

Abstract
The gene defect in Huntington’s disease (HD) may result in an impairment of energy metabolism. Malonate and 3-nitropropionic acid (3-NP) are inhibitors of succinate dehydrogenase that produce energy depletion and lesions that closely resemble those of HD. Oral supplementation with creatine or cyclocreatine, which are substrates for the enzyme creatine kinase, may increase phosphocreatine (PCr) or phosphocyclocreatine (PCCr) levels and ATP generation and thereby may exert neuroprotective effects. We found that oral supplementation with either creatine or cyclocreatine produced significant protection against malonate lesions, and that creatine but not cyclocreatine supplementation significantly protected against 3-NP neurotoxicity. Creatine and cyclocreatine increased brain concentrations of PCr and PCCr, respectively, and creatine protected against depletions of PCr and ATP produced by 3-NP. Creatine supplementation protected against 3-NP induced increases in striatal lactate concentrations in vivo as assessed by 1H magnetic resonance spectroscopy. Creatine and cyclocreatine protected against malonate-induced increases in the conversion of salicylate to 2,3- and 2,5-dihydroxybenzoic acid, biochemical markers of hydroxyl radical generation. Creatine administration protected against 3-NP-induced increases in 3-nitrotyrosine concentrations, a marker of peroxynitrite-mediated oxidative injury. Oral supplementation with creatine or cyclocreatine results in neuroprotective effects in vivo, which may represent a novel therapeutic strategy for HD and other neurodegenerative diseases.
Keywords: creatine, ATP, oxidative injury, 3-nitrotyrosine, 3-nitropropionic acid, Huntington
There is substantial evidence that impairment of energy production may play a role in the pathogenesis of neurodegenerative diseases (Albin and Greenamyre, 1992; Beal, 1992). Impaired energy production may lead to activation of excitatory amino acid receptors, increases in intracellular calcium, and the generation of free radicals (Beal, 1995). In Huntington’s disease (HD) there is reduced mitochondrial complex II–III activity in postmortem tissue (Gu et al., 1996; Browne et al., 1997) and increased cerebral lactate concentrations in vivo (Jenkins et al., 1993). Animal models of Huntington’s disease involve defects in energy production. Malonate and 3-nitropropionic acid are, respectively, reversible and irreversible inhibitors of mitochondrial complex II (succinate dehydrogenase), which produce striatal lesions similar to those of HD (Beal et al., 1993b; Greene et al., 1993; Brouillet et al., 1995). The pathogenesis of lesions produced by these compounds involves energy depletion, followed by activation of excitatory amino acid receptors and free radical production (Schulz et al., 1995a,b). The ensuing cell death involves both apoptosis and necrosis (Pang and Geddes, 1997; Sato et al., 1997).
Creatine kinase (CK) is a key enzyme involved in regulating energy metabolism in cells with intermittently high and fluctuating energy requirements, including the brain (Chen et al., 1995). The enzyme catalyzes the reversible transfer of the phosphoryl group from phosphocreatine (PCr) to ADP to generate ATP (for review, see Wallimann, 1992). Several cytoplasmic and mitochondrial isoforms have been identified, which along with the substrates creatine and PCr constitute an intricate cellular energy buffering and transport system, connecting sites of energy production with sites of energy consumption (Hemmer and Wallimann, 1993).
The mitochondrial isoform of creatine kinase (Mi-CK) is located at contact sites between the inner and outer membranes, where it is associated with porin (Wallimann et al., 1992; Brdiczka et al., 1994). Mi-CK can convert intramitochondrially produced ATP to PCr directly, which then gets transported to sites of energy consumption. The mitochondrial isoform is also coupled to oxidative phosphorylation via the adenine nucleotide translocator, and a functional coupling between the isoenzyme and porin has been postulated (Hemmer and Wallimann, 1993). A complex between porin and the adenine nucleotide translocator appears to play a role in the mitochondrial permeability transition, which is linked to both apoptotic and necrotic cell death (Beutner et al., 1997).
If energy impairment plays a critical role in the aforementioned animal models of Huntington’s disease, then compounds that increase the cerebral energy reserve might be neuroprotective. Both creatine and cyclocreatine are substrates for mitochondrial creatine kinase and have been shown to modulate rates of ATP production (Boehm et al., 1996). Cyclocreatine is the most kinetically active analog of creatine in the CK reaction, leading to formation of phosphocyclocreatine (PCCr). We therefore examined whether oral administration of either creatine or cyclocreatine could exert neuroprotective effects against malonate and 3-nitropropionic acid neurotoxicity. We also examined the ability of creatine and cyclocreatine to buffer malonate-induced ATP depletions and whether neuroprotective effects correlated with reduced free radical generation.
MATERIALS AND METHODS
We initially examined whether oral administration of creatine or its analog cyclocreatine could attenuate malonate lesions. Male Sprague Dawley rats (Charles River, Cambridge, MA) weighing 300–325 gm were used. Malonate, 3-nitrotyrosine, creatine, and 2,3- and 2,5-dihydroxybenzoic acid (DHBA) were obtained from Sigma (St. Louis, MO). Cyclocreatine was supplied by the Avicena Group and was synthesized as reported previously (Roberts and Walker, 1982). Creatine was administered orally to rats in their feed at doses of 0.25–3% in the diet. Cyclocreatine was administered at 0.25–1.0%. Controls received unsupplemented but otherwise identical diets. The compounds were administered for 2 weeks before intrastriatal administration of malonate and then for a further week before sacrifice. Malonate was dissolved in distilled, deionized water, and the pH was adjusted to 7.4 with 0.1 m HCl. Intrastriatal injections of 1.5 μl of malonate containing 3 μmol were made with a 10 μl Hamilton syringe fitted with a 26 gauge blunt-tipped needle, into the left striatum at the level of the bregma, 2.4 mm lateral to the midline, and 4.5 mm ventral to the dura as described previously. Animals were killed at 7 d after injection, and the brains were quickly removed and placed in ice-cold 0.9% saline solution. Brains were sectioned at 2 mm intervals. Slices were then placed posterior side down in 2% 2,3,5-triphenyltetrazolium chloride (TTC). Slices were stained in the dark at room temperature for 30 min and then removed and placed in 4% paraformaldehyde, pH 7.3. Lesions, noted by pale staining, were evaluated on the posterior surface of each section using a Bioquant 4 system by an experienced histologist blinded to experimental conditions. These measurements have been validated by comparing them with measurements obtained on adjacent Nissl-stained sections (Schulz et al., 1995a).
