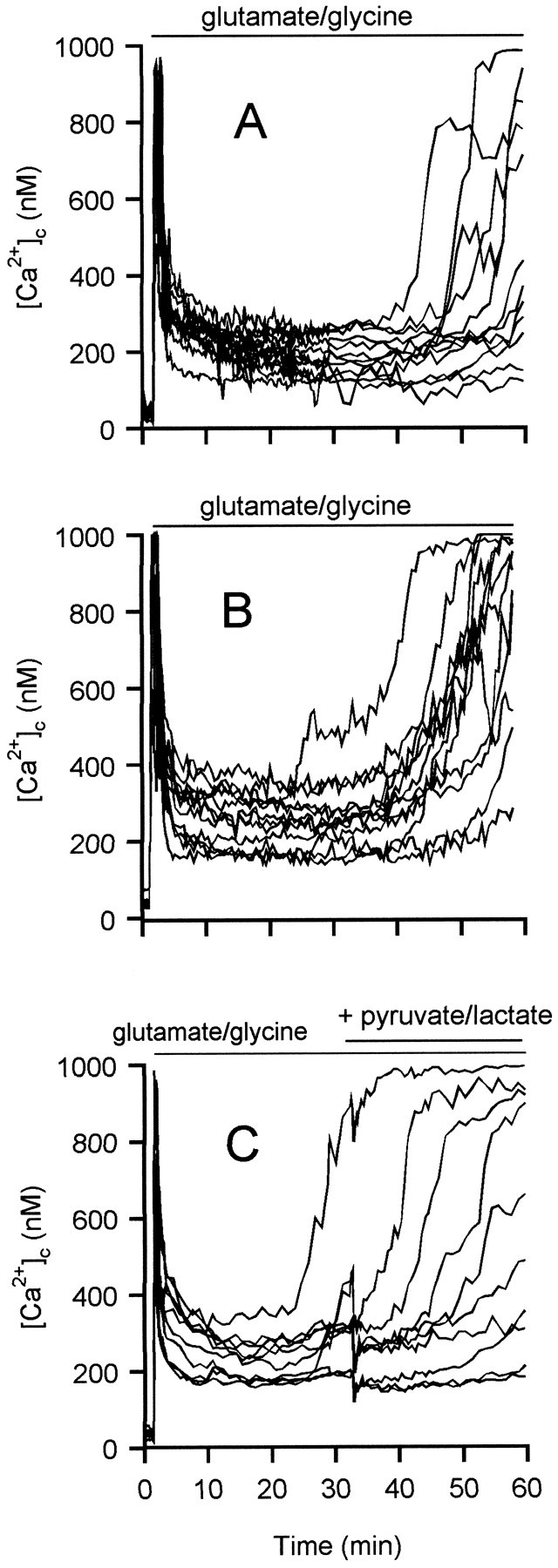
DCD occurs in the presence of nonglycolytic substrates and is not reversed by additional substrate. Granule cells were incubated in the absence of glucose and the presence of 10 mm lactate (A), 10 mmpyruvate (B), or 15 mm glucose (C). In each case, cells were exposed to 100 μm glutamate–10 μm glycine for the period indicated. C, At 30 min, 10 mm lactate plus 10 mm pyruvate were added to provide additional mitochondrial substrate. Oligomycin was not present in these experiments.
