

Abstract
The molecular changes responsible for inducing neuronal apoptosis are unknown. Rat cortical neurons were treated with x-irradiation 7 d after isolation to test for the role of DNA damage in neuronal death. The response of neurons to x-irradiation was compared with that of astrocytes that had been isolated 3 weeks earlier from newborn rats. At the time of irradiation, the neurons appeared well differentiated morphologically and were predominantly (90–95%) noncycling, based on flow cytometric analysis. There was a similar, linear increase in DNA double-strand breaks with increasing radiation dose in neurons and astrocytes. However, whereas doses as low as 2 Gy induced typical apoptotic changes in neurons, including nuclear fragmentation and/or internucleosomal DNA fragmentation, doses as high as 32 Gy caused little or no apoptosis in astrocytes. Radiation-induced apoptosis of neurons started 4–8 hr after irradiation, was maximal at 12 hr, and was dependent on dose up to 16 Gy. It was prevented when cycloheximide, a protein synthesis inhibitor, was added up to 6 hr after irradiation. In addition to their distinct apoptotic response, neurons rejoined radiation-induced DNA double-strand breaks more slowly than astrocytes. Treatment with benzamide to inhibit ADP-ribosylation and strand break repair increased apoptosis; splitting the dose of radiation to allow increased time for DNA repair decreased apoptosis. These data suggest that DNA damage may induce neuronal apoptosis, that the extent of damage may determine the degree of apoptosis induced, and that slow repair of damage may play a role in the susceptibility of neurons to apoptosis.
Keywords: rat, neuron, astrocyte, ADP-ribosylation, benzamide, DNA damage, apoptosis, x-irradiation
Neurons appear to be particularly susceptible to injury from insults to the CNS. However, the sequence of events leading from the initial insult to neuronal dysfunction and death has not been established in many cases. It is important to identify these events so that methods of preventing or reducing injury can be developed.
There are at least two mechanisms by which neurons can die, apoptosis and necrosis. Apoptosis commonly is distinguished from necrosis in that the former generally is considered to be an active process requiring synthesis of particular proteins responsible for cell killing (Villa et al., 1994). Furthermore, internucleosomal cleavage of DNA and nuclear fragmentation often accompanies apoptosis, whereas cellular swelling, membrane disruption, and random cleavage of the DNA typically accompany necrosis (Wyllie et al., 1980). Many insults, including cerebral ischemia (MacManus et al., 1993; Hill et al., 1995; Islam et al., 1995;Li et al., 1995; Linnik et al., 1995; Du et al., 1996), trauma (Rink et al., 1995), and exposure to glutamate receptor agonists (Pollard et al., 1994; Gillardon et al., 1995) or β-amyloid (Loo et al., 1993), appear to induce neuronal death via apoptosis. Neuronal apoptosis also appears to occur in the brains of individuals with Huntington’s disease (Portera-Cailliau et al., 1995) and in animal models of Parkinson’s disease (Mitchell et al., 1994).
The precise stimuli responsible for the induction of neuronal apoptosis remain unclear, but oxidative stress and the accompanying free radicals may contribute to the induction. Many neuropathological conditions that are characterized by neuronal apoptosis, including stroke, Alzheimer’s disease, and amyotrophic lateral sclerosis, are accompanied by oxidative stress (Bowling et al., 1993; Kumar et al., 1994; Gunasekar et al., 1995; Yan et al., 1995; Chan, 1996). Furthermore, downregulation of Cu–Zn-superoxide dismutase (SOD), a free radical scavenger, increased apoptosis of neuron-like PC12 cells (Troy and Shelanski, 1994), and increased concentrations of Cu–Zn-SOD in sympathetic neurons can delay apoptosis induced after withdrawal of nerve growth factor (Greenlund et al., 1995).
Numerous molecular changes can accompany free radical damage, including protein oxidation, lipid peroxidation, and DNA alterations (Dizdaroglu, 1992; Floyd and Carney, 1992; Buettner, 1993), but the specific changes that induce neuronal apoptosis after cerebral injury are unknown. However, there is considerable interest in the role of DNA damage in neurodegenerative processes (Chopp et al., 1996). We hypothesized that if DNA damage induces apoptosis in neurons, then x-rays, which cause DNA damage in part via the generation of free radicals (Marin and Bender, 1963; Munro, 1970; Hall, 1988; Bump and Brown, 1990), also should induce apoptosis in neurons. Furthermore, we postulated that if DNA damage were responsible for apoptosis, then the processing of that damage in neurons should differ from the processing in cells that are resistant to apoptosis. In the present study we used x-rays to generate DNA damage in neurons isolated from embryonic rat cortex and astrocytes isolated from newborn rat pups, and we compared and contrasted the response of these two cell types to the resultant damage.
MATERIALS AND METHODS
Cell isolation and cultivation. Primary neuronal cultures were established from the cerebral cortex of Sprague Dawley rats at day 15–17 of gestation, using a modification of a previously described method (Yu et al., 1986). Pregnant rats were killed by CO2 inhalation, followed by decapitation, and the uterus was removed aseptically. The fetuses were collected, and the cerebral cortices were removed carefully under a dissecting microscope and then pooled and minced into fragments <1 mm3. The tissues were digested for 6 min by placing them in 0.2% trypsin in Puck’s balanced salt solution that was free of Ca2+and Mg2+. Digestion was stopped by adding a solution of modified Eagle’s medium (MEM) containing 10% horse serum. The cells were centrifuged at 1000 × g for 10 min, and the resultant pellet was resuspended and passed through an 80 μm nylon filter. The cell filtrate was diluted in MEM with an additional 25 mmd-glucose and 2 mml-glutamine. The cells were plated into plastic dishes that had been precoated with poly-l-lysine (1 ml of 50 μg/ml solution per 10 cm2 dish) 24 hr earlier. The plating density was such that there was one brain for each 10 cm2 dish. At 1 d after plating, the cells were treated for 24 hr with 10 μm cytosine arabinoside (Ara-C) to remove actively dividing cells. The medium was changed the next day to remove any cells that had degenerated as a result of Ara-C treatment. The cells were fed with fresh medium 5 d after cell isolation. Experiments were performed 7 d after isolation. This method of isolation typically produces cultures that are >95% neurons on the basis of positive immunocytochemical staining for neuronal markers, neuron-specific enolase and neurofilament protein, and negative staining for an astrocytic marker, glial fibrillary acid protein (data not shown).
Primary astrocyte cultures were established from the cerebral cortex of newborn (postnatal day 1) rat pups as previously described (Yu et al., 1986). After decapitation, the cerebral cortices were removed aseptically and minced into fragments <1 mm3 in MEM with 20% fetal calf serum (astrocyte growth medium). The tissue was disrupted by mixing on a vortex for 1 min and sequentially was passed through an 80 and a 10 μm nylon filter. The plating density was such that each brain was plated into ∼30 of the 28 cm2dishes. Unlike the previous astrocyte isolation procedure (Yu et al., 1986), there was no treatment with dibutyryl cyclic AMP 2 weeks after isolation. The cells were plated into plastic dishes and fed with astrocyte growth medium that was changed every 3–4 d. Experiments were performed 3 weeks after isolation when the cultures were confluent. This method of isolation typically produces cultures that are >99% astrocytes, based on immunocytochemical staining for glial fibrillary acid protein (data not shown).
