

Abstract
AMPA and NMDA receptor channels are closely related molecules, yet they respond to glutamate with distinct kinetics, attributable to differences in ligand binding and channel gating steps (for review, seeEdmonds et al., 1995). We used two complementary approaches to investigate the number of functional binding sites on AMPA channels on outside-out patches from cultured hippocampal neurons. The activation kinetics of agonist binding were measured during rapid steps into low concentrations of selective AMPA receptor agonists and during steps from a competitive AMPA receptor antagonist, 6-cyano-7-nitro-quinoxaline-2,3-dione, into a saturating concentration of agonist. Both approaches revealed sigmoidal kinetics, which suggests that multiple agonist binding steps or antagonist unbinding steps are needed for channel activation. A kinetic model with two independent binding sites gave a better fit to the activation phase than models with one or three independent sites. A more refined analysis incorporating cooperative interaction between the two binding sites significantly improved the fits to the responses. The affinity of the first binding step was two to three times higher than the second step. These results demonstrate that binding of two agonist molecules are needed to activate AMPA receptors, but the two binding sites are not identical and independent. Because NMDA receptors require four ligand molecules for activation (two glycine and two glutamate; Benveniste and Mayer, 1991; Clements and Westbrook, 1991), it may be that some binding sites on AMPA receptors are functionally silent.
Keywords: glutamate receptors, AMPA receptors, cyclothiazide, hippocampus, ion channels, patch clamp
At most central excitatory synapses, release of glutamate activates AMPA and NMDA channels that are colocalized at postsynaptic sites (Bekkers and Stevens, 1989). These multisubunit receptors share significant structural homology (Nakanishi, 1992; Wisden and Seeburg, 1993; Hollmann and Heinemann, 1994). The colocalized receptors are presumably exposed to the same concentration and duration of free transmitter (Clements, 1996). Yet their kinetics, channel properties, and their role in synaptic transmission are quite distinct. NMDA receptors have slow kinetics (McBain and Mayer, 1994), whereas AMPA currents peak within a few hundred microseconds and decay in a few milliseconds (Colquhoun et al., 1992; Hestrin, 1992; Silver et al., 1992). These differences in receptor kinetics reflect both the transmitter binding steps and the channel gating steps of the channel activation process. For example NMDA receptors have a much higher affinity for l-glutamate than AMPA receptors (Patneau and Mayer, 1990), and the high affinity accounts in part for the longer duration of the NMDA receptor-mediated excitatory postsynaptic current (Lester and Jahr, 1992).
Kinetic studies of the AMPA receptor have focused on desensitization and agonist dissociation as factors in the decay phase of the response (Vyklicky et al., 1991; Colquhoun et al., 1992; Jonas and Sakmann, 1992; Trussell et al., 1993). The agonist binding steps have generally been examined under the steady-state conditions of classical pharmacological analysis, but such studies are contaminated by receptor desensitization. The analysis of receptor activation kinetics for native receptors can provide insights into the number of binding steps necessary to activate the channel, the cooperativity of agonist binding, as well as the stoichiometry and subunit interactions for NMDA and AMPA receptors. The ligand binding site on glutamate channels is homologous to the bacterial periplasmic binding proteins with the N terminus and M3–4 loop contributing to two lobes of the binding pocket (O’Hara et al., 1993; Kuryatov et al., 1994; Stern-Bach et al., 1994;Bennett and Dingledine, 1995; Laube et al., 1997). This receptor model suggests that each subunit may contain a ligand binding site. A model with more than two ligand binding sites on glutamate receptor channels is supported by activation kinetics of NMDA receptors that suggest binding of four molecules (two glutamates and two glycines) is necessary to open the channel (Benveniste and Mayer, 1991; Clements and Westbrook, 1991). Because AMPA receptors do not require a coagonist, one possibility is that four or five glutamate molecules can bind to the receptor; in fact a model with three binding sites has been proposed (Raman and Trussell, 1992).
We investigated the number of binding steps required for native AMPA receptor activation using rapid application methods on outside-out patches from cultured hippocampal neurons. Pre-equilibration with cyclothiazide (CTZ) was used to block AMPA receptor desensitization selectively (Patneau et al., 1993; Yamada and Tang, 1993). AMPA receptor activation was recorded after a step into agonist at a concentration at which agonist binding was rate-limiting or after a step from a saturating concentration of antagonist into a high concentration of agonist at which antagonist unbinding was rate-limiting. Ensemble average currents were fitted with kinetic models incorporating one, two, or three independent binding sites as well as models with two unequal or cooperative binding sites. Our results suggest that two molecules of agonist are necessary and sufficient for AMPA receptor activation, and that the binding steps may show negative cooperativity.
MATERIALS AND METHODS
Cell culture. Hippocampal neurons dissociated from neonatal Sprague Dawley rats were cultured on a glial feeder layer as described previously (Legendre and Westbrook, 1990). Growth medium contained 5% horse serum, 95% minimal essential medium (MEM; Life Technologies, Gaithersburg, MD), and a supplement including insulin, transferrin, selenium, triiodothyronine, progesterone, and corticosterone. Experiments were performed 3–5 d after plating (density, 5–20 × 103/cm2).
Drug delivery. During experiments, the neurons were continuously perfused with a bath solution containing (in mm): NaCl, 170; KCl, 2.5; HEPES, 10; glucose, 10; CaCl2, 2; and MgCl2, 1; pH was adjusted to 7.3 with NaOH, and osmolarity was adjusted to 325 mOsm. Two internal solutions were used for recording and contained (in mm): CsCl, 150; HEPES, 10; EGTA, 10; CaCl2, 1; MgCl2, 5; and Na-ATP, 5 or alternatively potassium gluconate, 130; HEPES, 10; Cs4-BAPTA, 10; Mg-ATP, 5; and MgCl2, 2. For both solutions, the pH was adjusted to 7.3 by CsOH (or KOH), and osmolarity was adjusted to 310–320 mOsm. The external control solution was the same as the bath perfusing solution, except that glucose was omitted, and the following were added (in μm): tetrodotoxin, 0.5; picrotoxin, 100; strychnine, 2;dl-2-amino-5-phosphonovaleric acid (dl-AP5), 100; and 7-chlorokynurenic acid, 1, to inhibit Na channels, γ-aminobutyric acid/glycine receptor channels, and NMDA receptor channels, respectively. Agonists were prepared as stock solutions and added at the indicated final concentration. All solutions were filtered (0.22 μm) before use.
For outside-out patch recording, drugs were delivered by a gravity-fed, four-pore (four-square pattern) Pyrex tube (Vitrodynamics, Mountain Lakes, NJ) with a tip diameter of 80 μm. For drug application, excised patches were brought into the flow of the control solution and then switched to the drug-containing solution by moving the delivery tube with a piezoelectric bimorph (Vernitron, Bedford, OH) or a piezo stack translator (P245.30; Physik Instrument, Waldbronn, Germany). Solution exchange rate was estimated at the end of each patch recording using changes in tip potential induced by a 2% dilution of the drug solution. The solution exchange time constant was 200–400 μsec. Stock solutions were prepared as indicated (stock concentration, solvent): CTZ (10 mm, DMSO), 7-chlorokynurenate (500 μm, alcohol), dl-AP5 (300 mm, H2O), 6-cyano-7-nitro-quinoxaline-2,3-dione (CNQX, 40 mm, DMSO), domoate (10 or 100 mm, H2O), kainate (10 mm, H2O),l-glutamate (100 mm, H2O), and quisqualate (10 mm, H2O). CTZ was generously provided by Lilly Research Laboratories (Indianapolis, IN). Other drugs were either purchased from Tocris Neuramin (Bristol, UK) or Cambridge Research Biochemicals (Northwich, UK).
Data acquisition. Patch currents were recorded using an Axopatch 1C amplifier (Axon Instruments, Foster City, CA). Pipettes for recording from excised outside-out patches were made of borosilicate glass and pulled in two steps. Inner tip diameter after fire polishing was 2–3 μm. Data were low-pass-filtered at 2 kHz, digitally sampled at 4–8 kHz using pClamp (Axon Instruments), and analyzed on a Macintosh computer using AxoGraph (Axon Instruments). Ensemble average responses were constructed from 50–100 drug applications lasting 100–250 msec at a holding potential of −50 mV. The average response at 0 mV was subtracted from the ensemble average to remove the artifact attributable to the voltage applied to the piezoelectric translator. To determine patch current onsets precisely, the open tip potential was recorded after patch rupture; the time when solution exchange reached 90% of maximum was then defined as t = 0. Collected data were analyzed off-line using a kinetic modeling program (Benveniste et al., 1990).
