

Abstract
In cultures of dissociated rat hippocampal neurons, persistent potentiation and depression of glutamatergic synapses were induced by correlated spiking of presynaptic and postsynaptic neurons. The relative timing between the presynaptic and postsynaptic spiking determined the direction and the extent of synaptic changes. Repetitive postsynaptic spiking within a time window of 20 msec after presynaptic activation resulted in long-term potentiation (LTP), whereas postsynaptic spiking within a window of 20 msec before the repetitive presynaptic activation led to long-term depression (LTD). Significant LTP occurred only at synapses with relatively low initial strength, whereas the extent of LTD did not show obvious dependence on the initial synaptic strength. Both LTP and LTD depended on the activation of NMDA receptors and were absent in cases in which the postsynaptic neurons were GABAergic in nature. Blockade of L-type calcium channels with nimodipine abolished the induction of LTD and reduced the extent of LTP. These results underscore the importance of precise spike timing, synaptic strength, and postsynaptic cell type in the activity-induced modification of central synapses and suggest that Hebb’s rule may need to incorporate a quantitative consideration of spike timing that reflects the narrow and asymmetric window for the induction of synaptic modification.
Keywords: synaptic modification, plasticity, hippocampal neurons, LTP, LTD, correlated-activity, spike timing, spiking, Hebb’s rule, Hebbian, target specificity, cell culture
Repetitive electrical activity can induce a persistent increase or decrease of synaptic efficacy in various parts of the nervous system, commonly referred to as long-term potentiation (LTP) and long-term depression (LTD), respectively (Bliss and Lφmo, 1973; Levy and Steward, 1983; Siegelbaum and Kandel, 1991;Bliss and Collingridge, 1993; Linden and Connor, 1995; Nicoll and Malenka, 1995). Conventional protocols for the induction of LTP and LTD generally involve repetitive presynaptic stimulation at various frequencies, in some cases coupled with a steady depolarization of the postsynaptic neuron. Such protocols have also been used in cultures of dissociated hippocampal neurons to induce LTP (Bekkers and Stevens, 1990; Arancio et al., 1995; Deisseroth et al., 1996; Tong et al., 1996) and LTD (Goda and Stevens, 1996; Fitzsimonds et al., 1997). In these previous studies, however, successful induction of LTP usually involved preincubation of the culture in tetrodotoxin or perfusion with zero Mg2+ solution during stimulation. Recent studies in cortical and hippocampal slices (Magee and Johnston, 1997; Markram et al., 1997) and in slice cultures (Debanne et al., 1998) have shown that back-propagating action potentials, when initiated at the appropriate time during repetitive synaptic activation, are effective in inducing persistent synaptic potentiation or depression. In contrast to the conventional protocol, these recent studies suggest that the timing of presynaptic and postsynaptic action potentials can play a decisive role in determining the type of synaptic modification. In the present study, we have fully characterized the dependence of synaptic modifications on the relative timing of presynaptic and postsynaptic spiking. Such quantitative study is of particular interest in view of the findings that precise spike timing may be used to encode information in neural networks (Hopfield, 1995; Mainen and Sejnowski, 1995; de Ruyter van Steveninck et al., 1997; Rieke et al., 1997). Our results showed that postsynaptic spiking that peaked within a time window of 20 msecafter synaptic activation resulted in LTP, whereas spiking within a window of 20 msec before synaptic activation led to LTD. A narrow transition zone of ∼5 msec existed between the potentiation and depression windows. Furthermore, we observed that the susceptibility of these synapses to potentiation strongly depended on their initial strength, with significant potentiation consistently observed only in synapses with initial amplitudes of evoked synaptic currents <500 pA. Finally, glutamatergic synapses made onto GABAergic neurons were not susceptible to modifications by the correlated presynaptic and postsynaptic activity, suggesting that target cell-specific mechanisms were involved in the induction of synaptic modifications.
MATERIALS AND METHODS
Cell culture. Low-density cultures of dissociated embryonic rat hippocampal neurons were prepared as described previously (Wilcox et al., 1994). Hippocampi were removed from embryonic day 18–20 (E18–20) rats and treated with trypsin for 15 min at 37°C, followed by washing and gentle trituration. The dissociated cells were plated at densities of 20,000–50,000 cells/ml on poly-l-lysine-coated glass coverslips in 35 mm Petri dishes. The plating medium was DMEM (BioWhittaker, Walkersville, MD) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone, Logan, UT), 10% Ham’s F12 with glutamine (BioWhittaker), and 50 U/ml penicillin–streptomycin (Sigma, St. Louis, MO). Twenty-four hours after plating, the culture medium was changed to the above medium containing 20 mm KCl. Both glial and neuronal cell types are present under these culture conditions. Cells were used for electrophysiological recordings after 8–14 d in culture.
Electrophysiology. Whole-cell perforated-patch recordings (Hamill et al., 1981; Horn and Marty, 1988; Rae et al., 1991) from two to three hippocampal neurons were performed simultaneously, using amphotericin B (Sigma) for perforation. The micropipettes were made from borosilicate glass capillaries (Kimax), with a resistance in the range of 2–4 MΩ. The pipettes were tip-filled with internal solution and then back-filled with internal solution containing 150 ng/ml amphotericin B. The internal solution contained the following (in mm): potassium gluconate 136.5, KCl 17.5, NaCl 9, MgCl2 1, HEPES 10, EGTA 0.2, pH 7.20. The external bath solution was a HEPES-buffered saline (HBS) containing the following (in mm): NaCl 145, KCl 3, HEPES 10, CaCl2 3, glucose 8, MgCl2 2, pH 7.30. The bath was constantly perfused with fresh recording medium at a slow rate throughout the recording, and all experiments were performed at room temperature. The neurons were visualized by phase-contrast microscopy with a Nikon inverted microscope. Recordings were performed with two or three patch clamp amplifiers (Axopatch 200B; Axon Instruments, Foster City, CA). Signals filtered at 5 kHz using amplifier circuitry were sampled at 10 kHz and analyzed using Axoscope software (Axon Instruments). Series resistance (10–30 MΩ) was always compensated at 80% (lag 100 μsec). Data were accepted for analysis only in cases in which the coefficient of variation of EPSCs or IPSCs during the control period did not exceed 0.4 and the input resistance (100–300 MΩ) remained constant throughout the experiment. For assaying synaptic connectivity, each neuron was stimulated at a low frequency (0.03–0.06 Hz) by 1 msec step-depolarization from −70 mV to +30 mV in voltage-clamp mode, and the responses from the other neurons as well as autaptic responses in the stimulated neuron itself were recorded. Under our recording conditions both EPSCs and IPSCs are inward currents at resting membrane potentials (−70 to −55 mV). The IPSCs had distinctly longer decay times and more negative reversal potentials than EPSCs (around −50 mV). Autaptic currents, when present, were observed after the 1 msec step-depolarization in voltage-clamp mode, with a relatively short onset latency of <3 msec. Consistent with previous reports (Wilcox et al., 1994; Fitzsimonds et al., 1997), EPSCs and IPSCs observed in these cultures were glutamatergic and GABAergic in nature, respectively. As shown in Figure 1, pharmacological studies indicated that EPSCs were mediated by the AMPA subtype of glutamate receptors, because they were blocked by 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μm) (RBI, Natick, MA); IPSCs were mediated by GABAA receptors, because they were blocked by bicuculline methiodide (10 μm) (RBI) but not affected by CNQX. For cell pairs that formed reciprocal and/or autaptic connections (see Fig.1), the cell types were identified by examining the time course and reversal potential of synaptic currents, in some cases further confirmed by pharmacological blockade of the transmitter receptors. In cell pairs that lacked reciprocal or autaptic connections, patch recordings on a third neuron that received input from one or both of the cells were performed, and cell-type identification was performed similarly.
