

Abstract
We have characterized paired-pulse facilitation atAplysia sensory neuron-to-motoneuron synapses. This simple form of very short-term synaptic plasticity displayed an unusual feature: it decreased dramatically with repeated testing. Synaptic depression at these synapses and this use-dependent decrease in paired-pulse facilitation occurred independently of each other. Paired-pulse facilitation was inversely correlated with the size of the initial synaptic connection and was absent at stronger synapses. The use-dependent decrease in paired-pulse facilitation occurred at the same rate at large synapses as at small synapses, although the initial paired-pulse facilitation at large synapses was substantially smaller. Rates of synaptic depression were also independent of initial synaptic strength. Paired-pulse facilitation was blocked by presynaptic EGTA injection, but not by postsynaptic EGTA or BAPTA injection. These results indicate that presynaptic Ca2+ influx plays a critical role in paired-pulse facilitation. However, the persistence of the decrease in paired-pulse facilitation for longer than 15 min suggests that Ca2+ from the first paired action potential produces facilitation via a modulatory mechanism rather than by summating with Ca2+ influx during the second paired action potential in activating the Ca2+binding sites that initiate exocytosis. This modulatory mechanism may not involve protein phosphorylation because paired-pulse facilitation was unaffected by the protein kinase inhibitors H7 and KN-62. These findings further suggest that release by the second paired action potential occurs at sites distinct from those that mediate release by the first action potential.
Keywords: synaptic plasticity, facilitation, synaptic depression, Aplysia, paired-pulse facilitation, calcium, phosphorylation
Paired-pulse facilitation is a very short-lived form of use-dependent synaptic plasticity observed at synapses across diverse phylogenetic groups (Katz and Miledi, 1968;Zengel et al., 1980; Zengel and Magleby, 1982; Atwood and Wojtowicz, 1986; Zucker, 1989; Fisher et al., 1997). In paired-pulse facilitation, transmitter release evoked by a second presynaptic action potential is enhanced when it follows a previous action potential within tens or hundreds of milliseconds. Because paired-pulse facilitation has been believed to represent a mechanistically simple presynaptic effect of residual Ca2+ (Katz and Miledi, 1968; Younkin, 1974;Regehr et al., 1994) and because its brief duration enables it to be induced repeatedly, this phenomenon is commonly monitored as an index of presynaptic alterations during more persistent and complex forms of synaptic plasticity. For example, more persistent enhancement of transmitter release may be accompanied by a decrease in paired-pulse facilitation (McNaughton, 1982; Muller et al., 1988; Staubli et al., 1990; Zalutsky and Nicoll, 1990). A decline in the paired-pulse ratio after the induction of some forms of long-term potentiation (LTP) in hippocampus is believed to suggest an increase in release probability. One way this might occur is if both of the paired action potentials access the same limited pool of releasable vesicles, so that an increase in release evoked by the first action potential would be accompanied by a decrease in release evoked by the second. Alternatively, if the probability of release by the second action potential is already very high, any substantial increase in the probability of release by the first action potential will result in a decrease in paired-pulse facilitation (Stevens and Wang, 1995), even though the amplitude of the second EPSP may remain unchanged. The paired-pulse ratio frequently increases with synaptic depression, either because less of the releasable pool is depleted by the first stimulus or because the probability of release by the second stimulus remains high.
The synaptic connections from mechanosensory neurons (SNs) to motoneurons (MNs) in Aplysia have provided an attractive and extensively studied system for analyzing a number of forms of associative and nonassociative synaptic plasticity, including post-tetanic potentiation (Walters and Byrne, 1984; Schacher et al., 1990; Goldsmith and Abrams, 1991; Clark and Kandel, 1993; Bao et al., 1997). However, paired-pulse facilitation has not been characterized at these synapses. In the present study we have found that paired-pulse facilitation at Aplysia SN synapses is an extremely labile phenomenon, which is altered dramatically by the previous history of testing. These results suggest that, at least in this system, paired-pulse facilitation is a more complex phenomenon than has been understood previously. In addition, these results provide new insights into both the process of synaptic depression at these SN synapses and the mechanisms underlying variability in the initial strength of these connections.
MATERIALS AND METHODS
Electrophysiology. Aplysia californica, weighing 70–200 gm [obtained from Alacrity (Redondo Beach, CA) or Marinus (Long Beach, CA)] were anesthetized by injection with isotonic MgCl2. Abdominal ganglia were removed, and the ventral surface of the left hemiganglion was desheathed in a 1:1 mixture of MgCl2 and artificial seawater. Ganglia were superfused at room temperature with high-divalent saline (6 × normal Ca2+; 1.6 × normal Mg2+) (Goldsmith and Abrams, 1991) to reduce spontaneous synaptic activity: (in mm) 328 NaCl, 10 KCl, 66 CaCl2, 88 MgCl2, and 10 Na-HEPES, pH 7.6, supplemented with nutrients [7 mm glucose, MEM essential and nonessential amino acids (0.2 × normal concentration; Life Technologies, Gaithersburg, MD), and MEM vitamin solution (0.7 × normal concentration; Life Technologies)]. This high-divalent saline did not alter synaptic strength; in synapses in which monosynaptic SN-to-MN connections were compared by switching back and forth between modified saline and normal saline after the synapses first were depressed to stable levels, mean EPSPs were 4.0 ± 1.2 mV in high-divalent saline versus 4.0 ± 1.1 mV in normal saline (n = 5).
Siphon SNs and LFS MNs were penetrated with single 12–20 MΩ microelectrodes filled with 2 m K-acetate and 0.4m KCl. During penetration, 0.5 nA hyperpolarizing current was injected to prevent SN firing. Injection of EGTA, BAPTA, or H7 was by passive diffusion from the recording pipette; compounds were diluted into 20 mm K-HEPES and 400 mm KCl, pH 7.3, and electrodes filled with this solution were beveled to 7–10 MΩ; control neurons were penetrated with beveled pipettes containing the same buffered saline. SN action potentials were elicited by the injection of 2 msec depolarizing current pulses. The membrane potential of postsynaptic MNs was maintained at 60 mV below the resting potential to prevent action potentials. After a synaptic connection was identified, the synapse was rested for a minimum of 10 min before an experimental protocol was begun. During experiments on synaptic depression, action potentials in SNs were elicited at a 15 sec interval. During paired-pulse stimulation two SN action potentials were elicited at a 50 msec interval. During paired-pulse and tetanic stimulation, the intensity of the injected depolarizing current was increased 20–40% above threshold to ensure that an action potential was elicited during each depolarizing pulse.
In experiments examining the effect of injection of EGTA, BAPTA, or H7, the synaptic stimulation protocols were begun 30, 30, or 40 min, respectively, after penetration to allow time for diffusion to presynaptic or postsynaptic regions of the SNs or MNs. KN-62 was dissolved in DMSO and was diluted to a final DMSO concentration of 0.1%; this concentration of DMSO had no detectable effect on synaptic connections. Ganglia were incubated 30 min in saline with 20 μm KN-62 to allow for penetration of the compound into neuropilar processes.
Data analysis. Data were acquired digitally with DT 2821 A–D interface (Data Translation, Marlboro, MA) and analyzed with Spike software (Hilal Associates, Englewood Cliffs, NJ). ANOVA was performed with the Systat data analysis package (SPSS, Chicago, IL). Effects over time with 5-hydroxytryptamine (5HT), with repeated paired-pulse testing, or with repeated tetanic stimulation were evaluated by an ANOVA with one repeated measure (trial number). Differences between groups in the time course of synaptic depression or of the decrease in the paired-pulse ratio were evaluated by a repeated measures ANOVA, testing trial × group interactions, followed by comparisons of the second-order polynomial trend. Effects of treatments on the paired-pulse ratio were evaluated with a two-tailed Student’st test or ANOVA.
