

Abstract
N-Ethylmaleimide–sensitive fusion protein (NSF) is a cytosolic protein thought to play a key role in vesicular transport in all eukaryotic cells. Although NSF was proposed to function in the trafficking of synaptic vesicles responsible for neurotransmitter release, only recently have in vivoexperiments begun to reveal a specific function for NSF in this process. Our previous work showed that mutations in aDrosophila NSF gene, dNSF1, are responsible for the temperature-sensitive paralytic phenotype in comatose(comt) mutants. In this study, we perform electrophysiological and ultrastructural analyses in three differentcomt alleles to investigate the function of dNSF1 at native synapses in vivo. Electrophysiological analysis of postsynaptic potentials and currents at adult neuromuscular synapses revealed that in the absence of repetitive stimulation,comt synapses exhibit wild-type neurotransmitter release at restrictive (paralytic) temperatures. In contrast, repetitive stimulation at restrictive temperatures revealed a progressive, activity-dependent reduction in neurotransmitter release incomt but not in wild type. These results indicate that dNSF1 does not participate directly in the fusion of vesicles with the target membrane but rather functions in maintaining the pool of readily releasable vesicles competent for fast calcium-triggered fusion. To define dNSF1 function further, we used transmission electron microscopy to examine the distribution of vesicles within synaptic terminals, and observed a marked accumulation of docked vesicles at restrictive temperatures in comt. Together, the results reported here define a role for dNSF1 in the priming of docked synaptic vesicles for calcium-triggered fusion.
Keywords: NSF, neurotransmitter release, Drosophila, comatose, synaptic vesicle, neuromuscular transmission
The molecular machinery underlying chemical synaptic transmission represents a fundamental element of neural function. One critical aspect of this process is the highly regulated and rapid form of exocytosis responsible for the release of neurotransmitter. A number of proteins has now been identified as components of the neurotransmitter release apparatus, and recent progress has begun to address their specific roles.
The N-ethylmaleimide–sensitive fusion protein (NSF) is a soluble, oligomeric ATPase that served as one of the primary starting points from which this progress has developed. NSF was identified as anN-ethylmaleimide (NEM)–sensitive factor required for Golgi transport in vitro (Wilson et al., 1989; Rothman, 1994) and was termed a fusion protein on the basis that NEM caused an accumulation of transport vesicles at target membranes (Orci et al., 1989). The biochemical connection to membranes was made by the elegant characterization of an NSF-containing protein complex including the soluble NSF attachment proteins (SNAPs) (Clary et al., 1990; Whiteheart et al., 1993) and their membrane receptors, SNAP receptors (SNAREs) (Söllner et al., 1993; Südhof, 1995). In parallel with these studies, genetic analysis of constitutive secretion in yeast (Schekman, 1992; Bennett and Scheller, 1993; Ferro-Novick and Jahn, 1994), together with biochemical and functional analysis of synaptic proteins (Huttner, 1993; Südhof, 1995), independently identified and characterized the NSF, SNAP, and SNARE proteins. Thus a convergence of results revealed a set of core proteins functioning in intracellular vesicle trafficking in perhaps all eukaryotes.
The SNARE hypothesis (Rothman, 1994; Rothman and Wieland, 1996) was developed to explain the docking and fusion of transport vesicles with their target membranes. According to this hypothesis, vesicle docking involves assembly of a protein complex including vesicle SNAREs (e.g., synaptobrevin) and target SNAREs (e.g., SNAP-25 and syntaxin). Because different SNAREs seemed to function at distinct stages of the secretory pathway in yeast (Ferro-Novick and Jahn, 1994), the specificity of vesicle targeting was attributed to these proteins. It was further proposed that after docking, the SNARE complex is joined by the cytosolic SNAP and NSF proteins to form a fusion particle. Finally, hydrolysis of ATP by NSF was proposed to disassemble or rearrange this structure in a subsequent step necessary for fusion. The SNARE hypothesis has provided an excellent working model and has been both supported and challenged by subsequent functional analysis of constitutive membrane trafficking (Mayer et al., 1996; Nichols et al., 1997; Ungermann et al., 1998).
Regarding the role of SNARE complexes in neurotransmitter release,in vivo work in Drosophila (Broadie et al., 1995;Schulze et al., 1995; Sweeney et al., 1995; Deitcher et al., 1998), inCaenorhabditis elegans (Nonet et al., 1998), and at the squid giant synapse (Hunt et al., 1994; O’Connor et al., 1997) has been informative. Disruption of individual vesicle or target SNAREs in these systems eliminated or reduced evoked neurotransmitter release. However, in apparent contrast to predictions of the SNARE hypothesis, ultrastructural analysis (Hunt et al., 1994; Broadie et al., 1995;O’Connor et al., 1997) indicated that these SNAREs, at least individually, are dispensable for specific targeting and docking of synaptic vesicles. Regarding the role of NSF and SNAPs, only recently have in vivo experiments begun to address their function in neurotransmitter release (DeBello et al., 1995; Schweizer et al., 1998). Our previous work has demonstrated that a DrosophilaNSF homolog, dNSF1, represents the comatose(comt) locus (Pallanck et al., 1995a). Temperature-sensitivecomt alleles were shown previously to exhibit rapid paralysis and a neurophysiological defect at elevated temperatures (Siddiqi and Benzer, 1976). Here we use electrophysiological and ultrastructural analysis in comt to define an in vivo role for the dNSF1 gene product in neurotransmitter release.
Parts of this work have been published in preliminary form (Kawasaki and Ordway, 1997, 1998; Ordway et al., 1998).
MATERIALS AND METHODS
Dissection
Male or female flies 1–4 d of age were anesthetized using CO2 and then mounted over a hole in an air tube and secured with wax as described (Koenig et al., 1989). Air was delivered to the tracheal system using an aquarium pump. In all experiments the chamber was perfused at 0.5 ml/min with recording solution (see Microelectrode recordings).