Fig. 1.

Glutamatergic and GABAergic connections between two hippocampal neurons in cell cultures. A, Synaptic currents recorded from a pair of interconnected glutamatergic neurons. Step depolarizations (+ 100 mV, 1 msec) were applied sequentially to each neuron while EPSCs were monitored in both neurons (Vc = −70 mV). The matrices depict sample EPSCs recorded in either neuron (R1 orR2) when neuron 1 (S1) and neuron 2 (S2) were stimulated sequentially (average of 5 consecutive events). The three matrices represent synaptic and autaptic currents before and after sequential addition of bicuculline (10 μm) and CNQX (10 μm) into the culture.Arrowheads indicate monosynaptic EPSCs that were the focus of the present study. Calibration: 100 pA, 10 msec.B, Synaptic currents recorded from one glutamatergic neuron (R1) and an interconnecting GABAergic neuron (R2). Stimulating the latter (S2) results in IPSCs (marked by *). Three matrices represent synaptic and autaptic currents before and after sequential addition of CNQX (10 μm) and bicuculline (10 μm). Calibration: 100 pA, 10 msec.
RESULTS
Glutamatergic and GABAergic synapses in hippocampal cultures
Cultures of dissociated rat hippocampal neurons were prepared from E18–20 rat embryos and used after 8–14 d in vitro. Dual-perforated whole-cell recordings were made simultaneously from pairs of neurons that had formed functional synaptic connections. In many cases, reciprocal and autaptic connections were detected. The nature of synaptic connections was determined by the time course, the reversal potential, and the sensitivity of synaptic currents to pharmacological reagents (see Methods and Materials). Figure1A,Billustrates recordings from two pairs of neurons in these cultures. In the first pair, both cells were glutamatergic: synaptic and autaptic currents were not affected by bicuculline, an antagonist of GABAA receptors, but they were completely abolished by CNQX, a specific blocker of the AMPA subtype of the glutamate receptors. In the second pair, cell 1 was glutamatergic, whereas cell 2 was GABAergic, as confirmed by the sensitivity of synaptic and autaptic currents produced by cell 2 to bicuculline but not to CNQX. In the present study, we used only glutamatergic connections for which the postsynaptic cell type had been identified.
Potentiation induced by positively correlated postsynaptic spiking
We first examined activity-induced modifications of synaptic connections between two glutamatergic neurons. Synaptic efficacy was assayed at regular intervals by test stimulation of the presynaptic neuron (in voltage clamp) at a low frequency (0.03–0.06 Hz). To induce synaptic changes, repetitive stimulation (60 pulses at 1 Hz) was applied to the presynaptic neuron while both cells were held in current-clamp to allow spiking. Figure2A depicts results from a synaptic connection in which the evoked EPSPs during the repetitive stimulation were capable of initiating action potentials in the postsynaptic cell (a “suprathreshold” connection). Measurements of the amplitude of EPSCs revealed a persistent increase in synaptic efficacy after the repetitive stimulation. In another example shown in Figure 2B, the amplitude of EPSPs was too small to trigger action potentials in the postsynaptic neuron (a “subthreshold” synapse). However, depolarizing current pulses were injected into the postsynaptic cell to initiate spiking 5 msec after the onset of each EPSP. Significant potentiation was also observed in this case. Figure 2C summarizes the data from 14 excitatory connections similar to that described in Figure2A,B, with initial EPSC amplitude ranging from 20 to 500 pA. Of the 14 cases, 13 showed persistent potentiation in the EPSC amplitude after the repetitive stimulation. In all of these cases, the postsynaptic action potential peaked within 15 msec after the onset of each EPSP during the repetitive stimulation. This condition is referred to hereafter as “positively correlated spiking” to characterize the temporal relationship of postsynaptic spiking with the synaptic input. Finally, when the same experiments were performed in the presence of d-AP-5 (25 μm), an antagonist of the NMDA subtype of glutamate receptors, no synaptic potentiation was induced (n = 5) (Fig. 2C). This dependence of synaptic potentiation on the activation of NMDA receptors is similar to that found for LTP in the CA1 region of the hippocampus and several other brain areas (Bliss and Collingridge, 1993; Malenka and Nicoll, 1993).
Fig. 2.