DCD is not prevented by inhibitors of the mitochondrial permeability transition
The mitochondrial permeability transition (MPT) is a permeabilization of the inner membrane to solutes up to 1500 Da, which can be observed with isolated mitochondria as a consequence of Ca2+ overload in the presence of phosphate under oxidizing conditions (Zoratti and Szabo, 1995; Vercesi et al., 1997). The MPT results in mitochondrial swelling, rupture of the outer membrane, and release of cytochrome c. In isolated mitochondria, the MPT is inhibited by cyclosporin A andN-methylval-4-cyclosporin (Nicolli et al., 1996) and by bongkrekic acid, which locks the adenine nucleotide translocator in the M-conformation (Halestrap and Davidson, 1990). Most of the experimental evidence for the MPT has come from liver or heart mitochondria or from cellular systems from hepatocytes and astrocytes (Kristal and Dubinsky, 1997). Figure 9 shows that preincubation of the cells with 1 μm cyclosporin A (Fig. 9B) or 1 μmN-methylval-4-cyclosporin (Fig.9C) for 5 min resulted in only a slight delay of the onset of DCD. Increasing the concentration (up to 10 μm) or preincubation time (up to 30 min) for the cyclosporins failed to produce more extensive protection (data not shown). Bongkrekic acid (20 μm preincubated for 5 min) also failed to delay DCD significantly (data not shown).
Fig. 9.

Cyclosporin A andN-methylval-4-cyclosporin fail to protect cells against delayed calcium deregulation. Cells were exposed to 100 μm glutamate–10 μm glycine in the absence of oligomycin. A, Control. Cells were preincubated with 1 μm cyclosporin A (B) or 1 μmN-methylval-4-cyclosporin (C) for 5 min before addition of glutamate–glycine.
DISCUSSION
The complexity of the interactions between mitochondria and the rest of the cell during acute glutamate excitotoxicity has to date hindered elucidation of the mechanism(s) responsible for DCD preceding neuronal necrosis (Tymianski et al., 1993). In this paper, we have separated three conditions under which mitochondria influence glutamate-evoked [Ca2+]c: during the initial response to glutamate, during acute energy deficit, and during DCD itself.
The acute subexcitotoxic [Ca2+]cresponse of granule cells to glutamate is attenuated when mitochondria are depolarized (Fig. 1) under conditions that do not deplete cytoplasmic ATP (Budd and Nicholls, 1996b). No effect on the entry of the Ca2+ surrogate Mn2+ can be detected (Fig. 3), and because both the total45Ca2+ content of the cells (Fig. 2) and [Ca2+]c elevation (Fig. 1) are decreased, this suggests that Ca2+ efflux from the cell is facilitated, allowing a kinetic balance between uptake and efflux to be achieved at a lower indicated [Ca2+]c. Because rotenone–oligomycin-induced depolarization also results in decreased KCl-evoked cytoplasmic elevations (Budd and Nicholls, 1996a), it can be concluded that mitochondria can influence the re-extrusion of Ca2+ entering via both NMDA receptors and voltage-activated ion channels and that extrusion can be activated during the rising phase of the fura-2 transient sufficiently rapidly to limit the peak [Ca2+]c.
The plasma membrane Na+ electrochemical gradient is decreased in the presence of glutamate as a consequence of plasma membrane depolarization and cytoplasmic Na+elevation (Kiedrowski et al., 1994). Under these conditions, Na+/Ca2+ exchange would be thermodynamically incapable of extruding Ca2+, and the plasma membrane Ca2+-ATPase must be considered the dominant pathway of Ca2+ extrusion from the cell (Khodorov et al., 1995). In addition to requiring ATP, the activity of this calmodulin-activated ion pump is highly dependent on the local Ca2+ concentration in its environment (Monteith and Roufogalis, 1995; Werth et al., 1996) and will sense the high subplasmalemmal Ca2+ concentration resulting from the activation of proximal ion channels (Monteith and Roufogalis, 1995;Marsault et al., 1997). The local Ca2+ in this domain, monitored by recombinant targeted aequorin, can exceed ten times the bulk [Ca2+]c during ion channel activation (Marsault et al., 1997). The mitochondria within the small granule cell somata are located close to the plasma membrane (Van-Vliet et al., 1989), and it is possible that they may compete for this local Ca2+, decreasing the activity of the Ca2+-ATPase and allowing the bulk cytoplasmic [Ca2+]c detected by the Ca2+ indicator to rise. Additionally, it is clear that any restriction in ATP synthesis by respiratory inhibition (Fig.6) or substrate limitation (Fig. 1E) enhances the cytoplasmic Ca2+ signals.
