

Abstract
Unilateral limb-bud removal (LBR) before the outgrowth of sensory or motor neurons to the leg of chick embryos was used to examine the role of limb (target)-derived signals in the development and survival of lumbar motoneurons and sensory neurons in the dorsal root ganglia (DRG). After LBR, motor and sensory neurons underwent normal initial histological differentiation, and cell growth in both populations was unaffected. Before their death, target-deprived motoneurons also expressed a cell-specific marker, the homeodomain protein islet-1. Proliferation of sensory and motor precursor cells was also unaffected by LBR, and the migration of neural crest cells to the DRG and of motoneurons into the ventral horn occurred normally. During the normal period of programmed cell death (PCD), increased numbers of both sensory and motor neurons degenerated after LBR. However, whereas motoneuron loss increased by 40–50% (90% total), only ∼25% more sensory neurons degenerated after LBR. A significant number of the surviving sensory neurons projected to aberrant targets in the tail after LBR, and many of these were lost after ablation of both the limb and tail. Treatment with neurotrophic factors (or muscle extract) rescued sensory and motor neurons from cell death after LBR without affecting precursor proliferation of either population. Activity blockade with curare failed to rescue motoneurons after LBR, and combined treatment with curare plus muscle extract was no more effective than muscle extract alone. Treatment with the antioxidantN-acetylcysteine rescued motoneurons from normal cell death but not after LBR. Two specific inhibitors of the interleukin β1 converting enzyme (ICE) family of cysteine proteases also failed to prevent motoneuron death after LBR. Taken together these data provide definitive evidence that the loss of spinal neurons after LBR cannot be attributed to altered proliferation, migration, or differentiation. Rather, in the absence of limb-derived trophic signals, the affected neurons fail to survive and undergo PCD. Although normal cell death and cell death after target deprivation share many features in common, the intracellular pathways of cell death in the two may be distinct.
Keywords: cell death, chick embryo, spinal cord, motoneurons, sensory neurons, target deprivation, trophic factors
The regulation of vertebrate neuronal development and survival by target-derived signals is a recurring conceptual theme in neurobiology that began with the demonstration by Shorey (1909) that the amputation of limb-bud targets of spinal sensory and motor neurons in the chick embryo results in the loss of those neurons that normally innervate the limbs. After removal of the forelimb bud on embryonic day (E)2, Shorey observed the absence of peripheral brachial nerves and a marked reduction in the size of the brachial ventral horn and dorsal root ganglia (DRG) on E6. She mistakenly attributed these deficits to a failure of neuronaldifferentiation and rejected the possibility that the limb normally promotes neuronal survival. Although later attempts by Hamburger, Levi-Montalcini, and others to repeat her experiment were successful in that a hypoplasia and loss of neurons were observed (for review, see Oppenheim, 1981), these studies led to a different interpretation of the cell loss. After several replications of the basic limb-removal experiment in the 1930s and 1940s (Oppenheim, 1981), it was finally concluded that limb-derived trophic signals regulate the motor and sensory neurons that survive and successfully innervate their synaptic targets. At about the same time, it was discovered that a proportion of motor and sensory neurons undergo a period of normal or naturally occurring programmed cell death (PCD), and this led to the realization that the cell loss after limb removal was merely an exaggeration of this normal process. This in turn led to the suggestion that the limb provides trophic signals that promote and maintain neuronal survival (Hamburger and Levi-Montalcini, 1949), which was an important conceptual framework (the neurotrophic hypothesis) for the eventual discovery of nerve growth factor (NGF) (Oppenheim, 1996a).
Despite the considerable evidence supporting the idea that only the later survival and not the proliferation, migration, or initial differentiation of limb-innervating neurons is affected by removal of peripheral targets (Hamburger, 1958; Carr and Simpson, 1978a,b;Oppenheim et al., 1978), reports have continued to appear arguing against this idea (Lanser and Fallon, 1984, 1987; Lanser et al., 1986;Phelan and Hollyday, 1991). A related issue that has emerged in the context of target removal experiments is the extent to which developing sensory and motor neurons (MNs) depend on target-derived trophic signals for their survival versus trophic support from other sources (e.g., afferents, glia, hormonal, paracrine–autocrine, etc). For example, the selective removal of afferent input to various developing neuronal populations increases PCD in the face of apparent normal interactions of these cells with sources of target-derived trophic support (Oppenheim, 1991; Linden, 1996). However, in the absence of target cells (muscle, skin) virtually all limb-innervating MNs and large numbers of sensory neurons undergo PCD (Carr and Simpson, 1978a,b; Oppenheim et al., 1978; Oakley et al., 1997). Therefore, if MNs do require nontarget-derived trophic support, for example, such support does not appear to be sufficient for their survival after limb-bud removal (LBR). Alternatively, the absence of targets may alter the ability of MNs to gain access to or respond to these other sources of trophic support. If, in fact, muscle-derived trophic support is both necessary and sufficient for normal MN survival, then it should be possible to rescue virtually all MNs from normal PCD and from PCD after target ablation by treatment with muscle extracts or with putative muscle-derived trophic molecules. Similarly, if the sensory neurons that are lost after LBR reflect the absence of target-derived trophic support, then they should also be rescued by treatment with the neurotrophins (NGF, BDNF, NT-3), trophic factors that are known to be required for the survival of these neurons (Snider, 1994). In this paper, we have attempted to reexamine the issue of what aspects of sensory and motor neuron development (e.g., proliferation, migration, differentiation, survival) are affected by peripheral target ablation and the extent to which treatment with putative target-derived trophic signals can compensate for target removal. In examining these issues, we return again to the use of the almost century-old model of LBR, which remarkably is still able to provide new insights into neuron-target interactions.
MATERIALS AND METHODS
LBR. Unilateral removal of the right LBR was performed on E2.5, which corresponds to stages 16–18 of the Hamburger–Hamilton stage series (Hamburger and Hamilton, 1951), using the surgical methods described previously (Chu-Wang and Oppenheim, 1978; Oakley et al., 1997). Only those embryos later observed to have acomplete absence of the leg and pelvic girdle were used for further analysis (see Fig. 1). Complete deletion of all limb musculature leads to a disruption of normal nerve patterns in the plexus of the lumbosacral region, with the more anterior segments (L1–2) forming thoracic-like patterns and the more posterior segments projecting aberrantly toward the tail (see below). Because unilateral LBR in the chick has no effect on the development of contralateral sensory or motor neurons (Oppenheim et al., 1978), the contralateral side was used as an internal control (together with sham LBR) for comparison with the effects of LBR on development and survival.
Fig. 1.
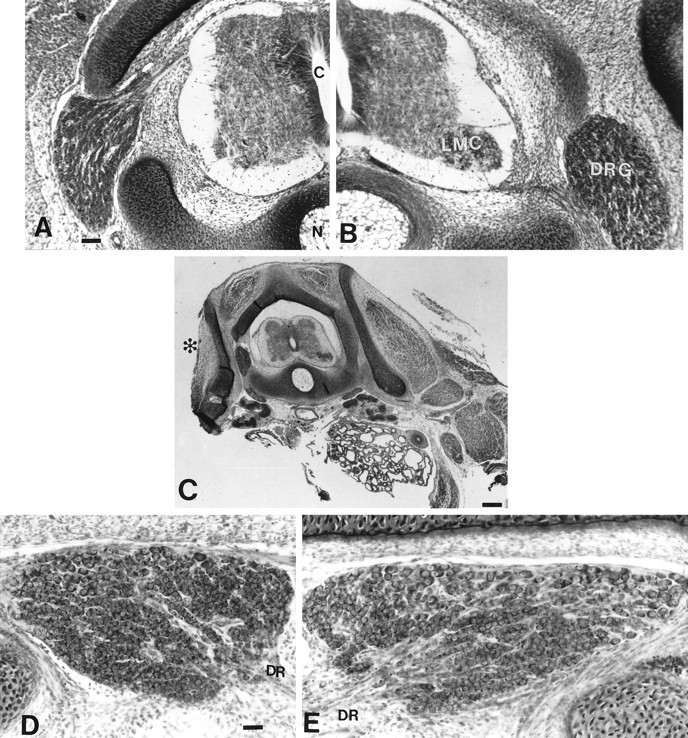
Transverse sections of the L3 spinal cord on E6 ipsilateral (A) and contralateral (B) to LBR. c, Central canal;DRG, dorsal root ganglion; LMC, lateral motor column; N, notochord. Note the almost complete absence of the LMC in A. Scale bar, 25 μm.C, Transverse section of E8 lumbar region. Note the complete absence of limb muscle (asterisk) ipsilateral to LBR. Scale bar, 100 μm. D, E, The L3 DRG on E8 ipsilateral (D) and contralateral to LBR. A comparison of D and E (also see Fig.2D,E) shows survival of many sensory neurons. Scale bar, 20 μm.
