

Abstract
Nerve growth factor (NGF) is a required differentiation and survival factor for sympathetic and a majority of neural crest-derived sensory neurons in the developing vertebrate peripheral nervous system. Although much is known about the function of NGF, the intracellular signaling cascade that it uses continues to be a subject of intense study. p21 ras signaling is considered necessary for sensory neuron survival. How additional intermediates downstream or in parallel may function has not been fully understood yet. Two intracellular signaling cascades, extra cellular regulated kinase (erk) and phosphatidylinositol-3 (PI 3) kinase, transduce NGF signaling in the pheochromocytoma cell line PC12. To elucidate the role these cascades play in survival and differentiation, we used a combination of recombinant adenoviruses and chemical inhibitors to perturb these pathways in sensory neurons from wild-type mice and mice deficient for neurofibromin in which the survival and differentiation pathway is constitutively active. We demonstrate that ras activity is both necessary and sufficient for the survival of embryonic sensory neurons. Downstream of ras, however, the erk cascade is neither required nor sufficient for neuron survival or overall differentiation. Instead, the activity of PI 3 kinase is necessary for the survival of the wild-type and neurofibromin-deficient neurons. Therefore, we conclude that in sensory neurons, NGF acts via a signaling pathway, which includes both ras and PI 3 kinase.
Keywords: NGF, p21 ras, PI 3 kinase, sensory neuron survival, recombinant adenovirus, neurotrophin signaling
The neurotrophin family of growth factors is required by a variety of neurons throughout the vertebrate nervous system. The neurotrophins and their receptors, the Trk family of receptor tyrosine kinases, play a critical role during development by providing both differentiation and survival signals to the maturing neurons (Segal and Greenberg, 1996; Snider and Silos-Santiago, 1996). Although the necessity of neurotrophins and Trks for neuron development has been well established using germ line mutations in mice (Conover and Yancopoulos, 1997), the intracellular signaling cascades used by the neurotrophins to exert their effects are not yet fully understood.
The majority of information concerning the intracellular signaling induced by the neurotrophins has been collected in the pheochromocytoma cell line PC12 cells, which, after exposure to nerve growth factor (NGF), differentiate to resemble sympathetic neurons (Tischler and Greene, 1975). NGF also acts as a survival factor for PC12 cells (Greene, 1978). Studies in PC12 cells have implicated the intermediates of the ras/extra cellular regulated kinase (erk) signaling cascade in both of these NGF-induced effects (Szeberenyi et al., 1990;Rukenstein et al., 1991; Wood et al., 1993; Cowley et al., 1994; Kaplan and Miller, 1997; Klesse et al., 1998). Activated ras induces differentiation of these cells in the absence of NGF (Bar-Sagi and Feramisco, 1985), whereas inhibition of ras signaling via dominant negative (DN) or function-blocking antibodies inhibits differentiation induced by NGF (Hagag et al., 1986; Szeberenyi et al., 1990). Other intermediates in this pathway function similarly (Wood et al., 1993; Cowley et al., 1994).
The role of the ras/erk cascade in NGF-induced survival of PC12 cells is less clear. Activated intermediates of ras, raf, and map/erk kinase (mek) are capable of supporting PC12 cell survival in the absence of serum or NGF. However, inhibition of this cascade does not inhibit cell survival promoted by NGF (Klesse et al., 1998). On the other hand, inhibition of phosphatidylinositol-3 (PI 3) kinase activity blocks NGF-induced PC12 cell survival (Yao and Cooper, 1995).
In primary neurons, ras activity has been implicated in neurotrophin-induced survival. Without neurotrophic support, wild-type embryonic sensory and sympathetic neurons will undergo a characteristic apoptosis (Edwards and Tolkovsky, 1994). Inhibition of ras prevents NGF from inducing survival in sensory (Borasio et al., 1993) and sympathetic neurons (Nobes and Tolkovsky, 1995), whereas an activated form of ras will promote survival (Borasio et al., 1989,1996; Nobes et al., 1996). Another important finding implicating ras signaling in neuron survival arose from the analysis of mice harboring a germ line mutation in the ras-GAP gene, NF1. NF1 is the causal gene of human Von Recklinghausen neurofibromatosis type 1, a heritable disorder associated with abnormalities in neural crest-derived tissues (for review, see Bernards, 1995). Neurons isolated from NF1-deficient embryos survive and differentiate in the absence of neurotrophin support (Vogel et al., 1995). Therefore, the deregulation of ras signaling results in neuron survival, likely via the activation of effectors downstream of ras.
In this study, we explore the role of two ras effector cascades, erk and PI 3 kinase, in sensory neurons. We generated recombinant adenoviruses that express each of the intermediates in the ras/erk pathway in their wild-type, DN, and constitutively active (CA) forms. We demonstrate that although ras activity is required and sufficient for promoting sensory neuron survival, the activities of raf, mek, and erk are not required for survival or overall differentiation. Instead, the activity of PI 3 kinase was necessary, together with ras, for NGF-induced survival of sensory neurons.
MATERIALS AND METHODS
Generation of recombinant adenovirus. The replication-defective recombinant adenoviruses that were generated are fully described elsewhere (Klesse et al., 1998). Signaling mutations that were used included the following: for ras, DN ras N17 (Feig and Cooper, 1988) and CA ras V12 (Tabin et al., 1982); for raf, DN raf with the kinase domain deleted (C4) (Bruder et al., 1992), CA raf with the N-terminal domain deleted (BXB) (Heidecker et al., 1990), and X raf with the kinase domain deleted and the ras binding site mutated (PM) (Zhang et al., 1993); for mek, CA mek with serines 217 and 221 mutated to glutamic acid, and DN mek with serine 217 (the first phosphorylation site) deleted (Cowley et al., 1994); for erk2, DN erk2 had mutations in the ATP binding domain (Robbins et al., 1993). The procedure for the generation of recombinant adenovirus is briefly as follows. Each of the above cDNA clones was inserted into the pAC-CMV vector. The vectors pJM17, containing the adenovirus 5 genome, and pAC-CMV (Gomez-Foix et al., 1992), containing the gene of interest, were cotransfected via calcium phosphate into HEK 293 cells. HEK 293 cells were maintained in Cellgro (Herndon, VA) DMEM supplemented with 10% heat-inactivated fetal bovine serum (Hyclone, Logan, UT), 1% penicillin–streptomycin (Life Technologies, Grand Island, NY), and 1% l-glutamine (Life Technologies) until a recombination event resulted in cell lysis. Recombinant adenoviruses were verified with Southern and Western analyses and clonally purified via at least one round of plaque purification. The infectivity of each recombinant adenovirus was determined by plaque assay. The biological activity of each of the generated recombinant adenoviruses was tested and verified in PC12 cells (Klesse et al., 1998).