Electrophysiological recordings. The fly was mounted laterally and dissected in recording solution to expose the lateral surface of one set of dorsal longitudinal flight muscles (DLMs) as well as the thoracic ganglion of the CNS. The posterior dorsal mesothoracic nerve, through which the DLM motor axons project from the thoracic ganglion to the muscle, was cut and pulled into a suction electrode for stimulation. Temperature was maintained at 20°C during dissection.
Ultrastructural analysis. The fly was mounted ventral side-up after the head, wings, and the second and third pairs of legs were removed. The fly was dissected to expose the sternal anterior rotator coxal muscles of the most anterior legs as described (Koenig et al., 1989). The same saline (recording) solution was used for these and the electrophysiology experiments.
Temperature control. Temperature control was achieved using a TC-202 temperature controller and PDMI microincubator (Medical Systems Corporation, Greenvale, NY). Temperature shifts from 20 to 36°C typically required ∼4.5 min.
Microelectrode recordings. These recordings were performed by conventional methods using an IX2–700 amplifier (Dagan Corporation, Minneapolis, MN) and glass microelectrodes filled with 3m KCl (∼30 MΩ). The recording solution consisted of (in mm): 128 NaCl, 2 KCl, 4 MgCl2, 1.8 CaCl2, 5 HEPES, and 36 sucrose. The pH was adjusted to 7.0 using NaOH. DLM resting potentials were typically −90 to −95 mV. Under the same recording conditions, two-electrode voltage clamp was performed using a TEV-200 amplifier (Dagan Corporation). In the voltage-clamp recordings, deviations from the command potential during recording of synaptic currents typically did not exceed 2 mV.
Electrophysiological data acquisition and analysis
Data were acquired on-line using a Power Macintosh 7500 computer, Pulse software (HEKA Elektronik, Lambrecht, Germany), and an ITC-16 laboratory interface (Instrutech Corporation, Great Neck, NY). Data were low-pass filtered at 3 kHz and acquired at 10–30 kHz. Stimulation was achieved using the Pulse program to trigger an S-900/S-910 stimulator (Dagan Corporation). Current measurements were performed in the data analysis program IGOR (WaveMetrics, Lake Oswego, OR). Peak values were obtained using cursor measurements, and total charge was determined using the area function. Microsoft Excel was used for data tabulation, graphing, and statistical analysis. By the use of an unpaired Student’s t test, statistical significance was assigned to comparisons with p values ≤ 0.05.
Transmission electron microscopy
After a 5 min incubation at the experimental temperature in saline solution (see Dissection), fixation was initiated by exposing the preparation to the same saline solution containing 2.5% paraformaldehyde and 1.5% glutaraldehyde (the primary fixative). Samples remained in primary fixative at room temperature for 1–3 hr and then overnight at 4°C. Further processing was performed using conventional methods. Briefly, fixed samples were processed in 2% aqueous osmium tetroxide and then in 2% aqueous uranyl acetate, dehydrated using an ethanol series, and embedded in Spurr resin (EM Sciences, Fort Washington, PA). Thin sections, ∼60 nm in thickness, were cut on an ultramicrotome and stained with uranyl acetate/lead citrate before viewing on a Jeol 1200EXII transmission electron microscope (TEM) housed at the Penn State University Electron Microscopy Facility.
TEM data analysis
Vesicle counts were performed directly on TEM negatives. Statistical significance was determined using an unpaired Student’st test. Significant differences were assigned to comparisons with p values ≤ 0.05. Data in the manuscript are presented as the mean ± SEM, and n represents the number of active zones analyzed. In all cases a minimum of three different preparations were examined. Images were acquired from photographic prints using a UMAX flatbed scanner and Adobe Photoshop software.
Drosophila lines and transformation rescue
All Drosophila lines used were cultured at 20°C. The three temperature-sensitive comt alleles used in this study contain point mutations leading to single amino acid changes. Missense mutations in comtST17 andcomtST53 have been reported previously (Pallanck et al., 1995a); comtTP7contains a missense mutation converting proline 398 to serine (R. Ordway and L. Pallanck, unpublished observations). Rescue experiments used a transgene construct containing a wild-type dNSF1 cDNA under the control of an hsp70 heat-shock promoter (Pallanck et al., 1995a). For rescue experiments, comt flies bearing a transgene were subjected to three heat shocks separated by 24 hr intervals (all heat shocks were at 38°C). A 15 min heat shock on the first day was followed by 30 min heat shocks on the second and third days. Recordings were obtained ∼24 hr after the last heat shock. In the case of the comtST53 rescue experiments, the third heat shock was omitted. As is the case for temperature-sensitive paralytic behavior (Pallanck et al., 1995a), controls in which comt flies lacking the transgene were heat-shocked demonstrated that phenotypic rescue was entirely dependent on the presence of the transgene.
RESULTS
Intracellular recordings from the dorsal longitudinal flight muscles (DLMs) were used to monitor postsynaptic potentials elicited by direct stimulation of the DLM motor axon. Typically the postsynaptic potential resulting from neurotransmitter activation of ligand-gated receptors triggers a DLM action potential (Fig.1). As reported previously, a progressive reduction in neurotransmitter release produces a graded reduction and eventual loss of this action potential as the underlying postsynaptic potential is reduced (Koenig et al., 1983).
Fig. 1.

Reversible reduction of the postsynaptic potential in comtST17 as a result of repetitive stimulation at a restrictive temperature. Recordings are of DLM postsynaptic potentials elicited by direct stimulation of the DLM motor axon. Each panel shows superimposed traces representing the first 100 responses generated during a 1 Hz stimulation train. In the case ofcomtST17 at 36°C, every eighthtrace is shown. The stimulation trains at the restrictive temperature were initiated after 3 min at 36°C. Recovery of comtST17 is shown 60 min after return to 20°C. For both wild type (WT) andcomtST17, data at 36°C and at 20°C recovery were obtained from the same cell.
