

Abstract
Mitochondrial injury has been implicated in ischemic neuronal injury. Mitochondria, producing adenosine triphosphate by virtue of electron flow, have been shown to be both the sites of superoxide anion (O2−) production and the target of free radical attacks. We evaluated these mechanisms in an in vivo cerebral ischemia model, using mutant mice with a heterozygous knock-out gene (Sod2 −/+) encoding mitochondrial manganese superoxide dismutase (Mn-SOD).Sod2 −/+ mice demonstrated a prominent increase in O2− production under normal physiological conditions and in ischemia, as evidenced by specific oxidation of a fluorescent probe, hydroethidine, reflecting decreased activity of Mn-SOD. A mitochondrial viability assay that used rhodamine 123, which is accumulated by transmembrane potential of viable mitochondria, demonstrated accelerated development of mitochondrial injury. This rapid progress of ischemic injury resulted in exacerbation of infarct size and hemisphere enlargement, causing advanced neurological deficits but without altering DNA fragmentation induction. The present study suggests that O2−overproduced in a mitochondrial compartment, when uncoupled from antioxidant defenses, induces impairment of mitochondrial function and causes exacerbation of cerebral infarction after ischemia.
Keywords: cerebral ischemia, oxidative stress, manganese superoxide dismutase, superoxide anion, mitochondrial injury, DNA fragmentation
Oxygen radicals have been implicated in the pathogenesis of ischemic neuronal injury (Smith et al., 1984;Kontos, 1985). These oxygen radicals induce lipid peroxidation, protein oxidation, and DNA damage, causing both acute and chronic neuronal injury. Among oxygen radicals, superoxide anion (O2−) plays a key role in the oxidative chain reaction, producing a highly reactive oxidant. These free radicals are processed by antioxidant enzymes and scavengers, including superoxide dismutases (SODs), glutathione peroxidase, and catalase. With the use of transgenic mice overexpressing copper-zinc SOD (CuZn-SOD), it has been demonstrated that CuZn-SOD (SOD1), a cytosolic antioxidant isoenzyme, plays a protective role in transient focal ischemia–reperfusion and in traumatic brain injury (Kinouchi et al., 1991; Yang et al., 1994; Mikawa et al., 1996), whereas cerebral infarction after permanent focal ischemia is not reduced (Chan et al., 1993). These results suggest that CuZn-SOD plays a protective role mainly during reperfusion after ischemia. Furthermore, recent evidence indicates that CuZn-SOD, as well as Bcl-2, has an inhibitory property in the apoptotic process via an antioxidant pathway (Kane et al., 1993;Greenlund et al., 1995; Kondo et al., 1997b).
Manganese SOD (Mn-SOD, SOD2) is an isoenzyme of SOD localized to the mitochondria. The mitochondria provide ATP for cellular homeostasis, function by means of oxidative phosphorylation and electron transport, and are also the sites of O2−production in ischemia as well as under normal physiological conditions. In NMDA receptor-mediated excitotoxicity, which is an important process of ischemic neuronal injury, mitochondrial production of oxygen radicals has been demonstrated to be a key event, as well as arachidonic cascade and the nitric oxide pathway (Dugan et al., 1995). Despite the mitochondrial detoxification, overproduced oxygen radicals, once uncoupled from this defense, induce oxidative stress. Mitochondria also are suggested to be the targets of free radical attacks, which impair ATP generation that causes energy depletion and increases free radicals (Hillered and Chan, 1989; Coyle and Puttfarcken, 1993).
Knock-out mutant mice with target-disrupted Sod2 genes encoding Mn-SOD have been established (Li et al., 1995b). We have demonstrated that infarct size is greater in the knock-out mutants than in the wild-type mice after mild focal ischemia–reperfusion but is not different after moderate ischemia–reperfusion, despite early progression of ischemic injury (Mikawa et al., 1995; Chan et al., 1996;Kondo et al., 1997a). We also have demonstrated that deficiency of Mn-SOD triggers neuronal cell death via both necrosis and apoptosis after ischemia–reperfusion in the mutant knock-outs (Kondo et al., 1997a). Although mitochondria are being recognized as the targets for oxidative stress and their mode of dysfunction can affect the outcome of cell death either by apoptosis or necrosis (Ankarcrona et al., 1995), it is still unknown whether Mn-SOD is neuroprotective in permanent ischemia without restoration of cerebral blood flow supplying oxygen as a substrate for oxygen radicals.
The present study was designed to investigate the role of Mn-SOD and mitochondrial injury in ischemia to clarify whether mitochondria with decreased Mn-SOD activity could be vulnerable targets of O2− relative to normal mitochondria.
MATERIALS AND METHODS
Permanent focal ischemia. The Sod2 gene knock-out mutants (heterozygous) with CD1/SV129 background were backcrossed with CD1 for at least five generations (F5), and littermate wild-type mice with the identical genetic background as Sod2heterozygous mice (3-month-old males, 35–40 gm) were subjected to permanent focal cerebral ischemia. Focal ischemia was induced by intraluminal middle cerebral artery (MCA) occlusion with a nylon monofilament suture (Yang et al., 1994).
Animals were anesthetized with 2.0% isoflurane in 30% O2/70% N2O. The rectal temperature was controlled at 37 ± 0.5°C with a homeothermic blanket (Homeothermic Blanket System, Harvard Apparatus, South Natick, MA) and a heating pad. A skin incision was made on the midline of the ventral neck. The left external carotid artery was exposed, and its branches were electrocoagulated. An 11 mm 5-0 monofilament nylon suture (Dermaron 1756-41, Devis & Geck, Manati, Puerto Rico) was introduced from the left external carotid artery stump into the left internal carotid artery. The skin incision was sutured. These surgical procedures were performed within 10 min. After surgery, the animals were taken care of in individual cages at 20°C.
To determine both the anatomical and physiological background of the ischemia, we evaluated cerebral vasculature and change of regional cerebral blood flow (rCBF) in both groups of animals. The vasculature was evaluated by carbon black injection (K. Murakami T. Kondo, and P.H. Chan, unpublished observations). After anesthesia with ketamine (200 mg/kg) and xylazine (10 mg/kg), the mice were killed by transcardial perfusion with 200 ml of 10 U/ml heparin in saline and 200 ml of 3.7% formaldehyde in PBS. Carbon black in an equal volume of 20% gelatin in H2O was injected from the ascending aorta. The brain was removed and fixed in 3.7% formaldehyde in PBS for 24 hr. The cerebral vasculature was observed with a dissecting microscope (Stemi 2000C, Zeiss, Oberkochen, Germany).