Kinetic modeling procedures. The modeling program was based on a chemical reaction scheme describing agonist binding to one or more receptors and subsequent transitions of the channels to open and desensitized states. Agonist responses were initially fitted to an identical, independent binding site scheme as follows:
where A is agonist, R is the receptor,AnR* is the open state of the channel,D is a desensitized state, and n reflects the number of agonist binding steps. The transition rates are defined as follows: kb is the binding rate, kuis the unbinding rate, β is the opening rate, α is the closing rate, and kd and kr are the desensitization and resensitization rates, respectively. For responses to quisqualate, β and α of the AMPA channels were fixed to 1000 and 500 sec−1, respectively, giving the model channels a peak open probability (Po) of ∼0.7 and mean open time of 2 msec (Hestrin, 1992; Kullmann, 1993). For responses to domoate, the β and α were both fixed to 1000 sec−1, respectively, giving the model channels a peak open probability of ∼0.5 and mean open time of 1 msec. The lowerPo for domoate corresponds to its lower efficacy relative to quisqualate (Patneau and Mayer, 1990). For steps into quisqualate and domoate, the unbinding and binding rates were free parameters to allow for possible effects of CTZ on agonist binding (Patneau et al., 1993). For the antagonist unbinding experiments, the ratio of ligand unbinding rate to binding rate for kainate was constrained to microscopic Kd values for a two-agonist site model based on measurements by Patneau and Mayer (1990). The CNQX binding rate was arbitrarily set to 40 sec−1, because it played no role in the response kinetics when stepping from equilibrium concentrations of CNQX. However, the CNQX unbinding rate was a free parameter.
For a given set of rate constants, the model iteratively calculated the evolution of the number of channels in each state. It predicted the current time course after agonist application from the evolution of the number of channels in the open state. The free rate constants in the model were varied using a simplex algorithm to minimize the sum of squared errors (SSE) between the experimental current time course and the model prediction. For most data sets the predominant noise source was not channel noise, so the SSE was not weighted during the fitting procedure. After each fit, the SSE was recalculated over a subregion of the response, and this value was divided by the SD of the noise in a period immediately preceding the response. This provided a measure of the goodness-of-fit that was independent of basal recording noise. Statistical comparisons between subregion SSEs were made using paired t tests; for multiple comparisons, repeated measures ANOVA with Tukey’s post hoccomparison test was used (p < 0.05 considered significant). For each patch, the ratio was calculated between the subregion SSE for a given model and the subregion SSE for the two identical independent binding site model. This ratio provided a measure of the relative accuracy of each model.
RESULTS
We examined the activation kinetics of native non-NMDA receptor responses in cultured hippocampal neurons. Although significant heterogeneity exists in the subunit composition of AMPA and kainate responses on hippocampal neurons (Geiger et al., 1995; Ruano et al., 1995; Fleck et al., 1996), we limited our observations to responses in pyramidal-shaped neurons that had linear I–Vplots and strongly desensitizing, cyclothiazide-sensitive responses. These criteria are consistent with AMPA receptor-mediated EPSCs in cultured hippocampal pyramidal neurons (Forsythe and Westbrook, 1988;Mennerick and Zorumski, 1995; Diamond and Jahr, 1995).
To determine the functional stoichiometry of ligand binding to AMPA receptors, we examined the activation kinetics evoked by fast agonist application to outside-out patches. The low-affinity agonistsl-glutamate and AMPA were not useful for these experiments, because the forward reaction rate (kb × [agonist]) at the concentrations required to elicit robust responses (>200 μm) was too fast to allow resolution of the rising phase of the response. Thus we used the high-affinity agonist quisqualate. However, as shown in Figure1A, fast desensitization of AMPA receptors activated by quisqualate develops before agonist binding reaches equilibrium, making it difficult to follow the underlying time course of agonist–receptor interactions. At 200 μm quisqualate, the response rise time for this patch was fast (0.9 msec, 20–80%), consistent with rapid delivery of agonist (Fig. 1A, inset). The response desensitized rapidly in the continued presence of agonist (double exponential, 4.4 and 14 msec time constants, 98.5% desensitization for this patch). At a lower concentration of quisqualate (2 μm), activation showed an apparent delay at the foot of the response, and the maximum rate of rise was not achieved until 2–3 msec. However, the rise time of the response was rapid (2.5 msec, 20–80%). Assuming a binding rate of 5–10 μm−1sec−1 similar to NMDA receptors (Clements and Westbrook, 1991), the response evoked by 2 μm quisqualate should take >100 msec to reach equilibrium. Thus the rapid rise time reflects the onset of fast desensitization. To eliminate desensitization, we used the benzothiadiazine diuretic cyclothiazide (Patneau et al., 1993; Yamada and Tang, 1993). Figure 1B shows that CTZ reduced desensitization in a dose-dependent manner. In the continuous presence of 100 μm CTZ, a step into quisqualate produced a response with residual desensitization of <15 ± 5% and a much longer time constant of 70 msec (n = 9). The use of cyclothiazide permitted evaluation of the activation kinetics of AMPA receptors at lower concentrations of quisqualate, at which the onset of the response was limited by agonist binding.
Fig. 1.
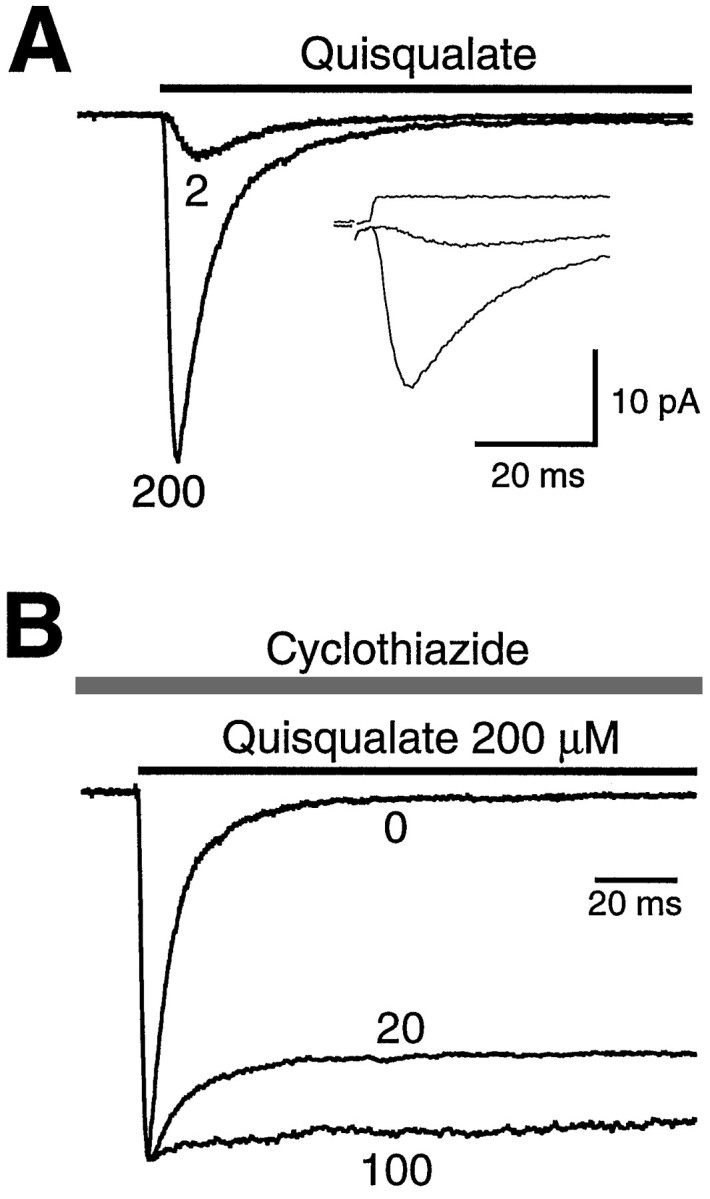
AMPA receptor activation kinetics can be unmasked when desensitization is blocked by CTZ. A, Quisqualate was applied to an outside-out patch from a cultured hippocampal neuron using fast solution exchange. Holding potential was −50 mV. Ensemble averages of currents evoked by quisqualate (2 and 200 μm) are shown. The response to 200 μm quisqualate desensitized rapidly (first time constant, 4.4 msec). The response to 2 μm quisqualate had a rise time of 2.5 msec (20–80%), much faster than the expected rise time of receptor occupancy at this concentration (∼100 msec; see Results), thus masking the kinetics of the agonist binding steps. Inset, The solution delivery as determined from open tip recordings after patch rupture for this patch is superimposed above the ensemble average current. Calibration for inset was 10 msec and 20 pA. B, The ensemble average response to 200 μm quisqualate was recorded in the continuous presence of CTZ (0, 20, or 100 μm). Pre-equilibration with CTZ removed AMPA receptor desensitization in a dose-dependent manner. In the presence of 100 μm CTZ, desensitization was <10% in this patch.
Binding of two agonist molecules is required for AMPA receptor activation
Ensemble averages of the current evoked by stepping into quisqualate (2.5, 5, 10, and 300 μm) in the presence of 100 μm CTZ are illustrated in Figure2A. Peak amplitudes were normalized to facilitate comparison. The rise time of the response was dose-dependent, confirming that agonist binding was the rate-limiting process at lower concentrations. The slower responses had a sigmoidal activation time course, suggesting that several steps are required for channel activation. To determine the number of activation steps, we tested three different models with one, two, or three identical independent agonist binding sites. The models incorporated closed, open and desensitized states. The unconstrained reaction rates (number of binding sites, kb,ku, kd, andkr) were adjusted to fit the recorded quisqualate responses optimally (see Materials and Methods).