More than 125 embryos that met the criteria for complete LBR (listed above) were used in the experiments described below. Control eggs and embryos underwent all of the same manipulations as the LBR group except for actual removal of the limb-bud (sham LBR). In a separate group of embryos both the right limb-bud and tail-bud were removed at the same time.
Quantitative analysis of neuron numbers and size. Control and LBR embryos were removed from the shell, decapitated, and immediately placed in either Carnoy’s or Bouin’s fixative. After fixation for 6–24 hr, the lumbosacral or thoraco–lumbosacral region, including vertebra and adjacent DRG, was dissected and processed for paraffin embedding, sectioned serially at 6–12 μm, and stained with either thionin or hematoxylin–eosin as described previously (Chu-Wang and Oppenheim, 1978). Motoneurons were counted in every tenth section through the entire lumbar enlargement, and DRG cells were counted in every fifth section through the third lumbar segment (L3) according to a previously described, reliable counting method (Clarke and Oppenheim, 1995). Pyknotic MNs and DRG cells were counted in the same sections used for healthy (surviving) cell counts (Clarke and Oppenheim, 1995). We used only those DRG cells (L3) and MNs that met the above criteria for cell counts and estimated cell size from measures of nuclear diameter using a previously described method in which nuclear diameter has been shown to be proportional to soma diameter (Oppenheim et al., 1992).
Distribution of peripheral nerves. In an attempt to examine possible aberrant projection patterns of peripheral sensory and motor nerves after LBR, a whole-mount procedure was used for visualizing peripheral axons. Whole chick hindlimb preparations were stained with TUJ1, a monoclonal antibody that recognizes a neural-specific form of β-tubulin (Lee et al., 1990; Easter et al., 1993), using a modification of the whole-mount procedure of Dent et al., (1989). E10 embryos were washed in PBS, decapitated, and eviscerated, and the thoracic and lumbosacral regions of the spinal cord were exposed via ventral laminectomy. After the spinal cord and sympathetic chains were removed, the embryos were hemisected along the midline, and the hindlimbs were isolated. These preparations were fixed and permeabilized for 4 hr in a 4:1 mixture of methanol/dimethyl sulfoxide (Dent’s fixative) at −20°C and bleached in 10% H2O2 (in Dent’s fixative) for 1–2 d at room temperature. The tissue was rehydrated through a graded methanol series and washed extensively in PBS. After they were blocked for 2 hr in Tris-buffered saline containing 0.4% Triton X-100 (Sigma, St. Louis, MO) and 20% horse serum, the hindlimbs were incubated for 18 hr in affinity-purified TUJ1 (1 μg/ml in blocking buffer) at 4°C. After five washes in PBS containing 0.02% Tween 20 (PBST) (USB, Cleveland, OH), the preparations were incubated in an HRP-conjugated secondary antibody (3 μl/ml in blocking buffer) (Jackson ImmunoResearch, West Grove, PA) for 18 hr at 4°C. After three washes in PBST and three washes in 0.1 m phosphate buffer, the tissue was preincubated for 30–60 min in phosphate buffer containing 0.5 mg/ml diaminobenzidine (DAB) and 5 mg/ml nickel ammonium sulfate. The DAB reaction was initiated by adding 3% H2O2 (10 μl/ml) and run for ∼30 min with gentle agitation. The hindlimbs were then washed in PBS, completely dehydrated through a graded methanol series, and cleared by soaking in a 1:2 mixture of benzyl alcohol/benzyl benzoate. All washes and incubations were performed with gentle shaking. The TUJ1 antibody was a generous gift from Dr. Anthony Frankfurter (University of Virginia).
In addition to the β-tubulin labeling, we also analyzed the pattern of peripheral axons originating from lumbar level DRG by injecting these ganglia with a lipophilic dye. After ventral laminectomy, all of the lumbar DRG on both sides were pressure-injected with 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI; 5 mg/ml in 90% ethanol, 10% dimethyl sulfoxide) (Molecular Probes, Eugene, OR) (see Honig and Hume, 1986) using broken micropipettes. These preparations were then fixed in 4% paraformaldehyde for 18 hr at room temperature, transferred to fresh fixative, and held at 37°C for 2–3 weeks.
Proliferation. To examine possible changes in the proliferation of cells in the spinal cord and DRG after LBR or trophic factor treatment (see below), bromodeoxyuridine (BrdU) (Sigma) was used as a marker of cells entering S-phase of the mitotic cycle. After a 2–4 hr pulse of a nontoxic dose of BrdU (10 μg) administered onto the chorioallantois through a window in the shell, embryos were killed, staged, fixed (Carnoy’s), and embedded in paraffin, and serial sections (6 μm) were labeled with an antibody against BrdU (Sigma) as described (Oakley et al., 1997). Because reductions in both MNs and sensory neurons after LBR first occur after E4, E3.5 was the earliest age examined for changes in BrdU labeling. BrdU-labeled cells with a complete and distinct nuclear membrane were counted in every other section through the L3 region of the spinal cord and in the L3 DRG. For the analysis of BrdU incorporation in the L3 DRG of embryos treated with trophic factors (see below), a 2–4 hr pulse of BrdU was delivered at the same time as the final treatment with trophic factor on E5 (stage 26–27). We estimate that at least 20–30% of all dividing cells are labeled by this procedure (Oakley et al., 1997).
Trophic factor and drug treatments. Control and LBR embryos were treated with various neurotrophic molecules that included NGF, neurotrophin-3 (NT-3), brain-derived neurotrophic factor (BDNF), insulin-like growth factor 1 (IGF-1), ciliary neurotrophic factor (CNTF), basic fibroblast growth factor (bFGF), glial cell line-derived neurotrophic factor (GDNF), and hepatocyte growth factor–scatter factor (HGF–SF). Except for NGF, which was purified from mouse salivary gland and generously provided by Eugene Johnson (Washington University), all of the other factors were human recombinant molecules generously provided by Amgen (Thousand Oaks, CA) (NT-3, BDNF, bFGF, CNTF), Cephalon (IGF-1), and Genentech (San Francisco, CA) (HGF–SF). Embryos were treated daily with optimal doses (5–10 μg) of each factor on the basis of previously published studies of sensory and MN survival (Neff et al., 1993; Oppenheim et al., 1993, 1995, 1997) by administration onto the chorioallantois. In addition, separate groups of control and LBR embryos were treated with tissue extracts (150 μg) from E10 chick embryo skeletal muscle (CMX) or brain (CBX) that were prepared according to previously described methods (Oppenheim et al., 1988, 1993; Johnson et al., 1995). Control embryos received equal volumes of saline, bovine serum albumin, or cytochrome C.