Dorsal root ganglion cultures. Embryos were isolated from killed females, and the embryonic stage was verified. An appropriate number of dorsal root ganglia were dissected from each embryo using watchmaker’s forceps and electrolytically sharped Tungsten needles in HBSS (Life Technologies) with 1% penicillin–streptomycin (Life Technologies). Routinely, two to three ganglia were isolated per plate for survival assays, whereas 25–30 ganglia were isolated per plate for Western analysis. Ganglia were washed once with Dulbecco’s PBS (DPBS) (Life Technologies) without calcium and magnesium and were trypsinized with 0.05% trypsin (Life Technologies) in DPBS for 13 min at 37°C. Trypsinized ganglia were washed twice with serum-containing neuron media (F14; Life Technologies), 5% heat-inactivated fetal bovine serum (Hyclone), 5% heat-inactivated horse serum (JRH Biologicals, Lenexa, KS), 0.5% penicillin–streptomycin (Life Technologies), and 0.5%l-glutamine (Life Technologies) to inactivate the trypsin. Ganglia were dissociated to single cells with a fire-polished Pasteur pipette and were added dropwise to 5 × 5 mm grided Nunclon (Roskilde, Denmark) tissue culture plates coated with polyornithine (Sigma, St. Louis, MO) and laminin (Life Technologies). Where indicated, NGF (7S; Life Technologies) was added to the culture at 10 ng/ml. Neurons were maintained in a humidified incubator at 37°C with 4% CO2. Four hours after initial plating, recombinant adenovirus was added to the cultures at 2 × 105 pfu. Neurons were counted every 24 hr on an Olympus Optical (Tokyo, Japan) IMT-2 inverted microscope under phase contrast. In each experiment, 500–800 neurons were counted per plate. For NGF-blocking experiments, cells were maintained as above for 48 hr. After initial counts were collected, 1 ml of neuron media was replaced, and anti-NGF antibodies (Boehringer Mannheim, Indianapolis, IN) were added to the culture at 25 ng/ml. For inhibitor experiments, the chemical inhibitors were added after the first counting of the neurons, 24 hr after initial plating. LY294002 (Sigma), wortmannin (Sigma), and PD98059 (New England Biolabs, Beverly, MA) were added at the indicated concentrations. DMSO (Sigma), the carrier for each of the inhibitors, was added in identical amounts as a control. All statistics were performed using the Sigma Stat program (Jansen Scientific).
Western analysis. Neurons used for Western analysis were cultured as described above. Twenty to 30 dorsal root ganglia from an embryo were dissociated and plated on a single 35 mm Nunclon tissue culture dish. Four hours after initial plating, the neurons were infected as indicated with 2 × 106 pfu. Twenty-four hours after plating, the neurons were lysed in 300 μl of NP-40 lysis buffer (150 mm NaCl, 50 mm Tris, pH 8.0, and 1% NP-40) containing protease inhibitors (2 μg/ml aproptin, 1 μg/ml pepstatin, 2 μg/ml leupeptin, and 100 μg/ml PMSF). Neuron lysates from each plate were concentrated to 30 μl with Amicon (Beverly, MA) microconcentrators. The concentrated lysates were separated on a 12% polyacrylamide gel and transferred to nitrocellulose. Blots were blocked with 3% nonfat milk in PBS with 0.1% Tween 20. The blots were probed as indicated and developed using the SuperSignal Ultra chemiluminescence kit (Pierce, Rockford, IL). Quantification analysis was performed using the Bio-Rad (Hercules, CA) phosphoimager and Molecular Analyst software. Antibodies used included anti-phosphorylated erk (Promega, Madison, WI), anti-erk (Santa Cruz Biotechnology, Santa Cruz, CA), and goat anti-rabbit (Santa Cruz Biotechnology).
RESULTS
Generation of recombinant adenoviruses
We generated recombinant adenoviruses that express each of the intermediates of the ras/erk signaling cascade in their wild-type, DN, and CA forms (see Materials and Methods) (Klesse et al., 1998). The replication-defective adenovirus expression system has been ideal for use in our primary sensory neuronal cultures, because it is capable of infecting postmitotic cells and exhibits minimal in vitrotoxicity. Each recombinant adenovirus was verified by Southern blot analysis, and the expression levels were tested via Western analysis (data not shown). Clonal stocks of each recombinant adenovirus were isolated and titered using plaque assays on HEK 293 cells. We routinely obtained recombinant adenoviruses with titers in the range of 108–109 pfu/ml.
We first tested the ability of recombinant adenovirus to infect and express genes in primary sensory neuronal cultures. Figure1 demonstrates the infection of neurons isolated from embryonic day 13.5 (E13.5) dorsal root ganglia with a β-galactosidase-expressing recombinant adenovirus. By infecting with 2 × 105 pfu/culture (an approximate multiplicity of infection of 200 virus particles per cell), we routinely observed expression in 90–95% of the cells in culture, with no observable cytotoxicity (Fig. 1D). Adenovirus-mediated toxicity was only observed when cultures were infected with greater than 1 × 107 pfu or when maintained beyond 6 d (data not shown).
Fig. 1.
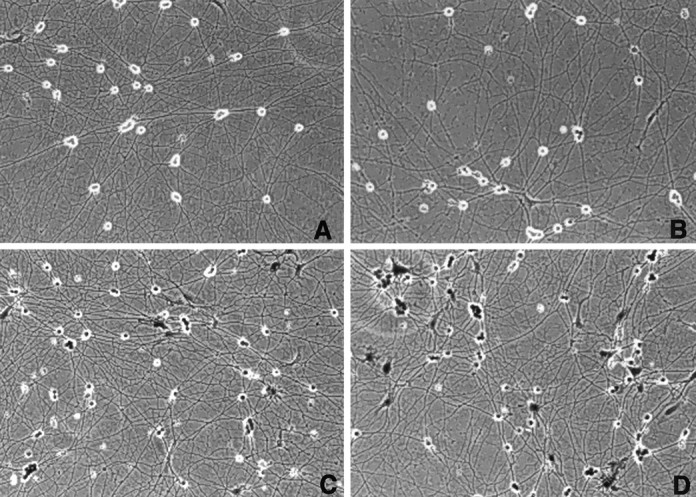
Populations of E13.5 dorsal root ganglia neurons infected with a recombinant adenovirus expressing β-galactosidase.A, No virus infection; B, 1 × 104 pfu; C, 5 × 104 pfu; D, 1 × 105 pfu.