Fig. 2.
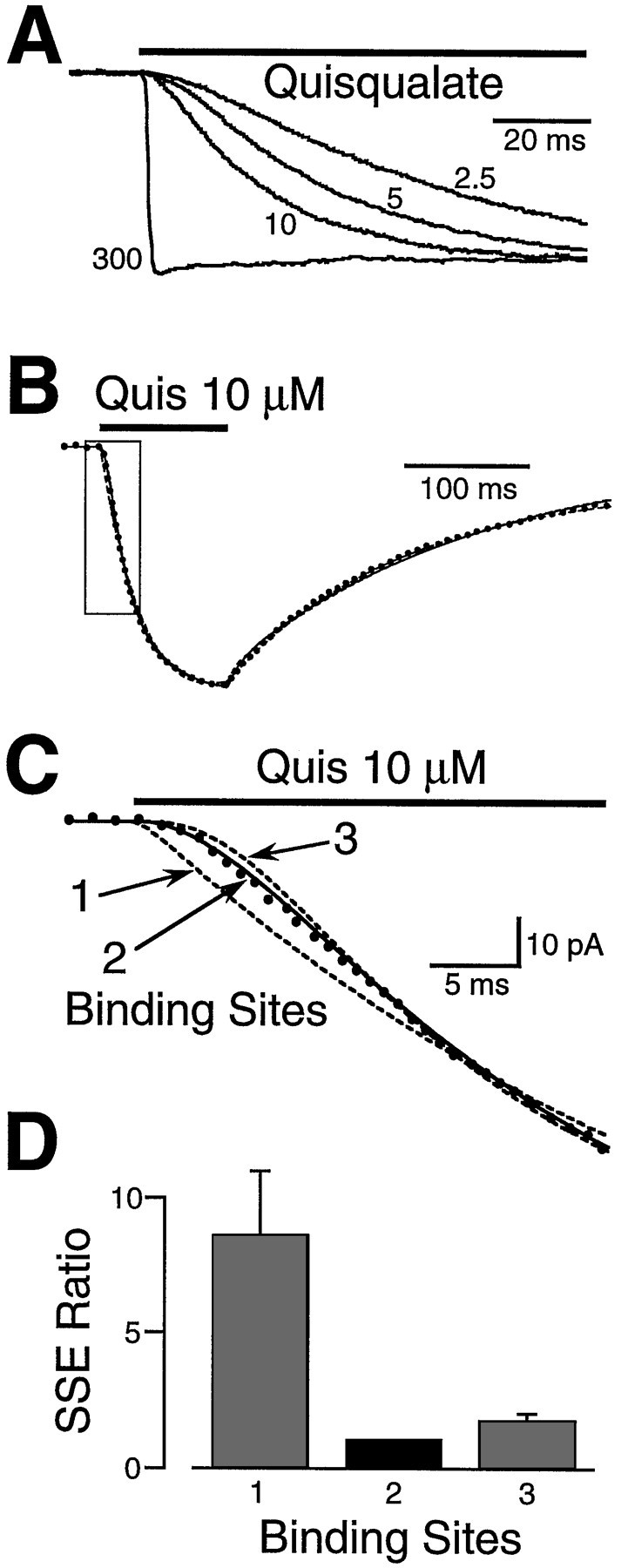
Quisqualate activation kinetics reveal that binding of two molecules of agonist are necessary for AMPA receptor activation. A, Quisqualate (2.5, 5, 10, and 300 μm) was applied to outside-out patches in the continuous presence of CTZ (100 μm) to block desensitization. Ensemble averages from four different patches are shown, and their peak amplitudes are normalized to facilitate comparison. The rise times of the responses are concentration-dependent, which suggests that the binding of quisqualate is the rate-limiting process at lower concentrations. The rising phase was clearly sigmoidal at low quisqualate concentrations, suggesting the presence of more than one binding step. B. The response to a 100 msec application of quisqualate (Quis; 10 μm) (dots) was fitted with three different kinetic models incorporating one, two, or three binding sites (lines). All three models gave a good fit late in the response when quisqualate binding was complete. However, the fits diverged during the rising phase (box). C, An enlargement of theboxed area in B, highlighting the early part of the rising phase. The optimally fitted transients are shown asdashed lines for the one- and three-site models and asolid line for the two-site model. D, The SSE was calculated between a fitted transient and the data over the rising phase of the response. The SSEs for the one- and three-site models were divided by the SSE for the two-site model. The mean SSE ratio for the responses from 13 patches are shown in thehistogram. Error bars indicate SEM. The two-site model gave the best fit to the activation phase of the response.
Not surprisingly, all three models produced a good fit to the data late in the response when agonist binding was complete (Fig.2B). To assess the quality of the fit during the critical period when quisqualate was binding to the receptors, the SSE between the data and the fitted transient was calculated over a restricted interval during the rising phase of the response (5 μm, first 100 msec; 10 μm, first 50 msec). The two- and three-site models gave a much better fit to the rising phase than the one-site model (Fig. 2C). The SSE for the two-identical-independent-site model was smaller than for other models by a factor of 9 ± 2 (one-site) and 1.7 ± 0.3 (three-site) (average ratio, n = 13) (Fig. 2D). The two-site model gave the best fit in 12 of 13 responses, and the SSEs of the three-site model were significantly larger than for the two-site model. This result suggests that two functional ligand binding sites are present per AMPA channel.
The microscopic binding and unbinding rates of quisqualate to the AMPA receptor were 4.6 ± 0.3 μm−1sec−1 and 30 ± 3 sec−1 (n = 13), corresponding to a microscopic Kd of 6.5 μm at each binding site. The binding rate is very similar to the binding rate ofl-glutamate to the NMDA receptor (Clements and Westbrook, 1991). Because the high value of Po and the prominent desensitization (in the absence of CTZ) increase the macroscopicKd, our average rate parameters predict a steady-state EC50 for quisqualate of ∼3.6 μm. This value is in relatively good agreement with the measured steady-state EC50 for quisqualate (0.9 μm; Patneau and Mayer, 1990). The best fit desensitization and resensitization rates were 4.3 ± 0.8 and 49 ± 19 sec−1 (n = 11), consistent with the small degree of macroscopic desensitization in CTZ.
The estimated number of binding steps is not affected by cyclothiazide
CTZ could alter the functional stoichiometry of the AMPA channel. For example, CTZ could restrict access to one or more agonist binding sites or alter the channel energetics so that fewer agonist binding events are required to open the channel. Domoate produces little or no desensitization when it activates AMPA channels and has a relatively high affinity (Patneau and Mayer, 1990). Thus we tested whether CTZ affects the functional stoichiometry by applying domoate in the presence or absence of CTZ. In the absence of CTZ, the rise time of ensemble average responses to domoate was dose-dependent (Fig.3A), consistent with that observed for quisqualate. Responses to domoate were fitted using three different kinetic models containing one, two, or three identical independent binding sites (Fig. 3B,C). Model parameters were optimized as above, and the goodness of fit was assessed over the rising phase as domoate was binding to the receptors (5 μm, first 150 msec; 10 μm, first 100 msec; 20 μm, first 40 msec). In the absence of CTZ, the SSEs for the two-site model were smaller than for other models by factors of 15 ± 4 (one-site) and 2.1 ± 0.3 (three-site) (average ratio, n = 6) (Fig. 3D). The two-site model gave the best fit in every case, and the SSEs for this model were significantly smaller than for the three-site model (n= 6). When CTZ was added, the peak responses were larger as expected, but the activation kinetics were not altered. In the presence of CTZ, the SSEs for the two-site model were smaller than for other models by factors of 14 ± 3 (one-site) and 1.9 ± 0.4 (three-site) (average ratio, n = 13) (Fig. 3D). The two-site model gave the best fit in 10 of 13 patches, and the SSEs for this model were significantly smaller than for the three-site model. The domoate binding and unbinding rates in the presence of CTZ were: binding, 2.2 ± 0.2 μm−1sec−1; and unbinding, 37.3 ± 4 sec−1 (n = 13), corresponding to a Kd of 16.9 μm. These results suggest that CTZ does not interfere with the number of binding sites required for channel activation.
Fig. 3.