To measure the amplitude of the second EPSP produced by paired-pulse stimulation, we calculated the postsynaptic response to a single action potential as follows: the first EPSP measured (with only a single SN spike) was scaled to match the amplitude of EPSP1 and then temporally aligned with EPSP1. This scaled single EPSP then was subtracted from the overall response to paired-pulse stimulation to yield the amplitude of EPSP2 alone.
RESULTS
Paired-pulse ratio at SN synapses decreases dramatically with repeated testing
In our initial studies, we hoped that measurements of paired-pulse facilitation would provide simple insights into mechanisms underlying more persistent forms of plasticity at Aplysia SN synapses. However, when we stimulated siphon SNs to fire pairs of spikes at a 50 msec interpulse interval, we observed a high level of variability in the paired-pulse ratio, much of which occurred within individual synaptic connections. Because SN synapses displayed paired-pulse depression as well as paired-pulse facilitation, we present results as the paired-pulse ratio (PPR) instead of presenting data on facilitation above control,
| Equation 1 |
where EPSP1 and EPSP2 are the synaptic responses to the first and second paired action potentials, respectively. Thus, a paired-pulse ratio <1 corresponds to paired-pulse depression and a paired-pulse ratio >1 corresponds to paired-pulse facilitation.
When synapses were stimulated repeatedly to fire pairs of action potentials, a striking pattern emerged: the paired-pulse ratio declined abruptly after the first test trial (Fig.1A,B). During the initial test of the paired-pulse ratio, we observed facilitation in the majority of synapses (mean initial PPR = 1.90 ± 0.22;n = 61), whereas with subsequent tests we usually observed paired-pulse depression (the second time that the paired-pulse ratio was tested, mean PPR = 0.74 ± 0.08; n= 39). This decrease in the paired-pulse ratio with repeated testing was highly significant, both with a single repeated test at a 15 min interval (p < 0.002; two-tailed, pairedt test; n = 6) and with a series of 15 tests at 15 sec intervals (F13,416 = 23.72;p < 0.001; n = 33; repeated measures ANOVA) (Fig. 1C). A limited number of experiments were conducted at several different interpulse intervals. With intervals of 30 and 100 msec, paired-pulse facilitation was present in the first trial and then disappeared with repeated testing in a qualitatively similar manner. With an interpulse interval of 1 sec, minimal paired-pulse facilitation was observed.
Fig. 1.

Persistent, use-dependent decrement in paired-pulse facilitation at SN synapses. Synaptic connections from siphon SNs to LFS MNs were recorded during paired-pulse stimulation of the presynaptic SN at a 50 msec interval. A, Example of paired-pulse stimulation at an intertrial interval (ITI) of 15 sec. Note that in the first paired-pulse trial, EPSP2 is substantially larger than EPSP1, whereas in the second paired-pulse trial, the two synaptic responses are comparable in size; by the eighth paired-pulse trial, EPSP2 is much smaller than EPSP1. B, Example of paired-pulse stimulation at an ITI of 15 min. Note that after the first paired-pulse test the paired-pulse facilitation is still reduced dramatically when tested 15 min later. (The amount of synaptic depression at 15 min in this example, to 70% of initial amplitude, is somewhat larger than was seen on average.) C, Group data for paired-pulse tests at ITIs of 15 sec and 15 min. For the longer ITI, only two tests were conducted. With both intervals, the paired-pulse ratio decreased significantly with repeated testing (for the 15 sec ITI: F13,416 = 23.72,p < 0.001, n = 33, repeated measures ANOVA; for the 15 min ITI: p < 0.002, two-tailed paired t test, n = 6). Note that, by the second trial with a 15 min interval, the paired-pulse ratio decreased almost as steeply as with a 15 sec interval. The difference in the paired-pulse ratios during the first trial probably reflects the small sample size for the 15 min ITI combined with the substantial variability in initial paired-pulse ratios.
The paired-pulse ratio decreases with increasing strength of the initial synaptic connection
When we examined the relationship between the initial paired-pulse ratio and the amplitude of EPSP1 at SN synapses, we observed an inverse correlation between initial synaptic strength and paired-pulse facilitation (“initial” refers to the time at which the paired-pulse ratio was first tested). For example, 88% of the synaptic connections that were initially <8 mV displayed facilitation (i.e., PPR > 1.1) the first time that they were tested with paired pulses (mean PPR = 2.75 ± 0.32; n = 34), whereas only 22% of the synaptic connections that were initially > 8 mV showed paired-pulse facilitation (mean PPR = 0.83 ± 0.07; n = 27) (Fig.2A). There was not, however, an inverse relationship between EPSP2 and EPSP1. When synapses were first tested, the amplitude of EPSP2 was independent of EPSP1 (Fig.2B); at synapses that previously had been depressed by repeated activation with single spikes (see below), EPSP2 was positively correlated with EPSP1(Fig. 2D). Thus, when synaptic connections of different strengths were compared, there was no tendency for greater release by the first stimulus to result in a decrease in release by the second stimulus. This lack of an inverse relationship between releases by the two paired action potentials has been observed in studies at hippocampal synapses where the probability of release by the second stimulus was either independent of, or positively correlated with, the probability of release by the first stimulus (Stevens and Wang, 1995;Dobrunz et al., 1997). Interestingly, the time course of decline of the paired-pulse ratio with repeated testing was nearly identical at large and small SN synapses (Fig. 3).
Fig. 2.

Relationship between the paired-pulse ratio and the amplitude of EPSP1. The paired-pulse ratio (A, C) and the amplitude of EPSP2 (B, D) are plotted versus the amplitude of EPSP1 for nondepressed synapses (A, B) and depressed synapses (C, D). Both sets of data are for the first trial in which a synapse was tested with paired stimulation. Paired-pulse data on nondepressed synapses were obtained 10–15 min after a synapse first was identified. Synaptic depression was achieved first by stimulating an SN to fire 15 single action potentials at a 15 sec ITI; this protocol reduced synaptic strength to an average of 32.8 ± 1.6% of the initial amplitude (see Fig.4A for data on the time course of depression for these same synapses). The paired-pulse test was 15 sec after the fifteenth SN spike. For A and C, the data were fit with hyperbolic functions of the form a +b/x, where x is the amplitude of EPSP1 (solid lines). InC, the dotted curve is the hyperbolic fit for nondepressed synapses from A; the dashed curve is the curve from A transformed by multiplying x by the average depression ratio (i.e., 0.328). In B and D, the data were fit with straight lines with a slope of 0.18 (with a correlation coefficient of 0.18) for the nondepressed synapses (B) and with a slope of 0.71 (with a correlation coefficient of 0.61) for the depressed synapses (D). Note that, whereas there is an inverse correlation between the paired-pulse ratio and the amplitude of EPSP1, the amplitude of EPSP1 is not negatively correlated with EPSP2.
Fig. 3.

Paired-pulse ratios for large and small synapses decline at the same rate with repeated testing. SN synapses were tested with paired-pulse stimulation at a 15 sec ITI. Note that, although the average initial paired-pulse ratio at synapses >8 mV (large synapses) was more than twofold smaller than the average initial paired-pulse ratio at synapses <8 mV (small synapses), the paired-pulse ratio at large synapses showed comparable decrement with repeated testing. The average initial EPSP amplitude was 4.7 ± 0.4 mV (n = 21) and 13.1 ± 1.1 mV (n = 12) for the small synapses and large synapses, respectively; these two groups of synapses initially had paired-pulse ratios of 2.60 ± 0.49 and 1.13 ± 0.26, respectively. Data are from the same experiments as in Figure 1C. Note that the paired-pulse ratios for the large and small synapses are plotted on two different scales. The time course of the decrease in paired-pulse ratios did not differ significantly between the two groups (F1,31 = 2.19; p = 0.149; repeated measures ANOVA testing trial × initial size interaction; comparison of the second-order polynomial trend).