Role of the erk cascade in sensory neuron survival
Wild-type E13.5 DRG neurons require neurotrophic support for appropriate survival and differentiation both in vivo andin vitro (Ernsberger and Rohrer, 1988). The majority of neurons in the DRG at this stage require NGF for survival (Snider and Silos-Santiago, 1996). Previous studies have implicated the necessity of p21 ras in sensory neuron survival (Borasio et al., 1993). One of the best characterized effector pathways downstream of ras is the erk cascade, which includes the serine–threonine kinase raf, the tyrosine–threonine kinase mek, and the serine–threonine kinase erk. This pathway has been widely implicated in transducing differentiation and survival signals induced by NGF-activated TrkA receptors in PC12 cells (Kaplan and Stephens, 1994). To identify the role of the ras/erk cascade in neurotrophin signaling, we first tested the recombinant adenoviruses that express DN forms of the ras/erk intermediates (DN adenoviruses). The DN forms of ras, raf, and mek were all capable of inhibiting erk phosphorylation when sensory neurons were cultured in the presence of NGF (Fig. 2). To control against effects induced by the recombinant adenovirus system, an inactive and inert form of raf, X raf, was used (Zhang et al., 1993). Neurons infected with X raf had levels of erk phophorylation comparable to uninfected neurons (Fig. 2). Unlike the other DN molecules, the DN erk2-expressing adenovirus is itself phosphorylated but can be distinguished from the endogenous erk 2, because it migrates at a higher molecular mass (Fig. 2). Although the DN adenoviruses inhibited signaling through the erk cascade, as measured by erk phophorylation, only DN ras blocked the survival of the DRG neurons in the presence of NGF. This effect was comparable to the action of NGF-neutralizing antibodies (Fig.3A). Of the DRG neurons infected with DN ras, 40.1 ± 5.9% remained alive 72 hr after initial plating and infection compared with 42.4% ± 2.31 in cultures with the NGF-blocking antibody. In contrast, 100 ± 1.5% of the neurons survived in the noninfected controls in the presence of NGF. Neurons infected with X raf (99.1 ± 2.4%), DN mek (98.3 ± 3.9%), and DN erk2 (98.7 ± 6%) exhibited neuronal survival comparable to noninfected controls (Fig. 3A). DN raf-infected neurons exhibited only a slight inhibition of survival (87.7 ± 2.1%; p = 0.048). Overall, DN ras expression had a highly significant effect on the survival of neurons (p = 0.0001). Not only did neurons infected with the DN raf, DN mek, or DN erk2 survive, they exhibited differentiation characteristics that were indistinguishable from uninfected neurons (Fig. 3B). These results indicate that the activities of raf, mek, and erk are not required for survival or overall differentiation of primary embryonic sensory neurons. Ras activity, however, is necessary for neuron survival.
Fig. 2.

DN intermediate-expressing adenoviruses inhibit erk phophorylation in E13.5 sensory neurons. DRG neurons were cultured in the presence of 2 × 106 pfu adenovirus and 10 ng/ml NGF for 24 hr. All cultures in the presence of serum-containing growth media, except the lane −serum, which is cultured in the presence of NGF and F14 alone. Top panel is probed with a phospho-specific erk antibody.Middle panel is probed with an erk antibody, which recognized phosphorylated and unphosphorylated forms. Quantitation analysis was done using the Bio-Rad phosphoimager and Molecular Analyst software.
Fig. 3.
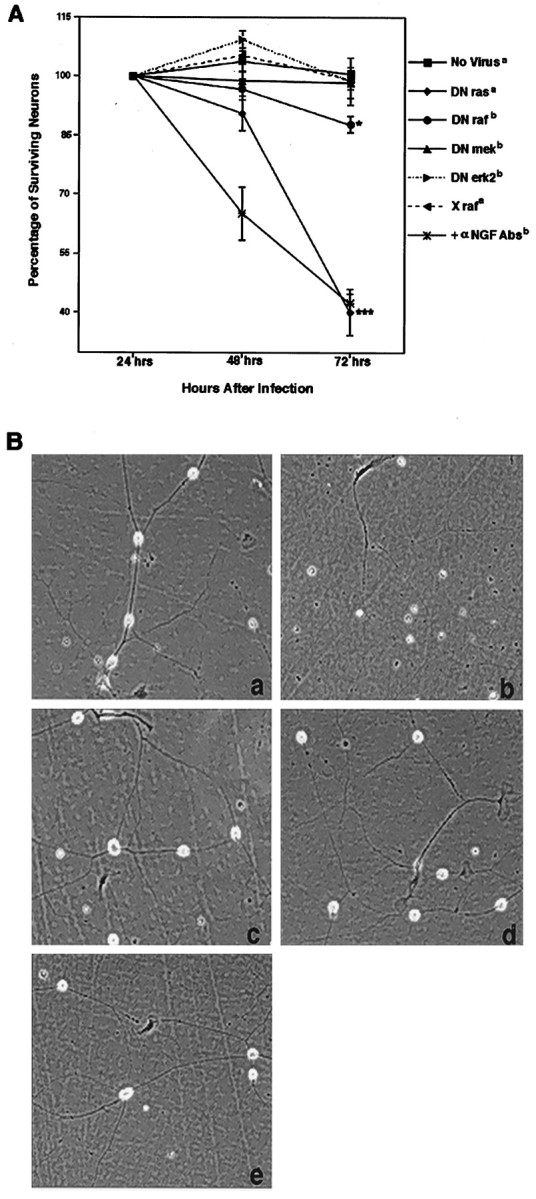
A, DN ras inhibits the survival of wild-type E13.5 DRG neurons in the presence of NGF. Neurons were cultured in the presence of 10 ng/ml NGF and infected with 2 × 105 pfu of each adenovirus. Neurons were counted every 24 hr. *p = 0.046; ***p≤ 0.0001; five independent experiments were counted.a, n = 10; b,n = 6. B, DN adenoviruses do not inhibit neuronal differentiation. Neurons were cultured as above.a, X raf-infected; b, DN ras;c, DN raf; d, DN mek; e, DN erk2.