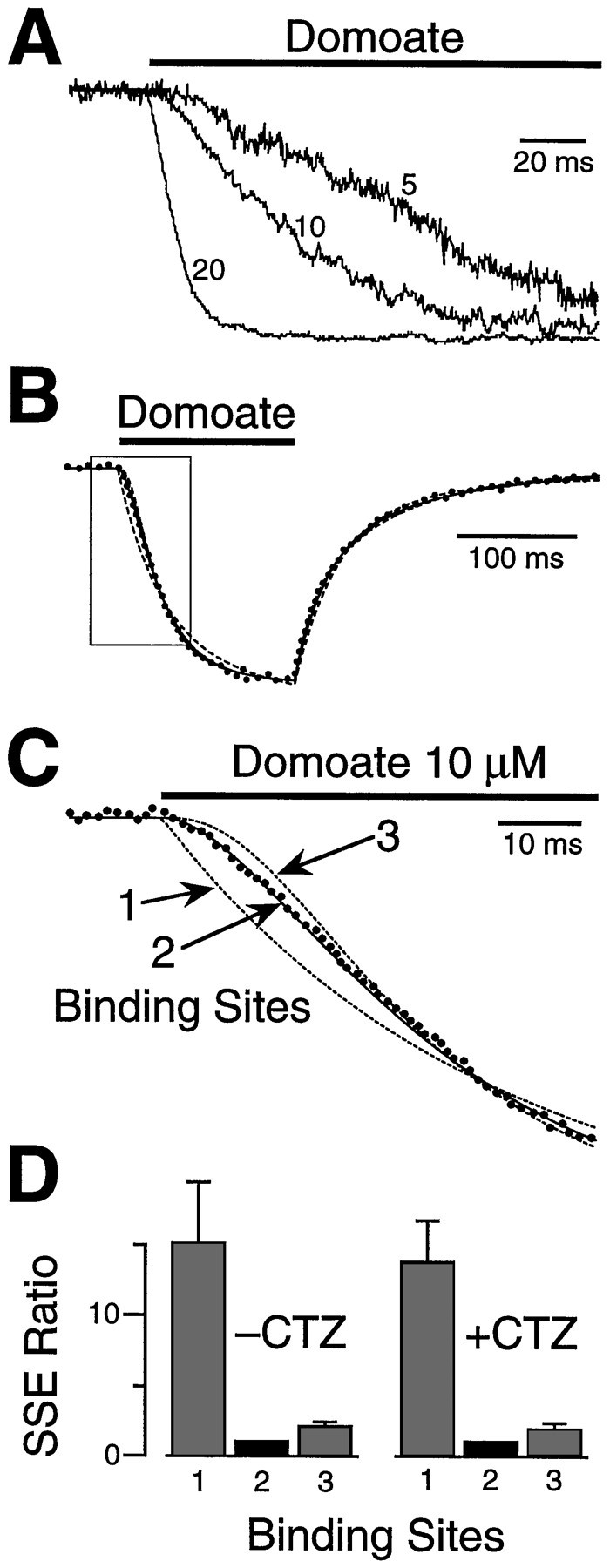
Activation of AMPA receptors by the nondesensitizing agonist domoate indicate that CTZ does not distort the estimated number of agonist binding steps. A, Domoate, (5, 10, and 20 μm) produced a nondesensitizing response in outside-out patches. Ensemble averages from three different patches are shown with their peak amplitudes normalized to facilitate comparison. As for quisqualate, the rise times of the responses were concentration-dependent, with sigmoidal activation at low agonist concentrations. B, The response to a pulse (150 msec) of domoate (10 μm) (dots) was fitted with three different kinetic models incorporating one, two, or three binding sites (lines). All three models gave a good fit late in the response when domoate binding was complete but diverged in the critical rising phase (box). C, An enlargement of the boxed area in B, highlighting the early part of the rising phase. The optimally fitted transients are shown as dashed lines for the one- and three-site models and a solid line for the two-site model. D, Domoate responses in the absence or presence of 100 μm CTZ were compared using the three kinetic models. The mean SSE in the absence of CTZ (−CTZ;n = 6) and presence of CTZ (+CTZ;n = 13) are shown in the histograms. As for quisqualate, the two-site model gave the best fit to the activation phase, and CTZ did not alter the stoichiometry of agonist binding.
Antagonist unbinding also predicts two sites
The stoichiometry of ligand binding to AMPA channels was reexamined using an independent approach based on the dissociation kinetics of a competitive antagonist (Benveniste and Mayer, 1991;Clements and Westbrook, 1994). Outside-out patches were pre-equilibrated with a saturating concentration of CNQX (20 μm) and then rapidly stepped out of CNQX and into a saturating concentration of kainate (1 mm). Because the activation phase after steps into kainate is complete in <1 msec (0.57 ± 0.04 msec; n = 9) (also see Patneau et al., 1993), the slower activation after a step out of CNQX and into kainate reflects the number of antagonist unbinding steps. As shown in Figure 4A, the rising phase of the response was sigmoidal and required >100 msec. Three different models containing one, two, or three identical independent antagonist binding sites were used to fit the entire time course of the recorded responses. The goodness of fit was assessed over the first 100 msec of the rising phase, while CNQX was unbinding from the receptors (Fig. 4B). The SSE for the two-site model was smaller than for other models by factors of 20 ± 6 (one-site) and 3.3 ± 1.2 (three-site) (average ratio, n = 12) (Fig. 4C). The two-site model gave the best fit in 9 of 12 patches, and the SSEs were significantly smaller than for the three-site model.
Fig. 4.
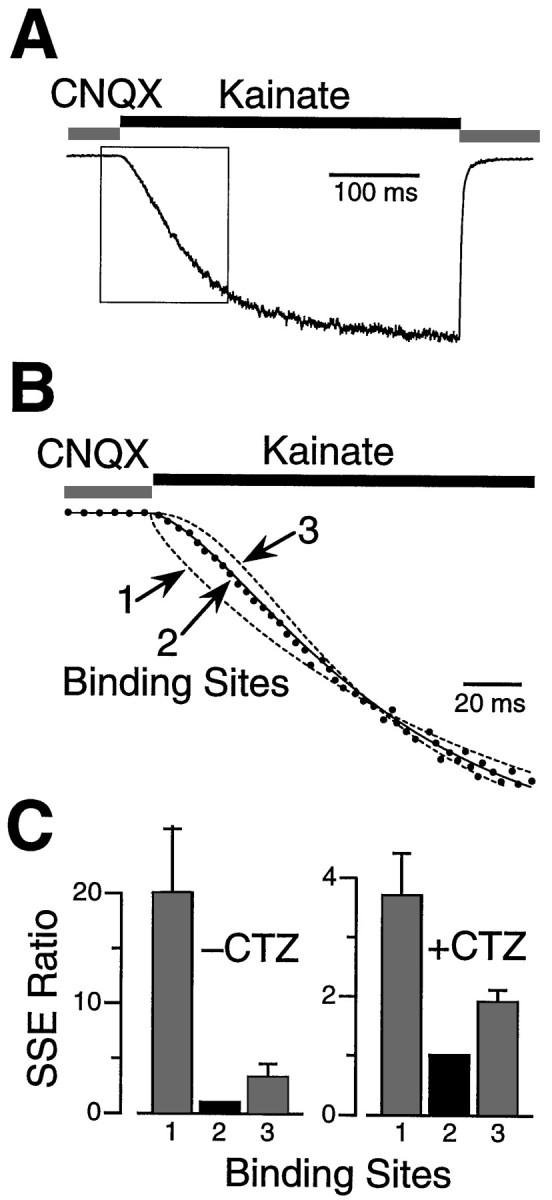
CNQX dissociation kinetics reveal that two molecules of antagonist bind to the AMPA receptor. A, An outside-out patch was stepped from a saturating concentration of CNQX (20 μm) into a saturating concentration of kainate (1 mm). CNQX dissociation is the rate-limiting process for activation of this response. The early part of the response (box) is sigmoidal, which suggests a multistep process.B, An enlargement of the boxed area inA shows the response (dots) fitted with three different kinetic models incorporating one, two, or threeantagonist binding sites (lines). All three models gave a good fit late in the response when CNQX unbinding was complete (data not shown). However, the fits diverged in the rising phase. The optimally fitted transients are shown as dashed lines for the one- and three-antagonist site models and asolid line for the two-antagonist site model.C, Responses after a step from CNQX into kainate were recorded in the absence or presence of 100 μm CTZ and fitted with the three kinetic models. As shown in the SSE ratios in thehistogram, the two-antagonist-site model gave the best fit to the activation phase of the response in the absence (−CTZ; n = 7) and in the presence (+CTZ; n = 12) of CTZ.
Although kainate or domoate activation of AMPA receptors showed little desensitization in whole-cell recording, we, as others, observed desensitization even with these “nondesensitizing” agonists during rapid application in outside-out patches (39 ± 3%, τ = 3.9 msec; n = 9). Thus to exclude the possibility that receptor desensitization during CNQX dissociation affected the fits, we repeated the experiments in the continuous presence of CTZ. The responses were fitted using the same three models. The SSE for the two-site model was again smaller than for other models by factors of 3.7 ± 0.7 (one-site) and 1.9 ± 0.2 (three-site) (average ratio, n = 7) (Fig. 4C). The two-site model gave the best fit in all seven patches, and the SSEs were significantly smaller than for the three-site model. Thus two different approaches, based on either agonist binding or antagonist unbinding, are consistent with two binding sites per channel.