Creatine or cyclocreatine was administered orally at a dose of 1% in feed to animals treated with 3-nitropropionic acid (3-NP). Controls received unsupplemented rat chow. 3-NP was diluted in water and adjusted to pH 7.4 with NaOH and administered at a dose of 10 mg/kg intraperitoneally every 12 hr for 9–11 d. Animals became acutely ill after 9–11 d. Because of variability in the times at which animals became ill, they were examined clinically 3 hr after the injections, and when an animal was acutely ill one animal from each group was killed, regardless of whether it was on a control diet or a creatine-supplemented diet (Schulz et al., 1995a). Nine to 10 animals were examined in each group. Animals were killed after showing acute illness, and striatal lesion volume was assessed by TTC staining as described above. Four additional animals that received 1% creatine and four controls were perfused with 4% buffered paraformaldehyde. They were then cryoprotected, and brains were sectioned at 50 μm intervals on a freezing microtome. Sections were double-stained using a combined technique for NADPH histochemistry with Nissl (Schulz et al., 1995a).
We investigated the effects of oral supplementation with 1% creatine for 2 weeks on increases in striatal lactate concentrations producedin vivo by 3-nitropropionic acid. Male Sprague Dawley rats weighing 250–300 gm were administered 3-nitropropionic acid at a dose of 33 mg/kg intravenously and were imaged 2.5–3.0 hr later. Seven control and six creatine-fed animals were examined. Nuclear magnetic resonance (NMR) imaging experiments were performed at 4.7 tesla on a General Electric Omega chemical shift imager (CSI) with a 35 mm bird cage. Lactate signal intensity was measured using a rapid two-dimensional (w,y) water-suppressed CSI sequence with a preparatory inversion pulse for lipid suppression based on the difference in T1 values between brain metabolites and lipids with repetition time of 2200 msec, inversion time of 208 msec, echo time of 272 msec, NA of 8, and 16 phase-encode steps (Jenkins et al., 1996). This sequence allows spatial localization in one dimension of the important brain metabolites lactate, N-acetylaspartate (NAA), glutamate, glutamine, creatine, and choline. Lactate measurements from the acute lesions and NAA signal intensity were measured by extracting spectra from the two-dimensional files corresponding to the lines that project along the superior–inferior axis in the rat brain from coronal slices. Because NAA is unaltered during the first few hours after 3-NP lesioning, lactate signal intensity of the lines projected through the striatum was measured and normalized to the NAA signal intensity in the unaffected regions of the brain by direct integration of the extracted spectra. The NAA concentration was assumed to be 7 mm. The effects of feeding with 1% cyclocreatine on brain PCr and ATP levels were examined using 31P magnetic resonance spectroscopy at baseline and 1, 1.5, 2, and 6 weeks compared with normal controls (Koroshetz et al., 1997).
To investigate the effects of 3-NP on energy metabolites further, we administered 1% creatine in the diet and 1% cyclocreatine compared with a normal diet. In the group fed with creatine we also examined the effect of 3-NP compared with a control diet. Eight to 10 animals were examined in each group. Animals were treated with 3-NP at a dose of 10 mg/kg intraperitoneally every 12 hr until they developed symptoms, and then they were killed 2 hr later by the freeze-clamp technique in pairs (either a control or a creatine-fed animal depending on which initially became ill). Creatine, lactate, PCr, inosine monophosphate (IMP), AMP, nicotinamide adenine dinucleotide (NAD), GDP, ADP, and ATP were measured using a modification of published procedures (Bernocchi et al., 1994).
The apparatus consisted of a Perkin-Elmer (Norwalk, CT) gradient HPLC pump, a Waters Associates (Milford, MA) 409 multiple UV wavelength detector, and a Shimadzu 501 integrator. Frozen striatal tissue was dissected on a freezing cold plate (−20°C) and placed in 0.4m perchloric acid (10 μl/mg wet weight), homogenized, and centrifuged. The supernatant was neutralized with 25 μl of 2m K2CO3 added to 200 μl of the supernatant and recentrifuged. Supernatants were then stored at −80°C until injected. Standards were prepared in 0.4 mperchloric acid at concentrations of 10 μm for creatine, cyclocreatine, PCr, and IMP, 5 μm for NAD, ADP, and ATP, and 200 μm for lactate (based on tissue concentrations). Samples were separated on a 15 cm 3 μm Nikko Bioscience C18 HPLC column (ESA, Inc., Chelmsford, MA) at a flow rate of 1 ml/min using a gradient. Buffer A was 25 mmNa2HPO4 with 100 mg/l tetrabutylammonium (TBA), pH 5.5, whereas buffer B was 200 mmNaH2PO4 with 100 mg/l TBA, pH 4.0, and 10% acetonitrile. The gradient was 100% buffer A for 0–5 min, 100% buffer A to 100% buffer B for 5–25 min, and 100% buffer B for 25–34 min. Samples were monitored at 214 nm for 0–9 min, 260 nm for 9–25 min, and 214 nm for 25–34 min. The retention times were (in min): creatine, −1.8; cyclocreatine, −2.0; lactate, −3.1; PCr (PCCr), −5.4; IMP, −13.8; GDP, −17.2; AMP, −18.4; NAD, −19.0; ADP–GTP, −21.0; and ATP, −23.4. The ADP and GTP peaks did not resolve, and the combined peak was calculated using the ADP standard. All standards were linear over a 100-fold concentration range.
The salicylate hydroxyl radical-trapping method was used for measuring levels of hydroxyl radicals in striatal tissue after malonate injections. Eight animals in each group were fed either a normal diet, a 1% creatine-supplemented diet, or a 1% cyclocreatine-supplemented diet for 2 weeks before intrastriatal malonate, as described above. Animals were injected with 200 mg/kg salicylate intraperitoneally just before the malonate injections and were killed 1 hr later. The striata were then dissected rapidly from a 2-mm-thick slice and placed in 0.25 ml of chilled 0.1 m perchloric acid. Samples were subsequently sonicated, frozen rapidly and thawed, and centrifuged twice. An aliquot of supernatant was analyzed by HPLC with 16-electrode electrochemical detection (Beal et al., 1990). Salicylate, 2,3- and 2,5-DHBA, tyrosine, and 3-nitrotyrosine were measured electrochemically by oxidation at 840, 240, 120, 600, and 840 mV, respectively, with retention times of 20.5, 9.4, 6.3, 10.5, and 18.2 min, respectively. The data were expressed as the ratio of 2,3- and 2,5-DHBA to salicylate to normalize the DHBA concentrations for differing brain concentrations of salicylate. Similarly, 3-nitrotyrosine levels were normalized to tyrosine levels. We also examined the effects of 1% creatine supplementation for 2 weeks on 3-NP-induced increases in 3-nitrotyrosine levels. Male Sprague Dawley rats were treated with 3-NP at a dose of 20 mg/kg intraperitoneally and then killed at 3 hr. Ten animals were examined in each group. The striata were dissected and placed in chilled 0.1 m perchloric acid. 3-Nitrotyrosine and tyrosine concentrations were measured by HPLC with electrochemical detection as above.