Ras is sufficient for survival of sensory neurons
Because ras activity appeared to be a critical component of neurotrophin signaling, we next tested whether activity of ras was sufficient for neuron survival. DRG neurons were initially cultured in the presence of NGF and infected with adenoviruses expressing CA forms of either ras, raf, or mek (CA adenoviruses). Forty-eight hours after infection, function-blocking antibodies to NGF were added to the neuronal cultures to neutralize residual NGF activity. As demonstrated in Figure 4, only CA ras was capable of supporting neuron survival in the presence of NGF-blocking antibodies. CA raf and CA mek were unable to support neuron survival above the baseline observed in mock-infected or X raf-infected controls. Therefore, ras activity is not only necessary but is sufficient for maintaining sensory neuron survival. The activities of the downstream ras effectors raf, mek, and erk do not appear to play a role in DRG neuron survival.
Fig. 4.

CA ras is sufficient for DRG neuron survival in the presence of NGF function-blocking antibodies. DRG neurons were cultured in the presence of NGF (10 ng/ml) and 2 × 105 pfu of recombinant adenovirus for 24 hr. Neuron media was then replaced, and 25 ng/ml NGF-blocking antibodies were added. Neurons were counted for the next 2 d.n = 7; ***p = 0.0003.
NF1−/− neurons do not require Erk activity for survival
DRG neurons derived from mice homozygous for a germ line mutation of the NF1 gene (Brannan et al., 1994) do not require neurotrophin support for survival and differentiation (Vogel et al., 1995). Neurofibromin, the protein product of NF1, has a ras-GAP activity, i.e., it negatively regulates ras signaling by stimulating GTP hydrolysis (Ballester et al., 1990; Xu et al., 1990). We have postulated previously that the lack of neurofibromin in NF1−/− neurons results in a continuous level of activated ras (Vogel et al., 1995). Because ras is sufficient for neuron survival (Fig. 4), this continuous level of ras activity may result in neurotrophin-independent survival. To ascertain the level of ras activity in NF1 mutant neurons, we indirectly examined activity by measuring the level of activation of known downstream effectors, the erk kinases. As demonstrated in Figure5, B and C, in the absence of neurotrophins, DRG neurons derived from NF1-deficient embryos exhibit elevated erk phosphorylation levels that are comparable to wild-type neurons in the presence of NGF. Thus, ras signaling is active in NF1-deficient neurons in the absence of neurotrophins. We next infected these neurons with the DN adenoviruses to determine whether NF1-deficient neurons require activity of the ras/erk cascade for survival. Figure 5A demonstrates that, consistent with data obtained with wild-type neurons in the presence of NGF, the activities of raf, mek, and erk were not required for neuron survival. In contrast to wild-type neurons, DN ras did not inhibit the survival of NF1 mutant neurons (Fig. 5A). We find, however, that in contrast to wild-type neurons in which we observed complete inhibition of erk phophorylation with DN ras (Figs. 2,5B,C), DN ras only slightly inhibits erk phosphorylation in NF1-deficient neurons (Fig.5B,C). Thus, the DN ras adenovirus is unable to block ras activity in NF1-deficient neurons. This inability of DN ras to completely block erk phosphorylation or survival is likely because of the nature of the DN ras mutation (N17) (see Discussion).
Fig. 5.

A, DN adenoviruses do not inhibit survival of NF1-deficient DRG neurons. E13.5 DRG neurons were cultured in the absence of NGF. p values calculated in comparison to X raf-infected neurons: no virus, p = 0.3155; DN ras, p = 0.395; DN raf,p = 0.6683; DN mek, p = 0.931; and DN erk2, p = 0.1739. n = 9.B, DN ras only slightly inhibits erk phosphorylation in NF1−/− sensory neurons. NF1−/− neurons were cultured in the presence of 2 × 106 pfu and no NGF. Wild-type neurons were cultured in the presence of 10 ng/ml NGF. Top panel is probed with an anti-phosphorylated erk antibody.Bottom panel is probed with an antibody that recognizes both phosphorylated and unphosphorylated erk. C, Quantitation of erk levels in the above Western analysis. Analysis performed using Molecular Analyst (Bio-Rad) software.Dark trace depicts relative phophorylated erk levels, and light trace represents control erk levels.
We also tested the effects of the CA adenoviruses on NF1-deficient neurons. Because NF1−/− neurons do not require neurotrophins for survival, the addition of NGF function-blocking antibodies to the cultures had no significant effect on survival (data not shown). None of the CA adenoviruses altered the survival of NF1−/− neurons compared with uninfected cultures or X raf-infected controls (data not shown). These results were expected, however, because the ras/erk pathway is already activated, even in the absence of NGF stimulation (Fig. 5B).
PI 3 kinase activity is required for wild-type and NF1 mutant sensory neuron survival
Because ras was required and sufficient for neuron survival but the activities of raf, mek, and erk were not, we tested another ras effector, PI 3 kinase, for a role in sensory neuron survival. PI 3 kinase has previously been reported to interact specifically with ras-GTP and become activated (Rodriguez-Viciana et al., 1994, 1996). The PI 3 kinase cascade, which includes the serine–threonine kinase Akt, has recently been implicated in anti-apoptotic signaling in several cell types, including sympathetic neurons (Crowder and Freeman, 1998). To perturb PI 3 kinase signaling, we tested two widely used chemical inhibitors, wortmannin and LY294002. At low concentrations, each of these PI 3 kinase inhibitors blocked survival of wild-type neurons in the presence of NGF- (Fig.6A) and NF1-deficient neurons in the absence of NGF (Fig. 6B). For instance, 10 μm of LY294002 reduced survival of wild-type neurons to 34% (p = 0.008) and survival of NF1−/− to 14.8% (p = 0.0002). Neither of the PI 3 kinase inhibitors perturbed erk phosphorylation, except at high concentrations (above 100 μm for LY294002), indicating that they acted specifically (data not shown). We also used an inhibitor specific for mek signaling, PD98059. In agreement with the data obtained using the DN adenoviruses, the mek inhibitor did not affect the survival of wild-type (Fig.6A) or NF1−/− sensory neurons (Fig.6B). The mek inhibitor had limited biological activity and was able to inhibit erk phosphorylation for 12 hr after addition (data not shown). Control experiments in which the inhibitors were re-added at 12 hr intervals gave identical results. Thus, we confirm that erk activity is not necessary for neuron survival. Instead, PI 3 kinase activity, along with ras, is required.