The two binding sites show negative cooperativity
Although fits to the two identical independent antagonist binding sites were superior to either one or three sites, there were small but systematic deviations from the observed responses during the critical rising phase. Thus we tested several models incorporating a more detailed description of antagonist dissociation. As agonist and antagonist binding rates usually fall within a limited range (2–20 μm−1sec−1), it is often assumed that the ligand binding rate is “diffusion-limited.” In this view of receptor ligand interaction, affinity is determined predominantly by the ligand unbinding rate. If two sites on a channel have different affinities, this implies a difference in the ligand unbinding rate at the two sites. Similarly, if binding to one site on a channel alters the affinity of the other site (cooperativity), this will be expressed through a change in the unbinding rate at the second site. Thus, antagonist dissociation kinetics would be expected to provide a more sensitive test for cooperative interactions between binding sites than agonist binding kinetics (Clements and Westbrook, 1994). Thus we tested the ability of more detailed kinetic models to fit the response after a step from CNQX into a saturating concentration of kainate.
The new models were variations on the two-identical-independent (II) binding site model, which produced the best fit in the first round of analysis. In one variant, the binding sites were independent and had the same antagonist binding rates but different antagonist unbinding rates. This model describes two different, independent (DI) sites (Fig.5A). In the second variant, the two sites were identical, but antagonist binding to the first site altered the antagonist unbinding rate at the second site. This model describes two identical, cooperative (IC) sites (Fig. 5A). For steps from CNQX into kainate, the IC model gave a better fit to the activation phase than the II model (Fig. 5B). In contrast, the DI model did not give a significantly better fit, although it had the same number of free parameters as the IC model. The ratio between the SSE for the II model and for other models was 0.85 ± 0.06 (IC) and 1.01 ± 0.04 (DI) (average ratio, n = 12) (Fig. 5C). The IC model gave the best fit in 10 of 12 patches with SSEs that were significantly smaller than for the II model. Similar results were obtained in the presence of CTZ (Fig.5C). The optimum rate constants for the IC model revealed negative cooperativity in the binding of CNQX to AMPA receptors. When both sites were occupied the unbinding rate from the first site was 29 ± 2 sec−1 and from the second site was 16 ± 1 sec−1 (n = 12). Faster CNQX unbinding rates were observed in the presence of CTZ. When both sites were occupied, the unbinding rate from the first site was 75 ± 5 sec−1 and from the second site was 33 ± 5 sec−1 (n = 6). The faster unbinding of CNQX in CTZ is consistent with the right shift in the CNQX dose–response curve in the presence of CTZ (128 ± 5.8 to 573 ± 45 μm, respectively, n = 4–10). Thus, occupancy of one site on the AMPA receptor by CNQX reduced the affinity at the second site by approximately half.
Fig. 5.
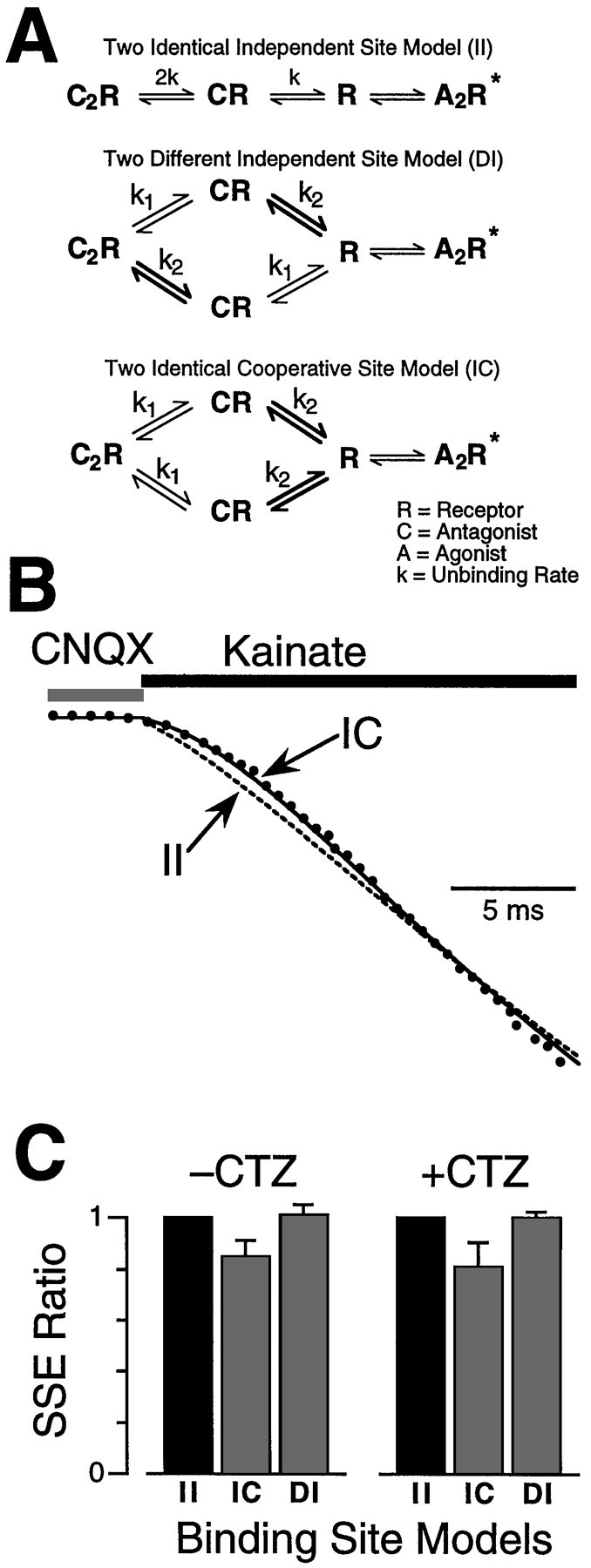
The two antagonist binding sites on the AMPA receptor exhibit negative cooperativity. A, The reaction schemes describe three ways in which the competitive antagonist, CNQX, can unbind from the AMPA receptor. In all three schemes, theleft state shows a receptor (R) with two molecules of competitive antagonist (C) bound to it. The first scheme describes unbinding from two identical independent (II) antagonist sites. Two routes are possible; antagonist unbinds first from site 1, or first from site 2. These two equivalent paths have been collapsed into a single path for simplicity. In the collapsed scheme, the unbinding rate for the first step (2k) is twice as fast as the binding rate for the second step (k). The second scheme describes unbinding from two different independent (DI) sites. The unbinding rate from site 1 (k1) is different from the unbinding rate from site 2 (k2), but there is no interaction between the sites. The third scheme describes unbinding from two identical cooperative (IC) sites. The rate of the first unbinding step from either site 1 or 2 is the identical (k1), but this step alters the rate of the second unbinding step (k2) via a cooperative interaction. When both sites have been vacated by antagonist, the subsequent steps leading to channel activation are so fast under the present experimental conditions that they can be combined into a single kinetic transition with no loss of accuracy. Thus a single step is shown for the transition from the vacant receptor (R) to active channel with two molecules of agonist bound (A2R*).B, The response (dots) after a step from CNQX (20 μm) into kainate (1 mm) was fitted with the three different kinetic models shown in A. All three models gave a good fit late in the response when CNQX unbinding was complete (not shown) but diverged in the rising phase. The optimal fits for the II model (dashed line) and the IC model (solid line) are shown. The IC model provided a better fit than either the II model or the DI model (data not shown). C, The SSEs for the DI andIC models were divided by the SSE for theII model. The cooperative model (IC) gave the best fit to the activation phase of the response and was not affected by the presence of CTZ (100 μm). SSE ratios for seven responses in the presence of CTZ and 12 responses in the absence of CTZ are shown.
The II, IC, and DI models were adapted to describe agonist binding. The rate constants were optimized to fit the response to quisqualate in 100 μm CTZ over the entire time course of the response (Fig.6A). As outlined above, dissociation (deactivation) kinetics are more sensitive to cooperative interactions between binding sites than are binding kinetics. Thus SSEs between the data and the fitted transients were calculated during deactivation (Fig. 6B). Both the DI and IC models gave significantly better fits to the quisqualate deactivation than the II model. The ratios between the SSE for the II model and for other models were 0.54 ± 0.14 (IC) and 0.53 ± 0.14 (DI) (average ratio, n = 9) (Fig. 6C). There was no significant difference between the fit obtained with the IC or DI models, but the agonist deactivation data do provide further evidence that the agonist binding sites on the AMPA receptor are not identical and independent.
Fig. 6.