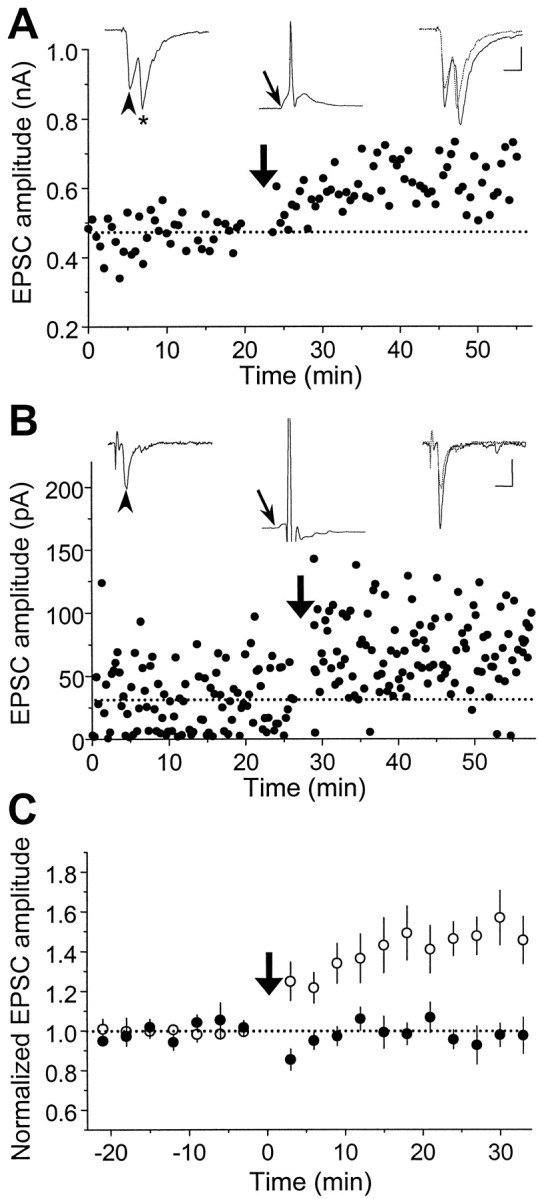
Synaptic potentiation induced by repetitive presynaptic stimulation with “positively correlated postsynaptic spiking.” A, Results obtained from a pair of glutamatergic neurons in hippocampal culture. Data points depict the amplitude of monosynaptic EPSCs induced by test stimuli (0.03 Hz,Vc = −70 mV) before and after repetitive stimulation of the presynaptic neuron (60 pulses at 1 Hz, marked by thethick arrow), with both neurons held in current clamp. Traces of EPSCs (average of 5–10 consecutive events) 5 min before (left) and 20 min after (right) the repetitive stimulation are shown above, with the 5 min trace (dashed line) superimposed onto the latter.Arrowheads mark the EPSCs being studied. * indicates a polysynaptic EPSC. The EPSP (with its onset time marked by thethin arrow) and the spike recorded during one cycle of the repetitive stimulation are depicted by the middle trace above. Note that each presynaptic stimulus was capable of initiating an action potential that peaked at ∼5 msec after the onset of the EPSP. Calibration: 200 pA, 10 msec for EPSCs; 40 mV, 10 msec for EPSPs.B, Results obtained from another pair of glutamatergic neurons. In this case, during each cycle of repetitive stimulation, the EPSP was subthreshold, and a depolarizing current pulse (2 nA, 2 msec) was injected into the postsynaptic neuron after the presynaptic activation to induce a spike that peaked at ∼5 msec after the onset of the EPSP. Calibration: 20 pA, 10 msec for EPSCs; 20 mV, 10 msec for EPSPs. C, Summary of all experiments with positively correlated postsynaptic spiking similar to that described inA and B in the absence (○,n = 14) or presence (•, n = 5) of d-AP-5 (25 μm). Data from all synaptic connections with initial EPSC amplitude smaller than 500 pA were included in the analysis. The amplitude of EPSCs from each experiment was grouped with a 3 min bin size and normalized to the mean value (dotted line) recorded before the repetitive stimulation. Data points represent mean ± SEM. The mean percentage change in synaptic strength after induction was 48.4 ± 9.9% (±SEM) and −2.3 ± 4.9% (±SEM) for experiments in the absence and presence of d-AP-5, respectively. Percentage change of each experiment was calculated from the mean EPSC amplitude 20–30 min after the induction protocol. When compared with the baseline value before induction, significant potentiation was observed in the absence of d-AP-5 (p < 0.001, t test) but not in the presence ofd-AP-5 (p > 0.1,t test).
Depression induced by negatively correlated postsynaptic spiking
To further examine the effects of postsynaptic spiking associated with repetitive presynaptic stimulation on synaptic efficacy, postsynaptic spiking was initiated by repetitive injection of depolarizing current pulses before the activation ofsubthreshold synaptic inputs. Recording from a pair of glutamatergic neurons is shown in Figure3A. Repetitive initiation of postsynaptic action potentials that peaked at 6 msec before the onset of EPSPs resulted in a persistent reduction in the EPSC amplitude. In addition, we observed synaptic depression of autaptic or polysynaptic connections made onto the same neuron that was repetitively stimulated. Figure 3B depicts one such case in which the action potential peaked at 5 msec before the onset of the autaptic EPSP during the repetitive stimulation. Figure 3C summarizes the results of all experiments in which the postsynaptic action potential peaked at 3–30 msec before the onset of each subthreshold EPSP, a condition referred to hereafter as “negatively correlated spiking” (n = 12, including both synaptic and autaptic connections). In contrast, when a similar set of experiments was performed in the presence of d-AP-5 (25 μm), no obvious synaptic depression was observed (n = 5) (Fig. 3C). The activation of NMDA receptors is therefore required for the induction of synaptic depression, similar to that found for LTD in the CA1 region of hippocampus (Dudek and Bear, 1992; Mulkey and Malenka, 1992).
Fig. 3.