The decrease in the paired-pulse ratio occurs independently of synaptic depression
Aplysia mechanosensory neuron synapses display marked homosynaptic depression with repeated activation at intervals of several minutes or shorter (Castellucci and Kandel, 1974; Byrne, 1982). The decrease in paired-pulse ratio with repeated testing at a 15 sec intertrial interval might suggest a change caused by synaptic depression. However, several lines of evidence indicate that the use-dependent decrease in the paired-pulse ratio occurs independently of synaptic depression. First, with the longer, 15 min, interval, there was minimal depression (EPSP1 at the time of the second test was 93.8% of EPSP1 at the time of the first test), and yet there was also a dramatic decrease in the paired-pulse ratio (to 47 ± 8% of the initial value, from 1.53 ± 0.10 to 0.71 ± 0.13) (see Fig. 1). Second, when the paired-pulse ratio was tested repeatedly at a 15 sec intertrial interval, the change in the paired-pulse ratio was much more dramatic and rapid than the depression of EPSP1 (data not shown). Third, to test directly whether the paired-pulse ratio decreases as a result of homosynaptic depression, we examined SN synapses after they first had been depressed with a series of 15 single stimuli at a 15 sec interval to, on average, 32.8 ± 1.6% of their initial amplitude (Fig.4A). When the paired-pulse ratio was first tested, 15 sec after the fifteenth single action potential, the paired-pulse ratio (2.23 ± 0.38;n = 30) was as large as the paired-pulse ratio at naive, nondepressed synapses (1.90 ± 0.22; n = 61) (see also Fig. 4B, in which the paired-pulse ratios at nondepressed and depressed synapses are separated according to the size of initial connection). Thus, repeated stimulation with single spikes did not decrease paired-pulse facilitation.
Fig. 4.
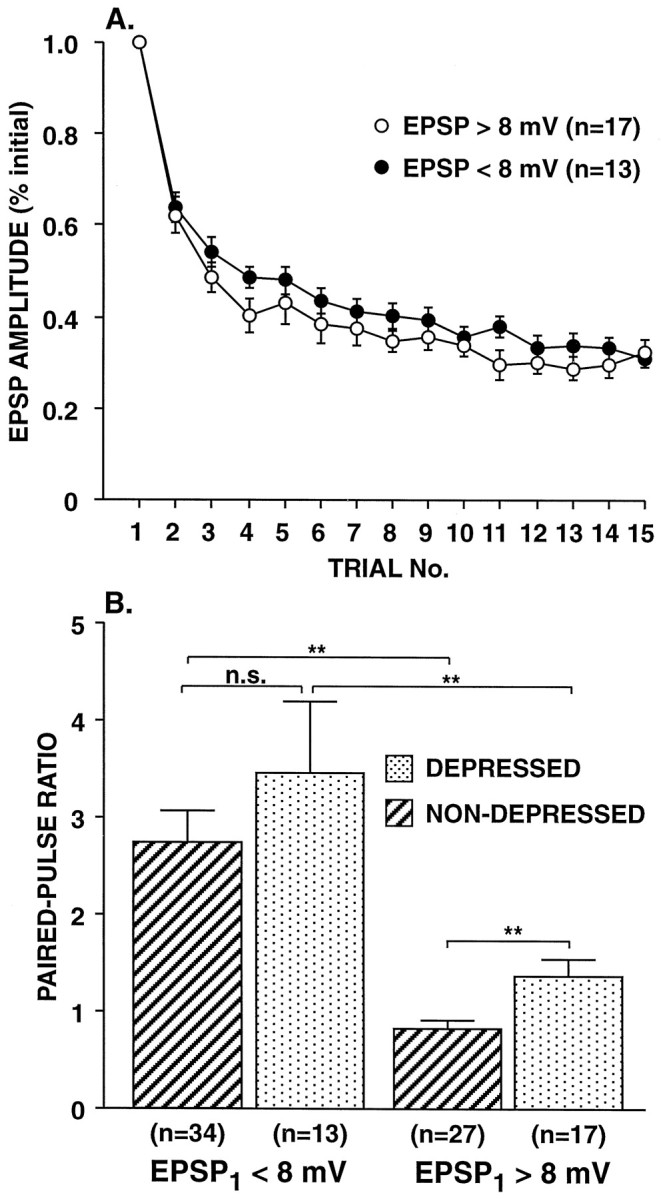
Effect of previous synaptic depression on the initial paired-pulse ratio. A, Time course of synaptic depression for large and small synapses. Synapses were depressed by stimulating SNs to fire single action potentials at a 15 sec ITI, and the paired-pulse ratio was then tested after the fifteenth trial. The mean amplitude of the EPSPs on trial number 15 was 32.8 ± 1.6% of the initial value. Note that the large (>8 mV) and small synapses (<8 mV) depressed at the same rate. The time course of synaptic depression did not differ significantly between the two groups (F1,28 = 1.34; p= 0.258; repeated measures ANOVA testing trial × initial size interaction; comparison of the second-order polynomial trend).B, Group data for paired-pulse ratios of large and small synapses tested once, either without depression (NON-DEPRESSED) or 15 sec after a series of 15 single action potentials (DEPRESSED). Both before and after depression, the large synapses (>8 mV) differed significantly from the small synapses (<8 mV) in their paired-pulse ratios (p < 0.001 for nondepressed synapses andp < 0.01 for depressed synapses; two tailedt test). After synaptic depression the paired-pulse ratio for large synapses increased significantly (p < 0.01; two tailed ttest); in contrast, despite comparable synaptic depression, there was no significant change in the paired-pulse ratio of small synapses. The average initial EPSP amplitudes were 14.8 ± 1.1 mV (n = 27) and 4.4 ± 0.3 mV (n = 34) for the large and small synapses, respectively, in the nondepressed group; for synapses in the depressed group, the average EPSP amplitudes before depression were 16.7 ± 1.5 mV (n = 17) and 5.7 ± 0.4 mV (n = 13) for the large and small synapses, respectively. Depressed synapse data are from the same synapses as inA.
Depression reduces EPSP2 along with EPSP1
Although synaptic depression with single action potentials did not reduce the paired-pulse ratio, the absolute amplitude of EPSP2 did decrease. The decrease in EPSP2 can be detected by comparing Figure 2, B and D. It is also evident that EPSP2 decreased with depression at small synapses (with initial amplitudes <8 mV) because, despite a threefold decrease in the size of EPSP1, there was not a significant increase in the paired-pulse ratio (Fig.4B). In the case of large synapses, although an increase in the paired-pulse ratio was observed after depression (PPR = 1.38; Fig. 4B), it was substantially less than what would be predicted if EPSP2 had remained constant [initial PPR ÷ (EPSP1-depressed/EPSP1-initial) gives a predicted PPR for large synapses after depression of 2.53 (where EPSP1-initial was determined without paired-pulse stimulation)].
Does synaptic depression change large synapses into small synapses?