Change in rCBF was evaluated in both groups of animals with a laser Doppler flowmeter (LASERFLO BPM2, Vasomedic, St. Paul, MN). The laser Doppler probe was placed on the cranial window (made by using a dental drill) above the MCA territory cortex (0.5 mm posterior and 4 mm lateral from the bregma). The rCBF was monitored continuously from 10 min before until 30 min after the ischemia induction. Residual rCBF during MCA occlusion was calculated as (ischemia rCBF/ pre-ischemia rCBF) × 100% and compared between knock-out mutants and wild-type animals.
In situ detection of O2−production. The spatial production of O2− in cerebral ischemia was investigated by the method of in situ detection of oxidized hydroethidine (HEt) (Bindokas et al., 1996; Kondo et al., 1997a). HEt is oxidized to ethidium (Et) selectively by O2−, but not by other reactive oxygen species such as hydrogen peroxide, hydroxyl radical, or peroxynitrite (Bindokas et al., 1996). HEt solution (200 μl; stock solution of HEt, 100 mg/ml in dimethylsulfoxide, diluted to 1 mg/ml with PBS) was administered intravenously 15 min before ischemia induction. In a preliminary kinetic study after the fluorescence of HEt and Et, we have determined that HEt rapidly disappeared in the serum and penetrated into the brain shortly after intravenous injection, whereas Et remained mainly in the serum. In the brain of intravenously HEt-injected animals, fluorescence was assessed microscopically at Ex = 355 nm and Em > 415 nm for HEt detection, or at Ex = 510–550 nm and Em > 580 nm for Et detection. Animals were killed at 1 or 4 hr after the ischemia induction by transcardial perfusion as described above. After post-fixation in 3.7% formaldehyde for 2 hr, brain sections 50 μm in thickness at the level of the anterior commissure were placed on a glass slide, using a vibratome. These sections were observed with a microscope (Axioplan, Zeiss) under fluorescent light (HBO W/2, Zeiss). Photomicrographs of the fluorescent microscopy were taken in both the ischemic and nonischemic hemispheres, and the intensity and expression patterns of the oxidized HEt were observed and compared between wild-type and knock-out mutant mice.
Evaluation of mitochondrial injury. To assess mitochondrial viability in ischemic and nonischemic brain tissue, we used rhodamine 123 (Rh 123), a cell-permeant cationic fluorescent probe selectively accumulated by transmembrane potential in viable mitochondria. Animals were perfused with ice-cold PBS and were killed at 4 and 24 hr after ischemia. The brains were removed immediately after decapitation and frozen in 2-methyl butane at −20°C. Frozen brain sections at the level of the anterior commissure (20 μm in thickness) were used quantitatively to evaluate and compare mitochondrial injury between the wild-type and knock-out mutant mice. The frozen sections were dried at room temperature and were incubated with 2.0 × 10−3 μm Rh 123 (Molecular Probes, Eugene, OR) and with 2.5 × 10−3 μg/ml Hoechst 33258 (Molecular Probes) in PBS for 15 min. After being fixed in 3.7% formaldehyde in PBS for 15 min, the sections were rinsed in ddH2O and mounted with glycerol. Furthermore, to confirm that the mitochondrial accumulation of Rh 123 in fact is caused by the mitochondrial transmembrane potential, we tested agents that have been shown to disrupt the mitochondrial membrane potential, valinomycin (potassium ionophore; Sigma, St. Louis, MO) and 2,4-dinitrophenol [DNP (proton ionophore); Sigma], for their effects on the probe–mitochondrial interaction (Johnson et al., 1981). The brain sections were pretreated with 5 ng/ml valinomycin or 1.0 × 10−3m DNP for 60 min at room temperature before the incubation with Rh 123. The control sections were incubated with PBS. Then the effects of these inhibitors were determined.
We also determined whether brain slices under freezing conditions would reduce the Rh 123 uptake. The animals were anesthetized deeply with methoxyflurane and were perfused with ice-cold saline. The animals (n = 6) were killed, and the brains were removed and immersed in ice-cold PBS. Brain sections of 200 μm were cut in ice-cold PBS on glass slides and were dried at room temperature for 5 min. The brain sections of three animals were frozen at −80°C for 1 hr, and the slices from the other one-half of the animals were kept in ice-cold PBS for the same amount of time. Both the frozen and control sections were incubated with Rh 123 and Hoechst at the same concentrations as just described. Sections were fixed with 3.7% formaldehyde in PBS for 30 min, followed by a 5 min wash with ddH2O. The sections were mounted on slides with aqueous mounting medium.
The sections were observed with a microscope under fluorescent light. Photomicrographs were taken with high-powered magnification (400×) in the lateral caudoputamen of both the ischemic and nonischemic hemispheres. These photomicrographs were taken by double exposure to Rh 123 and Hoechst 33258. Total cells and Rh 123-positive cells were quantified on the photomicrographs by a blinded investigator. The ratio of Rh 123-positive cells to total cells was calculated and compared between wild-type and knock-out mutant mice.
Evaluation of neurological deficits and histological outcome.Neurological deficits of the experimental animals were graded on a scale of 0–5 as described by Yang et al. (1994), with minor modification, before death at 24 hr. It also was confirmed whether the dead mice died of cerebral ischemia or surgical complications such as subarachnoid hemorrhage caused by the mechanical damage of blood vessels by the inserted suture.
Frozen brain sections taken at 500 μm intervals by a cryostat were stained with cresyl violet. The unstained area was measured on each section by an image analysis system; then the infarct volume was calculated by multiplying the unstained area by the distance (Swanson et al., 1990; Swanson and Sharp, 1994). Hemisphere enlargement was also calculated and expressed as ipsilateral hemisphere volume/contralateral hemisphere volume × 100%.
In situ detection of DNA-fragmented cells. DNA fragmentation after permanent focal cerebral ischemia was determined by the TUNEL (terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling) method in both groups of animals. Frozen brain sections at the anterior commissure were dried at room temperature and fixed with 3.7% formaldehyde for 45 min. Endogenous peroxidase was inactivated with 60 mm hydrogen peroxide and 100 mm sodium azide for 30 min. After the slides were washed with PBS, they were immersed in terminal deoxynucleotidyl transferase buffer (Life Technologies, Gaithersburg, MD) at room temperature for 15 min and incubated with terminal deoxynucleotidyl transferase (Life Technologies) and biotin-16-uridine-5′-triphosphate (Boehringer Mannheim, Indianapolis, IN) at 37°C for 60 min. The reaction was stopped by washing with 6 mm sodium citrate and 60 mm sodium chloride for 30 min. Then the slides were incubated with 2% bovine serum albumin in PBS. After the slides were washed with PBS, the sections were incubated with avidin–biotin–horseradish peroxidase (ABC kit, Vector Laboratories, Burlingame, CA) for 30 min at room temperature, and staining was visualized with 3 mm 3,3′-diaminobenzidine tetrahydrochloride and 18 mm hydrogen peroxide in PBS. After being rinsed in ddH2O, the sections were stained with methyl green for 10 min and then were dehydrated and mounted.