Statistical comparisons were made by unpaired Student’s ttest or by one-way ANOVA followed by Fisher’s protected least significant difference test for post hoc comparisons.
RESULTS
In initial pilot experiments we found that oral supplementation with both creatine and cyclocreatine protected against striatal malonate lesions. We then examined a dose–response curve for neuroprotection of both creatine and cyclocreatine against malonate-induced striatal lesions. As shown in Figure1, increasing doses of creatine from 0.25 to 3% administered for 2 weeks in the diet exerted dose-dependent neuroprotective effects against malonate-induced striatal lesions. Significant protection occurred with doses of 1 and 2% in the diet. There was less protection at 3% creatine, suggesting that a U-shaped dose response may occur with higher doses. Administration of cyclocreatine resulted in dose-dependent neuroprotective effects, which were significant at a dose of 1% cyclocreatine.
Fig. 1.

Effects of increasing doses of creatine (CRT) and cyclocreatine (Cyclo) on striatal lesions produced by 3 μmol of malonate. *p < 0.05; **p < 0.01 (ANOVA). Eight to 10 animals were used in each group.
We examined the effects of both creatine and cyclocreatine on subacute 3-NP neurotoxicity. Dietary supplementation with 1% creatine for 2 weeks resulted in a significant 83% reduction in lesion volume produced by 3-NP (Fig. 2). In contrast, animals treated with cyclocreatine became ill earlier and appeared to have an exacerbation of toxicity (data not shown). This striking neuroprotection was histopathologically confirmed using Nissl stains (Fig. 3). Lesions could not be detected in the creatine-fed animals, whereas the mean lesion volumes in the 3-NP-treated animals on control diets were 19.7 ± 5.1 mm3.
Fig. 2.

Effects of 1% creatine on lesions produced by subacute systemic administration of 3-NP. ***p < 0.001. Ten animals were used in each group.
Fig. 3.
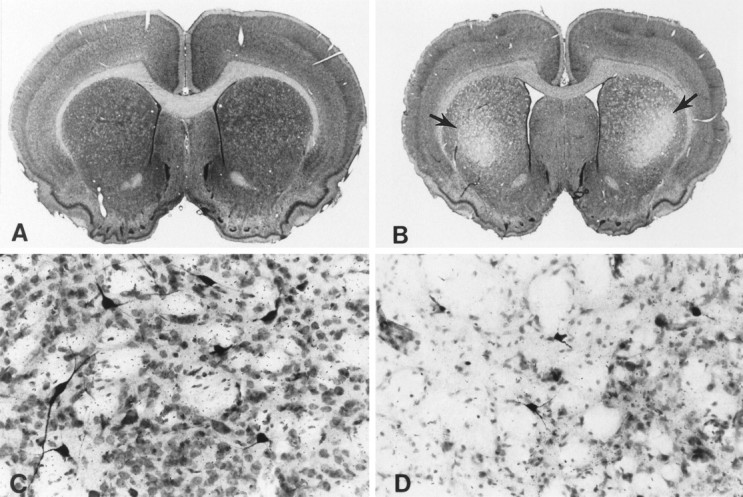
Nissl and NADPH–diaphorase double-stained coronal brain sections at the level of the striatum and anterior commissure from a creatine-fed rat (A, C) and a non-creatine-fed rat (B, D). Rats prefed 1% creatine with subsequent administration of 3-nitropropionic acid resulted in no demonstrable striatal lesions (A) or neuronal loss or alteration (C). In contrast, treatment with 3-nitroproprionic acid alone resulted in bilateral striatal lesions within the caudate–putamen (arrows) (B) with marked neuronal loss and relative preservation of NADPH-diaphorase neurons (dark-stained neurons) (D). Note the ventricular enlargement attributable to striatal atrophy in the 3-nitroproprionic acid-treated rat.
We investigated the mechanism of neuroprotection of creatine against malonate and 3-NP-induced neurotoxicity. We examined whether creatine can increase brain energy reserves and can prevent depletions of high-energy phosphate compounds induced by 3-NP. We measured creatine, lactate, PCr, GDP, AMP, NAD, ADP–GTP, and ATP levels after oral administration of 1% creatine (column 2) or 1% cyclocreatine (column 3) for 2 weeks compared with an unsupplemented diet (control) (Table1). The ADP–GTP peak did not resolve the two compounds and therefore reflects a combination of both. Creatine administration produced a significant increase in striatal PCr levels. Cyclocreatine resulted in significant reductions in creatine, which has been noted previously, presumably because of its own buildup in the brain. Cyclocreatine was phosphorylated efficiently by creatine kinase to yield PCCr, which builds up to levels almost 20-fold greater than PCr levels. Administration of 3-NP in controls (column 4) produced a significant decrease in striatal concentrations of creatine, PCr, GDP, AMP, NAD, ADP–GTP, and ATP, whereas there was a significant increase in lactate. The reductions in Cr, PCr, GDP, AMP, NAD, ADP–GTP, and ATP and the increase in lactate were attenuated significantly in animals fed for 2 weeks with 1% creatine (column 5).
Table 1.