Fig. 6.

Inhibitors of PI 3 kinase inhibit survival of DRG neurons. A, Wild-type neurons in the presence of 10 ng/ml NGF. n = 5; **p = 0.008; ***p ≤ 0.0001. B, NF1-deficient neurons in the absence of NGF. DMSO is used as a carrier for each of the indicated inhibitors. n = 4; *p = 0.01–0.05; ***p ≤ 0.0003.
CA ras is unable to support neuron survival in the presence of LY294002
Because CA ras was sufficient for survival of DRG neurons in the presence of anti-NGF antibodies, we next tested the ability of CA ras to promote neuron survival in the presence of the PI 3 kinase inhibitors. In both wild-type (Fig.7A) and NF1-deficient (Fig.7B) neurons, CA ras was not able to support neuron survival above controls in the presence of 10 μm LY294002. This inability of CA ras to support neuron survival when PI 3 kinase activity is blocked supports the model that PI 3 kinase acts downstream of ras in neurotrophin signaling in neurons.
Fig. 7.
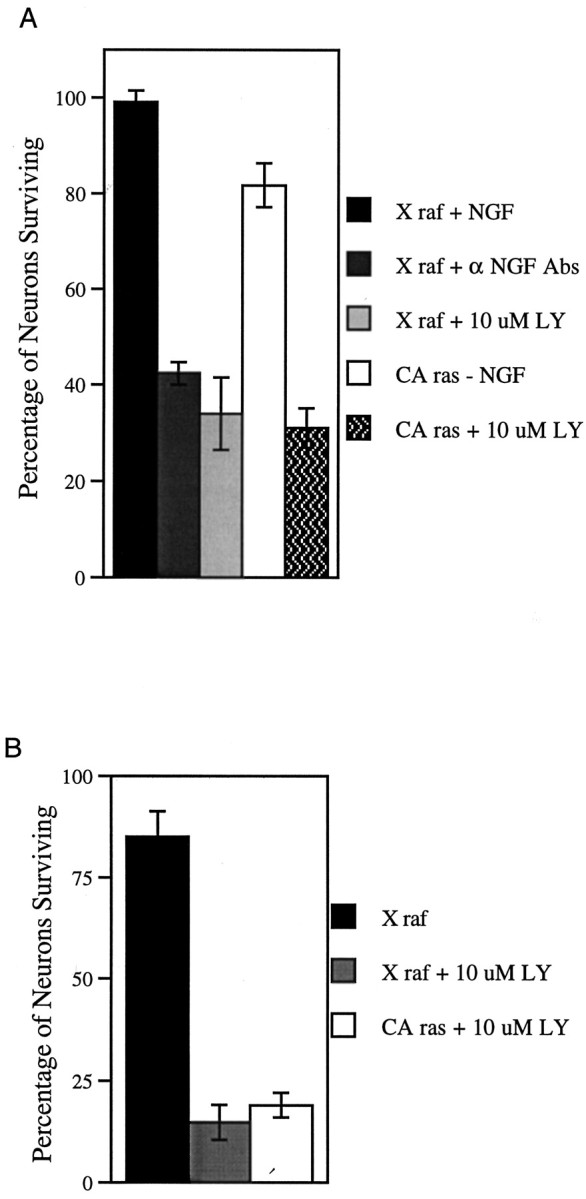
CA ras is not capable of promoting neuron survival in the presence of LY294002. A, Wild-type neurons infected with 2 × 105 pfu of recombinant adenovirus. X raf cultures in the presence of 10 ng/ml NGF. CA ras and X raf plus α NGF antibodies in the presence of 50 ng/ml function-blocking NGF antibodies. n = 4.B, NF1-deficient neurons infected with 2 × 105 pfu recombinant adenovirus in the absence of NGF. n = 3.
DISCUSSION
These studies identify ras and PI 3 kinase as requisite intracellular mediators of neurotrophin survival signaling in sensory neurons. The activity of ras was not only necessary for neuron survival but was also capable of supporting survival in the absence of NGF. By using recombinant adenoviruses, we were able to systematically analyze the endogenous role of each intermediate of the erk cascade in the survival and differentiation of sensory neurons. Our data indicate that none of these intermediates, raf, mek, or erk, are required for sensory neuron survival or overall differentiation. Recent reports using the mek inhibitor PD98059 in sympathetic neurons have indicated that mek activity also is not required for sympathetic neuron survival (Creedon et al., 1996; Virdee and Tolkovsky, 1996). The signaling cascade activated downstream of ras is less well defined. We demonstrate that PI 3 kinase activity is required for neuron survival. PI 3 kinase is already a known effector of ras signaling (Rodriguez-Viciana et al., 1994, 1996), and inhibition of PI 3 kinase blocks the ability of CA ras to promote neuron survival in the absence of NGF. These results implicate a signaling cascade that includes NGF/Trk activation of ras, which activates PI 3 kinase leading to neuron survival.
PI 3 kinase has been implicated in anti-apoptotic signaling in a number of cell types, including sympathetic neurons (Crowder and Freeman, 1998). One known effector of PI 3 kinase is Akt, a serine–threonine kinase also known as protein kinase B or Racα. Akt is activated in a PI 3 kinase-dependent manner in response to stimulation with a number of growth factors (Franke et al., 1995; Datta et al., 1996). Both PI 3 kinase and Akt have been shown recently to be required for the survival of several neuronal cell types, including sympathetic neurons stimulated with NGF and cerebellar neurons stimulated with IGF-1 (Dudek et al., 1997; Crowder and Freeman, 1998). Therefore, it is likely in sensory neurons that Akt is also activated in response to NGF in both a ras- and PI 3 kinase-dependent manner.
How PI 3 kinase and Akt induce survival of such a wide variety of neurons is not yet known but may be linked to the ability of Akt to interact with and phosphorylate the Bcl-2-related protein Bad (Datta et al., 1997; del Peso et al., 1997). Bad is a proapoptotic member of the Bcl-2 family and is postulated to act by binding to and neutralizing the anti-apoptotic Bcl-2-related proteins (Korsmeyer et al., 1993;Korsmeyer, 1995; Yang et al., 1995). Phosphorylation of Bad alters its ability to interact with these family members (Zha et al., 1996). In support of this model, it has been demonstrated that overexpression of Bcl-2 in sympathetic neurons promotes survival in the absence of NGF (Garcia et al., 1997).