At a permissive temperature (20°C), DLM action potentials incomtST17 were indistinguishable from those in wild-type flies and remained constant during 1 Hz stimulation (Fig. 1). At restrictive temperatures in comt (e.g., 36°C as shown), the first stimulus in the train also produced a wild-type action potential. However, subsequent stimuli revealed a striking phenotype at comt synapses under these conditions. During a 1 Hz stimulus train, a progressive reduction and then loss of the DLM action potential revealed the underlying postsynaptic potential, which continued to progressively decrease in amplitude. This activity-dependent reduction was reversible and, like temperature-sensitive paralysis, recovered slowly over a period of ∼60 min after return to 20°C (Fig. 1).
The above recordings extend the initial neurophysiological studies incomt (Siddiqi and Benzer, 1976) which used microelectrode recordings of DLM responses to central neural stimulation or simply compound extracellular recordings from the eye (electroretinograms or ERGs). However, to characterize the functional role of dNSF1, additional approaches are required. To address whether the reduction in the postsynaptic potential reflects a reduction in neurotransmitter release and to examine whether the kinetics of neurotransmitter release are altered in comt, two-electrode voltage-clamp analysis was performed to record DLM synaptic currents.
At 36°C in comtST17, the first stimulus elicited a synaptic current indistinguishable from wild type under the same conditions (Fig.2A). The peak current amplitudes in wild type and comtST17 were 2.37 ± 0.19 μA (n = 5) and 2.36 ± 0.16 μA (n = 5), respectively. Furthermore, time course measurements yielded essentially identical values for the time-to-peak current (0.76 ± 0.05 msec in wild type and 0.74 ± 0.04 msec in comtST17) and for the time for decay to half amplitude (0.40 ± 0.03 msec in wild type and 0.38 ± 0.03 msec in comtST17). Thus we conclude that the first stimulus in comtST17 at a restrictive temperature elicits neurotransmitter release at wild-type levels and with wild-type kinetics, indicating that regulated exocytosis and postsynaptic receptor function are normal under these conditions.
Fig. 2.

Activity-dependent reduction of the synaptic current in comtST17.A, Two-electrode voltage-clamp recordings of DLM synaptic currents at a restrictive temperature in wild type andcomtST17 are shown. Wild type andcomtST17 were exposed to 36°C for 10 and 9 min, respectively. Each panel shows representative traces from the first 100 responses elicited during a 1 Hz stimulation train. The holding potential was −80 mV. Arrows indicate stimulation of the motor axon; stimulation artifacts were removed. B, Peak amplitude measurements of DLM EPSCs are plotted as a function of time for 1 Hz stimulation trains initiated after 8–10 min at 36°C. The 0 time point is the beginning of the stimulus train. Each point represents the average amplitude ± SEM for four cells from four different preparations.
Although the first stimulus produced a wild-type response incomt, repetitive stimulation at a restrictive temperature caused a progressive reduction in the synaptic current incomtST17 but not in wild type (Fig.2A). Quantitation of this reduction was performed by comparing peak synaptic currents in wild-type andcomtST17 in response to 1 Hz stimulation at 36°C. These data, shown in Figure 2B, indicate that comt exhibits a reproducible and strictly activity-dependent decrease in the synaptic current. Because basic mechanisms of synaptic transmission seem to operate normally at a restrictive temperature in comt, the activity-dependent reduction in the synaptic current indicates that dNSF1 functions in maintaining neurotransmitter release during repetitive stimulation.
As seen in Figure 2A, the activity-dependent reduction in neurotransmitter release observed in comt was associated with a slowing of the kinetics of the synaptic current. At 36°C, the 50th response in a 1 Hz stimulus train exhibits a 62% decrease in current amplitude (Fig. 2B). Comparing the kinetics of the first (see above) and the 50th responses revealed a 12% increase in the time to peak (0.84 ± 0.02 msec for the 50th response; n = 5) and a 2.18-fold increase in the time for decay to half amplitude (0.83 ± 0.07 msec for the 50th response; n = 5). These changes in current kinetics raise the possibility that peak current measurements may not accurately represent changes in neurotransmitter release. Thus area measurements may be used to determine the total charge flowing during the synaptic current. To supplement the peak current measurements reported in Figure2B, we performed area measurements on the same currents. This method yielded a 37% decrease in the synaptic current in comtST17, from 2.00 ± 0.15 to 1.27 ± 0.18 nC of charge for the first and 50th responses, respectively (n = 5).
To address whether the change in the kinetics of the synaptic current in comt is specifically related to dNSF1 function or rather is generally characteristic of a reduction in neurotransmitter release, we examined a temperature-sensitive paralytic mutant in which neurotransmitter release is reduced by a different mechanism. At restrictive temperatures, shibire (shi) mutations disrupt synaptic vesicle endocytosis, resulting in depletion of synaptic vesicles and an activity-dependent reduction in neurotransmitter release (see Koenig et al., 1983, 1989).comt and shi currents were compared at 33°C because shi currents were greatly reduced at higher temperatures. As seen in Figure 3, a decrease in neurotransmitter release in shi produced a slowing of the synaptic current similar to that observed incomt. For the comtST17 andshiTS1 currents shown, the amplitude of the first response was 1.97 and 1.93 μA, the time to peak was 0.79 and 0.74 msec, and the time for decay to half amplitude was 0.43 and 0.48 msec, respectively. After neurotransmitter release was reduced to 0.79 μA in each case, the comtST17 andshiTS1 currents exhibited time-to-peak values of 0.86 and 0.86 msec and time for decay to half amplitude values of 0.75 and 0.72 msec, respectively. The similarity of these changes in current kinetics suggests that they are a general consequence of reduced neurotransmitter release and further suggests that dNSF1 activity does not influence the kinetics of synaptic vesicle fusion.
Fig. 3.

Reducing neurotransmitter release inshi mutants produces a slowing of synaptic current kinetics similar to that observed in comt. DLM synaptic currents were recorded in comtST17and shiTS1 during 1 Hz stimulation at 33°C. In comtST17, the first and 43rd responses are shown. In shiTS1, the first and 77th responses are shown.