We reported previously that DCD was strongly inhibited when mitochondria were depolarized before glutamate (Budd and Nicholls, 1996b), and the results reported in Figure 4 extend this by demonstrating that DCD is a function of the period for which the mitochondria are polarized. Thus, the correlation reported by Tymianski et al. (1993) between the duration of glutamate exposure and the subsequent extent of DCD may be more accurately restated as the duration for which mitochondria are Ca2+-loaded. An important control (Fig. 5) is that mitochondrial depolarization is protective, even in the presence of elevated external Ca2+, when the initial peak and plateau [Ca2+]c responses of rotenone–oligomycin-pretreated cells (Fig. 5B) are at least as great as those seen in the presence of oligomycin and normal extracellular Ca2+ (Fig. 4A). Thus, the protective effect of mitochondrial depolarization is not related to its ability to maintain lower cytoplasmic Ca2+ concentrations.
Because we and most other groups rely heavily on measurements of cytoplasmic free Ca2+, it is important to establish the distinction between DCD and the virtually ICD of cytoplasmic Ca2+ seen under conditions of acute energy limitation (Fig. 6) when Ca2+ extrusion from the cell will be inhibited. Glutamate-induced ICD can be seen in these cells during substrate limitation (Fig. 1E) and after the addition of protonophore (Fig. 6A) or respiratory chain inhibitors (Fig. 6B,C) in the absence of oligomycin, when ATP synthase reversal rapidly depletes cytoplasmic ATP (Budd and Nicholls, 1996b). Interestingly, although ICD is induced by glutamate addition, it is not reversed by NMDA receptor inhibition (Fig. 6D) unless the ATP synthase is inhibited (Fig. 6E,F). This suggests that ATP hydrolysis by the mitochondria may deplete cytoplasmic ATP to the extent that the ATP-requiring steps in glycolysis are unable to reactivate glycolysis (Erecinska et al., 1996). Significantly, after an initial enhancement caused by the Pasteur effect, glycolysis rapidly fails in rotenone-treated isolated nerve terminals (Kauppinen and Nicholls, 1986).
Whereas ICD can be reversed by restoring metabolic competence, DCD cannot be reversed by NMDA receptor inhibition in the presence (Fig.5A) or absence (Fig. 7) of oligomycin or by the addition of alternative substrates, such as lactate or pyruvate (Fig.8C). ATP generation by lactate-derived pyruvate is by oxidative phosphorylation; thus, DCD cannot be ascribed to failed glycolysis. Conversely, the ability to see DCD in the presence of oligomycin eliminates a failure of oxidative phosphorylation. Khodorov et al. (1996a) have reported no increase in the rate of Ca2+ entry into granule cells undergoing DCD, as determined by Mn2+-induced fura-2 quenching, and this is consistent with our failure to reverse or stop DCD by a cocktail of channel inhibitors (Fig. 7).
The MPT can be readily observed with isolated mitochondria incubated with Ca2+ in the presence of phosphate and is facilitated by oxidative stress and inhibited by adenine nucleotides, bongkrekic acid, oligomycin, and certain cyclosporin derivatives (for review, see Zoratti and Szabo, 1995; Bernardi and Petronilli, 1996). Evidence for the involvement of the MPT in intact cells is based primarily on the action of cyclosporin A. However, this immunosuppressant, in complex with its cyclophilin, is a potent inhibitor of calcineurin (Sabatini et al., 1997) and will result in a hyperphosphorylated state of a multiplicity of proteins. Thus, although cyclosporin A inhibits glutamate-induced necrosis and apoptosis in granule cells (Ankarcrona et al., 1996), this is attributable to calcineurin inhibition rather than inhibition of the MPT. We observed only a marginal retardation of DCD with cyclosporin A, the more selective N-methylval-4-cyclosporin, or bongkrekic acid and thus have no convincing evidence for a significant role for the MPT under these conditions. Specifically, DCD occurs in the presence of each of these agents (Fig. 9); thus, assuming their effectiveness in this preparation, DCD cannot be a manifestation of the permeability transition. It should also be borne in mind that oligomycin, which is present routinely in these experiments, is also an inhibitor of the MPT (Nicholls and Åkerman, 1982).