Neurofibromin, the protein product of NF1, has been hypothesized to function as a ras-GAP (Ballester et al., 1990; Xu et al., 1990). Neurofibromin has also been implicated, however, in ras-independent functions (Johnson et al., 1994). For instance, inDrosophila, the absence of NF1 had no effect on ras activity (The et al., 1997). Instead, the Drosophila NF1 mutant phenotype was rescued by activation of the cAMP/protein kinase A cascade (Guo et al., 1997; The et al., 1997). Our data are consistent with neurofibromin acting as a ras-GAP in neurotrophin signaling. An effector pathway of ras is activated in NF1-deficient neurons in the absence of neurotrophins. Also, the neurotrophin independence of the NF1 neurons can be mimicked in wild-type neurons by expression of a CA form of ras. Finally, inhibition of PI 3 kinase, another effector of ras, inhibits the ability of NF1-deficient neurons to survive in the absence of neurotrophin. Surprisingly, expression of DN ras in NF1 mutant neurons did not inhibit neuron survival. Western analysis of NF1-deficient neurons in the presence of the DN ras, however, demonstrates the inability of DN ras to completely block signaling to the erks. The inefficiency of DN ras is likely because of the nature of the N17 ras mutation used. N17 ras inhibits activation of effectors because of an increased affinity for GDP (Feig and Cooper, 1988). Therefore, N17 ras binds to activating exchange factors but does not itself become activated. However, in the absence of ras-GAP activity, any endogenous ras-GTP in the cell would be stabilized and immune to the N17 mutant effects. Apparently, the low level of endogenous ras activity that we detect indirectly via erk analysis is sufficient for signaling and, thus, neuron survival. Blocking ras activity directly in NF1−/− neurons with function-blocking Fab fragments inhibits neurotrophin-independent survival, indicating that ras activity is necessary for NF1 mutant neuron survival (K. S. Vogel, M. El-Afandi, L. F. Parada, unpublished observations). Overall, the data presented support a model in which neurofibromin acts a negative regulator of ras in sensory neurons.
Although NGF appears to activate similar signaling cascades in PC12 cells and primary neurons, these pathways do not appear to have the same function in each cell type. In contrast to PC12 cells, inhibition of the erk cascade did not appear to inhibit sensory neuron differentiation. The intermediates ras, raf, mek, and erk are all necessary and sufficient for neurite outgrowth, a hallmark of neuronal differentiation, in PC12 cells (Klesse et al., 1998). When the erk cascade is blocked in sensory neurons, however, no obvious inhibition or retraction of their neurites is observed. Therefore, in sensory neurons, NGF may mediate differentiation via a different signaling cascade. One possibility is PI 3 kinase, whose activity has been linked previously with extension and maintenance of neurites in PC12 cells (Kimura et al., 1994; Jackson et al., 1996). Rap1 signaling has also recently been implicated in PC12 cells in aspects of differentiation, such as the expression of neuronal-specific genes, including sodium channels (York et al., 1998). Because Rap1 is postulated to signal downstream of ras via Raf-B to the erks (York et al., 1998), it is an unlikely candidate based on our data, which shows no obvious effects on differentiation in the presence of DN forms of mek or erk. More detailed analysis may revel a partial role in of Rap1 in neuronal differentiation. Overall, the data presented indicates that perturbing signaling of the erk cascade does not affect morphological differentiation of sensory neurons.
The ability of CA ras to promote survival in the absence of NGF and the rapidity with which the neurons die in the presence of PI 3 kinase inhibitors (<24 hr) indicates that this signaling cascade may be the predominant effector of NGF-induced survival signals. Our results, however, do not preclude the presence of a signaling cascade that actively promotes apoptosis. For instance, in PC12 cells, the Jun kinase cascade (JNK) is necessary for apoptosis induced when NGF is withdrawn (Xia et al., 1995). This signaling cascade, or one similar, may also be required for neuronal apoptosis. In sympathetic neurons, inhibition of c-jun, the transcription factor activated by the JNK pathway, protects the neurons from NGF withdrawal-induced apoptosis (Ham et al., 1995). Besides the JNK cascade, the low-affinity neurotrophin receptor p75 has also been implicated in neuronal apoptosis signaling (Frade et al., 1996; Bredesen and Rabizadeh, 1997). p75 is a member of the tumor necrosis factor cytokine receptor family and has been demonstrated to activate the JNK cascade (Casaccia-Bonnefil et al., 1996). Whether p75 and/or JNK signaling is necessary for sensory neuron apoptosis is currently under investigation. It is possible that inhibition of the survival-inducing cascade (ras/PI 3 kinase) is not sufficient for neuronal apoptosis. Instead, neuronal programmed cell death may also require activation of a cell death-inducing pathway.
In summary, our data confirms the importance of ras signaling in sensory neurons and identifies PI 3 kinase as the downstream effector required for survival. These results were observed in both wild-type neurons and neurons deficient for neurofibromin, whose survival signaling is constitutively active. Our results also support the role of neurofibromin as a ras-GAP in neurotrophin signaling.
Footnotes
This work was supported by National Institute of Neurological Diseases and Stroke Grant ROI-NS34296, Department of Defense Grant DAMD17-97-1-7343, and American Paralysis Association Consortium. We thank M. Cobb (erk), S. Cowley (mek), L. Feig (ras), J. Frost (erk), C. Marshall (mek), U. Rapp (raf), and M. White (ras) for providing the cDNA contructs used in the generation of the recombinant adenovirus. pAC-CMV was provided by B. Gerard, and pJM17 was provided by C. Newgard. The β-galactosidase adenovirus was given to us by J. Hertz and J. Alcorn. We also thank Kristine Vogel for her expertise in the neuronal culture system.
Correspondence should be addressed to Dr. Luis F. Parada, Center for Developmental Biology, University of Texas, Southwestern Medical Center, Dallas, TX 75235-9133.