We wanted to know whether, after depression, initially large synapses show paired-pulse facilitation comparable to that of initially small synapses. We addressed this issue in three ways. First, we compared the paired-pulse ratio for initially large synapses after depression with the paired-pulse ratio for initially small synapses. The paired-pulse ratios of initially large synapses (>8 mV initial amplitude) increased significantly after the synapse had been depressed, from 0.83 ± 0.07 (n = 27) to 1.38 ± 0.18 (n = 17) (p < 0.01; Fig. 4B); however, the average PPR after depression of 1.38 did not approach the average PPR of small synapses, either before or after depression (Fig. 4B). In contrast, the paired-pulse ratio at smaller synapses (<8 mV initial amplitude) did not change significantly after depression (Fig.4B). Second, we fit hyperbolic functions to the paired-pulse ratio data from depressed and nondepressed synapses of the form PPR = a + b/x, wherex is the amplitude of EPSP1 (see Fig.2A,C). If after depression, large synapses simply became small synapses, then the hyperbola fit to the data from depressed synapses would be similar to the curve fit to the data from nondepressed synapses; this is not the case (see Fig. 2C). On the other hand, if the paired-pulse ratios of large synapses change little after depression, then we should be able to predict the paired-pulse ratios of depressed synapses by scaling the hyperbola fit to the data for nondepressed synapses, multiplying the xvalues by the depression ratio, 0.328. The hyperbola fit to the actual data from depressed synapses is intermediate between the nondepressed curve and the scaled curve and somewhat closer to the latter. Third, we considered only the EPSPs that were initially >8 mV but that depressed to <8 mV. Although the average amplitude for EPSP1 when first tested after depression was 4.3 ± 0.4 mV for this group, the average paired-pulse ratio was 1.42 ± 0.21 (n= 11). In contrast, the paired-pulse ratio for nondepressed synapses <8 mV was significantly greater, averaging 2.75 ± 0.32 (p < 0.05; two-tailed t test), although the average amplitude for EPSP1 when it was tested initially was comparable (4.4 ± 0.3 mV; n = 34). Thus, EPSP size, independently of initial synaptic strength, is not an adequate predictor of the paired-pulse ratio.
Use-dependent plasticity of paired-pulse facilitation provides evidence that a separate set of release sites contributes to EPSP2
In some experimental systems (e.g., Stevens and Wang, 1995), changes in the paired-pulse ratio may represent primarily a change in the probability of release evoked by the first stimulus. However, we observed a dramatic reduction in the release evoked by the second stimulus after a single paired-pulse trial, when there was minimal change in release evoked by the first. We consider here what synaptic organization is required for the observed decrease in the amplitude of EPSP2 with repeated testing that occurred independently of any substantial change in EPSP1. The number of vesicles released by the second of two paired action potentials can be expressed as:
| Equation 2 |
where m2 is the mean quantal content for EPSP2, n1 is the number of release sites available to contribute to EPSP1,P1b is the probability of release during the second action potential at these sites that mediate EPSP1, n2 is the number of release sites that constitute a hypothetical separate class that is activated exclusively by the second paired action potential, andP2 is the probability of release at these hypothetical release sites that contribute selectively to EPSP2. By definition, the n2 sites are not effectively activated by a single action potential and do not contribute substantially to EPSP1.P1b can be considered to be a function ofP1 and F1, whereP1 is the probability of release during the first action potential at the n1 sites, andF1 is a Ca2+-dependent facilitatory factor initiated by the first paired action potential that acts transiently to increase P1b (see Results below for evidence of Ca2+ dependence).P2 also is enhanced by a Ca2+-dependent facilitatory factor (F2); however, in the absence of the Ca2+ signal from the first action potential,P2 is very low.
In principle, EPSP2 could be simply a direct function of the same factors that control release by the first action potential (which together determine P1), in combination with the Ca2+-dependent facilitatory factor F1. If this were the case, the productn2 × P2 would be negligible, and the only significant release during the second action potential would occur at that same population of sites that can be effectively activated by the first action potential, i.e., then1 sites. Use-dependent changes in the paired-pulse ratio then would be explained entirely by changes in the Ca2+-dependent facilitatory factorF1 because changes in eithern1 or P1 would alter EPSP1. However, this facilitatory factorF1 is an unlikely site for the persistent use-dependent plasticity that was observed here because repeated single stimuli do not decrease paired-pulse facilitation (Fig. 4); ifF1 changed simply by being activated, then it would decrease incrementally during a series of single presynaptic spikes. This leaves the remaining two parameters,n2 and P2, either one or both of which could change in a manner that explains the use-dependent decrease in the paired-pulse ratio. Even if it is a decline in P2 that is responsible for the decrement in paired-pulse facilitation with repeated testing, there must exist a unique set of release sites that is accessed primarily by the second paired spike, with which P2 is associated. The second paired action potential also may activate some of the n1 sites; however, there must be very low-probability activation of these sites given that EPSP1can show little depression, whereas EPSP2 depresses dramatically or fails completely (e.g., Fig. 1A).
We do not know whether a single action potential evokes release at a low probability from the same sites that are accessed by the second paired action potential. We observed that EPSP2 was reduced in size during a series of single spikes that depress EPSP1. There are at least two ways that this could occur. During a single unpaired action potential there may be modest, very low-probability activation of the sites that contribute primarily to EPSP2 (the n2 sites). Alternatively, there may be a component of synaptic depression that occurs independently of release (Dobrunz et al., 1997), as suggested by the high-Mg2+ saline results of Castellucci (1974), for example, a depression of Ca2+ current.
Post-tetanic potentiation persists through repeated testing
Because paired-pulse facilitation has some similarities with post-tetanic potentiation (PTP), we asked whether PTP also disappeared after testing. Before examining PTP at each synapse, we repeatedly stimulated the SN at a 15 sec interval to depress the connection to a stable level to allow for comparisons of multiple episodes of PTP. The SN was then stimulated at 20 Hz for 2 sec to induce PTP. After five post tests at 15 sec intervals, the synapse was rested 10 min and the protocol was repeated. PTP decreased significantly with repeated testing (F2,12 = 7.37; p = 0.008; n = 7) (Fig. 5). Nevertheless, PTP remained highly significant even during the third episode (F9,54 = 6.40; p < 0.001; n = 7; repeated measures ANOVA, testing the trial effect for the last five pretests and the five posttests). Thus, unlike paired-pulse facilitation, PTP did not disappear after initial testing. A number of differences between PTP and paired-pulse facilitation, particularly in various aspects of their Ca2+ dependence, have been observed previously (e.g., Kamiya and Zucker, 1994; Fisher et al., 1997), which also suggests that facilitation is not simply a modest form of PTP.
Fig. 5.

PTP persists during repeated testing. PTP was tested three times at depressed SN-to-MN synapses. The synapses were depressed before tetanic stimulation to enable a comparison of sequential episodes of PTP under similar conditions. For each PTP episode, the synapses were depressed first by stimulating the SN to fire single spikes at a 15 sec interval; 15 sec after the sixteenth pretest the SN was stimulated at 20 Hz for 2 sec. At 15 sec after the tetanus, single stimuli were resumed for five posttests at a 15 sec interval. This sequence of pretests, tetanus, and posttests was repeated three times; the synapses were rested 10 min before each series of pretests. The mean amplitudes of the last three pretests during each episode (34.0 ± 6.0%, 30.9 ± 3.9%, and 26.8 ± 5.5%, expressed as a percentage of the amplitude of the first EPSP in the first sequence) were not significantly different (F2,12 = 1.46; p = 0.271). The amplitude of the EPSP 15 sec after the tetanus, which was the time of maximum potentiation, is expressed as a percentage of the mean EPSP amplitude during the last three pretests before the tetanus. PTP decreased significantly during the repeated episodes (F2,12 = 7.37; p = 0.008; n = 7); however, PTP remained highly significant throughout (for the 15 sec posttest in the third episode,p < 0.01; two-tailed ttest). (See Results for an analysis of the results of all five posttests).