Irradiation. Cultures were irradiated with an orthovoltage x-ray generator operating at 150 peak kilovoltage and 19.2 mA. Dose rate was 1.2 Gy/min except for the experiment on lactate dehydrogenase (LDH) release in which a dose rate of 3.5 Gy/min was used because of the high doses (up to 64 Gy) used in that experiment.
Drug treatment. Cycloheximide, benzamide, and aminobenzamide were all obtained from Sigma (St. Louis, MO). The drugs were dissolved at 100× the final concentration in water (cycloheximide) or ethanol (benzamide and aminobenzamide), passed through a 0.2 μm pore filter to remove any contamination by microorganisms, and added directly to the medium overlying the cells. An equal volume of sterile water or ethanol was added to cultures used as controls.
Measurement of nuclear fragmentation. Cells that had been plated into 24-well culture dishes were fixed in 3.7% buffered formalin and stained with 2 μg/ml of Hoechst Dye 33258 (Sigma) in PBS, pH 7.0, for 10 min. A single field from each well (50–200 cells) was selected at random from each well and photographed with epi-fluorescence. The fraction of cells within each well showing evidence of nuclear fragmentation was estimated from the photographs by an observer blinded to the treatment. Nuclear fragmentation was defined as the presence of two or more distinct nuclear lobes within a single cell.
Measurement of cell viability. Cell viability was measured on the basis of the retention of LDH, an intracellular enzyme, by viable cells and the release of LDH into the medium by damaged cells with increased membrane permeability. The amount of LDH within the medium overlying the cells and within the cells was measured as previously described (Gobbel et al., 1994). The fraction of LDH retained by the cells was determined by dividing the amount of intracellular LDH by the total amount (released plus intracellular) of LDH present in the culture.
Detection of internucleosomal fragmentation of DNA. Neurons and the overlying media from five Petri dishes (100 mm diameter) were placed in ice-cold Tris-buffered saline, pH 7.4, and spun at 500 × g. The pellet was incubated overnight in lysis buffer (10 mm Tris-HCl, 10 mm NaCl, 10 mmEDTA, and 1% SDS) with 300 μg/ml of proteinase K at 50°C. DNA was extracted with phenol:chloroform:isoamyl alcohol (25:24:1) and precipitated overnight at −20°C with 70% alcohol containing 0.3m sodium acetate and 10 mm MgCl2. The precipitate was centrifuged at 12,000 × g for 30 min at 4°C, dried, and suspended in TE buffer (10 mmTris-HCl with 1 mm EDTA, pH 8.0) containing 100 μg/ml of DNase-free RNase for 1 hr at room temperature. Then the DNA was reextracted as before. A 25 μl volume (∼20 μg of DNA) of each sample was separated electrophoretically (100 V for 2 hr) in a 1.8% agarose gel containing TBE buffer (4.5 mm Tris-borate and 1 mm EDTA) with ethidium bromide (2.5 μg/ml).
Cell cycle analysis. The fraction of cells in various phases of the cell cycle was determined by flow cytometry. Propidium iodide (PI) staining was used to quantify DNA content, and bromodeoxyuridine (BrdU) uptake was used to quantify the fraction of cells in S-phase. A 1 mm sterile solution of BrdU in PBS was added to the medium overlying the cells to a final concentration of 10 μm. For pulse-labeling experiments, the cells were collected 1 hr after BrdU addition by trypsinization (0.25% in saline with 0.02% EDTA) and then fixed in 70% ethanol. After centrifugation at 400 × g for 3 min, the cells were resuspended and incubated in 2.5 m HCl with 0.5% Triton X-100 for 30 min. After the cells were washed three times by suspension in wash buffer (0.5% Tween 20 in Ca2+- and Mg2+-free PBS), followed by centrifugation at 400 × g for 5 min, they were exposed to the primary antibody against BrdU (IU-4; Dako, Carpinteria, CA) diluted 1:500 in PBS with 0.5% Tween 20, 1.5% dried milk, and 0.02% sodium azide for 30 min at room temperature, and then washed twice. They were incubated in the dark at room temperature in a solution of PBS with 0.5% Tween 20, 1.5% dried milk, and 0.02% sodium azide that contained a 1:500 dilution of the secondary antibody, a goat anti-mouse antibody conjugated to fluorescein isothiocyanate (FITC; Sigma), for 30 min. After being washed twice, the cells were suspended in a solution of PI (10 μg/ml in PBS) for 30 min in the dark and then filtered through a 37 μm nylon mesh. The amount of BrdU incorporation and PI staining of 10,000 cells was analyzed under dual parameter (FITC vs PI) with a Becton Dickinson FACScan (Becton Dickinson, San Jose, CA). Raw data were refined by plotting the area of the PI fluorescence signal versus width and then selecting only the single cell population for further analysis. Cell cycle phase fractions were estimated by identifying and counting cell populations on a plot of FITC fluorescence versus PI fluorescence, using the CellQuest software package (Becton Dickinson).
A malignant glioma cell line, U251-MG, was used as a positive control for BrdU incorporation by cycling cells. U251-MG cells were maintained in MEM with 10% fetal calf serum and 1% nonessential amino acids. For this experiment BrdU (10 μm) was added to both neurons and U251-MG cells for 0, 1, 6, 12, 24, or 48 hr.
Measurement of DNA strand breaks. Induction and rejoining of DNA double-strand breaks were quantified by subjecting the DNA isolated from the cells, before and after irradiation, to pulsed-field gel electrophoresis (PFGE). Double-strand breaks reduce the molecular weight of the DNA and increase the efficacy with which the DNA can be eluted into an electrophoretic gel. Therefore, the extent of DNA double-strand breaks can be quantified on the basis of the percentage of DNA that is eluted into the gel.
DNA was isolated via a technique that does not involve handling of naked DNA and thus should minimize the artificial introduction of breaks. Cells were irradiated on ice to inhibit DNA repair during the induction of DNA damage. They were collected immediately after irradiation to measure induction of DNA damage or at various times after incubation at 37°C following irradiation to measure repair of DNA damage. The cells were harvested by scraping and were centrifuged at 500 × g for 5 min. The pellet was washed twice by resuspending it in Ca2+- and Mg2+-free PBS and repeating the centrifugation. DNA concentration in the suspension was adjusted to 1–7 μg/ml, based on the binding of the dye To-Pro-1 (Molecular Probes, Eugene, OR), as compared with its binding to a known amount of calf thymus DNA. Bound dye was measured by using a Perkin-Elmer spectrofluorometer (excitation wavelength, 515 nm; emission wavelength, 531 nm). Then the samples were mixed with an equal volume of 2% agar heated to 65°C and were injected into Tygon tubing and placed on ice. The hardened agar was extruded and cut into 5-mm-long plugs. To release the DNA, we placed the plugs overnight in a lysis solution of 1% N-lauroyl sarcosine and 0.1% proteinase K. Then the sample plugs were stored in buffer at 4°C.
Electrophoresis of the plugs was performed on a clamped homogeneous electric field (CHEF; Bio-Rad, Richmond, CA) unit at neutral pH at 14°C for 25 hr by using a pulse size of 60 V and pulse time of 75 min. The gels were stained with ethidium bromide (3 μg/ml in distilled and deionized H2O) and photographed, and the negatives were scanned with a densitometer (Molecular Dynamics, Sunnyvale, CA).