Finally, an informative aspect of the activity-dependent phenotype incomt is shown in Figure 4. Five Hertz stimulation at 36°C was used to markedly reduce the synaptic current in comtST17. Subsequently resting the synapse while maintaining the restrictive temperature resulted in complete recovery of the current; a second 5 Hz stimulus train then produced the same progressive reduction seen during the first train.
Fig. 4.

Recovery of the postsynaptic current at a restrictive temperature in the absence of stimulation. DLM postsynaptic currents were recorded from comtST17in response to two 5 Hz stimulation trains at 36°C. The stimulation trains were 200 pulses in length, and each panel shows representative traces superimposed. The first train was initiated after 21 min at 36°C. After 2 min and 20 sec at 36°C without stimulation, the second train was initiated.
To test the generality of the above phenotype and to verify that it can be attributed entirely to the dNSF1 mutations, we recorded DLM action potentials from two additional comt alleles, as well as fromcomt flies in which the temperature-sensitive paralytic phenotype has been rescued by transgenic expression of wild-type dNSF1 protein. Like in comtST17, bothcomtTP7 andcomtST53 exhibited wild-type action potentials at 20°C and a strictly activity-dependent reduction in the action potential and postsynaptic potential at the restrictive temperature (Fig. 5A). Unlike in comtST17, 1 Hz stimulation at 36°C did not produce a marked reduction in either of these alleles. At a higher stimulation frequency of 10 Hz, however, both alleles exhibited a clear reduction, whereas wild-type responses were constant under these conditions (data not shown, but see rescue data in Fig.5B). These results indicate that the activity-dependent reduction in neurotransmitter release results from a general loss of dNSF1 function rather than from any specific amino acid substitution in the dNSF1 protein. DLM recordings were also performed using flies in which a transgene expressing the wild-type dNSF1 protein was introduced into a comt mutant background. After transgenic expression of wild-type dNSF1 protein, the activity-dependent reduction in neurotransmitter release, like the temperature-sensitive paralytic behavior (Pallanck et al., 1995a), was dramatically rescued (Fig.5B). Although not shown, the synaptic phenotype incomtST17 was also completely rescued. These experiments demonstrate that the reduction in neurotransmitter release results entirely from mutations in the dNSF1 gene.
Fig. 5.

Reduction of the postsynaptic potential and transformation rescue in two additional comt alleles.A, DLM action potentials and postsynaptic potentials recorded from comtTP7 andcomtST53 during 10 Hz stimulation trains initiated after 7 min at 36°C. B, Recordings similar to those shown in A after transgenic expression of wild-type dNSF1 protein in acomtTP7 orcomtST53 genetic background. IncomtTP7 Rescue andcomtST53Rescue flies, the comt phenotype has been rescued by transgenic expression of wild-type dNSF1 protein. To emphasize the rescue, we performed recordings in B at 38°C; similar results were obtained at 36°C. In all four panels,traces representing the first 50 responses are shown. InA, every fourth trace is shown.
To determine whether the physiological defect in comt is associated with an alteration in the distribution of synaptic vesicles, ultrastructural studies were performed at neuromuscular synapses of the coxal muscles controlling the anterior legs. This preparation was used because it is much more favorable for ultrastructural analysis of neuromuscular synapses than the DLM (Koenig et al., 1989). Transmission electron microscopy was used to image synaptic terminals and the presynaptic membrane densities (active zones) that represent sites of synaptic vesicle docking and fusion. Synaptic terminals incomt and wild-type flies were examined at both a permissive temperature (20°C) and restrictive temperatures (33 or 36°C). At all temperatures, the general appearance of the comt and wild-type terminals, including the total number of synaptic vesicles, was quite similar. However, at restrictive temperatures, closer examination of active zones revealed that comt mutants exhibited a striking elevation in the number of docked vesicles (those in contact with the plasma membrane at active zones) compared with wild type (Figs. 6,7). For example, at 33°C the number of docked vesicles per active zone was 2.6-fold higher incomtTP7 [3.57 ± 0.39 (n = 14)] than in wild type [1.35 ± 0.32 (n = 17)]; similar results were obtained incomtST53 andcomtST17. At 36°C in comt, extreme accumulation of docked vesicles precluded accurate vesicle counts (Fig. 6). Thus a marked accumulation of docked vesicles occurred in comt at restrictive temperatures. The number of undocked synaptic vesicles located within 200 nm (four to five vesicle diameters) of the active zone was also examined, revealing a moderate but statistically significant increase incomtTP7 [12.62 ± 1.73 (n = 13)] relative to wild type [8.24 ± 1.11 (n = 17)] at 33°C.
Fig. 6.
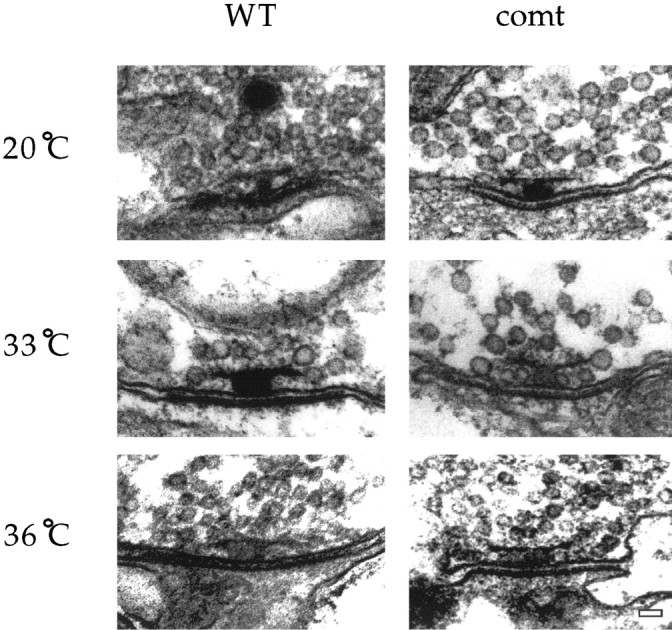
Docked vesicles accumulate incomt at elevated temperatures. TEM images of active zones from wild-type, comtTP7, andcomtST17 at permissive and restrictive temperatures. comtTP7images are shown at 20 and 33°C;comtST17 is shown at 36°C. Scale bar, 50 nm.