What then is the mechanism of DCD and by extension acute glutamate-induced neuronal necrosis? Failure of oxidative phosphorylation can be eliminated, because the phenomenon occurs after the same delay in oligomycin-treated cells (Budd and Nicholls, 1996b); this also argues against an inexorable rise in cellular ATP demand, because oligomycin-treated cells have a restricted ATP-generating capacity, relying entirely on glycolysis. Failure of glycolysis itself is inconsistent with the inability of the additional substrate lactate to rescue deregulating cells (Fig. 8). Activation of Ca2+ entry pathways is inconsistent with the failure of a cocktail of channel inhibitors to affect the final rise in [Ca2+]c (Fig. 7) and with the failure to observe enhanced Mn2+ quenching during DCD (Khodorov et al., 1996a). Release of mitochondrial Ca2+ stores caused by the MPT is inconsistent with the ineffectiveness of three inhibitors of the permeability transition (four including oligomycin) (Figure 9). A factor dependent on an elevation in [Ca2+]c can be eliminated, because cells with depolarized mitochondria withstand a high cytoplasmic Ca2+ without undergoing DCD (Fig.5). Almost by elimination, one is forced to focus on inhibition of the plasma membrane Ca2+-ATPase as a consequence of a process dependent on the duration for which the closely adjacent mitochondria are polarized and loaded with Ca2+(Fig. 4). It is notable that plasma membrane efflux can cope with Ca2+ released by depolarizing mitochondria after 5 min (Fig. 4C) but not after 35 min (Fig.4D).
Oxidative stress is an obvious candidate for this mechanism. Enhanced superoxide generation by glutamate-exposed neurons has been extensively reported (Lafon-Cazal et al., 1993; Culcasi et al., 1994; Gunasekar et al., 1995), although assays involving the generation of products whose fluorescence is dependent on ΔΨm (e.g., hydroethidine oxidation to ethidium monomer and dihydrorhodamine-123 oxidation to rhodamine-123) may be ambiguous under conditions that are associated also with mitochondrial depolarization (Budd et al., 1997). However, enhanced mitochondrial superoxide generation per se may be insufficient to induce DCD, because the protection afforded by the combination of oligomycin and antimycin A (Nicholls and Budd, 1998), which leads to enhanced superoxide generation (Budd et al., 1997), is similar to that seen with rotenone–oligomycin, which does not (R. F. Castilho and D. G. Nicholls, unpublished observations). Finally, although the plasma membrane Ca2+-ATPase has been reported to be very sensitive to oxidative damage (Zaidi and Michaelis, 1998), definitive proof of oxidative damage to the Ca2+-ATPase is still lacking.
Two additional studies have demonstrated the acute neuroprotective effect of mitochondrial depolarization. Sengpiel et al. (1998) have recently reported that rotenone–oligomycin protected primary rat hippocampal neurons from NMDA excitotoxicity, whereas Stout et al. (1998) observed that protonophore increased the cytoplasmic Ca2+ responses of rat forebrain neurons but decreased excitotoxicity. Thus, our observation may be applicable to a variety of neurons.
Footnotes
This work was supported by Wellcome Trust Grant 040237 and the Medical Research Council (research studentships to S.L.B. and M.W.W.). We thank Hazel Leith for expert technical assistance.
Correspondence should be addressed to D. G. Nicholls, Neurosciences Institute, Department of Pharmacology and Neuroscience, University of Dundee, Ninewells Medical School, Dundee DD1 9SY, Scotland, United Kingdom.
Dr. Hansson’s present address: Wallenberg Neuroscience Center, Department of Physiology and Neuroscience, Lund University, Soelvegatan 17, Lund S-223 62, Sweden.