The fraction of DNA eluted into the gel from the plug was used as an index of double-strand breaks. To calculate this index, we subtracted the background fluorescence of an adjacent part of the gel containing no DNA from the fluorescence of the plug and from the fluorescence of the DNA that elutes out of the plug and into the gel. The percentage of DNA remaining in the plug of the irradiated (Ir) samples was normalized to the percentage remaining in the plug of the unirradiated (Un) samples so that the percent retained in the plug after irradiation was given by the formula:
| Equation 1 |
The percentage eluted from the plug because of irradiation was then given as:
| Equation 2 |
Statistical analysis. The fraction of cells undergoing apoptosis or the fraction of LDH retained after the various treatments with radiation was compared by ANOVA with a statistical software package (SuperANOVA Statistical Software, Abacus Concepts, Berkeley, CA), and differences between individual treatment groups were examined with Scheffé’s test. The amount of radiation-induced apoptosis occurring in the presence of benzamide or aminobenzamide was compared with that in the control group at each radiation dose by a test of contrasts. Statistical significance was assigned at thep < 0.05 level.
RESULTS
Induction of DNA double-strand breaks
We first measured the extent of x-ray-induced DNA damage and determined whether there was a difference between astrocytes and neurons. The percentage of DNA eluted by pulsed-field gel electrophoresis increased with radiation dose (Fig.1). The increase was linear such that the percentage eluted approximately doubled for each twofold increase in dose. There was no significant difference in the percentage of DNA eluted out for astrocytes and neurons, indicating that there was no difference between these two cell types in the number of double-strand breaks induced by radiation.
Fig. 1.

The number of DNA double-strand breaks induced by x-irradiation was similar in astrocytes and neurons. Radiation-induced DNA double-strand breaks were quantified on the basis of the percentage of DNA isolated from neurons and astrocytes that eluted into the gel after pulsed-field electrophoresis. There was no significant difference (ANOVA; p > 0.10) between astrocytes and neurons. Each point represents the mean ± SEM of three independent cultures.
Nuclear and DNA fragmentation after irradiation
We next examined the morphological changes induced in astrocytes and neurons after irradiation. Although there were no apparent changes in neuronal or astrocytic cell morphology immediately after irradiation with 32 Gy, by 1 d later the vast majority of the neurons were shrunken, showed evidence of neurite beading, or were detached from the plate (Fig. 2B). In addition, there was an increase in nuclear fragmentation in irradiated neurons (Fig. 2D). There were no apparent changes in the astrocytes at that time (Fig. 2F). Astrocytes also were treated with 0, 2, 8, and 32 Gy and examined 6, 12, 24, 48, and 72 hr later to assure that nuclear fragmentation was not occurring in astrocytes at earlier or later times or in response to other doses than in the neurons. Although there did appear to be an increase in nuclear fragmentation 6–12 hr after irradiation, the incidence was extremely low (<0.5%), as compared with that occurring in the neurons. By 48–72 hr after irradiation, there were occasional (1–2%) astrocytes with swollen or disrupted nuclei, but there was no evidence of increased nuclear fragmentation.
Fig. 2.
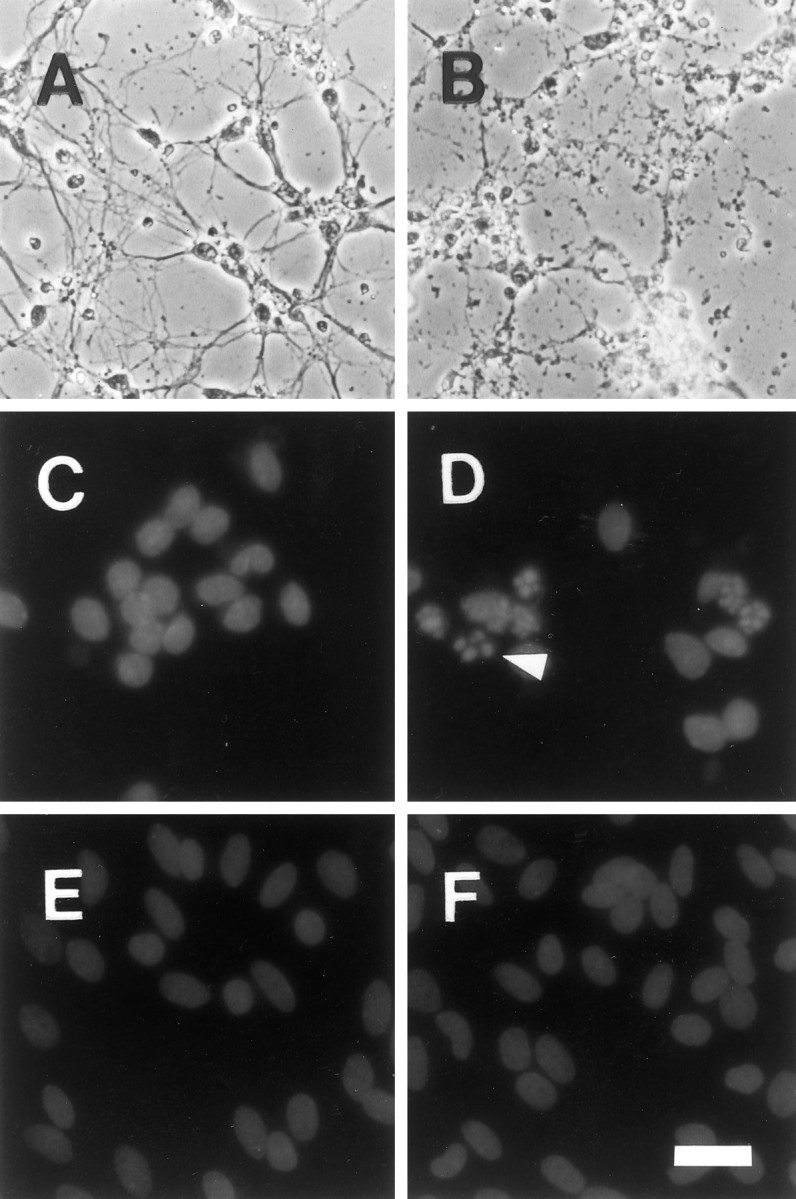
X-irradiation induced cellular degradation and nuclear fragmentation in neurons, but not in astrocytes. Phase-contrast microscopy revealed that irradiation of neurons was followed 24 hr later by neurite beading and cellular shrinkage (B) relative to unirradiated controls (A). In comparison, there were no cellular morphological changes observed in astrocyte cultures by phase-contrast microscopy (data not shown). Irradiation increased the percentage of cells with fragmented nuclei in neuron (D), but not in astrocyte (F), cultures relative to control, unirradiated neuron (C), and astrocyte (E) cultures. The arrowheadindicates a typical example of nuclear fragmentation. Several other apoptotic cells are visible within the irradiated neuronal cultures. Scale bar: A–D, 20 μm; E, F, 25 μm.
Neuronal DNA was isolated and subjected to conventional electrophoresis, and there was a clear ladder-like pattern in the DNA from the irradiated neurons (Fig. 3), indicating that radiation can induce internucleosomal DNA fragmentation, a common characteristic of apoptosis. Because it has been reported that Ara-C can induce apoptosis (Dessi et al., 1995), we tested whether the treatment of neurons with Ara-C during isolation was responsible for their response to radiation. Astrocytes were treated for 24 hr with 10 μm Ara-C and irradiated 1 week later with 32 Gy to parallel the treatment of the neuronal cultures. However, there was still no evidence of apoptosis in the astrocytes on the basis of morphology and Hoechst staining. Furthermore, when the neurons were not treated with Ara-C after isolation, the isolated cells still underwent nuclear fragmentation in response to irradiation, and the DNA isolated from these neurons still exhibited a ladder-like pattern when subjected to electrophoresis (data not shown).