Fig. 7.

Quantitation of docked vesicles in wild-type andcomtTP7 flies. Docked vesicles were counted as those in contact with the plasma membrane at the active zone. The number of active zones analyzed for each condition is as follows: for WT at 20°C, n = 9; for WT at 33°C, n = 17; forcomtTP7 at 20°C,n = 14; forcomtTP7 at 33°C,n = 14; and for comtTP7Rescue at 33°C, n = 15. Anasterisk denotes a value significantly different from that of WT at the same temperature.
Expression of wild-type dNSF1 protein in transgenic flies restored the number of docked vesicles at 33°C to wild-type levels (Fig. 7). Thus, as was the case for the electrophysiological phenotype, the accumulation of docked vesicles in comt was shown to result entirely from a deficit in dNSF1 function. Furthermore, the fact that multiple comt alleles exhibit similar ultrastructural phenotypes indicates that the accumulation of docked vesicles is a general consequence of disrupting dNSF1 activity.
DISCUSSION
The work presented here uses comt mutants, in which single gene mutations result in temperature-sensitive dNSF1 activity, to address the function of NSF at mature, native synapses in vivo. One key observation is that, at a restrictive temperature,comt exhibits wild-type neurotransmitter release in the absence of repetitive stimulation. A second important observation is that repetitive stimulation causes a progressive, activity-dependent reduction in neurotransmitter release under the same conditions.
At a restrictive temperature in the absence of repetitive stimulation,comt exhibits wild-type postsynaptic currents and potentials. Thus we infer that basic synaptic mechanisms including calcium signaling, the vesicle fusion process, and excitation of the postsynaptic membrane can operate normally under these conditions. Furthermore, the experiment showing recovery of the synaptic current during a rest period at a restrictive temperature (Fig. 4) indicates that this is true even when vesicles are maintained at a restrictive temperature when recruited to the readily releasable pool (those vesicles competent to fuse in response to an action potential). Thus persistence of normal fusion at a restrictive temperature cannot be explained by a protected pool of readily releasable vesicles in which the dNSF1 requirement was satisfied before temperature elevation. Finally, experiments observing the kinetics of neurotransmitter release in shi mutants suggest that the change in kinetics observed when synaptic currents are reduced in comt is secondary to the reduction in neurotransmitter release. Taken together, the above observations indicate that dNSF1 does not participate directly in vesicle fusion, an observation of particular interest given the initial characterization of dNSF1 as a fusion protein (Rothman, 1994).
A second important finding is that at restrictive temperatures incomt, repetitive stimulation causes a progressive, activity-dependent reduction in neurotransmitter release, indicating that dNSF1 functions in maintaining neurotransmitter release. Maintenance of release requires restoration of the readily releasable pool of synaptic vesicles. This is thought to occur by retrieval of vesicles from the plasma membrane by endocytosis, vesicle targeting and docking at active zones, and finally priming of docked vesicles leading to competence for fast calcium-triggered fusion (Pieribone et al., 1995). We do not attribute the comt phenotype to a defect in synaptic vesicle endocytosis, both because the rapid activity-dependent onset (<1 sec at 36°C; see Fig. 4) is more than an order of magnitude faster than estimates of the minimum time required for synaptic vesicle recycling (Betz and Wu, 1995) and because no general depletion of vesicles was seen in the ultrastructural studies. Thus the electrophysiological evidence indicates a role for dNSF1 either in the targeting and/or docking of synaptic vesicles or in the priming process after docking.
Ultrastructural analysis further defined the role of NSF by demonstrating a marked accumulation of docked synaptic vesicles at restrictive temperatures in comt. These results indicate that dNSF1 activity is not required for synaptic vesicle docking but rather plays an important role in the consumption of vesicles. Taken together, the electrophysiological and ultrastructural analyses reveal that dNSF1 functions in the priming of docked synaptic vesicles for fast calcium-triggered fusion.
Recovery of neurotransmitter release during a rest period at a restrictive temperature in comt indicates that the priming process is not completely blocked under these conditions, raising the interesting question of whether residual dNSF1 activity might account for residual priming. We have sought to address this issue by testing whether the most severe allele comtST17behaves as a genetic null mutation (exhibiting no residual activity) under restrictive conditions. Our results (data not shown) confirm this by showing that flies heterozygous forcomtST17 and a deletion that eliminates dNSF1 exhibit electrophysiological and behavioral phenotypes that are indistinguishable from those of comtST17homozygotes. Furthermore, we have also observed recovery at the higher restrictive temperature of 38°C. On the basis of these results, we favor a model in which dNSF1 is critical to, but not absolutely required for, the priming process. Regarding this possibility it is of interest that a second Drosophila NSF, dNSF2, has been identified and exhibits 84% amino acid identity to dNSF1 (Boulianne and Trimble, 1995; Pallanck et al., 1995b).
Several recent studies have examined NSF function in regulated secretion (Banerjee et al., 1996; Burgoyne et al., 1996; Schweizer et al., 1998). Informative work in semi-intact pheochromocytoma PC12 cells (Banerjee et al., 1996) indicated that the requirement for NSF function in dense-core granule fusion can be fulfilled at a distinct step after vesicle docking and preceding fusion. Although in this case neither the fast kinetics nor the activity dependence of release was examined, these results established a model in which NSF acts after docking in an ATP-dependent priming step preceding calcium-activated exocytosis. Additional support for models in which NSF does not participate directly in fusion is inferred from recent results showing that complementary vesicle and target SNAREs alone are sufficient to catalyze membrane fusion, although slowly, when reconstituted in lipid vesicles (Weber et al., 1998).