REFERENCES
- 1.Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, Collins F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63:851–859. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- 2.Bar-Sagi D, Feramisco JR. Microinjection of the ras oncogene protein into PC12 cells induces morphological differentiation. Cell. 1985;42:841–848. doi: 10.1016/0092-8674(85)90280-6. [DOI] [PubMed] [Google Scholar]
- 3.Bernards A. Neurofibromatosis type 1 and Ras-mediated signaling: filling in the GAPs. Biochim Biophys Acta. 1995;1242:43–59. doi: 10.1016/0304-419x(95)00003-x. [DOI] [PubMed] [Google Scholar]
- 4.Borasio GD, John J, Wittinghofer A, Barde YA, Sendtner M, Heumann R. ras p21 protein promotes survival and fiber outgrowth of cultured embryonic neurons. Neuron. 1989;2:1087–1096. doi: 10.1016/0896-6273(89)90233-x. [DOI] [PubMed] [Google Scholar]
- 5.Borasio GD, Markus A, Wittinghofer A, Barde YA, Heumann R. Involvement of ras p21 in neurotrophin-induced response of sensory, but not sympathetic, neurons. J Cell Biol. 1993;121:665–672. doi: 10.1083/jcb.121.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borasio GD, Markus A, Heumann R, Ghezzi C, Sampietro A, Wittinghofer A, Silani V. Ras p21 protein promotes survival and differentiation of human embryonic neural crest-derived cells. Neuroscience. 1996;73:1121–1127. doi: 10.1016/0306-4522(96)00084-x. [DOI] [PubMed] [Google Scholar]
- 7. Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, Buchberg AM, Jenkins NA, Parada LF, Copeland NG. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev 8 1994. 1019 1029[Erratum (1994) 8:2792]. [DOI] [PubMed] [Google Scholar]
- 8.Bredesen DE, Rabizadeh S. p75NTR and apoptosis: Trk-dependent and Trk-independent effects. Trends Neurosci. 1997;20:287–290. doi: 10.1016/s0166-2236(96)01049-1. [DOI] [PubMed] [Google Scholar]
- 9.Bruder JT, Heidecker G, Rapp UR. Serum-, TPA-, and Ras-induced expression from Ap-1/Ets-driven promoters requires Raf-1 kinase. Genes Dev. 1992;6:545–556. doi: 10.1101/gad.6.4.545. [DOI] [PubMed] [Google Scholar]
- 10.Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- 11.Conover JC, Yancopoulos GD. Neurotrophin regulation of the developing nervous system: analyses of knockout mice. Rev Neurosci. 1997;8:13–27. doi: 10.1515/revneuro.1997.8.1.13. [DOI] [PubMed] [Google Scholar]
- 12.Cowley S, Paterson H, Kemp P, Marshall CJ. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
- 13.Creedon DJ, Johnson EM, Lawrence JC. Mitogen-activated protein kinase-independent pathways mediate the effects of nerve growth factor and cAMP on neuronal survival. J Biol Chem. 1996;271:20713–20718. doi: 10.1074/jbc.271.34.20713. [DOI] [PubMed] [Google Scholar]
- 14.Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;18:2933–2943. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Datta K, Bellacosa A, Chan TO, Tsichlis PN. Akt is a direct target of the phosphatidylinositol 3-kinase. Activation by growth factors, v-src and v-Ha-ras, in Sf9 and mammalian cells. J Biol Chem. 1996;271:30835–30839. doi: 10.1074/jbc.271.48.30835. [DOI] [PubMed] [Google Scholar]
- 16.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 17.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 18.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine–threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 19.Edwards SN, Tolkovsky AM. Characterization of apoptosis in cultured rat sympathetic neurons after nerve growth factor withdrawal. J Cell Biol. 1994;124:537–546. doi: 10.1083/jcb.124.4.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ernsberger U, Rohrer H. Neuronal precursor cells in chick dorsal root ganglia: differentiation and survival in vitro. Dev Biol. 1988;126:420–432. doi: 10.1016/0012-1606(88)90151-0. [DOI] [PubMed] [Google Scholar]
- 21.Feig LA, Cooper GM. Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol Cell Biol. 1988;8:3235–3243. doi: 10.1128/mcb.8.8.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frade JM, Rodriguez-Tebar A, Barde YA. Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature. 1996;383:166–168. doi: 10.1038/383166a0. [DOI] [PubMed] [Google Scholar]
- 23.Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 24.Garcia I, Martinou I, Tsujimoto Y, Martinou J-C. Prevention of programmed cell death of sympathetic neurons by the bcl-2 proto-oncogene. Science. 1997;258:302–304. doi: 10.1126/science.1411528. [DOI] [PubMed] [Google Scholar]
- 25.Gomez-Foix AM, Coats WS, Baque S, Alam T, Gerard RD, Newgard CB. Adenovirus-mediated transfer of the muscle glycogen phosphorylase gene into hepatocytes confers altered regulation of glycogen metabolism. J Biol Chem. 1992;267:25129–25134. [PubMed] [Google Scholar]
- 26.Greene LA. Nerve growth factor prevents the death and stimulates the neuronal differentiation of clonal PC12 pheochromocytoma cells in serum-free medium. J Cell Biol. 1978;78:747–755. doi: 10.1083/jcb.78.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo HF, The I, Hannan F, Bernards A, Zhong Y. Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science. 1997;276:795–798. doi: 10.1126/science.276.5313.795. [DOI] [PubMed] [Google Scholar]
- 28.Hagag N, Halegoua S, Viola M. Inhibition of growth factor-induced differentiation of PC12 cells by microinjection of antibody to ras p21. Nature. 1986;319:680–682. doi: 10.1038/319680a0. [DOI] [PubMed] [Google Scholar]