Striatal energy metabolites in creatine- and cyclocreatine-fed animals (μmol/gm wet weight)
| Control | Creatine | Cyclocreatine | Control with 3-NP | Creatine with 3-NP | |
|---|---|---|---|---|---|
| Creatine | 9.07 ± 0.52 | 9.68 ± 0.43 | 5.60 ± 0.09* | 4.91 ± 0.76* | 7.88 ± 0.61** |
| Lactate | 3.08 ± 0.51 | 2.72 ± 0.33 | 3.40 ± 0.62 | 14.05 ± 3.02* | 8.75 ± 2.14*** |
| PCr, PCCr1-a | 1.64 ± 0.20 | 2.70 ± 0.29* | 53.5 ± 2.5* | 0.62 ± 0.16**** | 1.14 ± 0.17*** |
| GDP | 0.23 ± 0.02 | 0.24 ± 0.02 | 0.21 ± 0.01 | 0.07 ± 0.01* | 0.17 ± 0.02** |
| AMP | 0.91 ± 0.08 | 0.96 ± 0.06 | 0.79 ± 0.02 | 0.35 ± 0.06* | 0.65 ± 0.12***** |
| NAD | 1.06 ± 0.07 | 0.97 ± 0.05 | 0.91 ± 0.03 | 0.41 ± 0.09* | 1.01 ± 0.14** |
| ADP–GTP | 0.71 ± 0.03 | 0.79 ± 0.04 | 0.77 ± 0.02 | 0.23 ± 0.05* | 0.64 ± 0.07** |
| ATP | 1.23 ± 0.18 | 1.38 ± 0.17 | 1.50 ± 0.10 | 0.31 ± 0.10* | 0.99 ± 0.19***** |
n = 8–10 animals per group.
Values are PCr, except for after feeding with cyclocreatine, when they are PCCr.
*P < 0.001; ****P < 0.01 compared with control.
**P < 0.001; ***P < 0.05; *****P < 0.01 compared with control with 3-NP.
We also investigated the effects of creatine feeding on increases in striatal lactate concentrations induced by 3-NP in vivousing 1H magnetic resonance spectroscopy. Animals were fed with 1% creatine for 2 weeks before intravenous administration of 3-NP at a dose of 33 mg/kg. We adopted this route of administration because it resulted in more reproducible increases in lactate. As shown in Figure 4, there was a significant reduction in lactate/NAA ratios in animals fed with creatine. Representative chemical shift spectra are shown in Figure5. The effects of feeding 1% cyclocreatine on PCr and ATP levels were assessed by phosphorus magnetic resonance spectroscopy. Relative concentrations were calculated compared with external standards. We examined six animals fed with cyclocreatine and nine controls. Animals fed with cyclocreatine showed increases in ATP levels from 0.076 ± 0.009 at baseline to 0.115 ± 0.098 at 2–6 weeks (p < 0.05) and increases in PCr and PCCr from 0.057 ± .008 at baseline to 0.137 ± 0.098 at 2–6 weeks (p < 0.05).
Fig. 4.

Striatal lactate/NAA ratios in seven control and six creatine-fed animals 2.5–3.0 hr after intravenous administration of 3-NP. *p < 0.05.
Fig. 5.

Representative proton chemical shift spectra of a control and a creatine-fed rat after intravenous administration of 3-NP. Lactate is reduced, and creatine is increased in the creatine-fed rat.
A consequence of energy impairment produced by either malonate or 3-NP is the generation of free radicals, which appear to play a role in cell death, because both free radical scavengers and nitric oxide synthase inhibitors can attenuate the toxicity produced by either compound significantly (Greenamyre et al., 1994; Schulz et al., 1995a,b, 1996). In the present experiments we investigated the effects of oral administration of either 1% creatine or 1% cyclocreatine for 2 weeks on malonate-induced increases in DHBA/salicylate and 3-nitrotyrosine/tyrosine. Animals were killed 1 hr after the malonate injection. As shown in Figure 6, malonate produced significant increases in both 2,3- and 2,5-DHBA/salicylate, which were significantly attenuated in animals fed with either creatine or cyclocreatine. A significant increase in 3-nitrotyrosine/tyrosine was found in control animals, but smaller increases in 3-nitrotyrosine/tyrosine in the creatine- and cyclocreatine-fed animals did not reach significance. We also examined the effects of 1% creatine feeding for 2 weeks on increases in 3-nitrotyrosine/tyrosine levels produced by 20 mg/kg 3-NP at 3 hr after administration. As shown in Figure 7, increases in 3-nitrotyrosine/tyrosine were significantly decreased in the creatine-fed animals.
Fig. 6.

Production of 2,3- and 2,5-DHBA and 3-nitrotyrosine after intrastriatal injection of malonate in controls (Cont), animals fed with creatine, and those fed with cyclocreatine (Cyclo). **p < 0.01 compared with uninjected side; #p < 0.05 compared with DHBA elevation in controls. Eight animals were used in each group.
Fig. 7.

3-Nitrotyrosine levels after systemic administration of 3-NP to controls and creatine-fed animals. **p < 0.01 compared with saline-injected controls;##p < 0.01 compared with 3-NP-injected animals on a normal diet. Ten animals were used in each group.
DISCUSSION
There is substantial evidence that a secondary consequence of the gene defect in HD may be an impairment of energy metabolism. HD patients show weight loss despite a normal or increased caloric intake (O’Brien et al., 1990). Furthermore, we showed increased lactate concentrations in both the striatum and cerebral cortex of HD patients, as assessed using 1H magnetic resonance spectroscopy (Jenkins et al., 1993). We recently found a reduced PCr/Pi ratio in resting gastrocnemius muscle of HD patients (Koroshetz et al., 1997). In postmortem brain tissue there is decreased complex II–III activity in HD basal ganglia (Gu et al., 1996; Browne et al., 1997).
We and others found that intrastriatal injections of the reversible succinate dehydrogenase inhibitor malonate or systemic administration of the irreversible succinate dehydrogenase inhibitor 3-nitropropionic acid results in striatal lesions that closely mimic HD neuropathology (Beal et al., 1993a,b; Greene et al., 1993; Brouillet et al., 1995). There is sparing of striatal afferents and striatal NADPH–diaphorase interneurons, with a depletion of striatal projection neurons. In primates systemic administration of 3-NP produces the characteristic histopathological features of HD, as well as both a choreiform movement disorders and frontal-type cognitive deficits, which are typical clinical manifestations of HD (Brouillet et al., 1995; Palfi et al., 1996).
Both malonate and 3-NP produce energy defects in vivo, followed by activation of excitatory amino acid receptors and the generation of free radicals (Beal et al., 1993a,b; Greene et al., 1993;Greenamyre et al., 1994; Schulz et al., 1995a,b). If the initiating step in the pathological cascade is a depletion of cellular energy stores, then agents that can buffer cellular energy stores may be neuroprotective. The brain isoform of creatine kinase, along with the mitochondrial isoform and the substances Cr and PCr, constitute a system that seems to be critical in regulating energy homeostasis in the brain and other organs with high and fluctuating energy demands (Hemmer and Wallimann, 1993). The mitochondrial isoform is part of a complex of proteins that form an efficient, tightly coupled multienzyme energy channel, which generates and transports energy in the form of PCr, from the mitochondrial matrix to the cytoplasm. Creatine is an excellent stimulant for mitochondrial respiration, resulting in the generation of PCr (Kernec et al., 1996; O’Gorman et al., 1996).