TUNEL-labeled cells were quantified with a light microscope. These cells, displaying morphological features of apoptosis, were counted in the regions of interest, including the medial caudoputamen, piriform cortex, MCA territory cortex, border zone cortex, and lateral caudoputamen, by a blinded investigator who used high-powered magnification (400×). The number of TUNEL-labeled cells was expressed as per millimeter squared in each region and compared between the animals at 4 and 24 hr ischemia.
Statistical analysis. The grading score of neurological deficit outcome was analyzed with nonparametric statistics of the Mann–Whitney U test. The statistical significance of difference was evaluated by Student’s t test in infarct volume and hemisphere enlargement between wild-type and knock-out mutant mice and by ANOVA in the evaluation of mitochondrial injury and DNA fragmented cells between each time point and between mouse groups.
RESULTS
Cerebral vasculature and reduction of rCBF in focal cerebral ischemia
Because mice with different genotypes were used in the present study, although both groups originated from the same CD-1 strain, the cerebral vasculature and change of rCBF were analyzed to confirm that both anatomic and physiological backgrounds were the same in this study. Cerebral vasculature was determined by carbon black injection (Fig. 1A). The method used herein allowed us to determine the structure of major blood vessels in the brain. There was no remarkable difference between wild-type mice and knock-out mutants, not only in the structure of Willis’ circle but also in that of the MCA. The distribution of the MCA trunk and branch appeared to be anatomically identical.
Fig. 1.

A, Photomicrographs showing similarity in cerebral vasculature in wild-type (Wt) and knock-out mutant mice (Sod2 −/+), respectively. The origins of the MCA and the other major blood vessels involved in the Willis’ circle were almost the same in both animals. The distribution of the MCA territory was also similar between the two animals.B, Residual rCBF of the MCA territory cortex measured by laser Doppler flowmetry during MCA occlusion. The ischemia was induced at the same level in both wild-type and knock-out mutant mice.
Change of rCBF was measured indirectly by laser Doppler flowmetry, which showed a prominent reduction in rCBF in the ischemic cortex of both animal groups. As shown in Figure 1B, the rCBF was reduced to 9.7 ± 2.6% and 8.0 ± 1.0% of the baseline in wild-type and knock-out mutant mice, respectively, by MCA occlusion. There was no statistically significant difference in residual rCBF during ischemia between the two groups.
Production of O2− in cerebral ischemia
Production of O2− was determined by using HEt, a fluorescent dye oxidized to Et selectively by O2− at 1 and 4 hr after ischemia induction. Because the MCA was occluded permanently in the present study, HEt was administered 15 min before MCA occlusion. This fluorescent probe was distributed sufficiently, even to the brain tissue of the ischemic area, where the blood vessels were occluded. As previously observed by Kondo et al. (1997a), O2− production was shown by oxidized HEt signals as small particles in the cytosol, suggesting mitochondrial production of O2−. These signals were increased in knock-out mutants as compared with wild-type mice in the nonischemic area (Fig.2A,B). Some cells that are within the microvessel area showing the morphological characteristic of endothelial cells displayed strong signals under normal physiological conditions as well as in ischemia (Fig.2C,D). At 1 hr after ischemia induction, however, although the ischemic area showed a slightly increased mitochondrial expression of oxidized HEt signals relative to the nonischemic hemisphere, the difference was not marked between wild-type and knock-out mutant mice at this time point (Fig. 2E,F). The cellular and subcellular patterns of oxidized HEt signals appeared to be changed at 4 hr after ischemia. Some brain cells exhibited diffuse cytosolic expression of HEt fluorescent signals in the ischemic area (Fig.2G,H). The cytosol was filled with the diffuse HEt signals in these cells, and the shape of the cytosolic space, including dendrites, was represented by the fluorescent signal. This cytosolic pattern fluorescence of oxidized HEt was more intense and more frequently identified in knock-out mutants than in wild-type mice. Furthermore, these cells were observed more frequently in the cortex, the cortical penumbra in particular, than in the caudoputamen.
Fig. 2.
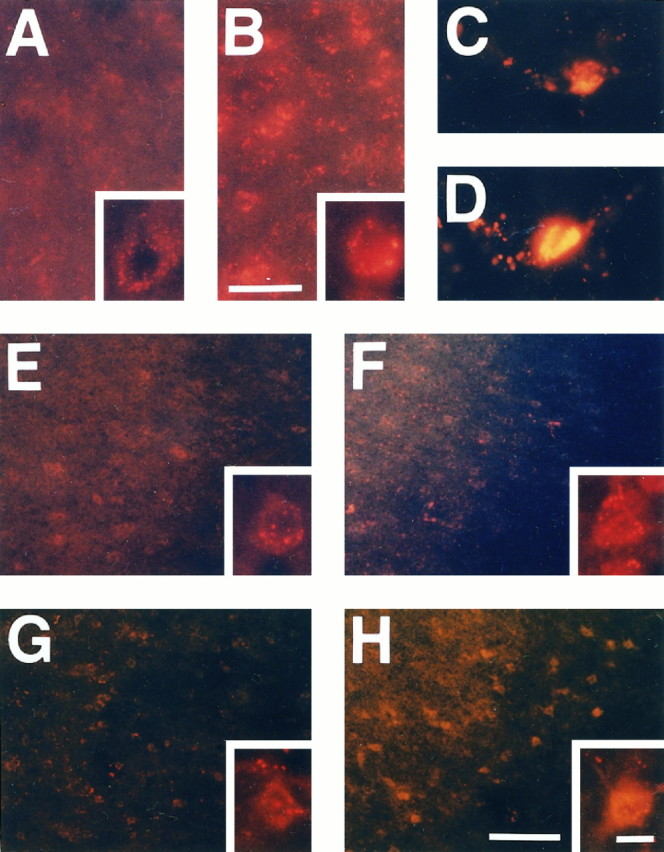
Representative photomicrographs showing increased production of O2− by the expression of oxidized HEt in Sod2 −/+ mouse brains. The basal level of O2− was increased even under normal physiological conditions in the knock-out mutant mice (B) as compared with wild-type animals (A), reflecting decreased activity of Mn-SOD. Under normal physiological conditions, HEt signals were detected as small particles in the cytosol, indicating mitochondrial production of O2−. These small particles were likely to be increased and intense in knock-out mutants. Endothelial cells demonstrated strong fluorescent signals rather than other cell populations under both normal (C) and ischemic conditions (D). E, F, Ischemic area showing increased production of O2− at 1 hr after ischemia induction. In the ischemic brain tissue, including the cortex and caudoputamen, these small red particles were increased slightly in both wild-type animals (E) and knock-out mice (F) at 1 hr. However, these HEt signals were likely to indicate not a cytosolic, but a mitochondrial, expression of O2−, and the difference was not remarkable between wild-type mice and knock-out mutants.G, H, At 4 hr, although a great deal of HEt was supposed to have been oxidized to Et, the intense and different subcellular pattern of the HEt signal was observed. Some cells in the ischemic area exhibited cytosolic expression of O2−. This fluorescent signal, which filled up the cytosolic space, displayed the shape of those cells. Furthermore, these cells with a cytosolic expression pattern of HEt were observed more frequently in theSod2 knock-out mutants (G) than in the wild-type mice (H), particularly in the penumbra area. Scale bars: 25 μm in A,B; 100 μm in E–H; 10 μm inC, D and the high magnification inA, B, E–H.