Another study has recently examined the in vivo role of NSF using injection of NSF peptides into the presynaptic terminal of the squid giant synapse (Schweizer et al., 1998). In this case two peptides were found to inhibit neurotransmitter release, presumably by competing for NSF binding partners. Inhibition was dependent on synaptic activity but did not appear to recover during a rest period despite ∼50% residual NSF activity estimated at the peptide concentration used. Ultrastructural analysis showed that the number of vesicles docked at active zones was increased slightly by the peptides, indicating that NSF is not required for vesicle docking. Finally, the reduction in neurotransmitter release was associated with a slowing of the rise and decay times of the synaptic current. In contrast to the results shown here, in this case the slowing was reported to be a specific consequence of inhibiting NSF activity. These observations led to a model in which NSF functions after docking in a manner that is dependent on vesicle turnover while also contributing to the kinetics of vesicle fusion.
A strength of the work presented here is that it benefits from knowledge of the specific gene product affected without concern either for nonspecific effects on other gene products or for complications arising from the presence of wild-type protein. In addition, transgenic expression techniques allow unequivocal demonstration that the phenotypes observed result specifically from mutations in dNSF1. Another unique aspect of this work is the clear separation of priming and fusion processes by comparison of wild type and comt at a restrictive temperature in the absence of repetitive stimulation. These experiments, together with the observation that slowing of the synaptic current in comt does not seem to be directly related to NSF function, indicate that dNSF1 does not participate directly in synaptic vesicle fusion.
Previous studies examining the role of NSF in vesicular trafficking have led to different biochemical models of NSF action (for review, seeHanson et al., 1997; Hay and Scheller, 1997). A role for NSF in catalyzing rearrangement of SNAREs leading to membrane fusion, as proposed in the SNARE hypothesis, is being reconsidered in light of an alternative model. This second model, derived in part from in vitro assays of homotypic vacuolar fusion in yeast (Ungermann et al., 1998), may provide an adequate and general explanation of the experimental observations to date. This model suggests that NSF is required only for the generation of free, activated SNAREs and does not directly participate in fusion. Despite the generality and appeal of this second model, additional work is required to establish whether either one of the above models is correct.
The molecular mechanisms of synaptic vesicle trafficking may be ideally investigated using biochemical and functional analyses in the same experimental system. Recent studies have pursued this objective by carefully characterizing a neural SNARE complex inDrosophila and analyzing the status of this complex incomt (Tolar and Pallanck, 1998). These studies show that exposure of comt to a restrictive temperature results in accumulation of an SDS-resistant SNARE complex and use subcellular fractionation to demonstrate that the accumulated complex is located on the plasma membrane (including docked vesicles). Thus the priming function of dNSF1 appears to involve disassembly or rearrangement of plasma membrane SNARE complex. Although these findings do not distinguish between the two biochemical models described above, they clearly represent significant progress in our understanding of NSF action.
The functional analysis reported here may also be interpreted in the context of either biochemical model. Two simple schemes presented in Figure 8 indicate that the biochemical action of NSF occurs either before or after fusion. Regardless of where NSF acts biochemically, in both models it should be clear that NSF action promotes the priming of docked vesicles for fast calcium-triggered fusion.
Fig. 8.
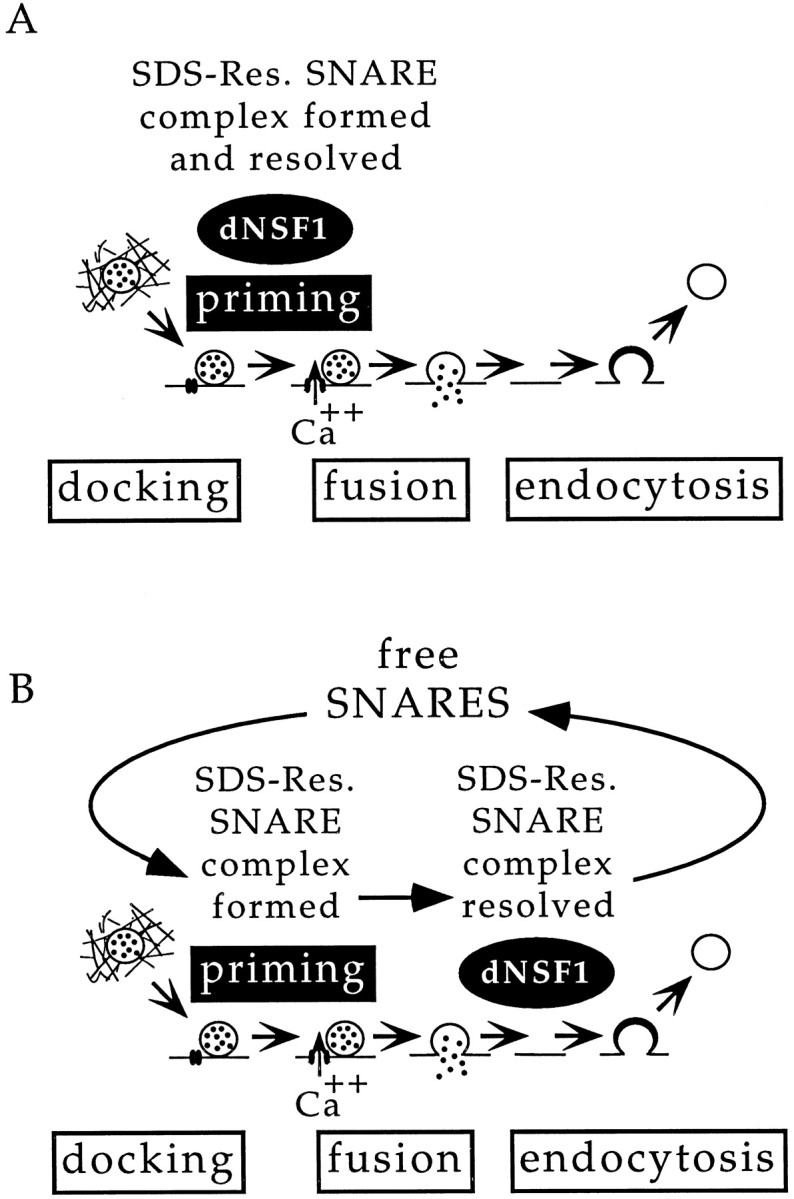
Possible models of dNSF1 action in neurotransmitter release. In each model the action of NSF promotes priming of docked vesicles for fast calcium-triggered fusion. Each model also shows the SDS-resistant SNARE (SDS-Res. SNARE) complex forming after vesicle docking. A, This complex is resolved or rearranged preceding fusion, leading to formation of a mature (primed) fusion apparatus. This model predicts that the accumulated SDS-resistant SNARE complex in comtwill be located on the plasma membrane in association with docked synaptic vesicles. B, The SDS-resistant SNARE complex participates in fusion and is resolved after the fusion process. In this case the role of dNSF1 is to provide free, activated SNAREs necessary for the priming of new vesicles. This model predicts that the SDS-resistant SNARE complex will accumulate on the plasma membrane or in recycled synaptic vesicles in comt. Biochemical demonstration that the complex accumulates on the plasma membrane incomt is consistent with either model (Tolar and Pallanck, 1998).