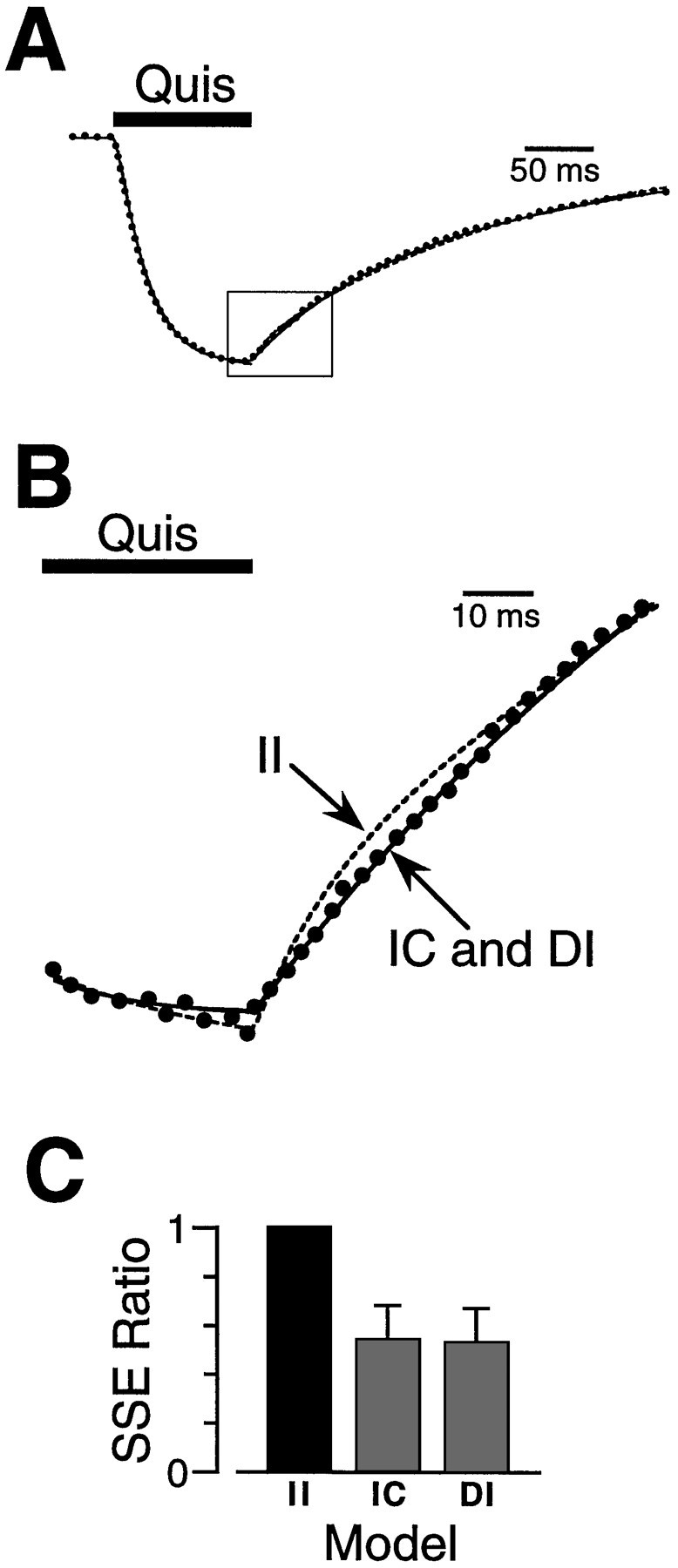
Quisqualate (Quis) deactivation kinetics show that the two agonist binding sites are unequal.A, The response to a pulse (100 msec) of quisqualate (10 μm) (dots) was fitted with three different kinetic models describing identical independent (II), different independent (DI), and identical cooperative (IC) agonist binding sites (lines). The fits diverged in the early part of the deactivation phase after the removal of agonist (box).B, An enlargement of the boxed area inA during agonist unbinding (deactivation). The optimally fitted transients are shown as a dashed line for theII model and solid lines for theDI and IC models. C, The mean SSE ratios for the responses from 13 patches are shown in thehistogram. The IC and DImodels gave better fits to the deactivation phase of the response than the II model.
DISCUSSION
Comparison between kinetic analysis and steady-state pharmacological analysis
Steady-state dose–response analysis as well as binding experiments have previously suggested the presence of more than one binding site on AMPA receptor channels, based on Hill coefficients ranging from 1.2 to 1.95 (Verdoorn and Dingledine 1988; Priestley et al., 1989; Trussell and Fischbach, 1989; Patneau and Mayer 1990; Jonas and Sakmann, 1992). Similar results have been obtained in dose–response analysis of the homomeric kainate receptor subunit glutamate receptor 6 (GluR6) and a nondesensitizing GluR3/6 chimera (Heckmann et al., 1996; Rosenmund and Stevens, 1996). In fact kinetic schemes attempting to describe AMPA channel behavior now use a model with two agonist binding sites (Jonas et al., 1993; Raman and Trussell, 1995; Häusser and Roth, 1997). These observations suggest a stoichiometry of at least two agonist binding sites per channel, but the presence of desensitization and cooperativity complicate steady-state measurements. Furthermore, in cochlear neurons, the possibility of more than two sites for AMPA channels has been suggested by limiting slope analysis (Raman and Trussell, 1992). These authors modeled a third low-affinity site to account for the roll off in the dose–response curve at very high agonist concentrations. The kinetic approach used here provides an alternative to steady-state pharmacological analysis by explicitly incorporating desensitization and cooperativity. The use of rapid application methods revealed a sigmoidal onset to the ensemble average current, providing unequivocal evidence that there is more than one binding site on AMPA channels. Fits of the ensemble average currents were best with a two-site model, but the difference between fits to the two- and three-site models, although statistically significant, was smaller than the difference with the one-site model. Thus the possibility of additional binding sites cannot be excluded completely (see below).
The reliability of our estimate for the number of binding sites depends on three major factors: any contamination of the activation phase by desensitization, the adequacy of the kinetic model, and possible effects of receptor heterogeneity on the number of agonist binding sites. We addressed the first problem with the use of CTZ, which greatly reduces AMPA receptor desensitization. Although the principle macroscopic effect of CTZ is the slowing of entry into the desensitized state, it also slows deactivation (Patneau et al., 1993), an effect that cannot be accounted for by changes in the rate into the desensitized state. In addition, CTZ also produces a left shift (approximately fourfold change in EC50) in the kainate dose–response curve (Patneau et al., 1993) (Y. Sahara, unpublished data) that has been modeled as an increase in agonist affinity along with a reduction in the entry into the desensitized state (Partin et al., 1996). Alternatively, CTZ could allow the channel to open from a desensitized state. In fact preliminary use of a model containing an “open desensitized” state provided good fits to the entire time course, including deactivation in the presence of CTZ (J. D. Clements, unpublished data). However, we obtained similar estimates for the number of binding sites for the nondesensitizing agonist domoate in the absence of CTZ, suggesting that cyclothiazide does not alter the number of functional agonist binding sites. This is supported by recent molecular analysis of GluR1, which indicated that the differential modulation by CTZ of the flip/flop splice variant depends on a single residue at position 750 (Partin et al., 1995,1996). Models of the glutamate binding site place the flip/flop domain in an external helix away from the proposed glutamate binding pocket (Sutcliffe et al., 1996).
To fit the activation time course, we used a simplified kinetic scheme for the AMPA receptor that incorporates previous measurements of transition rates between open, closed, and desensitized states. Although the simplified reaction scheme does not account for all experimental details of AMPA receptor activity (e.g., subconductance levels), similar schemes can accurately predict ensemble kinetic properties of ligand-gated channels under a wide range of experimental conditions (e.g., Jonas and Sakmann, 1992; Jonas et al., 1993; Diamond and Jahr, 1997). Wherever possible we constrained transition rates, e.g., between open and closed states, based on previous experimental results. Furthermore, the experiments were structured such that the binding steps were rate-limiting; thus possible inaccuracies in some of the rates would not have affected our results. For example the CNQX binding rate was irrelevant to the fits, because our measurements were made after CNQX had equilibrated at a high level of receptor occupancy. As an additional check on the accuracy of the model, the microscopic binding and unbinding rates generated by the kinetic model gave estimated steady-state EC50 values that were close to EC50 values measured for AMPA receptors on hippocampal neurons (Patneau and Mayer, 1990).
Stoichiometry and structure of AMPA channels
Glutamate receptor channels are heteromers consisting of four or five subunits with GluR1–4 coassembling to generate AMPA receptors (Wisden and Seeburg, 1993; Hollmann and Heinemann, 1994). There is considerable heterogeneity in the properties of AMPA receptors because of subunit composition, alternative splicing, and RNA editing (Sommer et al., 1990; Verdoorn et al., 1991; Burnashev et al., 1992; Lomeli et al., 1994; Mosbacher et al., 1994; Geiger et al., 1995;Swanson et al., 1997). Although this is an inevitable problem for all studies of native receptors, even with a single cell type such as CA1 pyramidal cells (e.g., Mackler and Eberwine, 1993), the CTZ sensitivity and linear I–V relationships of patch currents in our experiments are consistent with AMPA receptor heteromers containing edited GluR2 subunits. The high sensitivity to CTZ is also most consistent with flip-containing subunits (Fleck et al., 1996). Consistent with relatively uniform macroscopic channel behavior, the patch currents in our experiments were well fitted with a relatively simple kinetic model. However, patch-to-patch variability in response time course was greater than could be attributed to the recording noise, resulting in some scatter in the reaction rate constant estimates. Whether this was attributable to subunit heterogeneity or post-translational processing is unclear. However, the estimates for the number and cooperativity of binding sites were consistent from patch to patch, suggesting that receptor heterogeneity did not significantly affect our estimates of the number of binding sites per channel.