Mitochondrial injury was exacerbated in knock-out mutants
We evaluated the temporal change of Rh 123-accumulated cells to determine whether decreased activity of Mn-SOD causes mitochondrial susceptibility in knock-out mutants. Examination of the mitochondrial viability assay revealed vesicular accumulation of the cell-permeant cationic compound, Rh 123, in the brain cells. To confirm that Rh 123 accumulated in the mitochondria by the remaining transmembrane potential, we tested two agents for their effect on the probe–mitochondrial interaction. Pretreatment with these ionophores, which decrease the transmembrane potential of mitochondria, diminished this accumulation of Rh 123 as compared with the control that was treated with PBS (Fig. 3A). Small particles displayed by the accumulated Rh 123 were decreased by preincubation with both inhibitors. Furthermore, the intensity of vesicular fluorescent signals also was weakened by these agents, although background fluorescence was increased slightly. These results were observed both in the caudoputamen and in the cortex, suggesting that the transmembrane potential is likely to remain even in frozen brain tissue and that the small particles shown by the accumulated Rh 123 under fluorescence indicate the existence of viable mitochondria. We also tested the effects of freezing on Rh 123 staining. The Rh 123-positive cells were 75 ± 10% and 69 ± 3% (p > 0.05) for controls (without freezing) and for frozen slices, respectively.
Fig. 3.
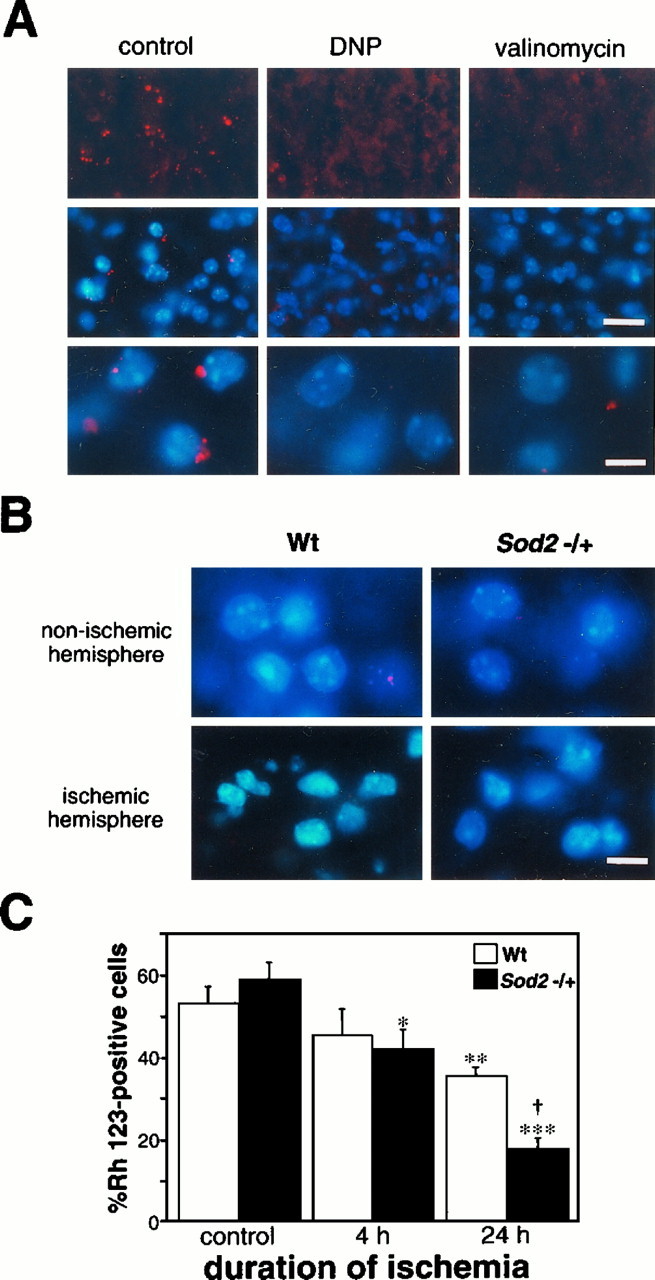
Evaluation of mitochondrial injury after ischemia. A, Representative microphotographs showing mitochondrial accumulation of the fluorescent probe, Rh 123, and the effects of mitochondrial membrane ionophores DNP and valinomycin.Top, middle, and bottomrows are photographs singly exposed to Rh 123, both doubly exposed to Rh 123 and counterstained by Hoechst 33258, and with high-powered magnification, respectively. Small red particles represent the fluorescent signals of Rh 123 accumulated by the transmembrane potential of mitochondria, suggesting the existence of viable mitochondria. This accumulation of Rh 123 was diminished by pretreatment with ionophores DNP and valinomycin. The vesicular fluorescent signals were observed to be weakened and decreased by the preincubation of the ionophores. Scale bars: 25 μm, top and middle; 10 μm,bottom. B, Fluorescent photomicrographs in both nonischemic (top) and ischemic hemispheres (bottom) of wild-type (Wt;left) and Sod2 knock-out mutant mice (right). Note that, in both wild-type and knock-out mutant mice, some cells were Rh 123-negative in the nonischemic area; Rh 123-positive cells also were observed in the ischemic brain tissue. Thus, not all of the cells without the small Rh 123 fluorescent particles necessarily indicate a mitochondrial injury. Scale bar, 10 μm. C, Ratio of the number of Rh 123-positive cells to that of total cells in the lateral caudoputamen of the ischemic hemisphere, a possible ischemic core, in the present ischemia model at 4 and 24 hr ischemia. Values are mean ± SE; *p < 0.05, **p < 0.01, and ***p < 0.001 versus control, ANOVA.†p < 0.05 versus wild-type, ANOVA. At 4 hr ischemia, the ratio of Rh 123-positive cells was inclined to decrease as compared with the control value, but no significant difference was observed between wild-type and knock-out mutant mice. However, these cells decreased in both groups at 24 hr ischemia and were significantly less in knock-out mutants than in wild-type mice.