Interaction of serotonin-induced facilitation with the paired-pulse ratio
Serotonin (5-hydroxytryptamine, 5HT) acts to increase release atAplysia SN synapses by at least two presynaptic mechanisms: modulation of K+ currents in the SNs, which results in increased Ca2+ influx, and modulation of either the exocytosis mechanism itself or the releasable pool (Byrne and Kandel, 1996). We asked whether 5HT-induced synaptic facilitation was accompanied by the expected decrease in the paired-pulse ratio, which is associated with several other forms of enhancement of transmitter release (e.g., McNaughton, 1982; Muller et al., 1988). Because the initial paired-pulse ratio at naive synapses is quite variable (see Fig. 2A) and because it decreases dramatically with testing, it was difficult to determine the effects of 5HT on the paired-pulse ratio at minimally stimulated synapses by using either between-synapse or within-synapse comparisons; therefore, we did not analyze 5HT-induced facilitation at rested synapses. Instead, we examined 5HT facilitation of moderately depressed synapses at which the paired-pulse ratio had been tested repeatedly. A 3 min exposure to 5HT produced reliable and maintained enhancement of EPSP1 that developed gradually and reached an amplitude more than twice the initial nondepressed amplitude (F9,72 = 10.66;p < 0.001; n = 9; repeated measures ANOVA) (Fig. 6A). In contrast, EPSP2 was facilitated only transiently (F9,72 = 3.40; p = 0.002) and did not approach its initial amplitude (Fig. 6B). During the period of initial facilitation, because EPSP2was increased in parallel with EPSP1, there was no significant change in the paired-pulse ratio, which remained below 15% of the initial value; however, when EPSP2 again declined, the paired-pulse ratio decreased to a very low level (∼5% of the initial PPR). Because after repeated testing EPSP2 was extremely small and often undetectable, it is not meaningful to compare quantitatively the facilitation of EPSP2 and EPSP1 nor to quantify the change in the paired-pulse ratio. Nevertheless, during the period of initial facilitation, there was no evidence that the paired-pulse ratio declined.
Fig. 6.

Effect of 5HT on EPSP1 and EPSP2 after the use-dependent decrease in the paired-pulse ratio. SN synapses were tested with paired stimuli 15 times at a 15 sec ITI, and 20 μm 5HT was then applied while the paired-pulse testing continued. EPSP amplitudes are expressed as a percentage of their initial amplitude when they were first tested with paired stimuli at the start of an experiment. Because of the very reduced and highly variable amplitude of EPSP2 after paired-pulse testing, the data plotted are the averages of three consecutive trials at each synapse, spanning 45 sec. The first data points shown (from −0.75 to −0.25 sec) are the means of the EPSPs in trials 13–15, just before the addition of 5HT. The subsequent data points are the means of trials 1–3, 4–6, and 7–9 after the onset of the 5HT exposure. Note that EPSP2 had shown dramatic (>10-fold) depression at the end of the 15 initial trials, whereas the depression of EPSP1 was extremely modest (to 82% of the initial value); the depression of EPSP1 was substantially less than depression with single action potentials (e.g., Fig.4A) because the paired stimulation produced burst-dependent protection from synaptic depression (Jiang and Abrams, 1997) (see Contribution of Ca2+ and Phosphorylation to Paired-Pulse Facilitation in Results). Note also that facilitation of EPSP1 by 5HT was maintained, whereas 5HT increased the amplitude of EPSP2 only transiently.
Why was the facilitation by 5HT of EPSP2 short-lived compared with the facilitation of EPSP1? It could be that, although 5HT effectively facilitated EPSP1 throughout the exposure, facilitation at the sites mediating EPSP2 was for some reason more transient. Alternatively, once the second spike again produced a moderate level of transmitter release in the presence of 5HT, there may be rapid additional depression of EPSP2; this could be a consequence of the same mechanism that produced profound depression of EPSP2 when it was initially tested by using the paired-pulse protocol. According to this proposal, whenever release is effectively activated at the sites mediating EPSP2, this release is depressed rapidly. This second alternative is attractive because it does not require a 5HT-induced facilitatory process that is unique to the second spike but, rather, results from the same dramatic depression of EPSP2 that is observed in the absence of 5HT.
Contribution of Ca2+ and phosphorylation to paired-pulse facilitation
The observation that during paired-pulse facilitation, Ca2+ influx during the first action potential was necessary for facilitation of the release evoked by the second action potential led to the residual Ca2+ hypothesis (Katz and Miledi, 1968; Younkin, 1974). In its simplest form, this model proposes that small amounts of residual Ca2+ summate with Ca2+ influx during a second action potential to increase exocytosis; such powerful, nonlinear summation could, in principle, occur because Ca2+-triggered release of transmitter apparently depends on the binding of approximately four Ca2+ ions (Zucker, 1989). However, several lines of evidence suggest that this early model might be too simple (e.g.,Yamada and Zucker, 1992; Van der Kloot, 1994; Winslow et al., 1994); thus, Ca2+ initiates facilitation via a mechanism that is not fully understood (Fisher et al., 1997).
We tested the effect on paired-pulse facilitation of presynaptic injection of EGTA. Because EGTA is a relatively slow Ca2+ buffer compared with the exocytosis process itself, in a number of systems presynaptic EGTA can block some forms of plasticity with either little effect or a modest effect on the release process itself (Adler et al., 1991; Winslow et al., 1994; Bao et al., 1997). After 30 min to allow for diffusion to synaptic sites, recording with 50 mm EGTA in the presynaptic electrode reduced EPSP amplitude by a highly variable amount (in 17 synapses in which measurable transmitter release persisted, EPSP amplitude decreased significantly to 50.93 ± 4.2% of the initial value, compared with a decrease to 89.4 ± 7.8% of the initial value for vehicle-injected SNs, n = 8, p < 0.001, two-tailed t test; in three other synapses, no EPSP was detectable after 30 min of EGTA injection). This reduction in transmitter release indicated that the chelator had diffused to presynaptic terminals. In these experiments, after EGTA injection, the paired-pulse ratio was reduced dramatically (PPR = 0.27 ± 0.04 for EGTA-injected SNs, n = 17, vs 1.44 ± 0.22 for vehicle-injected SNs, n = 8; p< 0.001; two-tailed t test) (Fig.7). Thus, presynaptic Ca2+ appears to be necessary for paired-pulse facilitation.
Fig. 7.
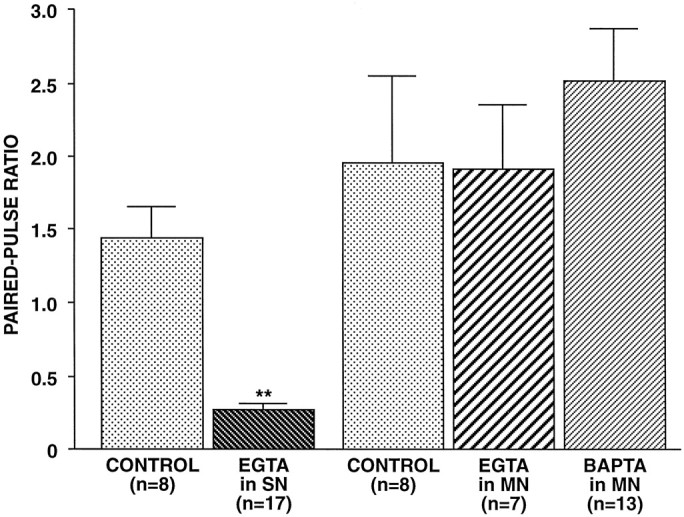
Effect of Ca2+ chelators injected pre- or postsynaptically on paired-pulse facilitation. Either EGTA (50 mm in pipette) was injected into the presynaptic SNs, or EGTA (100 mm in pipette) or BAPTA (200 mm in pipette) was injected into the postsynaptic MNs. Paired-pulse ratios were tested 30 min after penetration to allow the chelators to diffuse to presynaptic or postsynaptic regions of the neurons. Control neurons were injected with vehicle. Presynaptic EGTA significantly decreased the paired-pulse ratio, whereas postsynaptic EGTA or BAPTA had no significant effect (F4,48 = 8.96 and p < 0.001 for overall comparison among the five groups; pairwise comparisons revealed that EGTA-injected SNs were significantly different from vehicle-injected SN controls, p = 0.015, whereas EGTA-injected MNs and BAPTA-injected MNs were not significantly different from MN controls).