Fig. 3.

X-irradiation induced internucleosomal fragmentation in neuronal DNA. Electrophoretic separation of DNA from neurons irradiated with 32 Gy (32, middle lane) resulted in a ladder-like pattern of DNA fragments that are multiples of ∼200 bp in length. There was only slight evidence of a ladder-like pattern in the control 0 Gy neuronal cultures (0, left lane). A standard (STD, right lane) with DNA lengths that are multiples of 123 bp is shown for comparison.
Time and dose–response of radiation-induced apoptosis
To characterize the time course of the apoptotic response of neurons, we scored the fraction of apoptotic cells at various times after irradiation. There was an increase in apoptotic cells as early as 8 hr after irradiation, with the peak incidence occurring at ∼12 hr (Fig. 4A). By 24 hr, the fraction of cells displaying nuclear fragmentation had diminished. The time course was similar regardless of radiation dose (Fig.4A), but the percentage of cells undergoing apoptosis appeared to be dose-dependent. Although the magnitude of the differences between the individual dose groups varied depending on the time of evaluation, when the response over the entire 24 hr period was assessed (Sheffé’s test), there was a significant increase in apoptotic neurons after as little as 2 Gy, and the apoptotic fraction was significantly greater after 32 than after 2 Gy. When cells were treated with 10 μm cycloheximide 5–10 min before 8 Gy to inhibit protein synthesis, there was a significant reduction in the level of radiation-induced apoptosis (Fig. 4A). Cycloheximide was able to prevent radiation-induced apoptosis even when it was added up to 6 hr after irradiation (Fig. 4B), just before the onset of apoptosis (Fig. 4A).
Fig. 4.

Radiation-induced apoptosis was dependent on time after irradiation and radiation dose, and it was inhibited by cycloheximide. A, The fraction of cells in neuronal cultures undergoing apoptosis after irradiation was time- and dose-dependent (ANOVA; p < 0.001). The fraction of cells undergoing apoptosis began to increase between 4 and 8 hr after irradiation, peaked at ∼12 hr, and decreased by 24 hr. Cycloheximide (CHX; 10 μm added just before irradiation) significantly diminished the apoptotic response that followed 8 Gy (ANOVA; p < 0.001). Apoptotic fraction was determined by staining the cultures for nuclear fragmentation with Hoechst 33258 dye. Each point represents the mean ± SEM determined from four cultures, except that n= 3 at 8 hr after irradiation for the 8 and 32 Gy treatment groups.B, Cycloheximide (10 μm) prevented apoptosis even when added up to 6 hr after 8 Gy. Cycloheximide was added once at 0–10 hr after irradiation, and the fraction of neurons undergoing apoptosis was scored at 12 hr after irradiation in all cases. The apparent decrease in apoptosis in the control cultures treated with saline at >4 hr after irradiation likely was related to decreased adherence of apoptotic cells so that some cells were dislodged and lost into the medium during saline treatment. Eachpoint represents the mean ± SEM of four cultures.
Because the increase in apoptosis from 2 to 8 Gy 12 hr after irradiation was substantially greater than the increase from 8 to 32 Gy, we evaluated the apoptotic response to variations in radiation dose more carefully. We chose to use release of LDH as a measure of the proportion of cells undergoing apoptosis instead of using neuronal morphology because of our finding that many of the cells undergoing apoptosis detach from the plate. Because the neuronal morphology of detached cells cannot be determined, measures of the fraction of apoptosis based on neuronal morphology may underestimate the true amount of apoptosis induced. The fraction of apoptosis induced by irradiation did not increase continually with increasing dose on the basis of measurements of LDH retention and release. The fraction of LDH retained in the neuronal cultures at 24 hr after irradiation fell exponentially to ∼0.7 from 0 to 16 Gy (Fig.5) and then decreased at a much slower rate (from 0.7 to 0.6) with increasing radiation doses from 16 to 64 Gy. The decrease in LDH retention in response to radiation doses up to 32 Gy could be eliminated by the previous administration of cycloheximide (10 μm; data not shown), suggesting that the response was attributable to apoptosis.
Fig. 5.

The fraction of lactate dehydrogenase (LDH) retained intracellularly decreased exponentially at a relatively rapid rate from 0 to 16 Gy and then more slowly from 16 to 64 Gy. To compensate for LDH release in control cultures, we normalized the fraction of LDH retained after irradiation to the level in control (0 Gy) cultures. A double-exponential curve was fit to the data by minimizing the squared differences between the fit and the experimental data. The resultant equation was fraction LDH retained = 0.29e−0.204D + 0.74e−0.004D, where D was the radiation dose in Gy. Each point represents the mean ± SEM of four cultures.
Cell cycle analysis by flow cytometry
We used flow cytometry to determine whether the isolated neurons were postmitotic. Because most studies on apoptosis involve cycling cells, we wanted to know if the neurons used in the present study, which were isolated from embryonic tissue, might contain some primordial, dividing cells or neuronal precursors. Our analysis revealed that most of the isolated neurons were noncycling. The proportion of cells in S and G2/M combined was relatively low at 3.3 ± 0.7% (mean ± SD; n = 2) in unirradiated neuronal cultures. In comparison, the proportion of S and G2/M cells in the astrocyte cultures was 12.6 ± 0.6% (n = 2). Continuous labeling with BrdU for up to 48 hr also indicated that the neurons were predominantly noncycling. Whereas there was >90% labeling with BrdU in the U251-MG tumor cell line by 24 hr, only 2.5 and 8.1% of the neurons were labeled at 24 and 48 hr, respectively (Fig. 6). To determine whether irradiation might stimulate the neurons to enter into the cell cycle, possibly leading to mitotic catastrophe, we examined the neuronal cultures up to 12 hr after 32 Gy. However, there was relatively little change in the proportion of cells in the various phases of the cell cycle. At 2, 6, and 12 hr after irradiation the percentages of cells in S and G2/M remained relatively low at 5.8, 3.8, and 4.3%, respectively.
Fig. 6.

Bromodeoxyuridine labeled virtually all U251-MG cells but very few neurons. The percentage of U251-MG cells, a malignant glioma cell line, that incorporated bromodeoxyuridine (BrdU), increased rapidly, such that by 24 hr >90% of the cells were labeled with anti-BrdU antibodies as determined by flow cytometry. In contrast, there was very little evidence of BrdU incorporation by neurons, and only 8.1% of the cells showed evidence of BrdU incorporation even after 48 hr of exposure to 10 μm BrdU. Eachpoint represents the proportion of a sample of 10,000 cells.
DNA double-strand break rejoining after irradiation
We next determined whether there was a difference in how the astrocytes and neurons rejoined radiation-induced strand breaks, because the induction of damage could be important not only in the response to irradiation but also in the ability of the cell to respond to such damage. Neurons and astrocytes were irradiated with 32 Gy to produce a sufficient number of double-strand breaks and to allow us to detect accurately the changes in the number of breaks remaining at various times after irradiation. Although there was no significant difference in induction of DNA double-strand breaks in neurons, as compared with that in astrocytes (see Fig. 1), there was a difference in how the cells rejoined the breaks (Fig.7). In the astrocytes there was a decline in the percentage of DNA eluted as early as 5 min after irradiation, indicating that there was rejoining of double-strand breaks. In contrast, there was little or no evidence of double-strand break rejoining in the neurons until 6 hr after irradiation.