Substantial evidence supports a direct functional coupling of creatine kinase with Na+/K+ ATPase, neurotransmitter release, and in maintenance of membrane potentials and restoration of ion gradients before and after depolarization (Dunant et al., 1988; Hemmer and Wallimann, 1993). High-energy turnover and high creatine kinase concentrations have been found in those regions of the brain that are rich in synaptic connections, e.g., molecular layer of the cerebellum, glomerular structures of the granule layer, and the hippocampus (Kaldis et al., 1996). An important role of creatine kinase in the adult brain is supported by in vivo31P NMR transfer measurements showing the pseudo-first-order rate constant of the creatine kinase reaction (in the direction of ATP synthesis) (Corbett and Laptook, 1994). Creatine kinase flux correlates with brain activity, as measured by EEG, as well as the amount of 2-deoxyglucose phosphate found in the brain (Sauter and Rudin, 1993).
A novel strategy to improve brain energy stores is therefore to administer either creatine or an analog such as cyclocreatine, which leads to high-energy phosphagens and which potentially could sustain ATP production by the creatine kinase reaction. Previous studies in both the heart and skeletal muscle showed that cyclocreatine administration resulted in increased tissue levels of cyclocreatine and phosphocyclocreatine, delayed depletion of ATP levels under ischemic conditions, and delayed onset of ischemia-induced rigor (Griffiths and Walker, 1976; Annesley and Walker, 1980; Roberts and Walker, 1982; Turner and Walker, 1987; Elgebaly et al., 1994). Creatine had no effect on ischemia-induced ATP depletion in the heart, but it did protect against ATP depletion produced by arterial hypertension (Turner and Walker, 1985; Osbakken et al., 1992; Constantin-Teodosiu et al., 1995). In hippocampal slices creatine supplementation increased PCr levels, delayed synaptic failure, and ameliorated neuronal damage produced by anoxia (Whittingham and Lipton, 1981; Carter et al., 1995). Cyclocreatine administration in vivo increased brain concentrations of PCCr and appeared to buffer ATP stores (Woznicki and Walker, 1980).
In the present study we found that oral administration of both creatine and cyclocreatine produced dose-dependent neuroprotective effects against malonate lesions. The protection with creatine was diminished at the highest dose level, suggesting that there may be a U-shaped dose response at higher doses. We observed similar effects with MPTP-induced dopamine depletions (M. F. Beal et al., unpublished data). The best neuroprotection was that seen after subacute administration of 3-NP. A significant 83% reduction in lesion volume was observed in animals fed 1% creatine.
The mechanism of neuroprotection involves protection against depletions of both PCr and ATP. We found that administration of either creatine or cyclocreatine results in increased brain concentrations of PCr or PCCr, respectively. There was a trend toward increased ATP, as determined biochemically and by magnetic resonance spectroscopy. Increases in ATP levels are somewhat unexpected, because brain ATP levels are thought to be regulated tightly (Erecinska and Silver, 1989). Administration of 3-NP produced significant decreases in creatine, PCr, GDP, AMP, NAD, ADP–GTP, and ATP. These energy metabolites are also decreased by cerebral ischemia (Lazzarino et al., 1992) and by 3-NP in vitro (Erecinska and Nelson, 1994) and in vivo(Brouillet et al., 1993; Tsai et al., 1997). Creatine administration significantly protected against the decreases. Furthermore, creatine administration protected against 3-NP-induced increases of lactate, as assessed by 1H magnetic resonance spectroscopy in vivo. These findings therefore provide the first in vivo data that creatine administration can increase brain high-energy phosphate compounds and can protect against energy compromise produced by mitochondrial toxins. Decreases in NAD and ATP may be a consequence of both impaired mitochondrial function as well as activation of poly-ADP-ribose polymerase, which plays a role in neuronal cell death in vitro (Zhang et al., 1994). The ability of PCr to stimulate synaptic glutamate uptake and thereby to reduce extracellular glutamate may also play a role in the neuroprotective effects of creatine and cyclocreatine (Xu et al., 1996).
A secondary consequence of energy impairment is increased intracellular concentrations of calcium attributable to both impaired mitochondrial calcium buffering and activation of voltage-dependent NMDA excitatory amino acid receptors (Beal, 1992, 1995). This leads to increased free radical production by mitochondria as well as activation of neuronal nitric oxide synthase, which is calmodulin-dependent. This can lead to the generation of peroxynitrite, formed by the interaction of O⋅2 with NO⋅. Peroxynitrite can oxidize a intracellular molecules by a “hydroxyl radical”-like activity, and it also can lead to nitration of tyrosines (Beckman et al., 1990, 1992). We showed previously that both malonate and 3-NP result in increases in both hydroxyl radical activity, as well as 3-nitrotyrosine (Schulz et al., 1995a,b). Furthermore, both free radical scavengers and nitric oxide synthase inhibitors can attenuate both malonate and 3-NP neurotoxicity (Greenamyre et al., 1994; Schulz et al., 1995a,b, 1996).
Creatine kinase appears to be coupled directly or indirectly to energetic processes required for calcium homeostasis (Wallimann et al., 1992; Steeghs et al., 1997). Creatine pretreatment delayed increases in intracellular calcium produced by 3-NP in cortical and striatal astrocytes in vitro (Deshpande et al., 1997). Administration of creatine or cyclocreatine therefore may improve intracellular calcium buffering and may prevent free radical production by mitochondria. In the present experiments we found that administration of both creatine and cyclocreatine significantly attenuated malonate-induced increases in 2,3- and 2,5-DHBA/salicylate, markers of hydroxyl radical generation (Floyd et al., 1984). Increased 3-nitrotyrosine levels after malonate were also attenuated. Oral administration of creatine reduced 3-NP-induced increases in 3-nitrotyrosine significantly. These findings therefore suggest that improved energy buffering can act upstream to attenuate free radical generation, which is associated with cell death produced by mitochondrial toxins.