Based on the homology of the extracellular domain of glutamate receptor subunits with bacterial periplasmic binding proteins such as lysine–arginine–ornithine binding protein (LAOBP), it appears that each subunit has all the necessary domains for a functional binding site. The binding site of the glutamate channel subunits incorporates an LAOBP-like domain split between the N-terminal domain and the M3–M4 extracellular loop (O’Hara et al., 1993; Stern-Bach et al., 1994;Kuusinen et al., 1995). Thus whatever the exact subunit composition of the AMPA channels in our experiments, the number of agonist binding sites might be expected to equal the number of subunits. Such a hypothesis seems consistent with NMDA receptors, the subunits of which share homology with the AMPA subunits. In the case of NMDA receptors, the binding of four agonist molecules (two glutamates and two glycines) are required to activate the channel (Benveniste and Mayer, 1991;Clements and Westbrook, 1991, 1994). Furthermore, each NMDA channel contains two or three NR1 and two NR2 subunits (Behe et al., 1995;Premkumar and Auerbach, 1996) per channel, with glycine and glutamate binding to the NR1 and NR2 subunits, respectively (Kuryatov et al., 1994, Laube et al., 1997). This situation contrasts with the acetylcholine receptor family, in which the number of binding sites is less than the number of subunits, presumably because the two agonist binding sites reside at subunit–subunit interfaces (Karlin and Akabas, 1995; Smith and Olsen, 1995, but see Palma et al., 1996).
Are there more than two binding sites per channel?
Our finding that each AMPA channel has only twofunctional binding sites raises questions about the status of the presumed two or three additional binding sites. Either binding to these sites is restricted, or it does not measurably influence channel function (a “silent” binding site). Binding at some sites could be prevented by conformational constraints imposed by subunit–subunit interactions. For example, a subunit may need to be held in a “receptive” conformation by two neighboring subunits, both of which have to be in a “nonreceptive” conformation. Such subunit interactions could conceivably differ depending on subunit composition. For example, in a homomeric channel composed of GluR3/6 chimeric subunits, three conductance levels have been observed that are dependent on agonist concentration, implying a relationship between the conductance level and the number of binding sites (Rosenmund and Stevens, 1996).
An alternative explanation is that negative cooperativity between the binding sites could effectively preclude more than two binding events. When two sites are occupied, cooperative interactions could reduce the affinity of the remaining sites such that no significant agonist or antagonist binding occurs. Negative cooperativity is implied by our results, perhaps as an unavoidable consequence of allosteric interactions between binding sites on adjacent GluR subunits. For example, the binding of l-glutamate to NMDA receptors reduces the affinity of glycine binding, resulting in the phenomenon of glycine-dependent desensitization (Mayer et al., 1989). However, both quantitative and qualitative aspects of this explanation remain problematic. First, it was not possible in our experiments to distinguish unequivocally between models of agonist binding with two different independent or two identical cooperative sites. However, if the cooperative model is assumed, the estimated affinities for the two successive binding steps differ by a factor of 2–3 with a systematic tendency for a negative allosteric effect. If the twofold to threefold reduction in affinity from the first to second binding steps were simply extrapolated, then a third binding step should have been detectable. It is therefore necessary to assume a dramatic increase in negative cooperativity beyond the second binding step. This could be achieved if binding to adjacent subunits was less favored than binding to nonadjacent subunits. However, it seems surprising that negative cooperativity would be the same for different agonists and antagonists. Thus we favor the possibility that one or more binding sites are silent. Although the functional significance of such sites is unclear, they could influence more subtle aspects of receptor function such as desensitization or play a role in buffering of glutamate in the synaptic cleft.
Footnotes
This work was supported by United States Public Health Services Grant NS26494. J.D.C. was supported in part by a Queen Elizabeth II Fellowship from the Australian Research Council. We thank Jeff Volk for the preparation of the cell cultures. Cyclothiazide was generously provided by Eli Lilly Research Laboratories (Indianapolis, IN).
Correspondence should be addressed to Gary L. Westbrook, Vollum Institute, L474, Oregon Health Sciences University, Portland, OR 97201.
REFERENCES
- 1.Behe P, Stern P, Wyllie DJ, Nassar M, Schoepfer R, Colquhoun D. Determination of NMDA NR1 subunit copy number in recombinant NMDA receptors. Proc R Soc Lond [Biol] 1995;262:205–213. doi: 10.1098/rspb.1995.0197. [DOI] [PubMed] [Google Scholar]
- 2.Bekkers JM, Stevens CF. NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature. 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- 3.Bennett JA, Dingledine R. Topology profile for a glutamate receptor: three transmembrane domains and a channel-lining reentrant membrane loop. Neuron. 1995;14:373–384. doi: 10.1016/0896-6273(95)90293-7. [DOI] [PubMed] [Google Scholar]
- 4.Benveniste M, Mayer ML. Kinetic analysis of antagonist action N-methyl-d-aspartic acid receptors. Two binding sites each for glutamate and glycine. Biophys J. 1991;59:560–573. doi: 10.1016/S0006-3495(91)82272-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benveniste M, Clements J, Vyklicky L, Jr, Mayer ML. A kinetic analysis of the modulation of N-methyl-d-aspartic acid receptors by glycine in mouse cultured hippocampal neurones. J Physiol (Lond) 1990;428:333–357. doi: 10.1113/jphysiol.1990.sp018215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- 7.Clements JD. Transmitter timecourse in the synaptic cleft: its role in central synaptic function. Trends Neurosci. 1996;19:163–171. doi: 10.1016/s0166-2236(96)10024-2. [DOI] [PubMed] [Google Scholar]
- 8.Clements JD, Westbrook GL. Activation kinetics reveal the number of glutamate and glycine binding sites on the N-methyl-d-aspartate receptor. Neuron. 1991;7:605–613. doi: 10.1016/0896-6273(91)90373-8. [DOI] [PubMed] [Google Scholar]
- 9.Clements JD, Westbrook GL. Kinetics of AP5 dissociation from NMDA receptors: evidence for two identical cooperative binding sites. J Neurophysiol. 1994;71:2566–2569. doi: 10.1152/jn.1994.71.6.2566. [DOI] [PubMed] [Google Scholar]
- 10.Colquhoun D, Jonas P, Sakmann B. Action of brief pulses of glutamate on AMPA/kainate receptors in patches from different neurones of rat hippocampal slices. J Physiol (Lond) 1992;458:261–287. doi: 10.1113/jphysiol.1992.sp019417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diamond JS, Jahr CE. Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron. 1995;15:1097–1107. doi: 10.1016/0896-6273(95)90098-5. [DOI] [PubMed] [Google Scholar]
- 12.Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 1997;12:4672–4687. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edmonds B, Gibb AJ, Colquhoun D. Mechanisms of activation of glutamate receptors and the time course of excitatory synaptic currents. Annu Rev Physiol. 1995;57:495–519. doi: 10.1146/annurev.ph.57.030195.002431. [DOI] [PubMed] [Google Scholar]
- 14.Fleck MW, Bähring R, Patneau DK, Mayer ML. AMPA receptor heterogeneity in rat hippocampal neurons revealed by differential sensitivity to cyclothiazide. J Neurophysiol. 1996;75:2322–2333. doi: 10.1152/jn.1996.75.6.2322. [DOI] [PubMed] [Google Scholar]
- 15.Forsythe ID, Westbrook GL. Slow excitatory postsynaptic currents mediated by monosynaptic activation of NMDA receptors on cultured mouse central neurones. J Physiol (Lond) 1988;396:515–533. doi: 10.1113/jphysiol.1988.sp016975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Geiger JRP, Melcher T, Koh D-S, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 17.Häusser M, Roth A. Dendritic and somatic glutamate receptor channels in rat cerebellar Purkinje cells. J Physiol (Lond) 1997;501:77–95. doi: 10.1111/j.1469-7793.1997.077bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heckmann M, Bufler J, Franke C, Dudel J. Kinetics of homomeric GluR6 glutamate receptor channels. Biophys J. 1996;71:1743–1750. doi: 10.1016/S0006-3495(96)79375-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hestrin S. Activation and desensitization of glutamate-activated channels mediating fast excitatory synaptic currents in the visual cortex. Neuron. 1992;9:991–999. doi: 10.1016/0896-6273(92)90250-h. [DOI] [PubMed] [Google Scholar]