Using the present method, we determined and compared mitochondrial viability between wild-type and Sod2 knock-out mutant mice after ischemia. Ischemic cells demonstrated shrunken nuclei without an intense fluorescent signal of Rh 123, showing ischemic change lacking the mitochondrial membrane potential (Fig. 3B). These findings were not remarkably different between the animal groups. Figure 3C demonstrates the temporal pattern of mitochondrial injury development after ischemia in wild-type and knock-out mice. The ratio of Rh 123-positive cells in the nonischemic hemisphere was 53.1 ± 11.7% in wild-type mice and 58.9 ± 11.3% inSod2 knock-out mice. There was no significant difference between wild-type and knock-out mice in the nonischemic hemisphere. At 4 hr ischemia, a slight decrease of Rh 123-positive cells was observed; the ratio was 45.3 ± 12.6% (n = 4) in wild-type animals and 41.9 ± 9.8% (n = 4) in knock-out mice. The decrease of Rh 123-positive cells was significant in knock-out mice as compared with the control value (p < 0.05, ANOVA). This ratio was decreased remarkably at 24 hr: 35.5 ± 4.4 (n = 7) in wild-type mice and 17.8 ± 5.2% (n = 8) in knock-out mice. The ratio of Rh 123-positive cells to total cells was decreased significantly in both wild-type and knock-out mice relative to the control value (p < 0.01 in wild-type mice and p < 0.001 in knock-out mice, ANOVA), and mitochondrial injury was exacerbated significantly more in the knock-out mice than in the wild-type mice (p < 0.05, ANOVA).
Neurological deficit and histological damage were advanced in knock-out mutants
Among the surviving wild-type and knock-out mutant mice, a significant difference was not seen in neurological outcome at 24 hr ischemia. Higher mortality was seen in the knock-out mice as compared with the wild-type mice (12.5% in wild-type mice and 50.0% in knock-out mice). Although it was not statistically significant, there is a possibility that severely injured mice were already dead and thus have been excluded from the neurological evaluation. Therefore, autopsies were conducted to determine the cause of death; then the dead mice that were confirmed to have no surgical complications such as subarachnoid hemorrhage were assigned a score of 5 in the evaluation of neurological deficits. Dead mice showed severe brain swelling, and these findings are consistent with our previous data demonstrating that correlations existed between hemispheric enlargement and neurological deficits (Kondo et al., 1997a). The mean scores of wild-type and knock-out mice were 2.4 (n = 8) and 3.8 (n = 16), respectively, and neurological outcome was significantly exacerbated in the knock-out mice (p < 0.05, Mann–Whitney U test; Fig. 4).
Fig. 4.

Neurological deficit scores in wild-type (Wt) and knock-out mutant mice (Sod2 −/+) at 24 hr ischemia. Small circles andbars are the scores of each animal and mean scores, respectively. *p < 0.05, Mann–WhitneyU test. 0 = No observable neurological deficit, 1 = failed to extend right forepaw, 2 = circled to the right, 3 = fell to the right, 4 = could not walk spontaneously, and 5 = dead. Mean scores of knock-out mice were significantly higher than those of wild-type mice.
We next evaluated both the infarct size and hemisphere enlargement at 24 hr ischemia. Figure 5Ashows the typical findings of histology in wild-type and knock-out mice at 24 hr. The infarct area extended throughout the entire MCA territory, including both the caudoputamen and cortex in wild-type and knock-out mice. Infarct volume was 74.1 ± 27.4 mm3 (n = 7) in wild-type animals and 121.0 ± 31.2 mm3 (n = 8) in knock-out mice and was significantly greater in knock-out than in wild-type mice (p < 0.01, Student’st test; Fig. 5B). Furthermore, as shown in Figure5C, brain swelling, a possible index of ischemic brain edema, was also more severe in knock-out than in wild-type mice (p < 0.001, Student’s t test). Hemisphere enlargement was 17.9 ± 5.4% (n = 7) in wild-type and 34.3 ± 7.8% (n = 8) in knock-out mice.
Fig. 5.
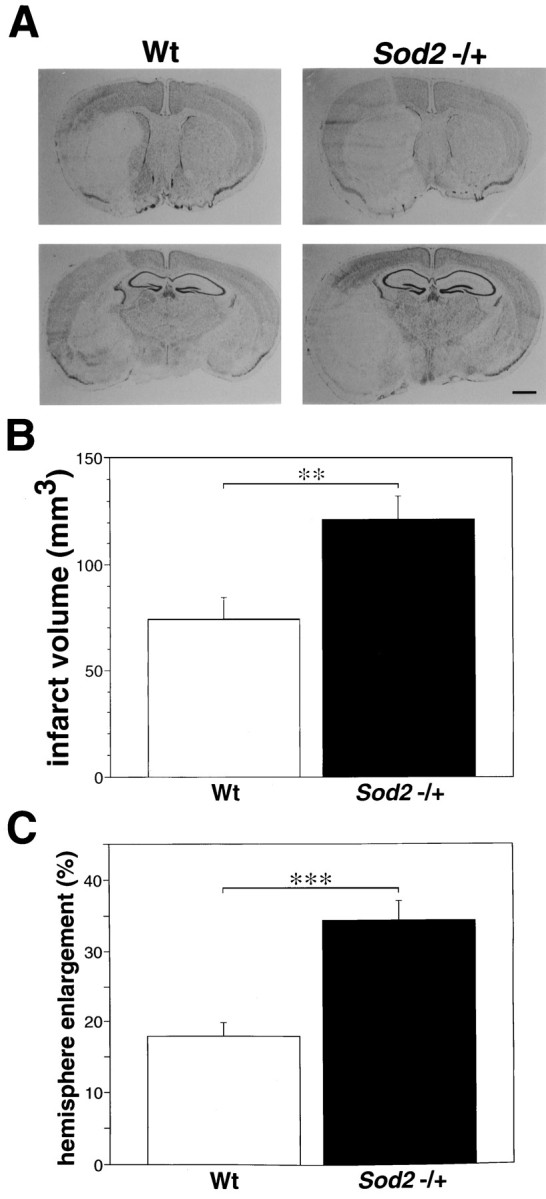
Histological analysis after 24 hr of MCA occlusion. A, Photomicrograph showing the histological changes after 24 hr of MCA occlusion in wild-type (Wt) and knock-out mutant mice (Sod2 −/+). The infarct area was localized in the caudoputamen and MCA territory cortex in both mice groups. However, cortical infarction extended to the boundary zone of the anterior cerebral artery territory, and brain swelling was extremely severe in the knock-out mutant mice. Also shown are infarct volume (B) and hemisphere enlargement (C) in wild-type and knock-out mutant mice at 24 hr ischemia. Values are mean ± SE; **p < 0.01 and ***p < 0.001, Student’s ttest. Cerebral infarction and hemisphere enlargement were significantly more severe in knock-out than in wild-type mice. Scale bar, 1 mm.