Fig. 7.

Rejoining of DNA double-strand breaks was slower in neurons than in astrocytes. The percentage of initial double-strand breaks remaining at 5, 60, and 360 min after 32 Gy was significantly lower in astrocytes than neurons (ANOVA; p < 0.001), indicating that astrocytes rejoin DNA strand breaks more rapidly than neurons. Each point represents the mean ± SEM of three cultures, except for the point representing neurons at 5 min, in which n = 2.
Effect of inhibition of ADP-ribosylation
We next determined if the induction of neuronal apoptosis by radiation might be related to the stimulation of ADP-ribosylation. DNA damage stimulates ADP-ribosylation, which can deplete ATP and NADPH and exacerbate neuronal injury (Zhang et al., 1994). Two inhibitors of ADP-ribosylation, benzamide and aminobenzamide, were added, and the percentage of neurons undergoing apoptosis after irradiation was determined. Treatment with benzamide, the more potent of the two inhibitors (Banasik et al., 1992), actually caused a significant increase in the number of neurons undergoing apoptosis in response to radiation (Fig. 8A), whereas aminobenzamide had no apparent effect. To determine whether the severity of injury because of radiation might be overcoming any protective effects of inhibition of ADP-ribosylation, we repeated the experiment with doses of 0, 1, 2, and 4 Gy and a single dose (0.5 mm) of benzamide and aminobenzamide. As before, only benzamide had an effect, and it significantly increased the number of neurons undergoing apoptosis in response to irradiation (Fig.8B). The effect of benzamide on radiation-induced neuronal apoptosis also was examined at 12 hr after irradiation, when the incidence of nuclear fragmentation appeared to be maximal (see Fig.4A). The neurons used in this experiment were not treated with Ara-C after isolation, because preliminary experiments examining LDH release and nuclear fragmentation after irradiation suggested that untreated cells were more sensitive to radiation, and we postulated that they also might be more sensitive to benzamide-induced alterations in radiation sensitivity. At 12 hr after 0, 2, 4, or 6 Gy the percentage of neurons that were not treated with benzamide and had fragmented nuclei was 1.5 ± 1.0%, 23.3 ± 6.2%, 45.3 ± 14.8%, and 46.9 ± 12.8% (mean ± SD; n= 6), respectively. Treatment with 0.5 mm benzamide did not appear to alter the percentage of cells with nuclear fragmentation in unirradiated cultures (1.6 ± 1.3%), but it did significantly increase (ANOVA; p < 0.025) the percentage of neurons undergoing nuclear fragmentation after 2, 4, or 6 Gy (33.1 ± 7.1%, 52.3 ± 9.7%, 55.2 ± 8.5%, respectively;n = 6).
Fig. 8.

Benzamide, an ADP-ribosylation inhibitor, exacerbated radiation-induced apoptosis of neurons. The percentage of cells undergoing apoptosis at 8 hr after 4–8 Gy (A) was increased significantly by benzamide (ANOVA; p < 0.005), but not by aminobenzamide. The significant effect of benzamide on radiation-induced apoptosis and lack of effect by aminobenzamide was also present when tested at lower radiation doses of 1–4 Gy (B; ANOVA;p < 0.05). Each bar represents the mean ± SEM of three (A) or four (B) cultures, except for controls inA (n = 6). From 100 to 200 cells per culture were examined for the presence of nuclear fragmentation typical of apoptosis. The same controls (0 mm drug) were used for both benzamide and aminobenzamide, and bars representing the mean ± SEM of the control values are repeated in bothleft and right graphs of Afor ease of comparison. An asterisk indicates that the fraction of apoptotic cells is significantly different from controls by a test of contrasts.
Effect of splitting radiation dose
We hypothesized that benzamide might exacerbate radiation damage by interfering with the ADP-ribosylation needed for DNA repair. Furthermore, if the slow rate of repair in neurons relative to astrocytes was contributing to the apoptotic response, then reducing the rate at which the radiation was delivered should decrease the amount of apoptosis. Neurons that had not been treated with Ara-C were irradiated with doses of 0, 1, 2, or 4 Gy that were split into two equal fractions delivered 4 hr apart. The percentage of LDH retained by these cells 22 hr after the second radiation dose was compared with the LDH retention of cells that had been irradiated with a single dose of 0, 1, 2, or 4 Gy either 22 or 26 hr earlier. There was no consistent or significant difference between the 22 and 26 hr controls (data not shown). However, in the cells in which the radiation dose was split by 4 hr to allow time for repair of the damage induced by the initial treatment, there was significantly less damage than in the 22 and 26 hr controls (Fig. 9).
Fig. 9.

When the radiation dose was split into equal fractions separated by 4 hr to allow for repair of the initial damage induced by the first dose, the fraction of lactate dehydrogenase (LDH) retained intracellularly after irradiation was increased significantly (ANOVA; p < 0.001). LDH retention was measured 22 hr after the second dose of irradiation for the split dose group, and it was measured both 22 and 26 hr after irradiation in the single dose group. There was no significant difference between the 22 and 26 hr dose groups, and the data were combined for the graph. The graph represents one result from three repetitions of this experiment. Each point represents the mean ± SEM of 12 cultures. An asteriskindicates that the fraction of LDH retained after a specific dose of radiation was significantly different between the single dose and the split dose groups.
DISCUSSION
Our data demonstrate that x-irradiation can induce apoptosis in noncycling neurons. This conclusion is based on the finding that radiation induced nuclear fragmentation typical of apoptosis. Furthermore, electrophoretic separation of DNA isolated from irradiated cells showed evidence of the ladder pattern that is associated with internucleosomal cleavage of DNA, a characteristic of apoptosis. Last, nuclear fragmentation after irradiation was abolished by cycloheximide, suggesting that the reaction to irradiation was an active process requiring de novo protein synthesis.
This is the first report to our knowledge to characterize the radiation-induced apoptotic response of neurons growing in vitro. There have been reports of radiation-induced apoptosis in the CNS, although the cell types undergoing apoptosis were not identified clearly. Ferrer (1992) noted that radiation-induced apoptosis occurred within the cerebral cortical gray matter of young (postnatal days 0–15), but not adult, rats, and it was suggested that both neurons and glia were involved. An earlier study of apoptosis within the developing rat forebrain suggested that the radiosensitive population consisted primarily of cells that were nearing cell division or recently had completed mitosis and were beginning to differentiate (Hicks et al., 1961). Our previous report (Bellinzona et al., 1996) demonstrated that radiation can induce apoptosis even in adult rats, but the apoptosis is confined to cells in the subependymal region, a mitotically active area that generates glial and neuronal progenitors (Lewis, 1969; Lois and Alvarez-Buylla, 1993). Our present results demonstrate that in vitro even neurons that are well differentiated with extensive neuritic processes undergo radiation-induced apoptosis.