The present studies demonstrate that oral administration of either creatine or cyclocreatine can buffer cellular ATP concentrations and can attenuate cell death in animal models that mimic the neuropathological and clinical phenotype of HD. By attenuating ATP depletion these compounds appear to prevent a pathological cascade, which leads to free radical generation and eventual cell death. Creatine administration is well tolerated in man and may have benefits in pathological conditions (Balsom et al., 1994; Dawson et al., 1995). Long-term administration to patients with gyrate atrophy of the choroid and retina prevented visual field constriction and resulted in improvement of muscle biopsy findings (Sipila et al., 1981). A pediatric patient with creatine deficiency in brain accompanied by an extrapyramidal movement disorder showed partial restoration of cerebral creatine concentrations and clinical improvement with oral creatine administration (Stockler et al., 1994, 1996). Creatine administration resulted in improvement in a patient with the mitochondrial disorder mitochondrial encephalopathy lactic acidosis and strokes (Hagenfeldt et al., 1994). The present findings suggest that treatment with creatine or its analogs might be a novel therapeutic strategy to slow or halt neurodegeneration in HD. Similar strategies might be effective in other neurodegenerative diseases in which defects in energy metabolism are implicated (Beal, 1992).
Footnotes
This work was supported by National Institutes of Health Grants NS35255, NS37102 (R.J.F.), NS32365, NS31579, NS16367, and NS35255 (M.F.B.). The secretarial assistance of Sharon Melanson is gratefully acknowledged.
Correspondence should be addressed to Dr. M. Flint Beal, Neurology Service, WRN 408, Massachusetts General Hospital, 32 Fruit Street, Boston, MA 02114.
REFERENCES
- 1.Albin RL, Greenamyre JT. Alternative excitotoxic hypotheses. Neurology. 1992;42:733. doi: 10.1212/wnl.42.4.733. [DOI] [PubMed] [Google Scholar]
- 2.Annesley TM, Walker JB. Energy metabolism of skeletal muscle containing cyclocreatine phosphate. J Biol Chem. 1980;255:3924–3930. [PubMed] [Google Scholar]
- 3.Balsom PD, Soderlund K, Ekblom B. Creatine in humans with special reference to creatine supplementation. Sports Med. 1994;18:268–280. doi: 10.2165/00007256-199418040-00005. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119–130. doi: 10.1002/ana.410310202. [DOI] [PubMed] [Google Scholar]
- 5.Beal MF. Aging, energy and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 6.Beal MF, Matson WR, Swartz KJ, Gamache PH, Bird ED. Kynurenine pathway measurements in Huntington’s disease striatum: evidence for reduced formation of kynurenic acid. J Neurochem. 1990;55:1327–1339. doi: 10.1111/j.1471-4159.1990.tb03143.x. [DOI] [PubMed] [Google Scholar]
- 7.Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman B. Neurochemical and histologic characterization of excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993a;13:4181–4191. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beal MF, Brouillet E, Jenkins B, Henshaw R, Rosen B, Hyman BT. Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J Neurochem. 1993b;61:1147–1150. doi: 10.1111/j.1471-4159.1993.tb03633.x. [DOI] [PubMed] [Google Scholar]
- 9.Beckman JS, Beckman TW, Chen J, marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beckman JS, Ischiropoulos H, Zhu L, van der Woerd M, Smith C, Chen J, Harrison J, Martin JC, Tsai M. Kinetics of superoxide dismutase- and iron-catalyzed nitration of phenolics by peroxynitrite. Arch Biochem Biophys. 1992;298:438–445. doi: 10.1016/0003-9861(92)90432-v. [DOI] [PubMed] [Google Scholar]
- 11.Bernocchi P, Ceconi C, Cargnoni A, Pedersini P, Curello S, Ferrari R. Extraction and assay of creatine phosphate, purine, and pyridine nucleotides in cardiac tissue by reversed-phase high-performance liquid chromatograph. Anal Biochem. 1994;222:374–379. doi: 10.1006/abio.1994.1505. [DOI] [PubMed] [Google Scholar]
- 12.Beutner G, Ruck A, Riede B, Brdiczka D. Complexes between hexokinase, mitochondrial porin and adenylate translocator in brain: regulation of hexokinase, oxidative phosphorylation and permeability transition pore. Biochem Soc Trans. 1997;25:151–157. doi: 10.1042/bst0250151. [DOI] [PubMed] [Google Scholar]
- 13.Boehm EA, Radda GK, Tomlin H, Clark JF. The utilization of creatine and its analogues by cytosolic and mitochondrial creatine kinase. Biochim Biophys Acta. 1996;1274:119–128. doi: 10.1016/0005-2728(96)00018-7. [DOI] [PubMed] [Google Scholar]
- 14.Brdiczka D, Kaldis P, Wallimann T. In vitro complex formation between the octamer of mitochondrial creatine kinase and porin. J Biol Chem. 1994;269:27640–27644. [PubMed] [Google Scholar]
- 15.Brouillet E, Jenkins BG, Hyman BT, Ferrante RJ, Kowall NW, Srivastava R, Roy DS, Rosen BR, Beal MF. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochem. 1993;60:356–359. doi: 10.1111/j.1471-4159.1993.tb05859.x. [DOI] [PubMed] [Google Scholar]
- 16.Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci USA. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MMK, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–651. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 18.Carter AJ, Muller RE, Pschorn U, Stransky W. Preincubation with creatine enhances levels of creatine phosphate and prevents anoxic damage in rat hippocampal slices. J Neurochem. 1995;64:2691–2699. doi: 10.1046/j.1471-4159.1995.64062691.x. [DOI] [PubMed] [Google Scholar]
- 19.Chen L, Roberts R, Friedman DL. Expression of brain-type creatine kinase and ubiquitous mitochondrial creatine kinase in the fetal rat brain: evidence for a nuclear energy shuttle. J Comp Neurol. 1995;363:389–401. doi: 10.1002/cne.903630305. [DOI] [PubMed] [Google Scholar]