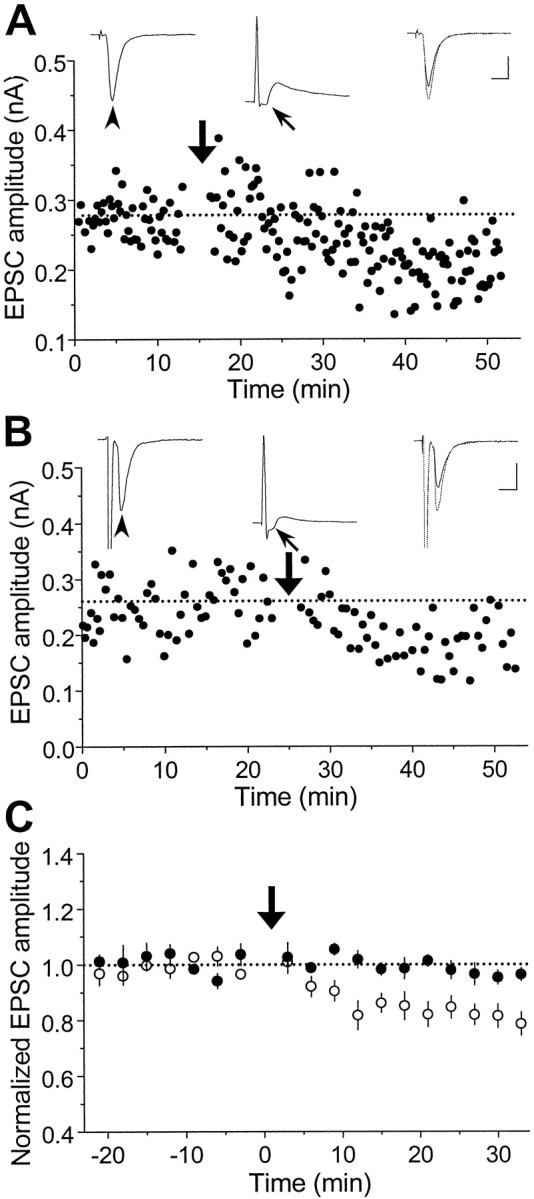
Synaptic depression induced by repetitive stimulation with “negatively correlated postsynaptic spiking” on subthreshold connections. A, Results from a pair of glutamatergic neurons that formed a subthreshold synaptic connection (similar to that in Fig. 2B). During each cycle of the repetitive stimulation (1 Hz, 60 sec, at the time marked by thearrow), a depolarizing current pulse was injected into the postsynaptic neuron to initiate an action potential that peaked at ∼6 msec before the onset of each EPSP. Calibration: 100 pA, 10 msec for EPSCs; 30 mV, 10 msec for EPSPs. B, An example of autaptic connections in which the action potential initiated by the current injection acted both presynaptically and postsynaptically. The interval between the onset of the autaptic response and the peak of the action potential was ∼5 msec. Calibration: same as in A. C, Summary of all experiments with negatively correlated postsynaptic spiking similar to that described in A and B in the absence (○,n = 12) or presence (•, n = 5) of d-AP-5 (25 μm). Data from all synaptic or autaptic connections with initial EPSC amplitude smaller than 1 nA were included in the analysis. Data points represent mean ± SEM. The mean percentage change in EPSC amplitude at 20–30 min after repetitive stimulation was −18.0 ± 3.2% (±SEM) and −2.5 ± 1.8% (±SEM) for experiments in the absence and presence ofd-AP-5, respectively. Significant depression was observed in the absence of d-AP-5 (p < 0.001, t test), but not in the presence ofd-AP-5 (p > 0.1,t test).
The same repetitive presynaptic and postsynaptic activation as described in Figure 3A were also applied tosuprathreshold connections that were strong enough to initiate postsynaptic spiking via synaptic activation. As shown in Figure 4A, initiating negatively correlated postsynaptic spiking by repetitive current injections 10 msec ahead of the repetitive synaptic activation did not result in depression in this type of synapses. Instead, synaptic potentiation was observed. The spikes initiated by synaptic activation had apparently exerted a dominant effect that prevented the depression attributable to the preceding spikes initiated by the current injection. Results from three experiments using this protocol are summarized in Figure 4B.
Fig. 4.

Effect of repetitive stimulation with negatively correlated postsynaptic spiking on suprathreshold connections.A, Results from an experiment similar to that described in Figure 3A, except that the synaptic activation was capable of initiating spiking of the postsynaptic neuron. The spike initiated by current pulse injection peaked at ∼10 msec before the onset of each EPSP during repetitive stimulation. Calibration: 100 pA, 10 msec for EPSCs; 40 mV, 10 msec for the EPSP. B, Summary of all experiments similar to that described inA. Data points represent mean ± SEM (n = 3). The mean percentage change in synaptic strength after induction was 31.9 ± 9.3% (±SEM). Significant potentiation was observed (p < 0.05,t test).
Dependence on synaptic strength
During the above study, we noted a consistent failure in the induction of synaptic potentiation for connections that had EPSCs of relatively large amplitudes. When the extent of synaptic potentiation, as indicated by the percentage change in the mean EPSC amplitude, was plotted against the mean EPSC amplitude before the repetitive positively correlated postsynaptic spiking, a clear inverse relationship was observed (Fig. 5). Because of the use of log scale for initial EPSC amplitude, the fitted line indicates that the percentage potentiation (by positively correlated spiking) and initial EPSC amplitude have a nonlinear but strongly inverse relationship. Potentiation occurred mostly in weak synapses, with initial EPSC amplitude <500 pA. We have also examined the correlation between the initial amplitude of EPSC and the extent of synaptic depression induced by negatively correlated spiking at subthreshold inputs. No significant correlation was observed for connections with initial EPSC amplitudes ranging from 20 to 900 pA (Fig. 5).
Fig. 5.

Dependence of synaptic modifications on the initial synaptic strength. The percentage change in the EPSC amplitude after the repetitive stimulation (1 Hz for 60 sec) was plotted against the initial mean amplitude of EPSCs. Open circlesrepresent data from synapses exposed to repetitive presynaptic stimulation with positively correlated postsynaptic spiking (data set includes those shown in Fig. 2C). Filled circles represent data from synapses exposed to repetitive presynaptic stimulation with negatively correlated postsynaptic spiking (data set includes those shown in Fig. 3C). Percentage changes were calculated from the average EPSC amplitude 20–30 min after the repetitive stimulation. Lines represent best fits with linear regression between the percentage change and the logarithm of initial EPSC amplitudes for positively correlated (r = −0.72, p = 0.00017) and negatively correlated (r = 0.037,p = 0.89) spiking, respectively.
Dependence on postsynaptic cell type
In a separate set of experiments, we have studied activity-induced modification of glutamatergic synapses onto GABAergic neurons (Fig.1B). As shown in Figure6A, repetitive stimulation of the presynaptic neuron (60 pulses at 1 Hz), coupled with postsynaptic spiking 6 msec after the onset of each EPSP, did not result in any change in synaptic efficacy. The summary of results from all similar experiments involving repetitive stimulation with positively correlated spiking of postsynaptic GABAergic neurons (with initial EPSC amplitude <500 pA) clearly indicated the absence of any synaptic change (Fig. 6C). The average percentage change in the EPSC amplitude 20–30 min after the repetitive stimulation was −0.3 ± 3.4% (SEM; n = 5), which was significantly different (p < 0.001;t test) from that found for cases using glutamatergic postsynaptic neurons (Fig. 2C) (48.4 ± 9.9%, SEM;n = 14). In another case (Fig. 6B), negatively correlated postsynaptic spiking was initiated (via current injection) in the postsynaptic GABAergic neuron 16 msec before the onset of each EPSP during repetitive stimulation (60 pulses at 1 Hz) of a “subthreshold” synaptic input. Again no change in synaptic efficacy was observed. The summary of all data on similar negatively correlated spiking of postsynaptic GABAergic neurons indicates a complete absence of synaptic depression (Fig. 6D). The average percentage change in EPSC amplitude 20–30 min after repetitive stimulation was −1.7 ± 2.0% (SEM; n= 4), which was significantly different (p < 0.001; t test) from that found for the corresponding cases with glutamatergic postsynaptic neurons (Fig. 4C) (−18.0 ± 3.2%, SEM; n = 12).