To examine the role of activity blockade on MN cell death, one group of LBR embryos was treated daily with suboptimal doses (1 mg) of the paralytic agent d-tubocurarine (Curare, Sigma) or with curare plus either optimal (150 μg) or suboptimal (75 μg) doses of CMX from E4 to E7 and killed on E7.5. These experiments were designed to test whether curare potentiates the effects of CMX on MN survival after LBR (Hory-Lee and Frank, 1996). Finally, to examine possible mechanisms of cell death after LBR, some embryos were treated either with the antioxidant N-acetylcysteine (NAC, Sigma) or with two different cell-permeable inhibitors of the interleukin β1 converting enzyme (ICE) family of cysteine proteases, Boc-aspartyl (OMc)-fluoromethylketone (BAF) (Enzyme Systems Products, Dublin, CA) and Ac-DEVD-CHO (Bachem Biosciences, King of Prussia, PA) on E4, E5, and E6. Both NAC and the ICE inhibitors were administered at doses (40 μg/d for ICE inhibitors; 30–60 mm d for NAC) previously shown to be optimal for inhibiting MN PCD on E8 in control embryos (Milligan et al., 1995; Li et al., 1997; M. Burek and R. Oppenheim, unpublished data). Healthy (surviving) or dying (pyknotic) sensory or motor neurons (or both) were counted after treatment with curare, NAC, BAF, and DEVD as described above.
Islet-1 immunocytochemistry. A monoclonal antibody (4D5) was used to examine the expression of a specific member of the islet family (islet-1) of LIM homeoproteins according to a previously described procedure (Yaginuma et al., 1996). Islet-1 is expressed by all classes of spinal MNs and is a reliable and specific marker for the early identification of MNs in the ventral spinal cord (Tuschida et al., 1994). This provided an independent means for assessing early MN differentiation after LBR. The number of islet-1-positive MNs per section was determined in the L3 segment both ipsi- and contralateral to the LBR on E4.5 (stage 25), before the onset of either normal or LBR-induced MN PCD.
RESULTS
Sensory and motor neurons differentiate normally after LBR
The differentiation of both sensory and motor neurons appeared normal after LBR (Figs. 1,2). Motoneuron size, based on nuclear diameter, was similar on the ipsi- and contralateral sides before or at the onset of cell death (data not shown). Although there was a small but statistically significant decrease in sensory neuron size ipsilateral to LBR on E5.5 when neurons in both the ventral-lateral (VL) and dorsal-medial (DM) regions were included, because most of the early dying sensory neurons are VL cells, this difference most likely reflects the greater depletion of the large VL cells at this time after LBR (Fig. 3). In fact, when measures of neuronal size were restricted to the DM region, no difference was found between the DRG ipsi- and contralateral to the LBR (data not shown). The number of MNs per section in L3 expressing the MN-specific marker islet-1 on E4.5 (Fig. 3) was also similar on the two sides [95 ± 11 (n = 4) ipsilateral vs 99 ± 13 (n = 4) contralateral]. In a previous paper (Oppenheim et al., 1978), MNs were also shown to undergo normal ultrastructural and biochemical differentiation after LBR. Additionally, chick DRG sensory neurons have also recently been shown to display a normalpattern of neurotrophin receptor (trk) expression after LBR (Oakley et al., 1997). Taken together, these data support the argument that the absence of peripheral targets does not affect the early differentiation of either sensory or motor neurons (but seeCampagna et al., 1997).
Fig. 2.
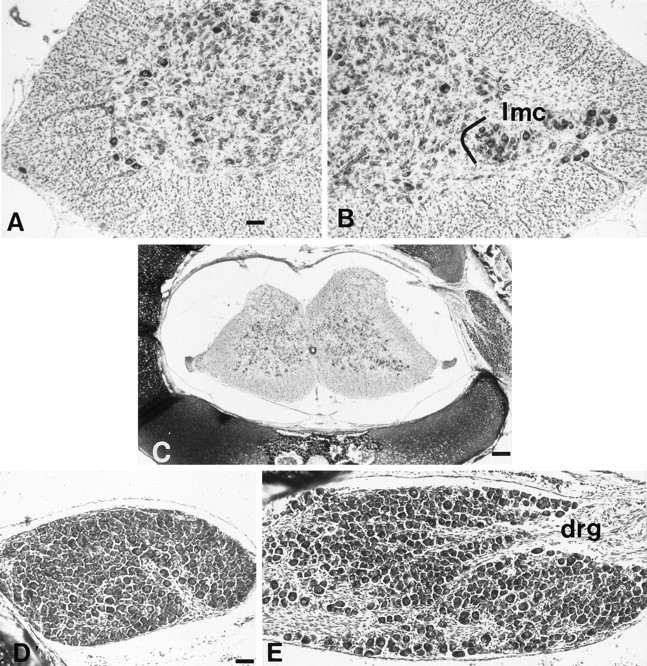
Transverse sections of the lumbarlmc on E18 ipsilateral (A) and contralateral (B) to LBR. Scale bar, 24 μm.C, Low-power photomicrograph of E18 lumbar spinal cord after LBR. Scale bar, 80 μm. D, E, The L3 DRG on E18 ipsilateral (D) and contralateral (E) to LBR. Scale bar (shown in Dfor D and E): 30 μm.
Fig. 3.
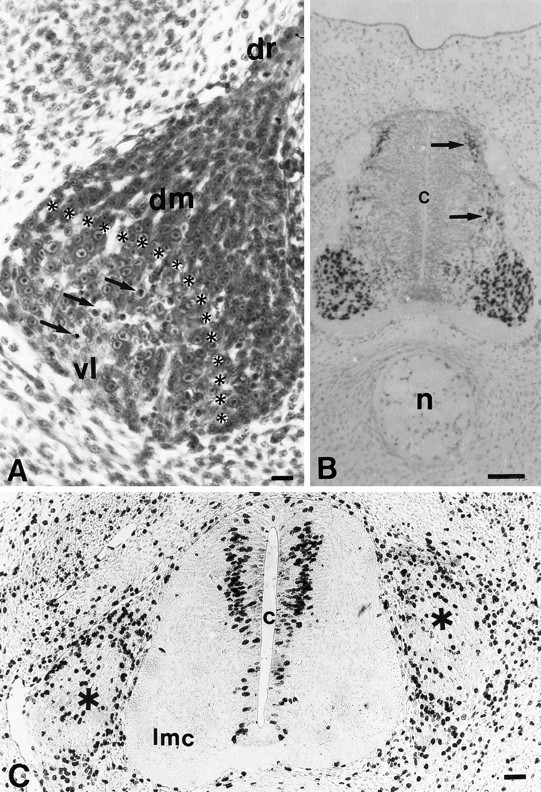
Transverse section (6 μm) of the L3 DRG ipsilateral to LBR on E5 (A). vl, Ventral-lateral; dm, dorsal-medial; dr, dorsal root; arrows, pyknotic cells in thevl region. Asterisks delineate the boundary between vl and dm. Scale bar, 10 μm. B, Islet-1 immunoreactivity of lumbar MNs on E4.5 after LBR (left side). A few immunopositive cells (interneurons?) are located in dorsal spinal cord (arrows). n, Notochord; c, central canal. Scale bar, 30 μm. C, BrdU immunoreactivity on E4.5 in lumbar spinal cord and DRG (asterisks) after LBR (left side). Note the larger number of immunopositive cells in the dorsal half of spinal cord. lmc, Lateral motor column. Scale bar, 30 μm.
The survival of sensory and motor neurons is differentially affected after LBR
Motoneurons
MNs in the lumbar spinal cord begin to undergo normal PCD on E5–5.5, and MNs continue to be lost until approximately E12 (Fig.4). After LBR, MN loss shows a similar trend beginning on E5 and continuing until E9–10. However, whereas normal PCD of MNs results in a loss of 50% of the initial population, after LBR >90% of the target-deprived MNs are lost. Dying pyknotic MNs are first observed on E5.5 in both the control (contralateral) and LBR (ipsilateral) lateral motor column (LMC) or ventral horn (Fig. 4,inset) and reach peak numbers on E7.5. However, the number of pyknotic cells in the ipsilateral LMC is significantly increased by LBR at all ages from E5.5 to E9.5.