- 29.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 30.Heidecker G, Huleihel M, Cleveland JL, Kolch W, Beck TW, Lloyd P, Pawson T, Rapp UR. Mutational activation of c-raf-1 and definition of the minimal transforming sequence. Mol Cell Biol. 1990;10:2503–2512. doi: 10.1128/mcb.10.6.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jackson TR, Blader IJ, Hammonds-Odie LP, Burga CR, Cooke F, Hawkins PT, Wolf AG, Heldman KA, Theibert AB. Initiation and maintenance of NGF-stimulated neurite outgrowth requires activation of a phosphoinositide 3-kinase. J Cell Sci. 1996;109:289–300. doi: 10.1242/jcs.109.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson MR, DeClue JE, Felzmann S, Vass WC, Xu G, White R, Lowy DR. Neurofibromin can inhibit Ras-dependent growth by a mechanism independent of its GTPase-accelerating function. Mol Cell Biol. 1994;14:641–645. doi: 10.1128/mcb.14.1.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaplan DR, Miller FD. Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol. 1997;9:213–221. doi: 10.1016/s0955-0674(97)80065-8. [DOI] [PubMed] [Google Scholar]
- 34.Kaplan DR, Stephens RM. Neurotrophin signal transduction by the Trk receptor. J Neurobiol. 1994;25:1404–1417. doi: 10.1002/neu.480251108. [DOI] [PubMed] [Google Scholar]
- 35.Kimura K, Hattori S, Kabuyama Y, Shizawa Y, Takayanagi J, Nakamura S, Toki S, Matsuda Y, Onodera K, Fukui Y. Neurite outgrowth of PC12 cells is suppressed by wortmannin, a specific inhibitor of phosphatidylinositol 3-kinase. J Biol Chem. 1994;269:18961–18967. [PubMed] [Google Scholar]
- 36.Klesse LJ, Meyers KA, Marshall CJ, Parada LF (1998) Nerve growth factor induces survival and differentiation through two distinct signaling cascades in PC12 cells. Oncogene, in press. [DOI] [PubMed]
- 37.Korsmeyer SJ. Regulators of cell death. Trends Genet. 1995;11:101–105. doi: 10.1016/S0168-9525(00)89010-1. [DOI] [PubMed] [Google Scholar]
- 38.Korsmeyer SJ, Shutter JR, Veis DJ, Merry DE, Oltvai ZN. Bcl-2/Bax: a rheostat that regulates an anti-oxidant pathway and cell death. Semin Cancer Biol. 1993;4:327–332. [PubMed] [Google Scholar]
- 39.Nobes CD, Tolkovsky AM. Neutralizing anti-p21ras Fabs suppress rat sympathetic neuron survival induced by NGF, LIF, CNTF and cAMP. Eur J Neurosci. 1995;7:344–350. doi: 10.1111/j.1460-9568.1995.tb01069.x. [DOI] [PubMed] [Google Scholar]
- 40.Nobes CD, Reppas JB, Markus A, Tolkovsky AM. Active p21Ras is sufficient for rescue of NGF-dependent rat sympathetic neurons. Neuroscience. 1996;70:1067–1079. doi: 10.1016/0306-4522(95)00420-3. [DOI] [PubMed] [Google Scholar]
- 41.Robbins DJ, Zhen E, Owaki H, Vanderbilt CA, Ebert D, Geppert TD, Cobb MH. Regulation and properties of extracellular signal-regulated protein kinases 1 and 2 in vitro. J Biol Chem. 1993;268:5097–5106. [PubMed] [Google Scholar]
- 42.Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 43.Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996;15:2442–2451. [PMC free article] [PubMed] [Google Scholar]
- 44.Rukenstein A, Rydel RE, Greene LA. Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J Neurosci. 1991;11:2552–2563. doi: 10.1523/JNEUROSCI.11-08-02552.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 46.Snider WD, Silos-Santiago I. Dorsal root ganglion neurons require functional neurotrophin receptors for survival during development. Philos Trans R Soc Lond B Biol Sci. 1996;351:395–403. doi: 10.1098/rstb.1996.0034. [DOI] [PubMed] [Google Scholar]
- 47.Szeberenyi J, Cai H, Cooper GM. Effect of a dominant inhibitory Ha-ras mutation on neuronal differentiation of PC12 cells. Mol Cell Biol. 1990;10:5324–5332. doi: 10.1128/mcb.10.10.5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tabin CJ, Bradley SM, Bargmann CI, Weinberg RA, Papageorge AG, Scolnick EM, Dhar R, Lowy DR, Chang EH. Mechanism of activation of a human oncogene. Nature. 1982;300:143–149. doi: 10.1038/300143a0. [DOI] [PubMed] [Google Scholar]
- 49.The I, Hannigan GE, Cowley GS, Reginald S, Zhong Y, Gusella JF, Hariharan IK, Bernards A. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science. 1997;276:791–794. doi: 10.1126/science.276.5313.791. [DOI] [PubMed] [Google Scholar]
- 50.Tischler AS, Greene LA. Nerve growth factor-induced process formation by cultured rat pheochromocytoma cells. Nature. 1975;258:341–342. doi: 10.1038/258341a0. [DOI] [PubMed] [Google Scholar]
- 51.Virdee K, Tolkovsky AM. Inhibition of p42 and p44 mitogen-activated protein kinase activity by PD98059 does not suppress nerve growth factor-induced survival of sympathetic neurones. J Neurochem. 1996;67:1801–1805. doi: 10.1046/j.1471-4159.1996.67051801.x. [DOI] [PubMed] [Google Scholar]
- 52.Vogel KS, Brannan CI, Jenkins NA, Copeland NG, Parada LF. Loss of neurofibromin results in neurotrophin-independent survival of embryonic sensory and sympathetic neurons. Cell. 1995;82:733–742. doi: 10.1016/0092-8674(95)90470-0. [DOI] [PubMed] [Google Scholar]
- 53.Wood KW, Qi H, D’Arcangelo G, Armstrong RC, Roberts TM, Halegoua S. The cytoplasmic raf oncogene induces a neuronal phenotype in PC12 cells: a potential role for cellular raf kinases in neuronal growth factor signal transduction. Proc Natl Acad Sci USA. 1993;90:5016–5020. doi: 10.1073/pnas.90.11.5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 55.Xu GF, Lin B, Tanaka K, Dunn D, Wood D, Gesteland R, White R, Weiss R, Tamanoi F. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell. 1990;63:835–841. doi: 10.1016/0092-8674(90)90149-9. [DOI] [PubMed] [Google Scholar]
- 56.Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 57.Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2006. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- 58.York RD, Yao H, Dillon T, Ellig CL, Eckert SP, McCleskey EW, Stork PJS. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature. 1998;392:622–626. doi: 10.1038/33451. [DOI] [PubMed] [Google Scholar]
- 59.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14–3-3 not BCL-X(L). Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 60.Zhang XF, Settleman J, Kyriakis JM, Takeuchi-Suzuki E, Elledge SJ, Marshall MS, Bruder JT, Rapp UR, Avruch J. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature. 1993;364:308–313. doi: 10.1038/364308a0. [DOI] [PubMed] [Google Scholar]