To clarify models of targeting and fusion mechanisms involving NSF, a number of important questions about the SDS-resistant SNARE complex remain to be addressed. What are the SDS-sensitive components and interactions of the complex in vivo? Does this complex represent or contribute to the primed fusion apparatus (postfusion disassembly) or rather to a precursor of it (prefusion disassembly)? Despite these questions, it is generally agreed that NSF-mediated disassembly or rearrangement of this complex is a key step. Determination of the precise timing and location of this event within the synaptic vesicle trafficking cycle in vivo will be essential to making connections between the priming function of NSF and the well-characterized biochemical interactions of these proteins.
Footnotes
This work was supported by National Science Foundation Grant IBN-9514485. We thank Missy Hazen for excellent technical assistance. We thank Leo Pallanck for communication and discussions of unpublished data and the Penn State University Electron Microscopy Facility for expert training and consultation.
Correspondence should be addressed to Dr. Richard W. Ordway, Department of Biology, The Pennsylvania State University, University Park, PA 16802.
REFERENCES
- 1.Banerjee A, Barry VA, DasGupta BR, Martin TFJ. N-Ethylmaleimide-sensitive factor acts at a prefusion ATP-dependent step in Ca2+-activated exocytosis. J Biol Chem. 1996;271:20223–20226. doi: 10.1074/jbc.271.34.20223. [DOI] [PubMed] [Google Scholar]
- 2.Bennett MK, Scheller RH. The molecular machinery for secretion is conserved from yeast to neurons. Proc Natl Acad Sci USA. 1993;90:2559–2563. doi: 10.1073/pnas.90.7.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betz WJ, Wu L-G. Kinetics of synaptic-vesicle recycling. Curr Biol. 1995;5:1098–1101. doi: 10.1016/s0960-9822(95)00220-x. [DOI] [PubMed] [Google Scholar]
- 4.Boulianne G, Trimble WS. Identification of a second homolog of N-ethylmaleimide-sensitive fusion protein that is expressed in the nervous system and secretory tissues of Drosophila. Proc Natl Acad Sci USA. 1995;92:7095–7099. doi: 10.1073/pnas.92.15.7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broadie K, Prokop A, Bellen HJ, O’Kane CJ, Schulze KL, Sweeney ST. Syntaxin and synaptobrevin function downstream of vesicle docking in Drosophila. Neuron. 1995;15:663–673. doi: 10.1016/0896-6273(95)90154-x. [DOI] [PubMed] [Google Scholar]
- 6.Burgoyne RD, Morgan A, Barnard RJO, Chamberlain LH, Glenn DE, Kibble AV. SNAPs and SNAREs in exocytosis in chromaffin cells. Biochem Soc Trans. 1996;24:653–657. doi: 10.1042/bst0240653. [DOI] [PubMed] [Google Scholar]
- 7.Clary DO, Griff IC, Rothman JE. SNAPs, a family of NSF attachment proteins involved in intercellular membrane fusion in animals and yeast. Cell. 1990;61:709–721. doi: 10.1016/0092-8674(90)90482-t. [DOI] [PubMed] [Google Scholar]
- 8.DeBello WM, O’Connor V, Dresbach T, Whiteheart SW, Wang SS-H, Schweizer FE, Betz H, Rothman JE, Augustine GJ. SNAP-mediated protein-protein interactions essential for neurotransmitter release. Nature. 1995;373:626–630. doi: 10.1038/373626a0. [DOI] [PubMed] [Google Scholar]
- 9.Deitcher DL, Ueda A, Stewart BA, Burgess RW, Kidokoro Y, Schwarz TL. Distinct requirements for evoked and spontaneous release of neurotransmitter are revealed by mutations in the Drosophila gene neuronal-synaptobrevin. J Neurosci. 1998;18:2028–2039. doi: 10.1523/JNEUROSCI.18-06-02028.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferro-Novick S, Jahn R. Vesicle fusion from yeast to man. Nature. 1994;370:191–193. doi: 10.1038/370191a0. [DOI] [PubMed] [Google Scholar]
- 11.Hanson PI, Heuser JE, Jahn R. Neurotransmitter release–four years of SNARE complexes. Curr Opin Cell Biol. 1997;7:310–315. doi: 10.1016/s0959-4388(97)80057-8. [DOI] [PubMed] [Google Scholar]
- 12.Hay JC, Scheller RH. SNAREs and NSF in targeted membrane fusion. Curr Opin Cell Biol. 1997;9:505–512. doi: 10.1016/s0955-0674(97)80026-9. [DOI] [PubMed] [Google Scholar]
- 13.Hunt JM, Bommert K, Charlton MP, Kistner A, Habermann E, Augustine GJ, Betz H. A post-docking role for synaptobrevin in synaptic vesicle fusion. Neuron. 1994;12:1269–1279. doi: 10.1016/0896-6273(94)90443-x. [DOI] [PubMed] [Google Scholar]
- 14.Huttner WB. Snappy exocytoxins. Nature. 1993;365:104–105. doi: 10.1038/365104a0. [DOI] [PubMed] [Google Scholar]
- 15.Kawasaki F, Ordway RW. A conditional neurophysiological defect in comatose; a Drosophila NSF mutant. Abstract of a paper presented at the Neurobiology of Drosophila meeting, September 1997. Cold Spring Harbor, NY; 1997. [Google Scholar]
- 16.Kawasaki F, Ordway RW (1998) A conditional neurophysiological phenotype in the CNS of comatose Abstract of a paper presented at the 39th Annual Drosophila Research Conference, Washington, DC, March 1998.