The finding that radiation induces apoptosis in neurons is unusual in that apoptosis generally is confined to proliferating tissues (Meikrantz and Schlegel, 1995). Our cell cycle analysis indicated that most of our neurons were postmitotic. The only other postmitotic cells that have been reported to undergo radiation-induced apoptosis are mature lymphoid cells (Yamada et al., 1981) and acinar cells of salivary glands (Stephens et al., 1991). We considered whether radiation-induced DNA damage might induce the neurons to reenter the cell cycle—many of the morphological features of apoptosis are similar to those present after mitotic catastrophe, which can occur when a cell is stimulated to undergo mitosis prematurely (Evan et al., 1995). Additionally, it has been reported that at least one factor involved in the cell cycle, cyclin D1, is upregulated during the programmed cell death of sympathetic neurons (Freeman et al., 1994). However, we found that, although BrdU labeling increased slightly from 3.3% before irradiation to 3.8–5.3% after 32 Gy, this increase was much less than the percentage of cells undergoing apoptosis in response to this dose of radiation (see Fig. 4A). These findings suggest that radiation probably does not stimulate neurons to reenter the cell cycle or at least not sufficiently to stimulate DNA synthesis.
The percentage of neurons undergoing apoptosis appeared to be dose-dependent up to ∼16 Gy. The fraction of nonapoptotic cells decreased at a relatively rapid rate from 0 to 16 Gy (see Fig. 5). An exponential decrease was expected because x-rays interact with matter stochastically so that the chances of a cell surviving generally should decrease exponentially as dose increases (Johns and Cunningham, 1983). However, the decrease in viability as dose increased from 16 to 64 Gy was comparatively small. Although the percentage of LDH retention never fell to 0%, preliminary experimental findings (data not shown) with a staining method that distinguishes viable from nonviable cells (Gobbel et al., 1994) indicate that doses of 32 Gy result in virtually complete cell killing.
X-rays generate free radicals (Hall, 1988) and cause cellular damage in a manner similar to other types of oxidative stress. Our data thus support previous reports showing that increased oxidative stress, including depletion of the antioxidant glutathione (Ratan et al., 1994) and administration of the pro-oxidant hydrogen peroxide (Whittemore et al., 1994), can lead to neuronal apoptosis. Exposure to hydrogen peroxide resulted in apoptosis only after a delay of 3–4 hr. Likewise, x-rays induced apoptosis only after a delay of >4 hr, suggesting that x-rays and hydrogen peroxide-induced oxidative stress may induce a similar set of cellular events leading to neuronal apoptosis.
Our finding that cycloheximide inhibits apoptosis even when added after irradiation (see Fig. 4B) suggests that some proteins needed for apoptosis are not synthesized until >6 hr after radiation exposure. However, Ratan and colleagues (Ratan et al., 1994) suggested that effects of cycloheximide on apoptosis might be attributable to decreased incorporation of cysteine into proteins and increased incorporation into glutathione, resulting in elevated glutathione levels and reduced damage from oxidative stress. Because free radicals generated by x-rays have half-lives on the order of nanoseconds (Hall, 1988), the protective effects of cycloheximide in our system cannot be attributed to effects of antioxidants on the initial radiation-induced oxidative burst.
The apoptotic response of neurons to x-rays was most likely a result of DNA damage, because there is considerable evidence that radiation-induced cell killing is attributable to such damage. For example, cells are much more sensitive to radiation targeted at the nucleus than to radiation targeted at the cytoplasm or plasma membrane (Marin and Bender, 1963; Munro, 1970). Furthermore, theoretical considerations suggest that it is unlikely that lipid or proteins would account for the response to x-rays, because doses up to 100 Gy would damage only ∼0.01% of these molecules (Bump and Brown, 1990). Our findings suggest that DNA damage may be a common pathway for the induction of apoptosis by oxidative stress. Other types of oxidative stress, such as ischemia and reperfusion, can cause apoptosis within the brain (MacManus et al., 1993; Hill et al., 1995; Islam et al., 1995; Li et al., 1995; Linnik et al., 1995; Du et al., 1996), and Liu et al. (1996) have demonstrated that cerebral ischemia and reperfusion can cause DNA alterations in the brain.
If DNA damage is responsible for the apoptotic response in neurons, then the genetic program induced by DNA damage and/or the generation and processing of the damage must be different in astrocytes. We focused in the present study on differences in the processing of DNA damage, specifically rejoining of DNA double-strand breaks, because the number of double-strand breaks remaining after irradiation is one of the best predictors of chromosomal aberrations (Carrano, 1979; Wlodek and Hittelman, 1988). Additionally, there is a direct correlation between the number of radiation-induced chromosomal aberrations and lethality (Hall, 1988). Although there was no apparent difference in the induction of DNA double-strand by x-rays in astrocytes versus neurons, neurons were significantly slower at rejoining the breaks. Furthermore, inhibiting ADP-ribosylation, which can slow the rejoining of DNA strand breaks (Ding et al., 1992; Palomba et al., 1995), exacerbated radiation-induced apoptosis. This finding contrasts with that of Zhang and colleagues, who reported that ADP-ribosylation inhibitors protected against neuronal injury induced by activation of NMDA receptors (Zhang et al., 1994). Their result suggested that DNA damage resulting from NMDA receptor activation leads to ADP-ribosylation, leading to depletion of ATP and NADPH. Our results suggest that in our system the deleterious effects on DNA repair resulting from inhibition of ADP-ribosylation may be greater than the benefits resulting from preventing a decrease in energy stores. This possibility would explain why splitting the dose of radiation, which should allow for the repair of sublethal events induced by the initial radiation, produced an apparent decrease in the number of cells undergoing apoptosis as quantified by LDH retention (Fig. 9). However, we cannot rule out the possibility that the first dose may have induced the cells to be resistant to subsequent doses.
Our results suggesting that neurons are slower at rejoining DNA strand breaks are supported by the findings of Wang and Wheeler, who reported that there was slower repair of single-strand breaks by rat cerebellar cells than by 9L brain tumor cells (Wang and Wheeler, 1978). Unfortunately, the use of whole tissue in that study made it impossible to verify whether the slow repair was attributable to the neurons or some other cell type. However, Hanawalt and colleagues reported that repair of cyclobutane dimers within genomic DNA after ultraviolet irradiation was slower in neuron-like PC12 cells induced to differentiate by nerve growth factor (Hanawalt et al., 1992). They also found that repair rates actually were enhanced in induced genes and decreased only in inactive genes, which suggests that a reduction in the overall transcription of the DNA in the differentiating cells might account for the relatively slow repair of the genome taken as a whole. If the repair rate of double-strand breaks within a region of DNA is dependent on transcriptional activity, then a generalized reduction in transcription within the neurons also might account for the slow repair in those cells. Unfortunately, there are no known methods to determine whether double-strand break repair is enhanced in actively transcribed regions of the genome. Another possible explanation for the decreased rate of repair in the neurons is that the activity of the so-called DNA-dependent protein kinase (DNA-PK), an enzyme required for rejoining of DNA double-strand breaks (Jeggo et al., 1995), might be decreased. Decreases in DNA-PK activity are associated with increases in sensitivity to radiation and also with defects in rejoining (Lee et al., 1995).
We cannot yet say definitively whether there is a connection between induction and repair of DNA damage and neuronal apoptosis. It is possible that rejoining may appear to be slow in neurons after irradiation because apoptotic cells may not repair double-strand breaks at all or because the DNA fragmentation associated with the apoptotic process may start very soon after irradiation. This possibility has been suggested as a potential cause of the slow repair of strand breaks in a lymphoblastic cell line, TK6, which is also subject to radiation-induced apoptosis (Olive and Banáth, 1993).