Fig. 4.

Lumbar motoneuron numbers in segments L1–8 (mean ± SD) ipsilateral (LBR, solid circle) and contralateral (open circle) to LBR. *p < 0.01; ** p < 0.001;t tests. Inset, Pyknotic lumbar motoneurons (mean ± SD) ipsilateral (LBR, solid circle) and contralateral (open circle) to LBR. * p < 0.05; ** p < 0.001;t tests. The SDs that were smaller than the symbols are not shown.
Sensory neurons
Although for convenience we have only systematically examined sensory neuron development and survival in the L3 DRG, both normal development and the effects of LBR appear similar in all limb-innervating DRG (our unpublished observations). In the normal contralateral L3 DRG, sensory neurons achieve peak numbers on E7.5, decrease by 31% between E7.5 and E9.5, and then decrease by an additional 25% between E9.5 and E15.5 (Fig.5). No further loss occurs between E15.5 and E18.5. From an examination of the number of healthy surviving DRG cells, it appears that normal PCD begins on E7.5. However, a separate analysis of pyknotic neurons shows that, in fact, dying cells can be observed as early as E4.5 and they exhibit two peaks, one on E6.5 and the second on E8.5 (Fig. 5, inset). After LBR, dying sensory neurons show a similar trend with two peaks, one on E5.5 and another on E8.5. However, in the LBR situation, there are significant increases in the number of dying cells on E4.5, E5.5, E7.5, and E8.5 compared with the contralateral control DRG (Fig. 5). In accord with a previous study of PCD in brachial (forelimb-innervating) DRG (Hamburger et al., 1981), we have also observed that the early period of cell death that peaks on E5.5 (LBR) or E6.5 (control) is restricted almost entirely to early differentiating sensory neurons in the VL region of the DRG, whereas the later period of DRG cell death, peaking on E8.5, is restricted to later differentiating neurons in the DM region of the DRG.
Fig. 5.

Sensory neuron numbers (mean ± SD) in the L3 DRG ipsilateral (LBR, solid circle) and contralateral (open circle) to LBR. * p < 0.01; ** p < 0.001; t tests.Inset, Pyknotic neurons (mean ± SD) in the L3 DRG ipsilateral (LBR, solid circle) and contralateral (open circle) to LBR. * p < 0.01; ** p < 0.001; t tests.
One major difference between the development of sensory and motor neurons in the chick embryo is that virtually all MNs are born (i.e., become postmitotic) before the onset of normal PCD (Hollyday and Hamburger, 1977), whereas sensory neurons continue to proliferate until approximately E7 (Carr and Simpson, 1978a,b), such that neuronal generation and degeneration in the DRG overlaps for ∼3 d. Accordingly, because new sensory neurons are being generated while at the same time others are dying, it is not until after E7.5, when neuron generation ceases but cell death continues, that a net loss of healthy surviving DRG cells is observed (Fig. 5). Because of this temporal overlap, it remains a formal possibility that LBR affects either sensory neuron generation or survival (or both). The effects of LBR on sensory neuron proliferation are described below.
Another difference between sensory and motor neurons is the extent of their survival after LBR. As described above, after LBR virtually all MNs undergo PCD. By contrast, in the same embryos, almost 50% of the peak number of sensory neurons present on E7.5 are maintained up to E18.5 in the apparent absence of all limb-derived trophic support. In fact, by E18.5, there are only 25% fewer sensory neurons in LBR versus control ganglia. This surprising result, which has also been reported in the earlier literature (Hamburger, 1934; Levi-Montalcini and Levi, 1942; Bueker, 1943, 1947; Hamburger and Levi-Montalcini, 1949), raises several important questions, which we address below.
LBR does not modify the proliferation of sensory or motor neurons after LBR
Although >95% of all lumbar MNs are generated before E4.5 (Hollyday and Hamburger, 1977; Burek et al., 1996) because after LBR MN numbers decrease relative to controls between E4.5 and E5.5 (Fig. 4), we cannot entirely exclude the possibility that LBR may affect the proliferation of late generated MNs. To examine this, we have used BrdU immunocytochemistry to label cells in the L3 segment of the spinal cord that are in S-phase of the cell cycle (Fig. 3). After a 2–4 hr pulse of BrdU, the total number of labeled cells was compared between the ipsilateral (LBR) and contralateral (control) sides of the spinal cord. As shown in Figure 6, at no time from E3.5 to E7.5 were any differences observed. Similar results were obtained when the analysis was confined to the ventral half (basal plate) of the spinal cord (data not shown). From these data, we conclude that up to E7.5 LBR is without effect on the generation of motoneurons (or of any other cell type) within the spinal cord.
Fig. 6.

BrdU-labeled cells (mean ± SD) in the lumbar spinal cord ipsilateral (LBR) and contralateral (CON) to LBR.
As noted above, and in sharp contract to MNs, the generation and degeneration of sensory neurons in the DRG overlaps between E4.5 and E7.5. An analysis of BrdU labeling of sensory neurons after LBR shows that, with the exception of E7.5, there were no differences in numbers of BrdU-labeled cells between ipsilateral and contralateral L3 DRG (Fig. 7). Because the generation of sensory neurons ceases on E7.5, later ages were not examined. Because the early peak in pyknotic cells (E5.5) is not accompanied by a difference in BrdU-labeled cells, this supports the idea that the loss of limb-derived signals does not cause the death of proliferating precursors but rather affects only differentiating neurons. However, because both normal and LBR-induced loss of sensory neurons occurs on E7.5 and later (Fig. 5), it is possible that the decreased number of ipsilateral BrdU-labeled cells at this time reflects an effect of target regulation on neuronal proliferation. Although we cannot completely exclude this, for reasons discussed in detail below, we think that it is considerably more likely that in fact this reflects an indirect effect on non-neuronal proliferation in the DRG caused by the increased PCD of sensory neurons after LBR.
Fig. 7.

BrdU-labeled cells (mean ± SD) in the L3 DRG ipsilateral (LBR) and contralateral (CON) to LBR. * p < 0.01;t test.
Sensory neurons make aberrant peripheral axonal projections after LBR
As noted above, after LBR virtually all MNs undergo degeneration, whereas there is only a 25% decrease in the number of sensory neurons that survive up to 1 week after cessation of the normal period of PCD on E12.5. In a previous paper, Bueker (1947) found a similar proportion of sensory neuron survival in adult chickens after embryonic LBR. There are several possible explanations for this rather remarkable ability of large numbers of sensory neurons to survive indefinitely in the apparent absence of their peripheral targets. (1) The surviving neurons may represent cells that normally project to nonlimb targets such as dorsal skin or cells that are visceral afferents. LBR would not be expected to deprive these neurons of their normal targets. (2) After LBR, many sensory neurons with normal limb targets may project their axons aberrantly to nonlimb targets where they receive sufficient trophic support, and (3) the surviving neurons may normally (or aberrantly after LBR) be maintained by autocrine- or paracrine-derived trophic support from neurons or non-neuronal cells in the DRG (Acheson et al., 1995). In an attempt to assess the first two possibilities, we have examined the axonal projections of surviving sensory neurons after LBR.