- 20.Constantin-Teodosiu D, Greenhaff PL, Gardiner SM, Randall MD, March JE, Bennett T. Attenuation by creatine of myocardial metabolic stress in Brattleboro rats caused by chronic inhibition of nitric oxide synthase. Br J Pharmacol. 1995;116:3288–3292. doi: 10.1111/j.1476-5381.1995.tb15137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corbett RJT, Laptook AR. Age-related changes in swine brain creatine kinase-catalyzed 31P exchange measured in vivo using 31P NMR magnetization transfer. J Cereb Blood Flow Metab. 1994;14:1070–1077. doi: 10.1038/jcbfm.1994.140. [DOI] [PubMed] [Google Scholar]
- 22.Dawson B, Cutler M, Moody A, Lawrence S, Goodman C, Randall N. Effects of oral creatine loading on single and repeated maximal short sprints. Aust J Sci Med Sport. 1995;27:56–61. [PubMed] [Google Scholar]
- 23.Deshpande SB, Fukuda A, Nishino H. 3-Nitropropionic acid increases the intracellular Ca2+ in cultured astrocytes by reverse operation of the Na+-Ca2+ exchanger. Exp Neurol. 1997;145:38–45. doi: 10.1006/exnr.1997.6457. [DOI] [PubMed] [Google Scholar]
- 24.Dunant Y, Loctin F, Marsal J, Muller D, Parducz A, Rabasseda X. Energy metabolism and quantal acetylcholine release: effects of botulinum toxin, 1-fluoro-2,4-dinitrobenzene, and diamide in the Torpedo electric organ. J Neurochem. 1988;50:431–439. doi: 10.1111/j.1471-4159.1988.tb02930.x. [DOI] [PubMed] [Google Scholar]
- 25.Elgebaly SA, Wei Z, Tyles E, Elkerm AF, Houser SL, Gillies C, Kaddurah-Daouk R. Enhancement of the recovery of rat hearts after prolonged cold storage by cyclocreatine phosphate. Transplantation. 1994;57:803–806. doi: 10.1097/00007890-199403270-00005. [DOI] [PubMed] [Google Scholar]
- 26.Erecinska M, Nelson D. Effects of 3-nitropropionic acid on synaptosomal energy and transmitter metabolism: relevance to neurodegenerative brain disease. J Neurochem. 1994;63:1033–1041. doi: 10.1046/j.1471-4159.1994.63031033.x. [DOI] [PubMed] [Google Scholar]
- 27.Erecinska M, Silver IA. ATP and brain function. J Cereb Blood Flow Metab. 1989;9:2–19. doi: 10.1038/jcbfm.1989.2. [DOI] [PubMed] [Google Scholar]
- 28.Floyd RA, Watson JJ, Wong PK. Sensitive assay of hydroxyl radical formation utilizing high pressure liquid chromatography with electrochemical detection of phenol and salicylate hydroxylation products. J Biochem Biophys Methods. 1984;10:221–235. doi: 10.1016/0165-022x(84)90042-3. [DOI] [PubMed] [Google Scholar]
- 29.Greenamyre JT, Garcia-Osuna M, Greene JG. The endogenous cofactors, thioctic acid and dihydrolipoic acid, are neuroprotective against NMDA and malonic acid lesions of striatum. Neurosci Lett. 1994;171:17–20. doi: 10.1016/0304-3940(94)90593-2. [DOI] [PubMed] [Google Scholar]
- 30.Greene JG, Porter RH, Eller RV, Greenamyre JT. Inhibition of succinate dehydrogenase by malonic acid produces an “excitotoxic” lesion in rat striatum. J Neurochem. 1993;61:1151–1154. doi: 10.1111/j.1471-4159.1993.tb03634.x. [DOI] [PubMed] [Google Scholar]
- 31.Griffiths GR, Walker JB. Accumulation of analog of phosphocreatine in muscle of chicks fed 1-carboxymethyl-2-iminoimidazolidine (cyclocreatine). J Biol Chem. 1976;251:2049–2054. [PubMed] [Google Scholar]
- 32.Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AHV. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 33.Hagenfeldt L, von Dobeln U, Solders G, Kaijser L (1994) Creatine treatment in MELAS. Muscle Nerve Oct:1236–1237. [PubMed]
- 34.Hemmer W, Wallimann T. Functional aspects of creatine kinase in brain. Dev Neurosci. 1993;15:249–260. doi: 10.1159/000111342. [DOI] [PubMed] [Google Scholar]
- 35.Jenkins B, Koroshetz W, Beal MF, Rosen B. Evidence for an energy metabolism defect in Huntington’s disease using localized proton spectroscopy. Neurology. 1993;43:2689–2695. doi: 10.1212/wnl.43.12.2689. [DOI] [PubMed] [Google Scholar]
- 36.Jenkins BG, Brouillet E, Chen Y-CI, Storey E, Schulz JB, Kirschner P, Beal MF, Rosen BR. Non-invasive neurochemical analysis of focal excitotoxic lesions in models of neurodegenerative illness using spectroscopic imaging. J Cereb Blood Flow Metab. 1996;16:450–461. doi: 10.1097/00004647-199605000-00011. [DOI] [PubMed] [Google Scholar]
- 37.Kaldis P, Hemmer W, Zanolla E, Holtzman D, Wallimann T. ’Hot spots’ of creatine kinase localization in brain: cerebellum, hippocampus and choroid plexus. Dev Neurosci. 1996;18:542–554. doi: 10.1159/000111452. [DOI] [PubMed] [Google Scholar]
- 38.Kernec F, Le Tallec N, Nadal L, Begue J-M, Le Rumeur E. Phosphocreatine synthesis by isolated rat skeletal muscle mitochondria is not dependent upon external ADP: a 31P NMR study. Biochem Biophys Res Commun. 1996;225:819–825. doi: 10.1006/bbrc.1996.1257. [DOI] [PubMed] [Google Scholar]
- 39.Koroshetz WJ, Jenkins BG, Rosen BR, Beal MF. Energy metabolism defects in Huntington’s disease and possible therapy with coenzyme Q10. Ann Neurol. 1997;41:160–165. doi: 10.1002/ana.410410206. [DOI] [PubMed] [Google Scholar]
- 40.Lazzarino G, Vagnozzi R, Tavazzi B, Pastore FS, Di Pierro D, Siragusa P, Belli A, Giuffre R, Giardina B. MDA, oxypurines, and nucleosides relate to reperfusion in short-term incomplete cerebral ischemia in the rat. Free Radic Biol Med. 1992;13:489–498. doi: 10.1016/0891-5849(92)90143-5. [DOI] [PubMed] [Google Scholar]
- 41.O’Brien CF, Miller C, Goldblatt D, Welle S, Forbes G, Lipinski B, Panzik J, Peck R, Plumb S, Oakes D, Kurlan R, Shoulson I. Extraneural metabolism in early Huntington’s disease. Ann Neurol. 1990;28:300–301. [Google Scholar]