Fig. 6.

Lack of synaptic modification for glutamatergic synapses onto GABAergic neurons. A, Results from an experiment performed in the same manner as that described in Figure2B except that the postsynaptic neuron was GABAergic. During each cycle of repetitive stimulation, the postsynaptic spike peaked at ∼5 msec after the onset of the EPSP. Calibration: 100 pA, 10 msec for EPSCs; 50 mV, 10 msec for EPSPs.B, Results from an experiment similar to that described in Figure 3A except that the postsynaptic neuron was GABAergic. Postsynaptic spiking was initiated 16 msec before the onset of each EPSP. Calibration: same as in A. C, D, Summary of all experiments with positively correlated (C) and negatively correlated (D) spiking similar to that described inA and B, respectively. The mean percentage change in the EPSC amplitude was −0.3 ± 3.4% (±SEM,n = 5) and −1.7 ± 2.0% (±SEM,n = 4), respectively, for C andD, indicating no significant synaptic change (p > 0.1, t test).
The critical window for synaptic modifications
To determine the precise timing required for repetitive correlated presynaptic and postsynaptic spiking to induce synaptic modifications, we further varied the time interval between the presynaptic stimulation and postsynaptic spiking, using the same protocol of repetitive stimulation. In these experiments we used only subthreshold connections on glutamatergic neurons with initial EPSC amplitude <500 pA. As shown in Figure 7, synaptic changes showed a strong but highly asymmetric dependence on spike timing. Potentiation was consistently induced when the postsynaptic spikes peaked within a time window of 20 msec after the onset EPSPs, whereas depression was induced when the spikes peaked within a window of 20 msec before the onset of EPSPs. The ability for correlated spiking to induce potentiation or depression decreases rapidly as the absolute value of spike timing increases, so that outside the 40 msec window, synaptic modification was essentially absent. A narrow transition range of ∼5 msec (∼Δt = 0) exists between maximal depression and the maximal potentiation at which the effect of correlated spiking showed large fluctuation.
Fig. 7.

Critical window for the induction of synaptic potentiation and depression. The percentage change in the EPSC amplitude at 20–30 min after the repetitive correlated spiking (60 pulses at 1 Hz) was plotted against the spike timing. Spike timing was defined by the time interval (Δt) between the onset of the EPSP and the peak of the postsynaptic action potential during each cycle of repetitive stimulation, as illustrated by the traces above. For this analysis, we included only synapses with initial EPSC amplitude of <500 pA, and all EPSPs were subthreshold for data associated with negatively correlated spiking. Calibration: 50 mV, 10 msec.
Dependence on Ca2+ channels
An immediate action of postsynaptic spiking is the opening of voltage-gated Ca2+ channels. We have thus tested the potential role of dendritic L-type Ca2+ channels (Bolshakov and Siegelbaum, 1994; B. R. Christie et al., 1995,1997; R. C. Christie et al., 1996; Magee and Johnston, 1995, 1997;Johnston et al., 1996; Deisseroth et al., 1998) in the induction of synaptic modifications induced by correlated spiking. Experiments were performed on subthreshold inputs onto glutamatergic neurons in the same manner as that described above, but with nimodipine (10 μm), an L-type Ca2+ channel blocker, added to the culture before the onset of the experiment. As shown in Figure 8, we observed a significant increase in EPSC amplitude (27.3 ± 8.6%, ± SEM;n = 4) after repetitive positively correlated spiking (60 pulses at 1 Hz). The extent of potentiation appears to be lower than that observed in the absence of nimodipine (48.4 ± 9.9%, ±SEM; n = 14), although the difference is not statistically significant (p = 0.068;t test). In contrast, no significant synaptic change in EPSCs was observed (−1.2 ± 1.3%, ± SEM; n = 7) after repetitive negatively correlated spiking. We thus conclude that Ca2+ influx through L-type Ca2+channels may contribute to, but is not necessary for, synaptic potentiation induced by positively correlated spiking. For depression induced by negatively correlated spiking, however, Ca2+ influx through L-type Ca2+channels is necessary.
Fig. 8.

Differential effects of nimodipine on the induction of LTP and LTD by correlated spiking. A, Results from an experiment similar to that shown in Figure2B except that the bath solution contained 10 μm nimodipine. During repetitive stimulation, the postsynaptic spike was initiated ∼5 msec after the onset of the EPSP. Calibration: 100 pA, 10 msec for EPSCs; 30 mV, 10 msec for EPSPs.B, Results from an experiment similar to that shown in Figure 3A except that the bath solution contained 10 μm nimodipine. During repetitive stimulation, postsynaptic spiking was initiated ∼5 msec before the onset of each EPSP. Calibration: 200 pA, 10 msec for EPSCs; 30 mV, 10 msec for EPSPs.C, Summary of experiments with positively correlated spiking similar to that in A. The mean percentage change in the EPSC amplitude was 27.3 ± 8.6% (±SEM,n = 4), which represents significant potentiation (p < 0.05, t test).D, Summary of experiments with negatively correlated spiking similar to that in B. The mean percentage change in the EPSC amplitude was −1.2 ± 1.3% (±SEM,n = 7), indicating no significant synaptic change (p > 0.2; t test).