To assess the effects of limb-bud deletion on the peripheral projections of sensory neurons, we used the TUJ1 antibody to label axons in whole mounts of chick hindlimbs at E10. In the absence of developing limb tissues, the normal pattern of peripheral projections was altered dramatically (Fig. 8). In all embryos examined (n = 5), neither the crural plexus nor the sciatic plexus developed normally (compare Fig. 8, A andB). Instead, the most anterior lumbar segments (L1 and L2) projected axons anteriorly into the thoracic body wall, whereas axons from the rest of the lumbar DRG joined together to project posteriorly to the tail region in a large nerve trunk (Fig. 8B) (also see Tosney and Landmesser, 1984). Within the tail region, most of these axons formed a novel plexus, with no obvious target (see below). A minority of these posteriorly directed axons joined the otherwise normal pudendal plexus (Fig. 8B). Examination of the pattern of skin innervation in LBR embryos showed that in all cases limb-bud deletion prevented the formation of the two major cutaneous nerves of the thigh: the lateral (compare Fig. 8, C andD) and medial (not shown) femoral cutaneous nerves. However, in all experimental limbs, the remaining skin was innervated nonetheless, mainly by branches of the dorsal rami of each spinal nerve (Fig. 8D). In addition, in all cases the more posterior regions of remaining skin also received innervation from some of the axons that formed the novel nerve plexus in the tail region of these embryos (Fig. 8D). To be certain that these novel nerve patterns were formed, in fact, by axons derived from lumbar sensory ganglia, we injected DiI into lumbar DRG to orthogradely label sensory axons. This procedure confirmed that the axons within the novel tail plexus and those that supplied the skin over the deletion site were derived from lumbar level DRG (n = 2; not shown).
Fig. 8.

Peripheral nerve patterns in whole mounts of normal and experimental limbs as revealed by staining with the TUJ1 antibody. A and B are focused on the medial aspect of each preparation to show nerve projection patterns;C and D are focused on the lateral aspect to show skin innervation in the same limbs. A, Normal pattern of peripheral nerve projections. Lumbar segments L1–L3 largely contribute axons to the crural plexus (c), which gives rise to many peripheral nerves of the limb including the prominent obturator nerve (o). Lumbar segments L3–L8 all contribute axons to the sciatic plexus (s). The more caudal segments contribute axons to the pudendal plexus (p). f, Femur.B, After limb-bud deletion, neither the crural plexus nor the sciatic plexus develops. L1 and L2 typically project axons anteriorly toward thoracic segments (arrowheads). L2–L8 all project axons posteriorly toward the tail, where they form a novel plexus (arrows) and contribute to the pudendal plexus (double arrowhead). Dashed lines indicate connections broken during processing; dr indicates dorsal roots. The spinal cord and sympathetic chains were removed for clarity. C, Normal patterns of skin innervation on the lateral surface of the thigh. The crural plexus (C) gives rise to the lateral femoral cutaneous nerve (lfc), which branches (arrowheads) to provide much of the cutaneous innervation of the lateral thigh. The more proximal skin receives axons from the dorsal rami (d) of each spinal nerve. f, Femur. D, After limb-bud deletion, the lateral femoral cutaneous nerve does not develop, and the remaining skin is innervated by branches from dorsal rami (d). Some axons also reach the skin over the deletion site from the novel plexus in the tail region (arrow). Scale bar, 1 mm.
These observations support the first two possibilities described above in that after LBR there is, in fact, a substantial projection of lumbar sensory axons to dorsal skin as well as aberrant projections to the tail that are never found in control embryos. If, in fact, the aberrant projections to the tail contribute to the survival of sensory neurons, then combined LBR plus tail ablation should result in an increased loss of sensory neurons compared with LBR alone. As shown in Figure9, there is significantly more cell loss in the L3 DRG (60%) after the combined ablation compared with LBR alone (40%). By comparison, tail-bud ablation alone had no effect on the survival of L3 DRG cells, and neither the combined ablation nor tail ablation alone had any effect on MN survival when compared with LBR. These results indicate that sensory neurons but not MNs are capable of deriving trophic support from the developing tail-bud.
Fig. 9.

The number of surviving motoneurons (MNs) and sensory neurons (mean ± SD) in L3 DRG on E9.5 after LBR (L), tail ablation (T), or L + T. *p< 0.01 (L + T vs L).Numbers in bars are sample size.
Specific trophic agents or tissue extracts rescue sensory and motor neurons from PCD after LBR
Motoneurons
In previous studies, we have shown that several different neurotrophic factors representing distinct gene families, as well as tissue extracts from CBX or CMX, can rescue sensory and motor neurons from normal PCD in the chick embryo in vivo (Oppenheim et al., 1988, 1991, 1993, 1995, 1997; Neff et al., 1993; Johnson et al., 1995). We have replicated some of those results here and have compared the effects of these same agents on cell death after LBR. Daily treatment of control embryos with the following agents (5–10 μg) from E6 to E9 promoted the survival of lumbar MNs: BDNF, IGF-1, GDNF, CNTF, HGF–SF, CMX, and CBX (Fig. 10), whereas bFGF, NT-3, and NGF were without effect (data not shown). By contrast, only CMX and GDNF were able to rescue a significant number of MNs from LBR-induced cell death (Fig.11). However, neither CMX nor GDNF were able to rescue all the MNs that die after LBR, although CMX was significantly more effective than GDNF. After LBR the number of surviving MNs on E7.5 was 7100 ± 815 saline, 12,300 ± 1877 CMX, and 8910 ± 1253 GDNF versus 15,516 ± 906 contralateral saline control. As shown in Figure 12, after LBR MNs are lost along the entire rostral-caudal extent of the lumbar enlargement (L1–8), and CMX rescued MNs in all but the most caudal region. Finally, CMX and GDNF were also the only factors that promoted normal MN survival on both the ipsilateral and contralateral (control) side after treatment from E5 to E7. This suggests that the other factors that were effective after treatment from E6 to E9 (Fig.10) act primarily after E7 during the middle tolater stages of the normal period of PCD of MNs (see Discussion). Hepatocyte growth factor was unique among all the factors tested in that it rescued MNs during normal cell death (E6–E10) and on the contralateral (control) side after LBR but failed to promote survival ipsilateral to LBR.
Fig. 10.

Lumbar motoneuron numbers (mean ± SD) on E10 after daily treatment of normal control embryos with saline (SAL) or different trophic agents from E6 to E9. *p < 0.001; t test (vsSAL). Numbers in bars = sample size (embryos). See Materials and Methods and Results.
Fig. 11.