Dr. Budd’s present address: Cerebrovascular and Neuroscience Institute, Harvard Medical School, 221 Longwood Avenue, Brigham and Women’s Hospital, Boston, MA 02115.
REFERENCES
- 1.Ankarcrona M, Dypbukt JM, Orrenius S, Nicotera P. Calcineurin and mitochondrial function in glutamate-induced neuronal cell death. FEBS Lett. 1996;394:321–324. doi: 10.1016/0014-5793(96)00959-3. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF. Mitochondria, free radicals, and neurodegeneration. Curr Opin Neurobiol. 1996;6:661–666. doi: 10.1016/s0959-4388(96)80100-0. [DOI] [PubMed] [Google Scholar]
- 3.Beal MF, Howell N, Bodis-Wollner I. Mitochondria and free radicals in neurodegenerative disease. Wiley-Liss; New York: 1997. [Google Scholar]
- 4.Bernardi P, Petronilli V. The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J Bioenerg Biomembr. 1996;28:131–138. doi: 10.1007/BF02110643. [DOI] [PubMed] [Google Scholar]
- 5.Bossy-Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Budd SL, Nicholls DG. A re-evaluation of the role of mitochondria in neuronal calcium homeostasis. J Neurochem. 1996a;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- 7.Budd SL, Nicholls DG. Mitochondrial calcium regulation and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996b;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- 8.Budd SL, Castilho RF, Nicholls DG. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cerebellar granule cells. FEBS Lett. 1997;415:21–24. doi: 10.1016/s0014-5793(97)01088-0. [DOI] [PubMed] [Google Scholar]
- 9.Courtney MJ, Lambert JJ, Nicholls DG. The interactions between plasma membrane depolarization and glutamate receptor activation in the regulation of cytoplasmic free calcium in cultured cerebellar granule cells. J Neurosci. 1990;10:3873–3879. doi: 10.1523/JNEUROSCI.10-12-03873.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Culcasi M, Lafon-Cazal M, Pietri S, Bockaert J. Glutamate receptors induce a burst of superoxide via activation of nitric oxide synthase in arginine-depleted neurons. J Biol Chem. 1994;269:12589–12593. [PubMed] [Google Scholar]
- 11.Dubinsky JM, Kristal BS, Elizondo-Fournier M. On the probabilistic nature of excitotoxic neuronal death in hippocampal neurons. Neuropharmacology. 1995;34:701–711. doi: 10.1016/0028-3908(95)00041-4. [DOI] [PubMed] [Google Scholar]
- 12.Duchen MR, Biscoe TJ. Relative mitochondrial membrane potential and [Ca2+]i in type I cells isolated from the rabbit carotid body. J Physiol (Lond) 1992;450:33–61. doi: 10.1113/jphysiol.1992.sp019115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erecinska M, Nelson D, Deas J, Silver IA. Limitation of glycolysis by hexokinase in rat brain synaptosomes during intense ion pumping. Brain Res. 1996;726:153–159. doi: 10.1016/0006-8993(96)00324-1. [DOI] [PubMed] [Google Scholar]
- 14.Friel DD, Tsien RW. An FCCP-sensitive Ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i. J Neurosci. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garthwaite G. In vitro neurotoxicity of excitatory acid analogues during cerebellar development. Neuroscience. 1986;17:755–767. doi: 10.1016/0306-4522(86)90043-6. [DOI] [PubMed] [Google Scholar]
- 16.Gunasekar PG, Kanthasamy AG, Borowitz JL, Isom GE. NMDA receptor activation produces concurrent generation of nitric oxide and reactive oxygen species: implication for cell death. J Neurochem. 1995;65:2016–2021. doi: 10.1046/j.1471-4159.1995.65052016.x. [DOI] [PubMed] [Google Scholar]