- 42.O’Gorman E, Beutner G, Wallimann T, Brdiczka D. Differential effects of creatine depletion on the regulation of enzyme activities and on creatine-stimulated mitochondrial respiration in skeletal muscle, heart, and brain. Biochim Biophys Acta. 1996;1276:161–170. doi: 10.1016/0005-2728(96)00074-6. [DOI] [PubMed] [Google Scholar]
- 43.Osbakken M, Ito K, Zhang D, Ponomarenko I, Ivanics T, Jahngen EGE, Cohn M. Creatine and cyclocreatine effects on ischemic myocardium: 31P nuclear magnetic resonance evaluation of intact heart. Cardiology. 1992;80:184–195. doi: 10.1159/000175002. [DOI] [PubMed] [Google Scholar]
- 44.Palfi S, Ferrante RJ, Brouillet E, Beal MF, Dolan R, Guyoi MC, Peschanski M, Hantraye P. Chronic 3-nitropropionic acid treatment in baboons replicates the cognitive and motor deficits of Huntington’s disease. J Neurosci. 1996;16:3019–3025. doi: 10.1523/JNEUROSCI.16-09-03019.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pang Z, Geddes JW. Mechanisms of cell death induced by the mitochondrial toxin 3-nitropropionic acid: excitotoxic necrosis and delayed apoptosis. J Neurosci. 1997;17:3064–3073. doi: 10.1523/JNEUROSCI.17-09-03064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts JJ, Walker JB. Feeding a creatine analogue delays ATP depletion and onset of rigor in ischemic heart. Am J Physiol. 1982;243:H911–H916. doi: 10.1152/ajpheart.1982.243.6.H911. [DOI] [PubMed] [Google Scholar]
- 47.Sato S, Gobbel GT, Honkaniemi J, Li Y, Kondo T, Murakami K, Sato M, Copin J-C, Chan PH. Apoptosis in the striatum of rats following intraperitoneal injection of 3-nitropropionic acid. Brain Res. 1997;745:343–347. doi: 10.1016/s0006-8993(96)01231-0. [DOI] [PubMed] [Google Scholar]
- 48.Sauter A, Rudin M. Determination of creatine kinase parameters in rat brain by NMR magnetization transfer: correlation with brain function. J Biol Chem. 1993;268:13166–13171. [PubMed] [Google Scholar]
- 49.Schulz JB, Matthews RT, Muqit MMK, Browne SE, Beal MF. Inhibition of neuronal nitric oxide synthase by 7-nitroindazole protects against MPTP-induced neurotoxicity in mice. J Neurochem. 1995a;64:936–939. doi: 10.1046/j.1471-4159.1995.64020936.x. [DOI] [PubMed] [Google Scholar]
- 50.Schulz JB, Matthews RT, Jenkins BG, Ferrante RJ, Siwek D, Henshaw DR, Cipolloni PB, Mecocci P, Kowall NW, Rosen BR, Beal MF. Blockade of neuronal nitric oxide synthase protects against excitotoxicity in vivo. J Neurosci. 1995b;15:8419–8429. doi: 10.1523/JNEUROSCI.15-12-08419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schulz JB, Henshaw DR, MacGarvey U, Beal MF. Involvement of oxidative stress in 3-nitropropionic acid neurotoxicity. Neurochem Int. 1996;29:167–171. doi: 10.1016/0197-0186(95)00122-0. [DOI] [PubMed] [Google Scholar]
- 52.Sipila I, Rapola J, Simell O, Vannas A. Supplementary creatine as a treatment for gyrate atrophy of the choroid and retina. N Engl J Med. 1981;304:867–870. doi: 10.1056/NEJM198104093041503. [DOI] [PubMed] [Google Scholar]
- 53.Steeghs K, Benders A, Oerlemans F, de Haan A, Heerschap A, Ruitenbeek W, Jost C, van Deursen J, Perryman B, Pette D, Bruckwilder M, Koudijs J, Jap P, Veerkamp J, Wieringa B. Altered Ca2+ responses in muscles with combined mitochondrial and cytosolic creatine kinase deficiencies. Cell. 1997;89:93–103. doi: 10.1016/s0092-8674(00)80186-5. [DOI] [PubMed] [Google Scholar]
- 54.Stockler S, Holzbach U, Hanefeld F, Marquardt I, Helms G, Requart M, Hanicke W, Frahm J. Creatine deficiency in the brain: a new, treatable inborn error of metabolism. Pediatr Res. 1994;36:409–413. doi: 10.1203/00006450-199409000-00023. [DOI] [PubMed] [Google Scholar]
- 55.Stockler S, Hanefeld F, Frahn J. Creatine replacement therapy in quanidinoacetate methyltransferase deficiency, a novel inborn error of metabolism. Lancet. 1996;348:789–790. doi: 10.1016/s0140-6736(96)04116-5. [DOI] [PubMed] [Google Scholar]
- 56.Tsai MJ, Goh CC, Wan YL, Chang C. Metabolic alterations produced by 3-nitropropionic acid in rat striata and cultured astrocytes: quantitative in vitro 1H nuclear magnetic resonance spectroscopy and biochemical characterization. Neuroscience. 1997;79:819–826. doi: 10.1016/s0306-4522(97)00015-8. [DOI] [PubMed] [Google Scholar]
- 57.Turner DM, Walker JB. Relative abilities of phosphagens with different thermodynamic or kinetic properties to help sustain ATP and total adenylate pools in heart during ischemia. Arch Biochem Biophys. 1985;238:642–651. doi: 10.1016/0003-9861(85)90210-3. [DOI] [PubMed] [Google Scholar]
- 58.Turner DM, Walker JB. Enhanced ability of skeletal muscle containing cyclocreatine phosphate to sustain ATP levels during ischemia following β-adrenergic stimulation. J Biol Chem. 1987;262:6605–6609. [PubMed] [Google Scholar]
- 59.Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ’phosphocreatine circuit’ for cellular energy homeostasis. Biochem J. 1992;281:21–40. doi: 10.1042/bj2810021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whittingham TS, Lipton P. Cerebral synaptic transmission during anoxia is protected by creatine. J Neurochem. 1981;37:1618–1621. doi: 10.1111/j.1471-4159.1981.tb06337.x. [DOI] [PubMed] [Google Scholar]
- 61.Woznicki DT, Walker JB. Utilization of cyclocreatine phosphate, an analogue of creatine phosphate, by mouse brain during ischemia and its sparing action on brain energy reserves. J Neurochem. 1980;34:1247–1253. doi: 10.1111/j.1471-4159.1980.tb09966.x. [DOI] [PubMed] [Google Scholar]
- 62.Xu CJ, Klunk WE, Kanfer JN, Xiong Q, Miller G, Pettegrew JW. Phosphocreatine-dependent glutamate uptake by synaptic vesicles. J Biol Chem. 1996;271:13435–13440. doi: 10.1074/jbc.271.23.13435. [DOI] [PubMed] [Google Scholar]
- 63.Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]