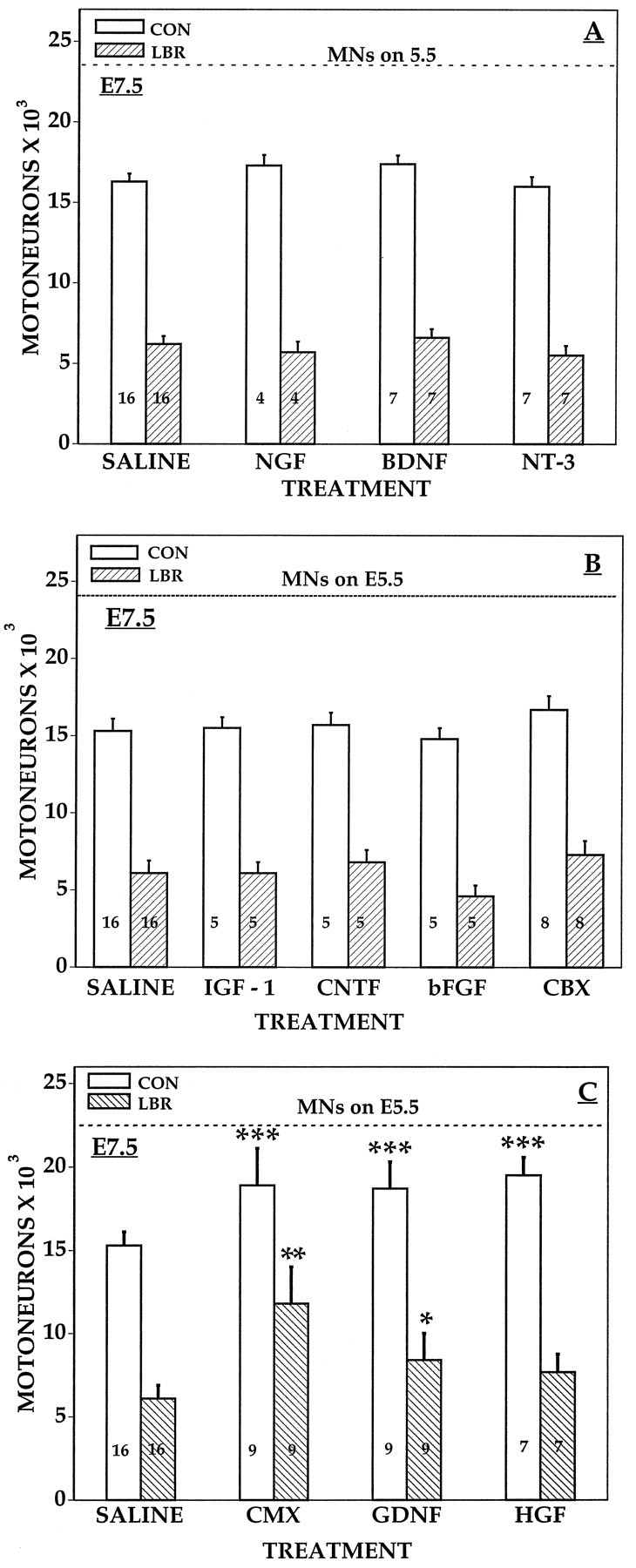
Lumbar motoneurons numbers (mean ± SD) on E7.5 ipsilateral (LBR) and contralateral (CON) to LBR after daily treatment with trophic factors from E4 to E7. *p < 0.05; **p < 0.001 (vs saline LBR); ***p < 0.0025 (vs saline CON); ttests. See Materials and Methods and Results.
Fig. 12.

The number (mean ± SD) of lumbar motoneurons on E7.5 ipsilateral (LBR) and contralateral (C) to LBR along the rostral-caudal axis after daily treatment with muscle extract (CMX) or saline (SAL) from E4 to E7. *p < 0.05; **p < 0.01; t tests. See Materials and Methods and Results.
Sensory neurons
As reported previously (Oppenheim et al., 1993), all three of the neurotrophins (NGF, BDNF, and NT-3) promote the survival of sensory neurons in the L3 DRG of control embryos after daily treatment (5–10 μg) from E5.0 to E9.0 (Fig. 13), whereas the other factors tested, including CMX and CBX, were ineffective (data not shown). Similarly, the neurotrophins were also the only factors tested that promoted the survival of sensory neurons after LBR. In the DRG of both the control and LBR animals, NGF rescued more neurons than either NT-3 or BDNF, and the combination of all three neurotrophins was more effective than each one alone. However, even the combination of neurotrophins failed to rescue all sensory neurons. This was true for embryos examined on either E7.5 (data not shown) or E10 (Fig. 13). Therefore, the incomplete rescue of sensory neurons on E10 is not likely caused by a failure of neurotrophins to sustain rescued neurons after E7.5. It is also important to note, however, that the combined treatment restored a normal complement of sensory neurons in the absence of any other limb-derived factors (i.e., there is not a significant difference between ipsilateral–combo vs saline contralateral). Because it is also clear that the combined treatment rescued more cells on the contralateral side, it seems likely that the combination of endogenous and exogenous neurotrophins is responsible for the increased survival in this situation.
Fig. 13.

The numbers of sensory neurons in the L3 DRG on E9.5 ipsilateral (LBR) and contralateral (CON) to LBR after daily treatment from E4 to E9 with neurotrophins. 1p < 0.001LBR versus CON;2p < 0.01 NGFversus SAL;3p < 0.01NGF versus SAL;4p < 0.01BDNF versus SAL;5p < 0.05BDNF versus SAL;6p < 0.05NT3 versus SAL;7p < 0.001COMBO versus SAL andp < 0.01 COMBO versusNGF, BDNF, or NT3;8p < 0.01COMBO versus NGF, BDNF, NT3. See Materials and Methods and Results.
To determine whether treatment with the neurotrophins (NGF, NT-3, BDNF) altered the proliferation of sensory neuron precursors after LBR, we examined BrdU incorporation on E5.5 (stage 27) after treatment with two doses of a single neurotrophin administered at stage 25 (E4.5) and stage 27 (E5.5). Although this is a period of active neurogenesis in the DRG (Carr and Simpson, 1978a,b), neither LBR nor neurotrophin treatment significantly altered the number of BrdU-labeled cells in the L3 DRG (Fig. 7, Table 1). From this, we conclude that changes in neuronal proliferation cannot account for the increased number of sensory neurons after treatment with NGF, NT-3, or BDNF.
Table 1.
Counts of BrdU-labeled cells (mean ± SD) in L3 DRG at E5.5
| Treatment | Contralateral (control) | Ipsilateral (LBR) |
|---|---|---|
| Sal | 217 ± 53 (4) | 258 ± 41 (4) |
| NGF | 239 ± 61 (4) | 240 ± 50 (4) |
| Sal | 256 ± 49 (4) | 224 ± 43 (4) |
| NT3 | 242 ± 45 (4) | 274 ± 52 (4) |
| Sal | 277 ± 57 (4) | 249 ± 48 (4) |
| BDNF | 260 ± 60 (4) | 245 ± 57 (4) |
Values in parentheses represent sample size. Sal, Saline.
Activity blockade does not promote MN survival after LBR
In previous studies, activity blockade with curare during the normal period of MN PCD was found to rescue most of the MNs (Pittman and Oppenheim, 1978, 1979), whereas curare treatment after LBR failed to prevent the target deprivation-induced loss of MNs. Because two recent studies have reported that curare plus CMX has additive effects on MN survival in vitro (Hory-Lee and Frank, 1996; Oppenheim et al., 1996), we have examined this same combination in vivo in the LBR model. As shown in Table2, either optimal or suboptimal doses of curare alone had no effect on MN survival after LBR, whereas in the same embryos an optimal dose of curare promoted MN survival on the contralateral control side. Furthermore, the combination treatment of curare + CMX was no more effective in rescuing MNs than CMX alone (LBR ipsilateral side) or of either CMX or curare alone on the contralateral control side (Table 2). These results indicate that in vivoactivity blockade with curare doesn’t potentiate the effects of CMX on the survival of either normal or peripherally deprived (LBR) MNs between E4 and E6.5.
Table 2.
Effects of CMX and curare on motoneurons (mean ± SD) on E7.5 after LBR
| Treatment | Ipsilateral (LBR) | Contralateral (control) |
|---|---|---|
| Saline | (A) 10,436 ± 970 (10) | (H) 17,506 ± 20112-b (10) |
| CMX (75 μg) | (B) 9197 ± 1009 (5) | (I) 17,562 ± 11432-b (5) |
| CMX (150 μg) | (C) 15,434 ± 16882-a (6) | (J) 19,704 ± 12542-c (6) |
| Curare (1 mg) | (D) 11,015 ± 1114 (6) | (K) 21,827 ± 12492-d (6) |
| Curare (2 mg) | (E) 10,178 ± 995 (5) | (L) 22,549 ± 13772-e (5) |
| CMX (75 μg) + curare (1 mg) | (F) 10,575 ± 1200 (5) | (M) 22,680 ± 10872-f (5) |
| CMX (150 μg) + curare (2 mg) | (G) 14,790 ± 1123 (5) | (N) 23,216 ± 12812-f (5) |
Values in parentheses represent sample size.
p < 0.01; (A vs C).
p < 0.001; (H, I vs A, B).
p < 0.05; (J vs H).
p < 0.05; (K vs H).
p < 0.01; (L vs H).
p = NS; (M, N vs J, K, L).