DISCUSSION
In vivo studies of rat hippocampus have shown that the temporal order of stimulation applied to the testing and conditioning pathways is crucial for the associative induction of synaptic modification (Levy and Steward, 1983). In rat brain slices and slice cultures, back-propagating action potentials initiated in the postsynaptic neuron by current injections at the soma in conjunction with presynaptic stimulation have been shown to induce persistent synaptic modifications (Magee and Johnston, 1997; Markram et al., 1997;Debanne et al., 1998). A synaptic input became potentiated when it preceded the back-propagating action potential by 10 msec and was depressed if it arrived at 10 msec after the action potential. In contrast, action potential initiated 100 msec before or after the synaptic input had no effect (Markram et al., 1997; Debanne et al., 1998). These earlier studies, however, did not provide a full description of the timing requirement. It was not clear, for example, whether synaptic input arriving 20 or 50 msec ahead of postsynaptic action potentials would become potentiated. The present results indicate a surprisingly narrow time window of 20 msec for either potentiation or depression to be induced. The dependence of synaptic modifications on spike timing is strikingly asymmetric and exhibits a sharp transition of 5 msec between depression and potentiation. A similar time window for activity-dependent synaptic modifications was also observed recently in a developing retinotectal system in vivo (Zhang et al., 1998). The effects of precise timing of spikes in the presynaptic and postsynaptic neurons may be used in neural networks to decipher information encoded in spike timing (Hopfield, 1995; Mainen and Sejnowski, 1995; de Ruyter van Steveninck et al., 1997; Rieke et al., 1997) and to store information relating to the temporal order of various synaptic inputs received by a neuron during learning and memory (Gerstner and Abbott, 1997; Mehta et al., 1997)
In these cultures we found that only weak synaptic connections are susceptible to synaptic potentiation by correlated spiking, with a “cutoff” amplitude of ∼500 pA. Larger EPSCs may represent either higher average sizes of evoked synaptic currents at individual synaptic contacts (boutons) made by the presynaptic neuron or a larger number of boutons, or both. If higher amplitude represents increased efficacy of individual boutons, then the existence of the cutoff amplitude for LTP induction may indicate that the machinery underlying the expression of synaptic potentiation has been saturated. For example, the probability of presynaptic vesicular fusion or the expression of new postsynaptic glutamate receptors may have reached the maximal level sustainable by the cell. Because synaptic inputs that contribute to the postsynaptic spiking fall into the “potentiation window” associated with the spikes, spontaneous spiking activity in these cultures may have continuously potentiated these synapses to a saturated level, resulting in failure in the induction of synaptic potentiation in older cultures.
The cellular basis that gives rise to the critical window for the induction of synaptic modifications remains to be determined. The involvement of NMDA receptors in both potentiation and depression suggests that elevation of cytosolic Ca2+ is critical in the induction process, similar to that for synapses in the CA1 region of the hippocampus (Nicoll and Malenka, 1995). Action potentials initiated during the critical time window after synaptic activation but before the dissociation of glutamate from the NMDA channel will lead to the opening of the channel (by removing the Mg2+ block) and a localized surge of cytoplasmic Ca2+ (Connor et al., 1994). This NMDA receptor-mediated Ca2+ influx may also act cooperatively with Ca2+ influx through the voltage-dependent Ca2+ channels to induce synaptic potentiation (Eilers et al., 1995; Yuste and Denk, 1995; Magee and Johnston, 1997). The finding of a reduced extent of synaptic potentiation in the presence of L-type Ca2+ channel blocker is consistent with the latter findings. Although the off-rate of glutamate from the NMDA receptor is much longer than 20 msec, the requirement of multiple Ca2+ binding in the activation of downstream effector molecules (e.g., calmodulin) could potentially sharpen the time window of synaptic modification. Alternatively, the dendritically expressed transient A-type K+ channels that can be inactivated by subthreshold EPSPs may also play a role by limiting the back-propagation of dendritic action potentials initiated outside the potentiation window (Hoffman et al., 1997). In the case of negatively correlated spiking, spike-induced Ca2+ elevation attributable to opening of Ca2+ channels before synaptic activation followed by a low-level Ca2+ elevation attributable to subthreshold synaptic activation may be responsible for the induction of synaptic depression. Indeed, blocking L-type Ca2+channels abolished the induction of LTD (Fig. 8). Interestingly, binding of glutamate to NMDA receptors is also required for the induction of LTD, although the membrane potential remained at a relatively negative level after the spike. Taken together, our results are consistent with the notion that spatial–temporal patterns of postsynaptic Ca2+ elevation are critical for the induction of synaptic changes (Lisman, 1989; Malenka et al., 1992;Neveu and Zucker, 1996). Finally, we noted that there was a conspicuous absence of short-term potentiation or depression in the present study. This can be accounted for by our use of low-frequency stimulation, because short-term potentiation or depression is known to result from changes in the presynaptic transmitter supply after high-frequency stimulation (Zucker et al., 1991).
The dependence of synaptic modifications on postsynaptic cell type has been observed in the Schaffer collateral (McMahon and Kauer, 1997) and the mossy fiber pathways (Maccaferri et al., 1998) in hippocampal slices. In both studies, the standard protocol of high-frequency stimulation that normally induces LTP at synapses onto pyramidal cells either had no effect or resulted in persistent depression of synapses onto interneurons. Our results showed that not only the induction of LTP is target-cell specific; similar target specificity also exists for the induction of LTD. The target specificity could result from differences in the postsynaptic molecular machinery underlying synaptic modifications. For example, both the α isoform of calcium/calmodulin-dependent protein kinase II (CaMK II α) and the Ca2+/calmodulin-dependent protein phosphatase 2B (calcineurin) appear to be absent in the postsynaptic densities of glutamatergic inputs onto GABAergic neurons in the cerebral cortex and hippocampus (Stevens et al., 1994; Liu and Jones, 1996, 1997;Sík et al., 1998). Interestingly, in parallel fiber synapses in the cerebellum-like electrosensory lobe of the mormyrid electric fish, where postsynaptic targets are GABAergic Purkinje-like cells, synaptic modifications can still be induced. However, the dependence on the temporal order of correlated presynaptic and postsynaptic spikes is opposite to that reported here (Bell et al., 1997).