Radiation-induced apoptosis of neurons should provide a useful model to better understand the molecular changes that result in apoptosis. Our current findings indicate that neurons process DNA damage differently than astrocytes do, which supports the role of DNA damage as a possible stimulus of neuronal injury and apoptosis. Further work is needed to determine the types of DNA damage that may act as stimuli, why the processing of DNA damage by neurons is slow, and the molecular mechanisms by which DNA damage is translated into a signal for apoptosis.
Footnotes
This work was supported by National Institutes of Health Grants NS 35782, CA 13525, NS 14543, NS 25372, and AG 08938.
Correspondence should be addressed to Dr. Glenn T. Gobbel, Health Sciences West 783, Box 0520, University of California, 505 Parnassus Avenue, San Francisco, CA 94143.
REFERENCES
- 1.Banasik M, Komura H, Shimoyama M, Ueda K. Specific inhibitors of poly(ADP-ribose) synthetase and mono(ADP-ribosyl) transferase. J Biol Chem. 1992;267:1569–1575. [PubMed] [Google Scholar]
- 2.Bellinzona M, Gobbel GT, Shinohara C, Fike JR. Apoptosis is induced in the subependyma of young adult rats by ionizing radiation. Neurosci Lett. 1996;208:163–166. doi: 10.1016/0304-3940(96)12572-6. [DOI] [PubMed] [Google Scholar]
- 3.Bowling AC, Schulz JB, Brown RH, Jr, Beal MF. Superoxide dismutase activity, oxidative damage, and mitochondrial energy metabolism in familial and sporadic amyotrophic lateral sclerosis. J Neurochem. 1993;61:2322–2325. doi: 10.1111/j.1471-4159.1993.tb07478.x. [DOI] [PubMed] [Google Scholar]
- 4.Buettner GR. The pecking order of free radicals and antioxidants: lipid peroxidation, alpha-tocopherol, and ascorbate. Arch Biochem Biophys. 1993;300:535–543. doi: 10.1006/abbi.1993.1074. [DOI] [PubMed] [Google Scholar]
- 5.Bump E, Brown J. Role of glutathione in the radiation response of mammalian cells in vitro and in vivo. Pharmacol Ther. 1990;47:117–136. doi: 10.1016/0163-7258(90)90048-7. [DOI] [PubMed] [Google Scholar]
- 6.Carrano AV. Chromosome aberrations and radiation-induced cell death. II. Predicted and observed cell survival. Mutat Res. 1979;17:355–366. doi: 10.1016/0027-5107(73)90007-9. [DOI] [PubMed] [Google Scholar]
- 7.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 8.Chopp M, Chan PH, Hsu CY, Cheung ME, Jacobs TP. DNA damage and repair in central nervous system injury: National Institute of Neurological Disorder and Stroke workshop summary. Stroke. 1996;27:363–369. doi: 10.1161/01.str.27.3.363. [DOI] [PubMed] [Google Scholar]
- 9.Dessi F, Pollard H, Moreau J, Ben-Ari Y, Charriaut-Marlangue C. Cytosine arabinoside induces apoptosis in cerebellar neurons in culture. J Neurochem. 1995;64:1980–1987. doi: 10.1046/j.1471-4159.1995.64051980.x. [DOI] [PubMed] [Google Scholar]
- 10.Ding R, Pommier Y, Kang VH, Smulson M. Depletion of poly(ADP-ribose) polymerase by antisense RNA expression results in a delay in DNA strand break rejoining. J Biol Chem. 1992;267:12804–12812. [PubMed] [Google Scholar]
- 11.Dizdaroglu M. Oxidative damage to DNA in mammalian chromatin. Mutat Res. 1992;275:331–342. doi: 10.1016/0921-8734(92)90036-o. [DOI] [PubMed] [Google Scholar]
- 12.Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischemia: a role for apoptosis? J Cereb Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Evan GI, Brown L, Whyte M, Harrington E. Apoptosis and the cell cycle. Curr Opin Cell Biol. 1995;7:825–834. doi: 10.1016/0955-0674(95)80066-2. [DOI] [PubMed] [Google Scholar]
- 14.Ferrer I. The effect of cycloheximide on natural and x-ray-induced cell death in the developing cerebral cortex. Brain Res. 1992;588:351–357. doi: 10.1016/0006-8993(92)91599-a. [DOI] [PubMed] [Google Scholar]
- 15.Floyd RA, Carney JM (1992) Free radical damage to protein and DNA: mechanisms involved and relevant observations on brain undergoing oxidative stress. Ann Neurol 32[Suppl]:S22–S27. [DOI] [PubMed]
- 16.Freeman RS, Estus S, Johnson EM. Analysis of cell cycle-related gene expression in postmitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 17.Gillardon F, Wickert H, Zimmermann M. Up-regulation of bax and down-regulation of bcl-2 is associated with kainate-induced apoptosis in mouse brain. Neurosci Lett. 1995;192:85–88. doi: 10.1016/0304-3940(95)11619-8. [DOI] [PubMed] [Google Scholar]
- 18.Gobbel GT, Chan TY-Y, Gregory GA, Chan PH. Response of cerebral endothelial cells to hypoxia: modification by fructose-1,6-bisphosphate but not glutamate receptor antagonists. Brain Res. 1994;653:23–30. doi: 10.1016/0006-8993(94)90367-0. [DOI] [PubMed] [Google Scholar]
- 19.Greenlund LJS, Deckwerth TL, Johnson EM. Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 20.Gunasekar PG, Kanthasamy AG, Borowitz JL, Isom GE. NMDA receptor activation produces concurrent generation of nitric oxide and reactive oxygen species: implication for cell death. J Neurochem. 1995;65:2016–2021. doi: 10.1046/j.1471-4159.1995.65052016.x. [DOI] [PubMed] [Google Scholar]
- 21.Hall EJ. Radiobiology for the radiologist, 3rd Ed. Lippincott; Philadelphia: 1988. [Google Scholar]
- 22.Hanawalt PC, Gee P, Ho L, Hsu RK, Kane CJ. Genomic heterogeneity of DNA repair. Role in aging? Ann NY Acad Sci. 1992;663:17–25. doi: 10.1111/j.1749-6632.1992.tb38644.x. [DOI] [PubMed] [Google Scholar]
- 23.Hicks SP, D’Amato CJ, Coy MA, O’Brien ED, Thruston JM, Foftes DL. Fundamental aspects of radiosensitivity: Brookhaven symposium on biology, number 14, pp 246–261. Brookhaven National Laboratory; Upton, NY: 1961. Migrating cells in the developing nervous system studied by their radiosensitivity and tritiated thymidine uptake. [PubMed] [Google Scholar]
- 24.Hill IE, MacManus JP, Rasquinha I, Tuor UI. DNA fragmentation indicative of apoptosis following unilateral cerebral hypoxia-ischemia in the neonatal rat. Brain Res. 1995;676:398–403. doi: 10.1016/0006-8993(95)00145-g. [DOI] [PubMed] [Google Scholar]
- 25.Islam N, Aftabuddin M, Moriwaki A, Hori Y. Detection of DNA damage induced by apoptosis in the rat brain following incomplete ischemia. Neurosci Lett. 1995;188:159–162. doi: 10.1016/0304-3940(95)11419-w. [DOI] [PubMed] [Google Scholar]
- 26.Jeggo PA, Taccioli GE, Jackson SP. Menage á trois: double-strand break repair, V(D)J recombination, and DNA-PK. BioEssays. 1995;17:949–957. doi: 10.1002/bies.950171108. [DOI] [PubMed] [Google Scholar]