Bao et al. (1997) demonstrated that a component of PTP at theseAplysia SN synapses depends on postsynaptic Ca2+. We tested the importance of postsynaptic Ca2+ in paired-pulse facilitation by recording from MNs with pipettes filled with 100 mm EGTA or 200 mm BAPTA. Postsynaptic injection of either of these Ca2+ chelators did not significantly affect paired-pulse facilitation (Fig. 7). The concentrations of these chelators injected in MNs were two to four times the concentration of EGTA injected presynaptically, which was sufficient to reduce release substantially; therefore, it is likely that the concentrations of EGTA and BAPTA that diffused to postsynaptic sites would have been adequate to interfere with any modulatory effects of Ca2+.
It recently has been suggested that Ca2+-dependent phosphorylation might mediate paired-pulse facilitation (Winslow et al., 1994). To test this possibility, we injected the broad-spectrum protein kinase inhibitor H7 into the presynaptic SN. H7 inhibits a number of kinases, including cAMP-dependent kinase, protein kinase C (PKC) (Hidaka et al., 1984), and calmodulin-dependent kinase II (CaMKII) (Waxham et al., 1993). Braha et al. (1990) observed that, atAplysia SN synapses, H7 applied extracellularly at a concentration of 200 μm significantly reduced the facilitation initiated by either cAMP-dependent kinase or PKC. We found that intracellularly injected H7 (50 mm in pipette) had no effect on the paired-pulse ratio (Fig.8A). In the majority of these experiments we verified that adequate concentrations of H7 diffused to the presynaptic terminals by examining an H7-sensitive form of synaptic plasticity that we had recently identified, burst-dependent protection from synaptic depression (Jiang and Abrams, 1997). Siphon SN synapses typically depress rapidly to less than one-half of their initial amplitude when activated repeatedly at an intertrial interval briefer than 1 min (e.g., Goldsmith and Abrams, 1991; Eliot et al., 1994); however, if the SNs fire brief bursts of several spikes, rather than single action potentials, these bursts almost completely protect the synapses from depression. (Burst-dependent protection is evident in Fig. 6 in the data for EPSP1, which, despite a series of 12 previous paired-pulse stimuli, had depressed only 15%.) In those paired-pulse experiments in which we subsequently examined synaptic depression when SNs were activated with bursts of four spikes, control synapses decreased minimally, to 87.7 ± 6.9% (n = 6) of their initial amplitude, whereas synapses from H7-injected SNs decreased significantly more, to 41.5 ± 9.4% (n = 6) of their initial size (p < 0.01).
Fig. 8.

Inhibition of protein phosphorylation with either H7 or KN-62 did not decrease paired-pulse facilitation.A, SNs were injected with H7 (50 mm in pipettes) for 40 min before testing. B, KN-62 (20 μm) was applied in the bath for 30 min before testing. Neither inhibitor significantly affected the PPR as compared with the controls.
The inhibition of CaMKII requires several-fold higher concentrations of H7 than does the inhibition of PKC (Hidaka et al., 1984; Waxham et al., 1993; Schweitzer et al., 1995; Sitges et al., 1995) [for example, in rat CNS protein kinase preparations, Neil Waxham (personal communication) determined Ki values for H7 of 8 μm for PKC and 20–32 μm for CaMKII]. Thus, it seemed possible that, although H7 reached sufficient concentrations at the presynaptic terminals of SNs after intrasomatic injection to inhibit PKC and to block burst-dependent protection, these concentrations may have been insufficient to inhibit CaMKII effectively. We therefore also conducted experiments with KN-62, a specific CaMKII inhibitor. Nakanishi et al. (1997) found atAplysia SN synapses that 10 μm KN-62, applied extracellularly for 15 min, blocked a component of short-term 5HT-induced synaptic facilitation. There was no reduction in the paired-pulse ratio after ganglia were incubated for 30 min with 20 μm KN-62 (Fig. 8B), indicating that paired-pulse facilitation does not depend on CaMKII.
DISCUSSION
We observed that paired-pulse facilitation at AplysiaSN synapses is a complex and plastic phenomenon that can be altered dramatically simply as a result of minimal paired-pulse testing. After a single paired-pulse trial, paired-pulse facilitation decreased abruptly and paired-pulse depression predominated for at least 15 min. This unusual plasticity of the paired-pulse ratio provides some novel insights into both paired-pulse facilitation and the regulation of synaptic strength at these SN connections.
The decrease in paired-pulse facilitation is independent of both synaptic depression and initial synaptic strength, suggesting that the second EPSP is mediated by a separate set of release sites
Three changes in synaptic strength appeared to occur independently of each other: (1) use-dependent decrease in paired-pulse facilitation, (2) synaptic depression, and (3) variation in initial synaptic size, which was inversely correlated with the initial paired-pulse ratio. Synaptic depression occurred without an associated decrease in paired-pulse facilitation; moreover, paired-pulse facilitation decreased dramatically with repeated testing, with little change in the amplitude of EPSP1. Synapses that were initially large had small paired-pulse ratios; however, the decrease in the paired-pulse ratio with repeated testing occurred at the same rates at initially small and initially large synapses (see Fig. 3), as did synaptic depression (see Fig. 4A). To account for these several aspects of synaptic strength changing independently, there must be at least three independent parameters that influence transmitter release during paired action potentials. The plasticity of EPSP2 argued for the existence of a separate set of release sites that are the primary loci mediating release evoked by the second spike. Such an independent set of release sites would provide the additional parameters, n2 andP2 (see Results), that would enable these three aspects of synaptic strength to vary independently. This separate set of release sites that is hypothesized to contribute selectively to EPSP2 could be part of a continuum of release sites with different probabilities of release, such that the probability of release at these sites during a single action potential is particularly low. Release at these sites may have a distinct dependence on Ca2+ compared with sites that release with a single spike, as is the case for release of neuropeptides (Whim and Lloyd, 1989; Peng and Zucker, 1993; Vilim et al., 1996). Thus, this second set of release sites may normally have a very low probability of release, which increases transiently when Ca2+ is elevated after the first paired spike. One explanation for the abrupt shift from paired-pulse facilitation to paired-pulse depression after a single pair of spikes is that, at this subset of release sites selectively activated by the second spike, the release process recovers very slowly.
Changes in the number of synaptic sites cannot account alone for the variability in the strength of SN synapses
Long-term changes in the strength of these SN synapses that are induced by both behavioral or cellular training are accompanied by changes in the number of sites of synaptic interaction, i.e., active zones and presynaptic varicosities (Bailey and Chen, 1988a,b; Martin et al., 1997). One might therefore predict that much of the variability in initial synaptic strength at SN synapses would be accounted for by differences in the number of synaptic contacts. If increased initial synaptic strength were primarily explained by an increased number of synaptic contacts that had, on average, the same properties as the varicosities between neurons with small synaptic connections, then we would expect that paired-pulse ratios would be similar at initially large and initially small synapses. However, the observation that paired-pulse facilitation showed a strong inverse correlation with initial synaptic strength suggests that active zones of initially large synapses have, on average, a higher probability of release than those of initially small synapses. Bailey and Chen (1983, 1989) observed that after long-term synaptic facilitation there is an increased number of vesicles associated with each active zone in SNs; such an increase in the number of docked vesicles could underlie an increase in release probability. Substantial heterogeneity in release probability has been described in hippocampal synapses where release sites of stronger synaptic connections tend to have higher release probabilities (Malinow and Tsien, 1990; Bolshakov and Siegelbaum, 1995; Dobrunz and Stevens, 1997). Moreover, hippocampal synapses with higher probabilities of release tend to have larger releasable pools, which suggests that the number of vesicles at the active zone may contribute to release probability (Dobrunz and Stevens, 1997; Murthy et al., 1997).