NAC and the ICE inhibitors BAF and DEVD do not promote MN survival after LBR
Previous in vitro studies have suggested that the loss of trophic support may induce a net increase in intracellular reactive oxygen species that could trigger PCD (Kane et al., 1993; Buttke and Sanstrom, 1994). In support of this idea, treatment with the antioxidant NAC has been shown to promote the survival of trophic factor-deprived sympathetic neurons and glial cells in vitro(Mayer and Noble, 1994; Ferrari et al., 1995; Greenlund et al., 1995). As shown in Figure 14, daily treatment of chick embryos with 30–60 mm NAC after LBR failed to prevent MN loss when embryos were examined on E6.5 (Fig. 14). Interestingly, in the same embryos, however, the small amount of normal PCD that occurs on the contralateral control side by E6.5 was significantly reduced by NAC as measured by a decrease in pyknotic MNs and an increase in surviving MNs.
Fig. 14.

Pyknotic motoneurons (mean ± SD) on E6.5–E7.0 ipsilateral and contralateral to LBR after treatment withNAC or saline (SAL) on E5 and E6 (A). *p < 0.01.B, Surviving healthy motoneurons (mean ± SD) on E6.5–E7.0 after NAC or saline treatment as noted above. *p < 0.01; t test. See Materials and Methods and Results.
In a recent paper (Milligan et al., 1995), we reported that treatment of embryos after LBR with the ICE protease inhibitors Ac-YVAD-CHO or Ac-YVAD-CMK failed to prevent MN death induced by limb removal, although these same inhibitors were effective in reducing the normal PCD of MNs between E6 and E10. To further examine the possibility that the death of MNs after LBR may be mediated by a non-ICE pathway, we have treated LBR embryos with two additional inhibitors of ICE family proteases, BAF and DEVD. Although both BAF and DEVD rescue MNs from normal PCD on E8 (Li et al., 1997), neither agent was able to prevent the MN death induced by LBR or the early cell death contralateral to LBR on E6.5. (Table 3). Taken together, these data on ICE inhibitors support the argument that MN death after LBR may be mediated by intracellular pathways distinct from those involved in normal PCD.
Table 3.
The number of healthy MNs (mean ± SD) after treatment with DEVD or BAF (40 μg)
| Treatment | Healthy MNs (E6.5) | |
|---|---|---|
| Control (CON) | LBR | |
| Saline | 16,734 (±617) | 10,1733-a (±738) |
| n = 7 | n = 7 | |
| DEVD | 15,800 (±831) | 85403-a (±1,843) |
| n = 5 | n = 5 | |
| BAF | 15,462 (±804) | 96343-a (±1,251) |
| n = 5 | n = 5 | |
p < 0.001; t test vs CON. See Materials and Methods.
DISCUSSION
One of the most striking features of the spinal cord of embryos after LBR is the reduction in the number of surviving sensory and motor neurons that would normally have innervated the missing limb. Because the surgical removal of the limb-bud is performed at a stage before the onset of peripheral axon projections by either sensory or motor neurons, this deficit cannot be attributed to axotomy-induced injury. Although it is not possible to entirely exclude an indirectsurgery-related effect, this seems highly unlikely, because even in the absence of surgery a similar phenotype is observed in the avian genetic mutant limbless (Lanser and Fallon, 1984, 1987; Oppenheim et al., 1990). Accordingly, the most reasonable explanation for the decreased number of motor and sensory neurons after LBR is the absence of critically important limb-derived signals. In this paper we have examined several different ways in which these putative limb-derived signals might influence the development of innervating neurons.
Sensory and motor neurons undergo normal differentiation after LBR
The decrease in numbers of phenotypically normal-appearing sensory neurons and MNs after LBR could be attributable, in principle, to several different factors, including altered proliferation, migration, differentiation, or survival. Previous studies have provided evidence against an effect of LBR on differentiation by showing that peripherally deprived MNs appear histologically normal at the light and electron microscopic level and project axons and undergo normal biochemical maturation (Hamburger, 1958; Oppenheim et al., 1978). In this paper, we have confirmed and extended these observations by showing that MN cell size is normal after LBR and that at least one MN specific marker, the islet-1 member of the LIM homeoprotein family (Tuschida et al., 1994), is expressed normally in peripherally deprived MNs before the onset of PCD. Taken together, these various lines of evidence support the argument that a failure of differentiation cannot account for the decreased numbers of surviving MNs after LBR. A similar argument can also be made for sensory neurons. With the appropriate controls (see Results), the size of sensory neurons in the DRG is unchanged after LBR. The histological differentiation of target-deprived sensory neurons was also indistinguishable from control neurons at the onset of PCD. Additionally, in a recent study it was found that despite the reduction in the number of sensory neurons, the topographic pattern of neurotrophin receptor expression of surviving DRG cells is normal after LBR (Oakley et al., 1997). These results indicate that LBR does not perturb the growth or early differentiation of sensory neurons.
Precursor proliferation of sensory and motor neurons is not controlled by the target
In the present study, LBR was performed at a stage before the completion of cell proliferation of the precursors of both spinal MNs and DRG sensory cells (Hollyday and Hamburger, 1977; Carr and Simpson, 1978a,b). Because at least one limb-derived neurotrophic factor, NT-3, has been implicated in the control of cell proliferation in avian peripheral ganglia (Kalchiem et al., 1992; Ockel et al., 1996), we have used BrdU incorporation to examine the proliferation of precursor cells in the spinal cord and DRG after LBR. Limb-bud deletion had no effect on the number of BrdU-labeled cells within the spinal cord at any time from E3.5 to E7.5. Therefore, changes in proliferation cannot account for the decreased number of MNs that result from LBR. This supports our observation that there are comparable numbers of islet-1-labeled MNs present in the LMC ipsi- and contralateral to LBR on E4.5, a stage when >95% of all MNs have become postmitotic (Hollyday and Hamburger, 1977). (It is also important to note that the presence of normal numbers of both sensory and motor neurons in the ipsilateral LMC and DRG on E4.5 rules out an effect of LBR on the migration of postmitotic MNs to the ventral horn or of neural crest cells to the DRG.)
Similar to MNs, no significant differences were found between the number of BrdU-labeled DRG cells ipsi- and contralateral to LBR from E4.5 to E6.5, a period of active neurogenesis in the DRG (Carr and Simpson, 1978a,b). Although the number of ipsilateral BrdU-labeled DRG cells was reduced by 37% on E7.5, this is after the major period of neurogenesis in the DRG and therefore most likely reflects an indirect effect of LBR on non-neuronal proliferation caused by increased neuronal loss (Carr and Simpson, 1978a,b). For example, the proliferation of non-neuronal Schwann cells is known to be controlled by neuronal-derived signals (Morrissey et al., 1995; Lemke, 1996). Because treatment of LBR embryos with NGF, NT-3, or BDNF also failed to alter the proliferation of sensory neuron precursors (also see Oakley et al., 1997), we conclude that neither the neuronal loss after LBR nor the increased numbers of sensory neurons after LBR plusneurotrophin treatment reflects an alteration of neuronal precursor proliferation.
Trophic factor treatment rescues sensory and motor neurons from PCD after LBR
Having excluded an effect of LBR on the proliferation, migration, and differentiation of sensory and motor neurons, we conclude that the major, if not the sole, effect of target deletion is on the maintenance and survival of these neuronal populations. The most reasonable explanation of this survival effect is the neurotrophic hypothesis; that is, that sensory and motor neurons compete for limiting amounts of target-derived trophic factors. In this scenario, if both normal and LBR-induced PCD is attributable to trophic factor deprivation, then it should be possible to rescue target-deprived neurons from PCD after LBR by treatment with those putative trophic factors thought to normally control the survival of sensory and motor neurons.