- 27.Johns HE, Cunningham JR. The physics of radiology, 4th Ed. Thomas; Springfield, IL: 1983. [Google Scholar]
- 28.Kumar U, Dunlop DM, Richardson JS. The acute neurotoxic effect of beta-amyloid on mature cultures of rat hippocampal neurons is attenuated by the anti-oxidant U-78517F. Int J Neurosci. 1994;79:185–190. doi: 10.3109/00207459408986079. [DOI] [PubMed] [Google Scholar]
- 29.Lee SE, Pulaski CR, He DM, Benjamin DM, Voss M, Um J, Hendrickson EA. Isolation of mammalian cell mutants that are x-ray-sensitive, impaired in DNA double-strand break repair and defective for V(D)J recombination. Mutat Res. 1995;336:279–291. doi: 10.1016/0921-8777(95)00002-2. [DOI] [PubMed] [Google Scholar]
- 30.Lewis PD. A quantitative study of cell proliferation in the subependymal layer of the adult rat brain. Exp Neurol. 1969;20:203–207. doi: 10.1016/0014-4886(68)90093-9. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Sharov VG, Jiang N, Zaloga C, Sabbah HN, Chopp M. Ultrastructural and light microscopic evidence of apoptosis after MCA occlusion in the rat. Am J Pathol. 1995;146:1045–1051. [PMC free article] [PubMed] [Google Scholar]
- 32.Linnik MD, Miller JA, Sprinkle-Cavallo J, Mason PJ, Thompson FY, Montgomery LR, Schroeder KK. Apoptotic DNA fragmentation in the rat cerebral cortex induced by permanent middle cerebral artery occlusion. Brain Res Mol Brain Res. 1995;32:116–124. doi: 10.1016/0169-328x(95)00069-5. [DOI] [PubMed] [Google Scholar]
- 33.Liu PK, Hsu CY, Dizdaroglu M, Floyd RA, Kow YW, Karakaya A, Rabow LE, Cui JK. Damage, repair, and mutagenesis in nuclear genes after mouse forebrain ischemia–reperfusion. J Neurosci. 1996;16:6795–6806. doi: 10.1523/JNEUROSCI.16-21-06795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lois C, Alvarez-Buylla A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc Natl Acad Sci USA. 1993;90:2074–2077. doi: 10.1073/pnas.90.5.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacManus JP, Buchan AM, Hill IE, Rasquinha I, Preston E. Global ischemia can cause DNA fragmentation indicative of apoptosis in rat. Neurosci Lett. 1993;164:89–92. doi: 10.1016/0304-3940(93)90864-h. [DOI] [PubMed] [Google Scholar]
- 37.Marin G, Bender MA. A comparison of mammalian cell-killing by incorporated H3-thymidine and H3-uridine. Int J Radiat Biol. 1963;7:235–244. doi: 10.1080/09553006314551151. [DOI] [PubMed] [Google Scholar]
- 38.Meikrantz W, Schlegel R. Apoptosis and the cell cycle. J Cell Biochem. 1995;58:160–174. doi: 10.1002/jcb.240580205. [DOI] [PubMed] [Google Scholar]
- 39.Mitchell IJ, Lawson S, Moser B, Laidlaw SM, Cooper AJ, Walkinshaw G, Waters CM. Glutamate-induced apoptosis results in a loss of striatal neurons in the parkinsonian rat. Neuroscience. 1994;63:1–5. doi: 10.1016/0306-4522(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 40.Munro TR. The relative radiosensitivity of the nucleus and cytoplasm of Chinese hamster fibroblasts. Radiat Res. 1970;42:451–470. [PubMed] [Google Scholar]
- 41.Olive PL, Banáth JP. Induction and rejoining of radiation-induced DNA single-strand breaks: “tail moment” as a function of position in the cell cycle. Mutat Res. 1993;294:275–283. doi: 10.1016/0921-8777(93)90010-e. [DOI] [PubMed] [Google Scholar]
- 42.Palomba L, Guidarelli A, Cattabeni F, Cantoni O. The effect of hydrogen peroxide/liter–histidine-induced DNA single- vs double-strand breaks on poly(ADP-ribose) polymerase. Eur J Pharmacol. 1995;291:167–173. doi: 10.1016/0922-4106(95)90139-6. [DOI] [PubMed] [Google Scholar]
- 43.Pollard H, Charriaut-Marlangue C, Catagrel S, Represa A, Robain O, Moreau H, Ben-Ari Y. Kainate-induced cell death in hippocampal neurons. Neuroscience. 1994;63:7–18. doi: 10.1016/0306-4522(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 44.Portera-Cailliau C, Hedreen JC, Price DL, Koliatsos V. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal model. J Neurosci. 1995;15:3775–3787. doi: 10.1523/JNEUROSCI.15-05-03775.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- 46.Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- 47.Stephens LC, Schultheiss TE, Price RE, Ang KK, Peters LJ. Radiation apoptosis of serous acinar cells of salivary and lacrimal glands. Cancer. 1991;67:1539–1543. doi: 10.1002/1097-0142(19910315)67:6<1539::aid-cncr2820670613>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 48.Troy CM, Shelanski ML. Down-regulation of copper/zinc superoxide dismutase causes apoptotic death in PC12 neuronal cells. Proc Natl Acad Sci USA. 1994;91:6384–6387. doi: 10.1073/pnas.91.14.6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Villa P, Miehe M, Sensenbrenner M, Pettman B. Synthesis of specific proteins in trophic factor-deprived neurons undergoing apoptosis. J Neurochem. 1994;62:1468–1475. doi: 10.1046/j.1471-4159.1994.62041468.x. [DOI] [PubMed] [Google Scholar]
- 50.Wang TS, Wheeler KT. Repair of x-ray-induced DNA damage in rat cerebellar neurons and brain tumor cells. Radiat Res. 1978;73:464–475. [PubMed] [Google Scholar]
- 51.Whittemore ER, Loo DT, Cotman CW. Exposure to hydrogen peroxide induces cell death via apoptosis in cultured rat cortical neurons. NeuroReport. 1994;5:1485–1488. doi: 10.1097/00001756-199407000-00019. [DOI] [PubMed] [Google Scholar]
- 52.Wlodek D, Hittelman WN. The relationship of DNA and chromosome damage to survival of synchronized x-irradiated L5178Y cells. I. Initial damage. Radiat Res. 1988;115:550–565. [PubMed] [Google Scholar]
- 53.Wyllie AH, Kerr JFR, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 54.Yamada T, Ohymama H, Kinjo Y, Watanabe M. Evidence for the internucleosomal breakage of chromatin in rat thymocytes irradiated in vitro. Radiat Res. 1981;85:544–553. [PubMed] [Google Scholar]
- 55.Yan SD, Yan SF, Chen X, Fu J, Chen M, Kuppusamy P, Smith MA, Perry G, Godman GC, Nawroth P, Zweier JL, Stern D. Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid beta-peptide. Nat Med. 1995;1:693–699. doi: 10.1038/nm0795-693. [DOI] [PubMed] [Google Scholar]
- 56.Yu ACH, Chan PH, Fishman RA. Effects of arachidonic acid on glutamate and γ-aminobutyric acid uptake in primary cultures of rat cerebral cortical astrocytes and neurons. J Neurochem. 1986;47:1181–1189. doi: 10.1111/j.1471-4159.1986.tb00738.x. [DOI] [PubMed] [Google Scholar]
- 57.Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]