- 17.Gunter KK, Gunter TE. Transport of calcium by mitochondria. J Bioenerg Biomembr. 1994;26:471–485. doi: 10.1007/BF00762732. [DOI] [PubMed] [Google Scholar]
- 18.Halestrap AP, Davidson AM. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem. 1997;272:3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- 20.Henneberry RC. Excitotoxicity as a consequence of impairment of energy metabolism: the energy-linked excitotoxic hypothesis. In: Beal MF, Howell N, Bodis-Wollner I, editors. Mitochondria and free radicals in neurodegenerative disease. Wiley-Liss; New York: 1997. pp. 111–143. [Google Scholar]
- 21.Isaev NK, Zorov DB, Stelmashook EV, Uzbekov RE, Kozhemyakin MB, Victorov IV. Neurotoxic glutamate treatment of cultured cerebellar granule cells induces Ca2+-dependent collapse of mitochondrial membrane potential and ultrastructural alterations of mitochondria. FEBS Lett. 1996;392:143–147. doi: 10.1016/0014-5793(96)00804-6. [DOI] [PubMed] [Google Scholar]
- 22.Kauppinen RA, Nicholls DG. Failure to maintain glycolysis in anoxic nerve terminals. J Neurochem. 1986;47:1864–1869. doi: 10.1111/j.1471-4159.1986.tb13100.x. [DOI] [PubMed] [Google Scholar]
- 23.Keelan J, Vergun O, Khodorov BI, Duchen MR. Calcium-dependent mitochondrial depolarization specific to toxic glutamate depolarization in cultured rat hippocampal neurones. J Physiol (Lond) 1998;506:75P. doi: 10.1111/j.1469-7793.1999.0451m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khodorov BI, Pinelis V, Golovina V, Fajuk D, Andreeva N, Uvarova T, Khaspekov L, Victorov I. On the origin of a sustained increase in cytosolic Ca2+ concentration after a toxic glutamate treatment of the nerve cell culture. FEBS Lett. 1993;324:271–273. doi: 10.1016/0014-5793(93)80132-e. [DOI] [PubMed] [Google Scholar]
- 25.Khodorov BI, Pinelis V, Vergun O, Storozhevykh T, Fajuk D, Vinskaya N, Arsenyeva E, Khaspekov L, Lyzin A, Isaev N, Andreeva N, Victorov I. Dramatic effects of external alkalinity on neuronal calcium recovery following a short-duration glutamate challenge: the role of the plasma membrane Ca2+/H+ pump. FEBS Lett. 1995;371:249–252. doi: 10.1016/0014-5793(95)00894-f. [DOI] [PubMed] [Google Scholar]
- 26.Khodorov BI, Fayuk DA, Koshelev SG, Vergun OV, Pinelis VG, Vinskaya NP, Storozhevykh TP, Arsenyeva EN, Khaspekov LG, Lyzhin AP, Isaev N, Victorov IV, Dubinsky JM. Effect of a prolonged glutamate challenge on plasmalemmal calcium permeability in mammalian central neurones. Mn2+ as a tool to study calcium influx pathways. Int J Neurosci. 1996a;88:215–241. doi: 10.3109/00207459609000616. [DOI] [PubMed] [Google Scholar]
- 27.Khodorov BI, Pinelis V, Vergun O, Storozhevykh T, Vinskaya N. Mitochondrial deenergization underlies neuronal calcium overload following a prolonged glutamate challenge. FEBS Lett. 1996b;397:230–234. doi: 10.1016/s0014-5793(96)01139-8. [DOI] [PubMed] [Google Scholar]
- 28.Khodorov BI, Pinelis VG, Storozhevykh T, Vergun OV, Vinskaya NP. Dominant role of mitochondria in protection against a delayed neuronal Ca overload induced by endogenous excitatory amino acids following a glutamate pulse. FEBS Lett. 1996c;393:135–138. doi: 10.1016/0014-5793(96)00873-3. [DOI] [PubMed] [Google Scholar]
- 29.Kiedrowski L, Wroblewski JT, Costa E. Intracellular sodium concentration in cultured cerebellar granule cells challenged with glutamate. Mol Pharmacol. 1994;45:1050–1054. [PubMed] [Google Scholar]