The general notion that correlated presynaptic and postsynaptic spiking is responsible for synaptic modifications is well known as “Hebb’s postulate of learning” (Hebb, 1949; Stent, 1973; Brown et al., 1990;Churchland and Sejnowski, 1992). Hebb’s original statement—“When an axon of a cell A is near enough to excite cell B or repeatedly or persistently takes part in firing it, some growth or metabolic change takes place in both cells such that A’s efficiency, as one of the cells firing B, is increased”—is remarkably accurate in describing qualitatively the synaptic potentiation induced by positively correlated spiking reported here. In the past few decades, this concept has been generalized and formulated mathematically into various “Hebbian rules” of synaptic modification (Bienenstock et al., 1982;Brown et al., 1990; Churchland and Sejnowski, 1992). Although the most commonly used ones are based on the statistical properties of presynaptic and postsynaptic activity (e.g., activity product, activity covariance, etc.) without considering the detailed temporal structure of the spike patterns, a number of formulations have indeed incorporated a factor to reflect the relative spike timing (Sutton and Barto, 1981; Tesauro, 1986; Klopf, 1988; Herz et al., 1989; Gerstner et al., 1993; Gerstner and Abbott, 1997). By introducing an asymmetric temporal window into the synaptic rule, a neural network can take advantage of this “built-in” causality/sequence detection mechanism to naturally implement a predictive function. This is especially clear in a network model of navigational map learning (Gerstner and Abbott, 1997), where the assumed rule of synaptic modification is very similar to what we have observed here (Fig. 7). Our results therefore provide both experimental support and crucial parameters for such interpretations of Hebb’s rule.
Footnotes
This work was supported by grants from National Institutes of Health (NS36999). G.B. is the recipient of a University of California President’s Postdoctoral Fellowship. We thank X.-y. Wang for cell culture preparations and L. Zhang and B. Berninger for helpful discussions.
Correspondence should be addressed to Mu-ming Poo, Department of Biology–0357, University of California at San Diego, La Jolla, CA 92093.
REFERENCES
- 1.Arancio O, Kandel ER, Hawkins RD. Activity-dependent long-term enhancement of transmitter release by presynaptic 3′,5′-cyclic GMP in cultured hippocampal neurons. Nature. 1995;376:74–80. doi: 10.1038/376074a0. [DOI] [PubMed] [Google Scholar]
- 2.Bekkers JM, Stevens CF. Presynaptic mechanism for long-term potentiation in the hippocampus. Nature. 1990;346:724–729. doi: 10.1038/346724a0. [DOI] [PubMed] [Google Scholar]
- 3.Bell CC, Han VZ, Sugawara Y, Grant K. Synaptic plasticity in a cerebellum-like structure depends on temporal order. Nature. 1997;387:278–281. doi: 10.1038/387278a0. [DOI] [PubMed] [Google Scholar]
- 4.Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 6.Bliss TV, Lφmo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol (Lond) 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- 8.Brown TH, Kairiss EW, Keenan CL. Hebbian synapses: biophysical mechanisms and algorithms. Annu Rev Neurosci. 1990;13:475–511. doi: 10.1146/annurev.ne.13.030190.002355. [DOI] [PubMed] [Google Scholar]
- 9.Christie BR, Eliot LS, Ito K, Miyakawa H, Johnston D. Different Ca2+ channels in soma and dendrites of hippocampal pyramidal neurons mediate spike-induced Ca2+ influx. J Neurophysiol. 1995;73:2553–2557. doi: 10.1152/jn.1995.73.6.2553. [DOI] [PubMed] [Google Scholar]
- 10.Christie BR, Schexnayder LK, Johnston D. Contribution of voltage-gated Ca2+ channels to homosynaptic long-term depression in the CA1 region in vitro. J Neurophysiol. 1997;77:1651–1655. doi: 10.1152/jn.1997.77.3.1651. [DOI] [PubMed] [Google Scholar]
- 11.Christie RC, Magee JC, Johnston D. The role of dendritic action potentials and Ca2+ influx in the induction of homosynaptic long-term depression in hippocampal CA1 pyramidal neurons. Learning Memory. 1996;3:169–169. doi: 10.1101/lm.3.2-3.160. [DOI] [PubMed] [Google Scholar]
- 12.Churchland PS, Sejnowski TJ. The computational brain. MIT; Cambridge, MA: 1992. [Google Scholar]
- 13.Connor JA, Miller LD, Petrozzino J, Muller W. Calcium signaling in dendritic spines of hippocampal neurons. J Neurobiol. 1994;25:234–242. doi: 10.1002/neu.480250304. [DOI] [PubMed] [Google Scholar]
- 14.Debanne D, Gahwiler BH, Thompson SM. Long-term synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J Physiol (Lond) 1998;507:237–247. doi: 10.1111/j.1469-7793.1998.237bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 16.Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- 17.de Ruyter van Steveninck RR, Lewen GD, Strong SP, Koberle R, Bialek W. Reproducibility and variability in neural spike trains. Science. 1997;275:1805–1808. doi: 10.1126/science.275.5307.1805. [DOI] [PubMed] [Google Scholar]
- 18.Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eilers J, Augustine GJ, Konnerth A. Subthreshold synaptic Ca2+ signalling in fine dendrites and spines of cerebellar Purkinje neurons. Nature. 1995;373:155–158. doi: 10.1038/373155a0. [DOI] [PubMed] [Google Scholar]
- 20.Fitzsimonds RM, Song HJ, Poo MM. Propagation of activity-dependent synaptic depression in simple neural networks. Nature. 1997;388:439–448. doi: 10.1038/41267. [DOI] [PubMed] [Google Scholar]
- 21.Gerstner W, Abbott LF. Learning navigational maps through potentiation and modulation of hippocampal place cells. J Comput Neurosci. 1997;4:79–94. doi: 10.1023/a:1008820728122. [DOI] [PubMed] [Google Scholar]
- 22.Gerstner W, Ritz R, Van Hemmen JL. Why spikes? Hebbian learning and retrieval of time-resolved excitation patterns. Biol Cybern. 1993;69:503–515. [PubMed] [Google Scholar]
- 23.Goda Y, Stevens CF. Long-term depression properties in a simple system. Neuron. 1996;16:103–111. doi: 10.1016/s0896-6273(00)80027-6. [DOI] [PubMed] [Google Scholar]
- 24.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 25.Hebb DO. The organization of behavior. Wiley; New York: 1949. [Google Scholar]