Synaptic depression alters a characteristic of synapses distinct from the synaptic property that influences initial synaptic strength
Large and small synapses displayed similar rates of synaptic depression. The observation that the rate of depression is independent of initial synaptic strength suggests that depression may result from the alteration of a synaptic property distinct from the property that underlies the difference in paired-pulse facilitation between large and small synapses; as suggested above, this difference in paired-pulse facilitation is likely to result from a difference in release probabilities. If release probability were to decrease during synaptic depression, the paired-pulse ratios of large synapses should, after depression, become comparable to the paired-pulse ratios of initially small synapses. However, depressing large synapses did not convert them to small synapses. Thus, large synapses must differ from small synapses in some factor that remains different even after short-term synaptic depression. If, as proposed above, initially large synapses have a greater percentage of high-probability release sites, then depression must result primarily from a reduction in the number of release sites rather than from the conversion of high-probability sites to low-probability sites. Consistent with this hypothesis, synaptic depression of small synapses was not accompanied by an increase in the paired-pulse ratio. If synaptic depression resulted from a decrease in the probability of release, then one would expect that it might be accompanied by a reciprocal increase in the paired-pulse ratio (e.g.,Mallart and Martin, 1968).
This proposed loss of release sites with depression could actually represent a series of abrupt changes in the probability of release at individual sites, such that individual sites become functionally inactive. Recently, Klein et al. (1997) have concluded, on the basis of quantal analysis, that synaptic depression at these same SN synapses may result from the switching of individual release sites from a high-probability release state to a very low-probability, or inactive, state, rather than from a gradual decrease in the probability of release at all sites. Such large changes in the probability of release of individual sites, such that the contributions of some sites become negligible, could be functionally equivalent to the decrease in the number of release sites during depression suggested by these paired-pulse ratio results.
Nature of the Ca2+ effect that underlies paired-pulse facilitation
The original “residual Ca2+” hypothesis proposed that, during paired-pulse facilitation, low levels of elevated Ca2+ persisting after the first spike summated with the Ca2+ increase during the subsequent spike to activate greater numbers of the Ca2+ binding sites that trigger exocytosis (e.g., Younkin, 1974). Alternatively, Ca2+ from the first spike could bind to a site separate from those that normally initiate vesicle release to modulate the exocytotic mechanism transiently, thereby increasing release by the second spike. Our findings argue against the traditional view that residual Ca2+ produces facilitation by more effectively activating those sites normally involved in triggering exocytosis, because persistent alteration of paired-pulse facilitation occurred with minimal change in release by the first spike. Any alteration related to Ca2+ binding to the sites that normally trigger release should have parallel effects on EPSP1 and EPSP2. Thus, Ca2+from the first spike may enhance release by modulating release probability, e.g., by altering vesicle docking or priming. This modulatory mechanism probably does not involve phosphorylation because the protein kinase inhibitors H7 and KN-62 did not affect facilitation. As mentioned above, it is possible that the release sites that primarily mediate EPSP2 require priming by Ca2+ from the first spike in order for release to be activated with a substantial probability by the second spike. The possibility that Ca2+ acts at heterogeneous sites in initiating release and facilitation was suggested previously by computer simulations (Yamada and Zucker, 1992).
Paired-pulse facilitation as an index of presynaptic function
If the second of two paired action potentials releases from a different set of sites than activated by the first action potential, then clearly plasticity at one set of release sites could occur independently of plasticity from a second set. This would make it difficult to use paired-pulse facilitation as a simple index of presynaptic function. These Aplysia SN synapses recover after activation unusually slowly. However, it is possible that, in other systems without such dramatic depression, release by the second paired spike could also occur at sites distinct from those that are activated by the first paired spike.
Footnotes
The experimental results that are cited were obtained with support from National Institutes of Health Grant MH 55880 to T.W.A. We thank Jonathan Cohen for critically reading and commenting on this manuscript and Marc Klein and Steve Siegelbaum for their helpful discussions of these findings. Allison Lin provided extensive assistance with data analysis and figures.
Correspondence should be addressed to Dr. Thomas W. Abrams, Department of Pharmacology, University of Maryland School of Medicine, BRB 4-002, 655 West Baltimore Street, Baltimore, MD 21201-1559.
REFERENCES
- 1.Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atwood HL, Wojtowicz JM. Short-term and long-term plasticity and physiological differentiation of crustacean motor synapses. Int Rev Neurobiol. 1986;28:275–362. doi: 10.1016/s0074-7742(08)60111-7. [DOI] [PubMed] [Google Scholar]
- 3.Bailey CH, Chen M. Morphological basis of long-term habituation and sensitization in Aplysia. Science. 1983;220:91–93. doi: 10.1126/science.6828885. [DOI] [PubMed] [Google Scholar]
- 4.Bailey CH, Chen M. Long-term memory in Aplysia modulates the total number of varicosities of single identified sensory neurons. Proc Natl Acad Sci USA. 1988a;85:2373–2377. doi: 10.1073/pnas.85.7.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bailey CH, Chen M. Long-term sensitization in Aplysia increases the number of presynaptic contacts onto the identified gill motor neuron L7. Proc Natl Acad Sci USA. 1988b;85:9356–9359. doi: 10.1073/pnas.85.23.9356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey CH, Chen M. Time course of structural changes at identified sensory neuron synapses during long-term sensitization in Aplysia. J Neurosci. 1989;9:1774–1780. doi: 10.1523/JNEUROSCI.09-05-01774.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bao JX, Kandel ER, Hawkins RD. Involvement of pre- and postsynaptic mechanisms in post-tetanic potentiation at Aplysia synapses. Science. 1997;275:969–973. doi: 10.1126/science.275.5302.969. [DOI] [PubMed] [Google Scholar]
- 8.Bolshakov VY, Siegelbaum SA. Regulation of hippocampal transmitter release during development and long-term potentiation [see comments]. Science. 1995;269:1730–1734. doi: 10.1126/science.7569903. [DOI] [PubMed] [Google Scholar]
- 9.Braha O, Dale N, Hochner B, Klein M, Abrams TW, Kandel ER. Second messengers involved in the two processes of presynaptic facilitation that contribute to sensitization and dishabituation in Aplysia sensory neurons. Proc Natl Acad Sci USA. 1990;87:2040–2044. doi: 10.1073/pnas.87.5.2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Byrne JH. Analysis of synaptic depression contributing to habituation of gill-withdrawal reflex in Aplysia californica. J Neurophysiol. 1982;48:431–438. doi: 10.1152/jn.1982.48.2.431. [DOI] [PubMed] [Google Scholar]