- 20.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 21.Jonas P, Sakmann B. Glutamate receptor channels in isolated patches from CA1 and CA3 pyramidal cells of rat hippocampal slices. J Physiol (Lond) 1992;455:143–171. doi: 10.1113/jphysiol.1992.sp019294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jonas P, Major G, Sakmann B. Quantal components of unitary EPSCs a the mossy fibre synapse on CA3 cells of the rat hippocampus. J Physiol (Lond) 1993;472:615–663. doi: 10.1113/jphysiol.1993.sp019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karlin A, Akabas MH. Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron. 1995;15:1241–1244. doi: 10.1016/0896-6273(95)90004-7. [DOI] [PubMed] [Google Scholar]
- 24.Kullmann DM. Quantal variability of excitatory transmission in the hippocampus: implications for the opening probability of fast glutamate-gated channels. Proc R Soc Lond [Biol] 1993;253:107–116. doi: 10.1098/rspb.1993.0088. [DOI] [PubMed] [Google Scholar]
- 25.Kuryatov A, Laube B, Betz H, Kuhse J. Mutational analysis of the glycine-binding site of the NMDA receptor: structural similarity with bacterial amino acid-binding proteins. Neuron. 1994;12:1291–1300. doi: 10.1016/0896-6273(94)90445-6. [DOI] [PubMed] [Google Scholar]
- 26.Kuusinen A, Arvola M, Keinänen K. Molecular dissection of the agonist binding site of an AMPA receptor. EMBO J. 1995;14:6327–6332. doi: 10.1002/j.1460-2075.1995.tb00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laube B, Hirai H, Sturgess M, Betz H, Kuhse J. Molecular determinants of agonist discrimination by NMDA receptor subunits: analysis of the glutamate binding site on the NR2B subunit. Neuron. 1997;18:493–503. doi: 10.1016/s0896-6273(00)81249-0. [DOI] [PubMed] [Google Scholar]
- 28.Legendre P, Westbrook GL. The inhibition of single N-methyl-d-aspartate-activated channels by zinc ions on cultured rat neurones. J Physiol (Lond) 1990;429:429–449. doi: 10.1113/jphysiol.1990.sp018266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lester RAJ, Jahr CE. NMDA channel behavior depends on agonist affinity. J Neurosci. 1992;12:635–643. doi: 10.1523/JNEUROSCI.12-02-00635.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lomeli H, Mosbacher J, Melcher T, Höger T, Geiger JRP, Kuner T, Monyer H, Higuchi M, Bach A, Seeburg PH. Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science. 1994;266:1709–1713. doi: 10.1126/science.7992055. [DOI] [PubMed] [Google Scholar]
- 31.Mackler SA, Eberwine JH. Diversity of glutamate receptor subunit mRNA expression within live hippocampal CA1 neurons. Mol Pharmacol. 1993;44:308–315. [PubMed] [Google Scholar]
- 32.Mayer ML, Vyklicky L, Jr, Clements JD. Regulation of NMDA receptor desensitization in mouse hippocampal neurons by glycine. Nature. 1989;338:425–527. doi: 10.1038/338425a0. [DOI] [PubMed] [Google Scholar]
- 33.McBain CJ, Mayer ML. N-Methyl-d-aspartate acid receptor structure and function. Physiol Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- 34.Mennerick S, Zorumski CF. Presynaptic influence on the time course of fast excitatory synaptic currents in cultured hippocampal cells. J Neurosci. 1995;15:3178–3192. doi: 10.1523/JNEUROSCI.15-04-03178.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mosbacher J, Schoepfer R, Monyer H, Burnashev N, Seeburg PH, Ruppersberg JP. A molecular determinant for submillisecond desensitization in glutamate receptors. Science. 1994;266:1059–1062. doi: 10.1126/science.7973663. [DOI] [PubMed] [Google Scholar]
- 36.Nakanishi S. Molecular diversity of glutamate receptors and implications for brain function. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- 37.O’Hara PJ, Sheppard PO, Thøgersen H, Venezia D, Haldeman BA, McGrane V, Houamed KM, Thomsen C, Gilbert TL, Mulvihill ER. The ligand-binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron. 1993;11:41–52. doi: 10.1016/0896-6273(93)90269-w. [DOI] [PubMed] [Google Scholar]
- 38.Palma E, Bertrand S, Binzoni T, Bertrand D. Neuronal nicotinic alpha 7 receptor expressed in Xenopus oocytes presents five putative binding sites for methyllcaconitine. J Physiol (Lond) 1996;15:151–161. doi: 10.1113/jphysiol.1996.sp021203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Partin KM, Bowie D, Mayer ML. Structural determinants of allosteric regulation in alternatively spliced AMPA receptors. Neuron. 1995;14:833–843. doi: 10.1016/0896-6273(95)90227-9. [DOI] [PubMed] [Google Scholar]
- 40.Partin KM, Fleck MW, Mayer ML. AMPA receptor flip/flop mutants affecting deactivation, desensitization, and modulation by cyclothiazide, aniracetam, and thiocyanate. J Neurosci. 1996;16:6634–6647. doi: 10.1523/JNEUROSCI.16-21-06634.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patneau DK, Mayer ML. Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-d-aspartate and quisqualate receptors. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patneau DK, Vyklicky L, Jr, Mayer ML. Hippocampal neurons exhibit cyclothiazide-sensitive rapidly desensitizing responses to kainate. J Neurosci. 1993;13:3496–3509. doi: 10.1523/JNEUROSCI.13-08-03496.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Premkumar LS, Auerbach A. Stoichiometry of recombinant NMDA receptor channels. Soc Neurosci Abstr. 1996;22:593. [Google Scholar]
- 44.Priestley T, Woodruff GN, Kemp JA. Antagonism of responses to excitatory amino acids on rat cortical neurones by the spider toxin, argiotoxin636. Br J Pharmacol. 1989;97:1315–1323. doi: 10.1111/j.1476-5381.1989.tb12594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raman IM, Trussell LO. The kinetics of the response to glutamate and kainate in neurons of the avian cochlear nucleus. Neuron. 1992;9:173–186. doi: 10.1016/0896-6273(92)90232-3. [DOI] [PubMed] [Google Scholar]
- 46.Raman IM, Trussell LO. The mechanism of α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionate receptor desensitization after removal of glutamate. Biophys J. 1995;68:137–146. doi: 10.1016/S0006-3495(95)80168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosenmund CR, Stevens CF. Single channel conductance of a AMPA-type glutamate channel is determined by agonist concentration. Biophys J. 1996;70:A251. [Google Scholar]
- 48.Ruano D, Lambolez B, Rossier J, Paternain AV, Lerma J. Kainate receptor subunits expressed in single cultured hippocampal neurons: molecular and functional variants by RNA editing. Neuron. 1995;14:1009–1014. doi: 10.1016/0896-6273(95)90339-9. [DOI] [PubMed] [Google Scholar]
- 49.Silver RA, Traynelis SF, Cull-Candy SG. Rapid-time-course miniature and evoked excitatory currents at cerebellar synapses in situ. Nature. 1992;355:163–166. doi: 10.1038/355163a0. [DOI] [PubMed] [Google Scholar]
- 50.Smith GB, Olsen RW. Functional domains of GABAA receptors. Trends Pharmacol. 1995;16:162–168. doi: 10.1016/s0165-6147(00)89009-4. [DOI] [PubMed] [Google Scholar]
- 51.Sommer B, Keinänen K, Verdoorn T, Wisden W, Burnashev N, Herb A, Köhler M, Takagi T, Sakmann B, Seeburg PH. Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science. 1990;249:1580–1585. doi: 10.1126/science.1699275. [DOI] [PubMed] [Google Scholar]
- 52.Stern-Bach Y, Bettler B, Hartley M, Sheppard PO, O’Hara PJ, Heinemann SF. Agonist selectivity of glutamate receptors is specified by two domains structurally related to bacterial amino acid-binding proteins. Neuron. 1994;13:1345–1357. doi: 10.1016/0896-6273(94)90420-0. [DOI] [PubMed] [Google Scholar]
- 53.Sutcliffe MJ, Wo ZG, Oswald RE. Three-dimensional models of non-NMDA glutamate receptors. Biophys J. 1996;70:1575–1589. doi: 10.1016/S0006-3495(96)79724-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swanson GT, Kamboj SK, Cull-Candy SG. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trussell LO, Fischbach GD. Glutamate receptor desensitization and its role in synaptic transmission. Neuron. 1989;3:209–218. doi: 10.1016/0896-6273(89)90034-2. [DOI] [PubMed] [Google Scholar]
- 56.Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-z. [DOI] [PubMed] [Google Scholar]
- 57.Verdoorn TA, Dingledine R. Excitatory amino acid receptors expressed in Xenopus oocytes: agonist pharmacology. Mol Pharmacol. 1988;34:298–307. [PubMed] [Google Scholar]
- 58.Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 1991;252:1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- 59.Vyklicky L, Jr, Patneau DK, Mayer ML. Modulation of excitatory synaptic transmission by drugs that reduce desensitization at AMPA/kainate receptors. Neuron. 1991;7:971–984. doi: 10.1016/0896-6273(91)90342-w. [DOI] [PubMed] [Google Scholar]
- 60.Wisden W, Seeburg PH. Mammalian ionotropic glutamate receptors. Curr Opin Neurobiol. 1993;3:291–298. doi: 10.1016/0959-4388(93)90120-n. [DOI] [PubMed] [Google Scholar]
- 61.Yamada KA, Tang C-M. Benzothiadiazides inhibit rapid glutamate receptor desensitization and enhance glutamatergic synaptic currents. J Neurosci. 1993;13:3904–3915. doi: 10.1523/JNEUROSCI.13-09-03904.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]