- 17.Koenig JH, Saito K, Ikeda K. Reversible control of synaptic transmission in a single gene mutant of Drosophila melanogaster. J Cell Biol. 1983;96:1517–1522. doi: 10.1083/jcb.96.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koenig JH, Kosaka T, Ikeda K. The relationship between the number of synaptic vesicles and the amount of transmitter released. J Neurosci. 1989;9:1937–1942. doi: 10.1523/JNEUROSCI.09-06-01937.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mayer A, Wickner W, Haas A. Sec18p (NSF)-driven release of Sec17p (α-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 20.Nichols BJ, Ungermann C, Pelham HRB, Wickner WT, Haas A. Homotypic vacuolar fusion mediated by t- and v-SNAREs. Nature. 1997;387:199–202. doi: 10.1038/387199a0. [DOI] [PubMed] [Google Scholar]
- 21.Nonet ML, Saifee O, Zhao H, Rand JB, Wei L. Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J Neurosci. 1998;18:70–80. doi: 10.1523/JNEUROSCI.18-01-00070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Connor V, Heuss C, DeBello WM, Dresbach T, Charlton MP, Hunt JH, Pellegrini LL, Hodel A, Bunger MM, Heinrich B, Augustine GJ, Schafer T. Disruption of syntaxin-mediated protein interactions blocks neurotransmitter secretion. Proc Natl Acad Sci USA. 1997;94:12186–12191. doi: 10.1073/pnas.94.22.12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orci L, Malhotra V, Amherdt M, Serafini T, Rothman JE. Dissection of a single round of vesicular transport: sequential intermediates for intercisternal movement in the Golgi stack. Cell. 1989;56:357–368. doi: 10.1016/0092-8674(89)90239-0. [DOI] [PubMed] [Google Scholar]
- 24.Ordway RW, Mattiuz AM, Kawasaki F (1998) Analysis of NSF function at neuromuscular synapses in comatose Abstract of a paper presented at the 39th Annual Drosophila Research Conference, Washington, DC, March 1998.
- 25.Pallanck L, Ordway RW, Ganetzky B. A Drosophila NSF mutant. Nature. 1995a;376:25. doi: 10.1038/376025a0. [DOI] [PubMed] [Google Scholar]
- 26.Pallanck L, Ordway RW, Ramaswami M, Chi WY, Krishnan KS, Ganetzky B. Distinct roles for N-ethylmaleimide-sensitive fusion protein (NSF) suggested by the identification of a second Drosophila NSF homolog. J Biol Chem. 1995b;270:18742–18744. doi: 10.1074/jbc.270.32.18742. [DOI] [PubMed] [Google Scholar]
- 27.Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P. Distinct pools of synaptic vesicles in neurotransmitter release. Nature. 1995;375:493–497. doi: 10.1038/375493a0. [DOI] [PubMed] [Google Scholar]
- 28.Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- 29.Rothman JE, Wieland FT. Protein sorting by transport vesicles. Science. 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- 30.Schekman R. Genetic and biochemical analysis of vesicular traffic in yeast. Curr Opin Cell Biol. 1992;4:587–592. doi: 10.1016/0955-0674(92)90076-o. [DOI] [PubMed] [Google Scholar]
- 31.Schulze KL, Broadie K, Perin MS, Bellen HJ. Genetic and electrophysiological studies of Drosophila syntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell. 1995;80:311–320. doi: 10.1016/0092-8674(95)90414-x. [DOI] [PubMed] [Google Scholar]
- 32.Schweizer FE, Dresbach T, DeBello WM, O’Connor V, Augustine GJ, Betz H. Regulation of neurotransmitter release kinetics by NSF. Science. 1998;279:1203–1206. doi: 10.1126/science.279.5354.1203. [DOI] [PubMed] [Google Scholar]
- 33.Siddiqi O, Benzer S. Neurophysiological defects in temperature-sensitive paralytic mutants of Drosophila melanogaster. Proc Natl Acad Sci USA. 1976;73:3253–3257. doi: 10.1073/pnas.73.9.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Söllner T, Whiteheart S, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 35.Südhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- 36.Sweeney ST, Broadie K, Keane J, Niemann H, O’Kane CJ. Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron. 1995;14:341–351. doi: 10.1016/0896-6273(95)90290-2. [DOI] [PubMed] [Google Scholar]
- 37.Tolar LA, Pallanck L. NSF function in neurotransmitter release involves rearrangement of the SNARE complex downstream of synaptic vesicle docking. J Neusci. 1998;18:10250–10256. doi: 10.1523/JNEUROSCI.18-24-10250.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ungermann C, Nichols BJ, Pelham HRB, Wickner W. A vacuolar v-t-SNARE complex, the predominate form in vivo and on isolated vacuoles, is disassembled and activated for docking and fusion. J Cell Biol. 1998;140:61–69. doi: 10.1083/jcb.140.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 40.Whiteheart SW, Griff IC, Brunner M, Clary DO, Mayer T, Buhrow SA, Rothman JE. SNAP family of NSF attachment proteins includes a brain-specific isoform. Nature. 1993;362:353–355. doi: 10.1038/362353a0. [DOI] [PubMed] [Google Scholar]
- 41.Wilson DW, Wilcox CA, Flynn GC, Chen E, Kuang W-J, Henzel WJ, Block MR, Ullrich A, Rothman JE. A fusion protein required for vesicle-mediated transport in both mammalian cells and yeast. Nature. 1989;339:355–359. doi: 10.1038/339355a0. [DOI] [PubMed] [Google Scholar]