- 11.Byrne JH, Kandel ER. Presynaptic facilitation revisited: state and time dependence. J Neurosci. 1996;16:425–435. doi: 10.1523/JNEUROSCI.16-02-00425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castellucci VF, Kandel ER. A quantal analysis of the synaptic depression underlying habituation of the gill-withdrawal reflex in Aplysia. Proc Natl Acad Sci USA. 1974;71:5004–5008. doi: 10.1073/pnas.71.12.5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark GA, Kandel ER. Induction of long-term facilitation in Aplysia sensory neurons by local application of serotonin to remote synapses. Proc Natl Acad Sci USA. 1993;90:11411–11415. doi: 10.1073/pnas.90.23.11411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- 15.Dobrunz LE, Huang EP, Stevens CF. Very short-term plasticity in hippocampal synapses. Proc Natl Acad Sci USA. 1997;94:14843–14847. doi: 10.1073/pnas.94.26.14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eliot LS, Kandel ER, Hawkins RD. Modulation of spontaneous transmitter release during depression and post-tetanic potentiation of Aplysia sensory motor neuron synapses isolated in culture. J Neurosci. 1994;14:3280–3292. doi: 10.1523/JNEUROSCI.14-05-03280.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fisher SA, Fischer TM, Carew TJ. Multiple overlapping processes underlying short-term synaptic enhancement. Trends Neurosci. 1997;20:170–177. doi: 10.1016/s0166-2236(96)01001-6. [DOI] [PubMed] [Google Scholar]
- 18.Goldsmith BA, Abrams TW. Reversal of synaptic depression by serotonin at Aplysia sensory neuron synapses involves activation of adenylyl cyclase. Proc Natl Acad Sci USA. 1991;88:9021–9025. doi: 10.1073/pnas.88.20.9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide-dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- 20.Jiang X-Y, Abrams TW. Protection from synaptic depression at the siphon sensory neuron synapses in Aplysia. Soc Neurosci Abstr. 1997;23:1332. [Google Scholar]
- 21.Kamiya H, Zucker RS. Residual Ca2+ and short-term synaptic plasticity. Nature. 1994;371:603–606. doi: 10.1038/371603a0. [DOI] [PubMed] [Google Scholar]
- 22.Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol (Lond) 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein M, Royer S, Coulson RL. Switching between high- and low-probability release states in synaptic plasticity at Aplysia sensorimotor synapses. Soc Neurosci Abstr. 1997;23:1332. [Google Scholar]
- 24.Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices [see comments]. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- 25.Mallart A, Martin AR. The relation between quantum content and facilitation at the neuromuscular junction of the frog. J Physiol (Lond) 1968;196:593–604. doi: 10.1113/jphysiol.1968.sp008525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin KC, Casadio A, Zhu H, E Y, Rose JC, Chen M, Bailey CH, Kandel ER. Synapse-specific, long-term facilitation of Aplysia sensory-to-motor synapses: a function for local protein synthesis in memory storage. Cell. 1997;91:927–938. doi: 10.1016/s0092-8674(00)80484-5. [DOI] [PubMed] [Google Scholar]
- 27.McNaughton BL. Long-term synaptic enhancement and short-term potentiation in rat fascia dentata act through different mechanisms. J Physiol (Lond) 1982;324:249–262. doi: 10.1113/jphysiol.1982.sp014110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muller D, Turnbull J, Baudry M, Lynch G. Phorbol ester-induced synaptic facilitation is different than long-term potentiation. Proc Natl Acad Sci USA. 1988;85:6997–7000. doi: 10.1073/pnas.85.18.6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murthy VN, Sejnowski TJ, Stevens CF. Heterogeneous release properties of visualized individual hippocampal synapses. Neuron. 1997;18:599–612. doi: 10.1016/s0896-6273(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 30.Nakanishi K, Zhang F, Baxter DA, Eskin A, Byrne JH. Role of calcium-calmodulin-dependent protein kinase II in modulation of sensorimotor synapses in Aplysia. J Neurophysiol. 1997;78:409–416. doi: 10.1152/jn.1997.78.1.409. [DOI] [PubMed] [Google Scholar]
- 31.Peng YY, Zucker RS. Release of LHRH is linearly related to the time integral of presynaptic Ca2+ elevation above a threshold level in bullfrog sympathetic ganglia. Neuron. 1993;10:465–473. doi: 10.1016/0896-6273(93)90334-n. [DOI] [PubMed] [Google Scholar]
- 32.Regehr WG, Delaney KR, Tank DW. The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapse. J Neurosci. 1994;14:523–537. doi: 10.1523/JNEUROSCI.14-02-00523.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schacher S, Montarolo P, Kandel ER. Selective short- and long-term effects of serotonin, small cardioactive peptide, and tetanic stimulation on sensorimotor synapses of Aplysia in culture. J Neurosci. 1990;10:3286–3294. doi: 10.1523/JNEUROSCI.10-10-03286.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schweitzer ES, Sanderson MJ, Wasterlain CG. Inhibition of regulated catecholamine secretion from PC12 cells by the Ca2+/calmodulin kinase II inhibitor KN-62. J Cell Sci. 1995;108:2619–2628. doi: 10.1242/jcs.108.7.2619. [DOI] [PubMed] [Google Scholar]
- 35.Sitges M, Dunkley PR, Chiu LM. A role for calcium/calmodulin kinase(s) in the regulation of GABA exocytosis. Neurochem Res. 1995;20:245–252. doi: 10.1007/BF00969539. [DOI] [PubMed] [Google Scholar]
- 36.Staubli U, Larson J, Lynch G. Mossy fiber potentiation and long-term potentiation involve different expression mechanisms. Synapse. 1990;5:333–335. doi: 10.1002/syn.890050410. [DOI] [PubMed] [Google Scholar]
- 37.Stevens CF, Wang Y. Facilitation and depression at single central synapses. Neuron. 1995;14:795–802. doi: 10.1016/0896-6273(95)90223-6. [DOI] [PubMed] [Google Scholar]
- 38.Van der Kloot W. Facilitation of transmission at the frog neuromuscular junction at 0°C is not maximal at time zero. J Neurosci. 1994;14:5722–5724. doi: 10.1523/JNEUROSCI.14-09-05722.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vilim FS, Cropper EC, Price DA, Kupfermann I, Weiss KR. Release of peptide cotransmitters in Aplysia: regulation and functional implications. J Neurosci. 1996;16:8105–8114. doi: 10.1523/JNEUROSCI.16-24-08105.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walters ET, Byrne JH. Post-tetanic potentiation in Aplysia sensory neurons. Brain Res. 1984;293:377–380. doi: 10.1016/0006-8993(84)91247-2. [DOI] [PubMed] [Google Scholar]
- 41.Waxham MN, Malenka RC, Kelly PT, Mauk MD. Calcium/calmodulin-dependent protein kinase II regulates hippocampal synaptic transmission. Brain Res. 1993;609:1–8. doi: 10.1016/0006-8993(93)90847-g. [DOI] [PubMed] [Google Scholar]
- 42.Whim MD, Lloyd PE. Frequency-dependent release of peptide cotransmitters from identified cholinergic motor neurons in Aplysia. Proc Natl Acad Sci USA. 1989;86:9034–9038. doi: 10.1073/pnas.86.22.9034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winslow JL, Duffy SN, Charlton MP. Homosynaptic facilitation of transmitter release in crayfish is not affected by mobile calcium chelators: implications for the residual ionized calcium hypothesis from electrophysiological and computational analyses. J Neurophysiol. 1994;72:1769–1793. doi: 10.1152/jn.1994.72.4.1769. [DOI] [PubMed] [Google Scholar]
- 44.Yamada WM, Zucker RS. Time course of transmitter release calculated from simulations of a calcium diffusion model. Biophys J. 1992;61:671–682. doi: 10.1016/S0006-3495(92)81872-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Younkin SG. An analysis of the role of calcium in facilitation at the frog neuromuscular junction. J Physiol (Lond) 1974;237:1–14. doi: 10.1113/jphysiol.1974.sp010466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]
- 47.Zengel JE, Magleby KL. Augmentation and facilitation of transmitter release. A quantitative description at the frog neuromuscular junction. J Gen Physiol. 1982;80:583–611. doi: 10.1085/jgp.80.4.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zengel JE, Magleby KL, Horn JP, McAfee DA, Yarowsky PJ. Facilitation, augmentation, and potentiation of synaptic transmission at the superior cervical ganglion of the rabbit. J Gen Physiol. 1980;76:213–231. doi: 10.1085/jgp.76.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]