- 26.Herz A, Sulzer B, Kuhn R, van Hemmen JL. Hebbian learning reconsidered: representation of static and dynamic objects in associative neural nets. Biol Cybern. 1989;60:457–467. doi: 10.1007/BF00204701. [DOI] [PubMed] [Google Scholar]
- 27.Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- 28.Hopfield JJ. Pattern recognition computation using action potential timing for stimulus representation. Nature. 1995;376:33–36. doi: 10.1038/376033a0. [DOI] [PubMed] [Google Scholar]
- 29.Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnston D, Magee JC, Colbert CM, Cristie BR. Active properties of neuronal dendrites. Annu Rev Neurosci. 1996;19:165–186. doi: 10.1146/annurev.ne.19.030196.001121. [DOI] [PubMed] [Google Scholar]
- 31.Klopf AH. A neuronal model of classical conditioning. Psychobiology. 1988;16:85–125. [Google Scholar]
- 32.Levy WB, Steward O. Temporal contiguity requirements for long-term associative potentiation/depression in the hippocampus. Neuroscience. 1983;8:791–797. doi: 10.1016/0306-4522(83)90010-6. [DOI] [PubMed] [Google Scholar]
- 33.Linden DJ, Connor JA. Long-term synaptic depression. Annu Rev Neurosci. 1995;18:319–357. doi: 10.1146/annurev.ne.18.030195.001535. [DOI] [PubMed] [Google Scholar]
- 34.Lisman J. A mechanism for the Hebb and the anti-Hebb processes underlying learning and memory. Proc Natl Acad Sci USA. 1989;86:9574–9578. doi: 10.1073/pnas.86.23.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu X, Jones EG. Alpha isoform of calcium-calmodulin dependent protein kinase II (CAM II kinase-alpha) restricted to excitatory synapses in the CA1 region of rat hippocampus. NeuroReport. 1997;8:1475–1479. doi: 10.1097/00001756-199704140-00030. [DOI] [PubMed] [Google Scholar]
- 36.Liu XB, Jones EG. Localization of alpha type II calcium calmodulin-dependent protein kinase at glutamatergic but not gamma-aminobutyric acid (GABAergic) synapses in thalamus and cerebral cortex. Proc Natl Acad Sci USA. 1996;93:7332–7336. doi: 10.1073/pnas.93.14.7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maccaferri G, Toth K, McBain CJ. Target-specific expression of presynaptic mossy fiber plasticity. Science. 1998;279:1368–1370. doi: 10.1126/science.279.5355.1368. [DOI] [PubMed] [Google Scholar]
- 38.Magee JC, Johnston D. Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Physiol (Lond) 1995;487:67–90. doi: 10.1113/jphysiol.1995.sp020862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- 40.Mainen ZF, Sejnowski TJ. Reliability of spike timing in neocortical neurons. Science. 1995;268:1503–1506. doi: 10.1126/science.7770778. [DOI] [PubMed] [Google Scholar]
- 41.Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- 42.Malenka RC, Lancaster B, Zucker RS. Temporal limits on the rise in postsynaptic calcium required for the induction of long-term potentiation. Neuron. 1992;9:121–128. doi: 10.1016/0896-6273(92)90227-5. [DOI] [PubMed] [Google Scholar]
- 43.Markram H, Lubke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- 44.McMahon LL, Kauer JA. Hippocampal interneurons express a novel form of synaptic plasticity. Neuron. 1997;18:295–305. doi: 10.1016/s0896-6273(00)80269-x. [DOI] [PubMed] [Google Scholar]
- 45.Mehta MR, Barnes CA, McNaughton BL. Experience-dependent, asymmetric expansion of hippocampal place fields. Proc Natl Acad Sci USA. 1997;94:8918–8921. doi: 10.1073/pnas.94.16.8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- 47.Neveu D, Zucker RS. Postsynaptic levels of [Ca2+]i needed to trigger LTD and LTP. Neuron. 1996;16:619–629. doi: 10.1016/s0896-6273(00)80081-1. [DOI] [PubMed] [Google Scholar]
- 48.Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- 49.Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- 50.Rieke F, Warland D, de Ruyter van Steveninck R, Bialek W. Spikes: exploring the neural code. MIT; Cambridge, MA: 1997. [Google Scholar]
- 51.Siegelbaum SA, Kandel ER. Learning-related synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1991;1:113–120. doi: 10.1016/0959-4388(91)90018-3. [DOI] [PubMed] [Google Scholar]
- 52.Sík A, Hájos N, Gulácsi A, Mody I, Freund TF. The absence of a major Ca2+ signaling pathway in GABAergic neurons of the hippocampus. Proc Natl Acad Sci USA. 1998;95:3245–3250. doi: 10.1073/pnas.95.6.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stent GS. A physiological mechanism for Hebb’s postulate of learning. Proc Natl Acad Sci USA. 1973;70:997–1001. doi: 10.1073/pnas.70.4.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stevens CF, Tonegawa S, Wang Y. The role of calcium-calmodulin kinase II in three forms of synaptic plasticity. Curr Biol. 1994;4:687–693. doi: 10.1016/s0960-9822(00)00153-6. [DOI] [PubMed] [Google Scholar]
- 55.Sutton RS, Barto AG. Toward a modern theory of adaptive networks: expectation and prediction. Psychol Rev. 1981;88:135–170. [PubMed] [Google Scholar]
- 56.Tesauro G. Simple neural models of classical conditioning. Biol Cybern. 1986;55:187–200. doi: 10.1007/BF00341933. [DOI] [PubMed] [Google Scholar]
- 57.Tong G, Malenka RC, Nicoll RA. Long-term potentiation in cultures of single hippocampal granule cells: a presynaptic form of plasticity. Neuron. 1996;16:1147–1157. doi: 10.1016/s0896-6273(00)80141-5. [DOI] [PubMed] [Google Scholar]
- 58.Wilcox KS, Buchhalter J, Dichter MA. Properties of inhibitory and excitatory synapses between hippocampal neurons in very low density cultures. Synapse. 1994;18:128–151. doi: 10.1002/syn.890180206. [DOI] [PubMed] [Google Scholar]
- 59.Yuste R, Denk W. Dendritic spines as basic functional units of neuronal integration. Nature. 1995;375:682–684. doi: 10.1038/375682a0. [DOI] [PubMed] [Google Scholar]
- 60.Zhang LI, Tao HW, Holt CE, Harris WA, Poo MM. A critical window for cooperation and competition among developing retinotectal synapses. Nature. 1998;395:37–44. doi: 10.1038/25665. [DOI] [PubMed] [Google Scholar]
- 61.Zucker RS, Delaney KR, Mulkey R, Tank DW. Presynaptic calcium in transmitter release and posttetanic potentiation. Ann NY Acad Sci. 1991;635:191–207. doi: 10.1111/j.1749-6632.1991.tb36492.x. [DOI] [PubMed] [Google Scholar]