DNA fragmentation is not altered by reduction of Mn-SOD in permanent focal cerebral ischemia
To determine the role of Mn-SOD in DNA fragmentation after ischemia, we evaluated the contribution of DNA fragmentation to ischemic neuronal injury with in situ TUNEL staining. TUNEL-labeled cells were distributed mainly in the inner boundary zones, such as the medial caudoputamen, at 24 hr ischemia in both wild-type and knock-out mutant mice (Fig.6A,B). These cells were densely labeled in the nuclei and showed characteristic features of morphology, nuclei condensation, cell shrinkage, and, occasionally, fragmentation of nuclei, suggesting apoptotic cells (Fig.6C,D). No morphological differences were observed between the cells of knock-out mutants and wild-type mice. These TUNEL-labeled cells were not observed in the nonischemic area. To determine the temporal pattern and anatomical distribution of DNA fragmented cells in ischemia, we counted TUNEL-labeled cells with the morphological features mentioned above in the five regions at 4 and 24 hr (Fig.6E). Quantitative analysis demonstrated that the DNA fragmentation was induced as early as 4 hr and greatly increased at 24 hr and that the contribution of DNA-fragmented cells to the ischemic injury was statistically the same level between wild-type and knock-out mutant mice. At 4 hr ischemia, a few cells were labeled in most of the regions, but not in the MCA area cortex of wild-type mice nor in the lateral caudoputamen of knock-out mutant mice. These TUNEL-labeled cells were dispersed throughout the entire ischemic lesion at 24 hr but frequently were observed at the inner boundary zone. These observations are consistent with previous reports (Li et al., 1995a; Murakami et al., 1997). However, no significant difference was seen in any regions between wild-type and knock-out mutant mice at 24 hr.
Fig. 6.

In situ detection of DNA fragmentation after ischemia by TUNEL staining. Photomicrographs show the distribution of DNA-fragmented cells labeled by TUNEL staining in the inner boundary zone in wild-type (Wt) (A) and knock-out mutant mice (Sod2 −/+) (B). TUNEL-labeled DNA fragmented cells were distributed mainly in the medial caudoputamen adjacent to the inner boundary. C, D, High-magnification photomicrographs showing morphological features of TUNEL-labeled cells. These cells represent cell shrinkage, nuclei condensation, or fragmentation (arrowhead). E, Temporal profile of DNA fragmentation in each region of ischemic area, the medial caudoputamen, piriform cortex, MCA territory cortex, penumbra area cortex, and lateral caudoputamen. DNA fragmentation was induced as early as 4 hr in both the wild-type and knock-out mutant mice. The TUNEL-labeled cells were dispersed throughout the entire ischemic area at 24 hr, particularly in the inner boundary zone. Quantitative analysis demonstrated no statistically significant difference in the induction of DNA fragmentation between wild-type and knock-out mice.CC, Corpus callosum; LV, lateral ventricle.
DISCUSSION
The brain is one of the most vulnerable organs to ischemia because of its high energy demand for cellular function and its relatively modest defense against secondary oxidative stress. Under normal physiological conditions, brain mitochondria generate ATP by virtue of effective electron transport in their respiratory chain for cellular maintenance and integrity. The biochemical function of mitochondria strongly depends on the membrane, which is one of the oxygen radical target sites. Disturbance of mitochondrial membrane integrity causes mitochondrial dysfunction, leading brain cells to energy failure. Therefore, energy failure because of mitochondrial dysfunction is likely to be an important factor in ischemic brain injury.
In the present study it was shown that the increased HEt oxidation, a possible index of O2− production, was increased in nonischemic areas as well as in ischemic areas inSod2 gene knock-out mutants, as compared with wild-type mice, reflecting decreased activity of mitochondrial Mn-SOD. This increased production of O2− was remarkable in the penumbra cortex, where blood flow is reduced to a critical level for neuronal cells to survive, but oxygen, as a substrate for reactive oxygen radicals, continued to be supplied. Furthermore, the mitochondrial injury progressed, as evidenced by the lack of transmembrane potential, and thus cerebral infarction and neurological deficit outcome were exacerbated in knock-out mutant mice. Therefore, these results suggest that mitochondria are the important sites at which oxygen radicals are produced under both normal and pathological conditions and that mitochondrial Mn-SOD plays a protective role as an antioxidant enzyme of mitochondrially produced O2− in cerebral ischemia.
However, caution must be taken when the results are interpreted. Although HEt has been shown to be a highly selective compound for superoxide radical reaction, it is unlikely to compete with Mn-SOD for superoxide, because the latter reaction is extremely fast. The only exception is that nitric oxide reacts with superoxide faster than Mn-SOD, but it is unlikely that HEt, an artificial exogenous agent, would react at the same rate as nitric oxide. However, it will be of special interest and importance to determine the rate constant of the HEt oxidation. On the other hand, HEt could be oxidized by oxidatively damaged mitochondrial electron transport proteins such as succinate dehydrogenase, the activity of which is known to be reduced inSod2 knock-out mutant mice (Li et al., 1995b).
In an in vivo study of cerebral ischemia, both the anatomical and physiological backgrounds of ischemia are likely to be important factors affecting the condition of ischemia. Recently, variations of the plasticity of Willis’ circle have been demonstrated to affect the infarct size after focal ischemia among some strains of mice (Barone et al., 1993). The analysis of cerebral vasculature showed similar structures of Willis’ circle and the MCA between the two animal groups. However, because the anatomical analysis is neither quantitative nor functional, the change in rCBF was investigated also. No significant difference was seen between wild-type and knock-out mutant mice, indicating that the ischemic condition induced by the present method was, in fact, the same level and did not contribute to the exacerbation of ischemic brain injury in Sod2 knock-out mutant mice. On the basis that the ischemic condition was the same level in both groups of animals, O2−production, mitochondrial injury, and the outcome of neurology and histology, including the involvement of DNA fragmentation, were evaluated to elucidate the role of Mn-SOD in permanent focal ischemia.