- 30.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 31.Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- 32.Kristal BS, Dubinsky JM. Mitochondrial permeability transition in the central nervous system: induction by calcium-cycling-dependent and -independent pathways. J Neurochem. 1997;69:524–538. doi: 10.1046/j.1471-4159.1997.69020524.x. [DOI] [PubMed] [Google Scholar]
- 33.Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- 34.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 35.Leyssens A, Nowicky AV, Patterson L, Crompton M, Duchen MR. The relationship between mitochondrial state, ATP hydrolysis, [Mg2+]i and [Ca2+]i studied in isolated rat cardiomyocytes. J Physiol (Lond) 1996;496:111–128. doi: 10.1113/jphysiol.1996.sp021669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 37.Manev H, Favaron M, Guidotti A, Costa E. Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death. Mol Pharmacol. 1989;36:106–112. [PubMed] [Google Scholar]
- 38.Marsault R, Murgia M, Pozzan T, Rizzuto R. Domains of high Ca2+ beneath the plasma membrane of living A7r5 cells. EMBO J. 1997;16:1575–1581. doi: 10.1093/emboj/16.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyata H, Silverman HS, Sollott SJ, Lakatta EG, Stern MD, Hansford RG. Measurement of mitochondrial free Ca2+ concentration in living single rat cardiac myocytes. Am J Physiol. 1991;261:H1123–H1134. doi: 10.1152/ajpheart.1991.261.4.H1123. [DOI] [PubMed] [Google Scholar]
- 40.Monteith GR, Roufogalis BD. The plasma membrane calcium pump—a physiological perspective on its regulation. Cell Calcium. 1995;18:459–470. doi: 10.1016/0143-4160(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 41.Nègre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Pénicaud L, Casteilla L. Role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J. 1997;11:809–815. [PubMed] [Google Scholar]
- 42.Nicholls DG, Budd SL. Mitochondria and neuronal glutamate excitotoxicity. Biochim Biophys Acta. 1998;1366:97–112. doi: 10.1016/s0005-2728(98)00123-6. [DOI] [PubMed] [Google Scholar]
- 43.Nicholls DG, Åkerman KEO. Mitochondrial calcium transport. Biochim Biophys Acta. 1982;683:57–88. doi: 10.1016/0304-4173(82)90013-1. [DOI] [PubMed] [Google Scholar]
- 44.Nicholls DG, Ferguson SJ. Bioenergetics 2. Academic; London: 1992. [Google Scholar]
- 45.Nicolli A, Basso E, Petronilli V, Wenger RM, Bernardi P. Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, a cyclosporin A-sensitive channel. J Biol Chem. 1996;271:2185–2192. doi: 10.1074/jbc.271.4.2185. [DOI] [PubMed] [Google Scholar]
- 46.Nicotera P, Leist M. Energy supply and the shape of death in neurons and lymphoid cells. Cell Death Differ. 1997;4:435–442. doi: 10.1038/sj.cdd.4400265. [DOI] [PubMed] [Google Scholar]
- 47.Novelli A, Reilly JA, Lysko PG, Henneberry RC. Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- 48.Park YB, Herrington J, Babcock DF, Hille B. Ca2+ clearance mechanisms in isolated rat adrenal chromaffin cells. J Physiol (Lond) 1996;492:329–346. doi: 10.1113/jphysiol.1996.sp021312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Petit PX, Susin SA, Zamzami N, Mignotte B, Kroemer G. Mitochondria and programmed cell death: back to the future. FEBS Lett. 1996;396:7–13. doi: 10.1016/0014-5793(96)00988-x. [DOI] [PubMed] [Google Scholar]
- 50.Richter C, Schweizer M, Cossarizza A, Franceschi C. Control of apoptosis by the cellular ATP level. FEBS Lett. 1996;378:107–110. doi: 10.1016/0014-5793(95)01431-4. [DOI] [PubMed] [Google Scholar]