Because HEt is oxidized continuously to Et by both normally and physiologically produced O2− and the oxidization of HEt was likely to reach the maximum within several hours after the injection, O2− production could be evaluated only in the early phase of ischemia. Oxidized HEt signals were seen as a small vesicular pattern under normal physiological conditions, and these vesicular signals were increased in knock-out mice even under normal physiological conditions. This finding may indicate that the mitochondria are the important sites of O2− production and that Mn-SOD plays an important role as an antioxidant, even under normal physiological conditions. However, considering that there was no evidence of oxidative injury in the brains of heterozygous knock-out mutants (Sod2 −/+) such as that seen in the hearts of homozygous knock-out mutants (Sod2 −/−) that died within several postnatal days (Li et al., 1995b), this increase in O2− production in knock-out mutant mice may not exceed the toxic threshold for neuronal injury. An intense and cytosolic signal of oxidized HEt was observed in some brain cells of the ischemic cortex, particularly in the cortical penumbra. These signals were observed more strongly and frequently in knock-out mutants than in wild-type mice. We first applied this fluorescent probe to detect O2− production in in vivo ischemia–reperfusion brain and observed that O2− production greatly progresses in the reperfusion state several hours after transient ischemia in mice (Kondo et al., 1997a). In consideration of these findings in both transient and permanent ischemia, O2−is likely to contribute to ischemic brain injury, but this might be more prominent in the penumbra during both permanent ischemia and the reperfusion state after transient ischemia when oxygen is effectively supplied as a substrate for O2−production. The present findings suggest that relatively increased O2−, which might be uncoupled from the antioxidant defense mechanism of mitochondria in knock-out mutant mice with decreased Mn-SOD, may exacerbate ischemic brain injury and result in the extension of the infarct area, especially in the penumbra.
Mitochondrial viability assay, which was evaluated by the integrity of mitochondrial membrane potential, demonstrated that the decreased level of Mn-SOD causes mitochondrial susceptibility in knock-out mutant mice. Rh 123 visualizes the mitochondria with normally inner negative potential at the membrane, which is generated by the electrochemical gradient of the proton (Johnson et al., 1981). Thus, Rh 123-negative cells are likely to be those that have lost mitochondrial viability. Accumulated Rh 123 exhibited red fluorescence as small vesicles around the nuclei, which were diminished by ionophores disrupting the potential. Interestingly, these vesicular patterns in Rh 123 fluorescent signals were similar to those observed in mitochondrial HEt expression. In ischemic brain tissue Rh 123-positive cells were decreased more prominently in knock-out than in wild-type animals.
Neurological and histological analyses also demonstrated significant exacerbation in knock-out mutants. Mortality was markedly high in knock-out mutant mice. Neurological deficits were exacerbated significantly in knock-out mutant mice relative to wild-type. Further, decreased activity of Mn-SOD also exacerbated histological outcome. Infarct volume and hemisphere enlargement were significantly larger in knock-out than in wild-type mice. In both mice groups the infarct area was observed in the entire MCA territory, including the caudoputamen and the frontoparietal cortex in the MCA area, after 24 hr of permanent MCA occlusion. However, because almost the whole caudoputamen was involved in the infarct area in both mice groups, enlargement of the infarct area might have resulted from the expansion of the infarct area to the cortical penumbra in knock-out mutants.
A recent study has demonstrated that mitochondrial function is required in the apoptotic process of neuronal death and that succession of the two distinct pathways to neuronal death, apoptosis or necrosis, is dependent on the preservation of mitochondrial function (Ankarcrona et al., 1995). We have demonstrated that ischemia–reperfusion injury was accelerated and that both necrosis and apoptosis contribute to the early progression of ischemia–reperfusion injury inSod2 knock-out mutant mice (T. Kondo, K. Murakami, and P.H. Chan, unpublished observations). Therefore, we determined whether occurrence of apoptotic cell death in ischemia alone was altered in knock-out mutant mice. DNA-fragmented cells labeled by TUNEL staining were quantified cautiously in five regions of interest, as described byCharriaut-Marlangue and Ben-Ari (1995), although whether TUNEL-labeled cells are exactly apoptotic cells is still controversial. However, although the distribution of DNA fragmentation was consistent with previous demonstrations (Li et al., 1995a; Murakami et al., 1997), no significant difference was seen between wild-type and knock-out mice in any regions. This insignificance could be attributable to the transient nature of apoptosis, which we might have missed during the course of the studies. This issue has been discussed in our previous studies involving CuZn-SOD knock-out mutant mice (Kondo et al., 1997b). We have demonstrated that DNA fragmentation was induced less in permanent focal ischemia than in transient focal ischemia followed by reperfusion and that severe ischemia, such as that induced in permanent MCA occlusion, might cause relatively increased necrotic cell death (Murakami et al., 1997). Recent evidence has shown that relatively intense insults, including a combination of hypoxia and substrate deprivation and exposure to high concentrations of NMDA, incline neuronal cell death to necrosis (Bonfoco et al., 1995; Copin et al., 1996). In the present study permanent ischemia induces such an intense ischemic insult that a significant difference could not be seen in the alternative apoptotic pathways to neuronal cell death between wild-type and knock-out mutant mice.
We have demonstrated previously that overexpressed CuZn-SOD plays a protective role in transient focal ischemia followed by reperfusion, but not in permanent focal ischemia (Chan et al., 1993; Yang et al., 1994). In permanent focal ischemia, cerebral blood flow is reduced severely to the level at which the ischemic brain yields to energy depletion; thus, neurons in the ischemic core rapidly die, whereas distant neurons in the penumbra remain viable. Therefore, an increased or decreased level of SOD is likely to play a protective or critical role, respectively, in the penumbra, but not in the ischemic region. However, considering the results obtained from the permanent focal ischemia studies conducted in both CuZn-SOD overexpressed transgenic mice and in Sod2 knock-out mutant mice herein, brain tissue in the penumbra might be exposed to very mild ischemia as compared with the ischemic core in permanent focal ischemia. Thus, knock-out mice brains might sustain oxidative injury by mitochondrially produced O2−, which would be detoxified instantly in wild-type brains, causing exacerbation of cerebral infarction after permanent focal ischemia. Another possibility for this discrepancy between the CuZn overexpresser and the Mn-SOD knock-out mutant mice after permanent focal cerebral ischemia may be attributable to the compartmentalizing role of this enzyme when oxygen delivery is cut off completely. It is likely that the mitochondrial compartment and the level of Mn-SOD are relatively sensitive to the lack of oxygen availability, which may lead to the increased ischemic injury.
Finally, we demonstrated that decreased Mn-SOD activity, in fact, causes increased oxidative stress and/or injury in cerebral ischemia and that mitochondria are both the sites of O2− production and the targets of free radical attacks. We conclude that mitochondrial Mn-SOD plays a protective role as an antioxidant defense in ischemic neuronal injury.
Footnotes
The present study was supported by National Institutes of Health Grants NS14543, NS25372, NO1 NS 5–2334, NS36147, and AG08938. We thank Dr. Charles J. Epstein, Dr. Ting-Ting Huang, and Elaine Carlson for their collaborative efforts in generating the mutant mice; Liza Reola and Bernard Calagui for their technical assistance; and Cheryl Christensen for her editorial assistance.
Correspondence should be addressed to Pak H. Chan, PhD, Neurosurgical Laboratories, Stanford University, 701B Welch Road, #148, Palo Alto, CA 94304.
REFERENCES
- 1.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 2.Barone FC, Knudsen DJ, Nelson AH, Feuerstein GZ, Willette RN. Mouse strain differences in susceptibility to cerebral ischemia are related to cerebral vascular anatomy. J Cereb Blood Flow Metab. 1993;13:683–692. doi: 10.1038/jcbfm.1993.87. [DOI] [PubMed] [Google Scholar]
- 3.Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-d-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA. 1995;92:7162–7166. doi: 10.1073/pnas.92.16.7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan PH, Kamii H, Yang G, Gafni J, Epstein CJ, Carlson E, Reola L. Brain infarction is not reduced in SOD-1 transgenic mice after a permanent focal cerebral ischemia. NeuroReport. 1993;5:293–296. doi: 10.1097/00001756-199312000-00028. [DOI] [PubMed] [Google Scholar]
- 6.Chan PH, Epstein CJ, Li Y, Huang T-T, Carlson E, Kondo T, Murakami K, Mikawa S, Chen SF, Reola L. Studies of neuronal injury mechanism in focal stroke using mitochondrial manganese superoxide dismutase-deficient mice. In: Krieglstein J, editor. Pharmacology of cerebral ischemia. Wissenschaftliche Verlagsgesellschaft; Stuttgart: 1996. pp. 573–579. [Google Scholar]
- 7.Charriaut-Marlangue C, Ben-Ari Y. A cautionary note on the use of the TUNEL stain to determine apoptosis. NeuroReport. 1995;7:61–64. [PubMed] [Google Scholar]
- 8.Copin J-C, Reola LF, Chan TYY, Li Y, Epstein CJ, Chan PH. Oxygen deprivation but not a combination of oxygen, glucose, and serum deprivation induces DNA degradation in mouse cortical neurons in vitro: attenuation by transgenic overexpressing of CuZn-superoxide dismutase. J Neurotrauma. 1996;13:233–244. doi: 10.1089/neu.1996.13.233. [DOI] [PubMed] [Google Scholar]
- 9.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 10.Dugan LL, Sensi SL, Canzoniero LM, Handran SD, Rothman SM, Lin TS, Goldberg MP, Choi DW. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-d-aspartate. J Neurosci. 1995;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Greenlund LJ, Deckwerth TL, Johnson EM., Jr Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 12.Hillered L, Chan PH. Brain mitochondrial swelling induced by arachidonic acid and other long chain free fatty acids. J Neurosci Res. 1989;24:247–250. doi: 10.1002/jnr.490240216. [DOI] [PubMed] [Google Scholar]
- 13.Johnson LV, Walsh ML, Bockus BJ, Lan BC. Monitoring of relative mitochondrial membrane potential in living cell by fluorescence microscopy. J Cell Biol. 1981;88:526–535. doi: 10.1083/jcb.88.3.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 15.Kinouchi H, Epstein CJ, Mizui T, Carlson E, Chen SF, Chan PH. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc Natl Acad Sci USA. 1991;88:11158–11162. doi: 10.1073/pnas.88.24.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kondo T, Li Y, Sato S, Murakami K, Copin J-C, Huang T-T, Epstein CJ, Chan PH. Subcellular localization of superoxide anions following focal cerebral ischemia and reperfusion. J Cereb Blood Flow Metab [Suppl] 1997a;17:5102. [Google Scholar]
- 17.Kondo T, Reaume AG, Huang T-T, Carlson E, Murakami K, Chen SF, Hoffman EK, Scott RW, Epstein CJ, Chan PH. Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation following transient focal cerebral ischemia. J Neurosci. 1997b;17:4180–4189. doi: 10.1523/JNEUROSCI.17-11-04180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kontos HA. George E. Brown memorial lecture. Oxygen radicals in cerebral vascular injury. Circ Res. 1985;57:508–516. doi: 10.1161/01.res.57.4.508. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Chopp M, Jiang N, Yao F, Zaloga C. Temporal profile of in situ DNA fragmentation after transient middle cerebral artery occlusion in the rat. J Cereb Blood Flow Metab. 1995a;15:389–397. doi: 10.1038/jcbfm.1995.49. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Huang T-T, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995b;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 21.Mikawa S, Li Y, Huang T-T, Carlson E, Chen S, Kondo T, Murakami K, Epstein CJ, Chan PH. Cerebral infarction is exacerbated in mitochondrial manganese superoxide dismutase (Sod-2) knock-out mutant mice after focal cerebral ischemia and reperfusion. Soc Neurosci Abstr. 1995;21:1268. [Google Scholar]
- 22.Mikawa S, Kinouchi H, Kamii H, Gobbel GT, Chen CF, Carlson E, Epstein CJ, Chan PH. Attenuation of acute and chronic damage following traumatic brain injury in copper, zinc-superoxide dismutase transgenic mice. J Neurosurg. 1996;85:885–891. doi: 10.3171/jns.1996.85.5.0885. [DOI] [PubMed] [Google Scholar]
- 23.Murakami K, Kondo T, Chan PH. Reperfusion following focal cerebral ischemia alters distribution of neuronal cells with DNA fragmentation in mice. Brain Res. 1997;751:160–164. doi: 10.1016/s0006-8993(97)00029-2. [DOI] [PubMed] [Google Scholar]
- 24.Smith ML, Bendek G, Dahlgren N, Rosen I, Wieloch T, Siesjo BK. Models for studying long-term recovery following forebrain ischemia in the rat. 2. A 2-vessel occlusion model. Acta Neurol Scand. 1984;69:385–401. doi: 10.1111/j.1600-0404.1984.tb07822.x. [DOI] [PubMed] [Google Scholar]
- 25.Swanson RA, Sharp FR. Infarct measurement methodology [letter; comment]. J Cereb Blood Flow Metab. 1994;14:697–698. doi: 10.1038/jcbfm.1994.88. [DOI] [PubMed] [Google Scholar]
- 26.Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume [comments]. J Cereb Blood Flow Metab. 1990;10:290–293. doi: 10.1038/jcbfm.1990.47. [DOI] [PubMed] [Google Scholar]
- 27.Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]