- 51.Sabatini DM, Lai MM, Snyder SH. Neural roles of immunophilins and their ligands. Mol Neurobiol. 1997;15:223–239. doi: 10.1007/BF02740635. [DOI] [PubMed] [Google Scholar]
- 52.Savidge JR, Bristow DR. Routes of NMDA- and K+-stimulated calcium entry in rat cerebellar granule cells. Neurosci Lett. 1997;229:109–112. doi: 10.1016/s0304-3940(97)00435-7. [DOI] [PubMed] [Google Scholar]
- 53.Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate excitotoxicity. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schurr A, Payne RS, Miller JJ, Rigor BM. Glia are the main source of lactate utilized by neurons for recovery of function posthypoxia. Brain Res. 1997;774:221–224. doi: 10.1016/s0006-8993(97)81708-8. [DOI] [PubMed] [Google Scholar]
- 55.Scorrano L, Petronilli V, Bernardi P. On the voltage dependence of the mitochondrial permeability transition pore—a critical appraisal. J Biol Chem. 1997;272:12295–12299. doi: 10.1074/jbc.272.19.12295. [DOI] [PubMed] [Google Scholar]
- 56.Sengpiel B, Preis E, Krieglstein J, Prehn JHM. NMDA-induced superoxide production and neurotoxicity in cultured rat hippocampal neurons: role of mitochondria. Eur J Neurosci. 1998;10:1903–1910. doi: 10.1046/j.1460-9568.1998.00202.x. [DOI] [PubMed] [Google Scholar]
- 57.Simpson PB, Challiss RAJ, Nahorski SR. Divalent cation entry in cultured rat cerebellar granule cells measured using Mn2+ quench of fura-2 fluorescence. Eur J Neurosci. 1995;7:831–840. doi: 10.1111/j.1460-9568.1995.tb01070.x. [DOI] [PubMed] [Google Scholar]
- 58.Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- 59.Tan S, Sagara Y, Schubert D. Glutamate toxicity: an unusual pathway of cell death. Soc Neurosci Abstr. 1997;23:542. [Google Scholar]
- 60.Thayer SA, Miller RJ. Regulation of the intracellular free calcium concentration in single rat dorsal root ganglion neurones in vitro. J Physiol (Lond) 1990;425:85–115. doi: 10.1113/jphysiol.1990.sp018094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van-Vliet BJ, Sebben M, Dumuis A, Gabrion J, Bockaert J, Pin JP. Endogenous amino acid release from cultured cerebellar neuronal cells: effect of tetanus toxin on glutamate release. J Neurochem. 1989;52:1229–1239. doi: 10.1111/j.1471-4159.1989.tb01870.x. [DOI] [PubMed] [Google Scholar]
- 63.Vercesi AE, Kowaltowski AJ, Grijalba MT, Meinicke AR, Castilho RF. The role of reactive oxygen species in mitochondrial permeability transition. Biosci Rep. 1997;17:43–52. doi: 10.1023/a:1027335217774. [DOI] [PubMed] [Google Scholar]
- 64.Werth JL, Usachev YM, Thayer SA. Modulation of calcium efflux from cultured rat dorsal root ganglion neurons. J Neurosci. 1996;16:1008–1015. doi: 10.1523/JNEUROSCI.16-03-01008.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.White RJ, Reynolds IJ. Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J Neurosci. 1995;15:1318–1328. doi: 10.1523/JNEUROSCI.15-02-01318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.White RJ, Reynolds IJ. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang J, Liu XS, Bhalla K, Kim CN, Ibrado AM, Cai JY, Peng TI, Jones DP, Wang XD. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 68.Zaidi A, Michaelis ML. Sensitivity of synaptic membrane Ca-ATPase to oxidative stress. J Neurochem. 1998;70:S26A. [Google Scholar]
- 69.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]