Although a number of different trophic factors and tissue extracts have been shown to promote the survival of avian MNs in vitro andin vivo (Oppenheim et al., 1993; Oppenheim, 1996b), of these only CMX and GDNF were effective in rescuing MNs after LBR. CMX was able to reduce the MN loss after LBR from 71 to 40% compared with saline-treated controls. Because this substantial rescue effect was attained after treatment with only one daily dose of CMX from E4 to E7, it seems possible that a different treatment paradigm (e.g., a continuous source of exogenous CMX) might be able to rescue all of the MNs that die after LBR (see Oppenheim et al., 1993). However, at present we cannot exclude the possibility that some MNs in this situation die for reasons other than target-derived trophic factor deprivation. It is important to point out here, however, that neither CMX nor any trophic factor so far tested has been able to rescue all of the MNs that undergo normal PCD between E6 and E10 (Oppenheim, 1996b). Therefore, the failure of CMX to rescue all MNs after LBR is not unique. The failure of several trophic factors to rescue MNs after LBR despite their ability to promote survival when administered during the normal period of MN PCD from E6 to E9 (Oppenheim et al., 1993) could be interpreted as supporting the argument that normal and LBR-induced MN death are controlled by entirely different mechanisms. Although we cannot entirely exclude this, we favor the idea that this may reflect a developmental regulation of trophic factor dependencies. In support of this, we have shown previously that BDNF only promotes normal MN survival when administered during later (E8–E10) versus earlier (E5–E7) stages of the normal cell death period, and this effect is correlated with increased MN expression of trkB after E7 (McKay et al., 1996). The present results with HGF are interesting in that unlike any other factor tested, HGF rescued MNs contralateral but not ipsilateral to LBR after treatment from E4 to E7. HGF also rescued MNs from normal cell death between E6 and E10.
Treatment of embryos after LBR with one or more of the neurotrophins (NGF, NT-3, BDNF) rescued sensory neurons in the DRG from both normal and LBR-induced cell death. In accord with the relative proportion of DRG neurons known to express one of the trk receptors (Oakley et al., 1997), NGF (trkA) rescued more neurons than either NT-3 (trkC) or BDNF (trkB), and the combination of all three neurotrophins rescued more sensory neurons than any single factor. Because of the temporal overlap between sensory neuron proliferation and PCD, it is not possible to determine what proportion of the cells that undergo normal PCD are rescued in this situation. However, if one compares the peak number of sensory neurons present in the control L3 DRG on E7.5 (∼16,000) with the number present on E10 (>14,000) after the combination neurotrophin treatment (vs 10,000 in control embryos), it is clear that the neurotrophins are remarkably effective. As was the case with MNs, however, the neurotrophins were not able to rescue all sensory neurons in either the normal control or target-deprived (LBR) DRG. After LBR, the combination neurotrophin treatment resulted in the survival of 44% more sensory neurons compared with the saline control.
The failure of trophic factors to rescue all sensory or motor neurons after LBR (or during normal PCD) can most likely be attributed to less than optimal supplies of the exogenous (or endogenous) factors or to a failure to test other known or novel target-derived factors that may be required by these neuronal populations. The first possibility is supported by the fact that exogenous neurotrophins rescue more cells in the contralateral versus ipsilateral (LBR) DRG (see Results). This suggests that the trophic factor levels attained by exogenous treatment alone are probably not saturating or optimal. Furthermore, in a recent report exogenous NT-3 was shown to rescue virtually all trkC+ sensory neurons in the DRG after LBR (Oakley, 1997). Therefore, in this situation an exogenous neurotrophin was able to fully compensate for the loss of target-derived NT-3 (also see Hamburger and Yip, 1984).
Aberrant sensory neuron projections after LBR
After LBR, virtually all MNs undergo PCD, whereas 75% of control numbers of sensory neurons survive for as long as 1 week after the cessation of normal PCD in the apparent absence of their peripheral targets. This rather striking difference between the response of sensory and motor neurons to LBR has been noted previously (Levi-Montalcini and Levi, 1942; Bueker, 1947) and was attributed to the survival of a subpopulation of DRG neurons with normal projections to nonlimb targets (e.g., to the viscera or dorsal trunk skin and musculature). Other possible explanations include aberrant projections to other targets, such as the tail, or autocrine–paracrine trophic support of some sensory neurons (Acheson et al., 1995). We have observed sensory projections to both the dorsal trunk (control and LBR) and the tail (LBR only), and a combined LBR and tail ablation results in an increased loss (33%) of sensory neurons on E9.5 compared with LBR alone. Therefore, some but not all of the surviving sensory neurons after LBR appear to be supported by aberrant projections to ectopic targets in the tail. It seems likely that the tail-bud may be a source of NGF. Most of the DRG neurons that survive after LBR are trkA+ and trkC− (Oakley et al., 1997). If the tail was a source of BDNF (or of other factors that promote MN survival), then MN survival should also be affected after LBR and tail-bud deletion. Because developing (vs adult) sensory neurons are apparently not dependent on autocrine–paracrine-derived trophic support (Acheson et al., 1995), it seems most likely that the residual sensory neurons surviving after ablation of both the limb and tail represent subpopulations that project to nonlimb targets such as dorsal trunk and viscera or to the skin covering the site of the LBR.
Activity blockade and MN survival after LBR
In a previous paper we reported that activity blockade with curare rescues MNs from normal PCD but is without effect on MN death after LBR (Pittman and Oppenheim, 1979). From this evidence, we concluded that activity blockade rescues MNs by a peripheral (neuromuscular) mechanism rather than by acting centrally within the spinal cord (also see Oppenheim et al., 1997). Recently, however, this conclusion has been challenged by the suggestion that curare rescues MNs by binding to neuronal versus muscle nicotinic acetylcholine receptors (nAChR) (Hory-Lee and Frank, 1996). On the basis of anin vitro study, Hory-Lee and Frank (1996) report that although curare itself does not promote the survival of cultured MNs, it does potentiate the effects of CMX. This raises the possibility that the reason curare fails to rescue MNs from LBR in vivo is the absence of some critical potentiating signal from the limb muscle targets of MNs. To test this, we treated embryos with combinations of curare and CMX after LBR. However, curare + CMX was no more effective than CMX alone in rescuing MNs from LBR. When considered together with other evidence that supports a peripheral site of action of activity blockade (Oppenheim et al., 1996, 1997), we think that it is unlikely that neuromuscular blocking agents promote MN survival by binding to neuronal nAChRs in the CNS.
MN death after LBR is not affected by NAC, BAF, or DEVD
Previous studies have reported that after trophic factor deprivation cultured neurons and glia can be rescued from PCD by treatment with NAC (Mayer and Noble, 1994; Ferrari et al., 1995;Greenlund et al., 1995), and we have found a similar effect of NAC on cultured chick MNs after CMX deprivation (C. Milligan, S. Wang, and R. W. Oppenheim, unpublished data). As shown here, NAC also promotes normal MN survival in vivo, but a similar dose fails to prevent MN loss after LBR. Interestingly, MNs undergoing normal PCD on the contralateral control LMC of these same embryos are rescued by NAC. Although the mechanism by which NAC prevents normal MN death is unknown, it does not appear to result from increased intracellular glutathione, because this effect of NAC is not blocked by the potent and specific glutathione inhibitor buthionine sulfoximine (M. Burek and R. W. Oppenheim, unpublished data).
Considerable evidence now supports the important role of ICE family proteases in the PCD of neurons (Schwartz and Milligan, 1996). We previously reported that two peptide inhibitors of ICE, Ac-YVAD-CHO and Ac-YVAD-CMK, rescued chick MNs from normal cell death in vitro and in vivo but were ineffective in preventing MN death after LBR (Milligan et al., 1995). In this paper we have examined the effects of BAF and DEVD, two additional inhibitors of ICE family proteases (Graybill et al., 1994; Thornberry and Molineaux, 1995;Deshmukh et al., 1996). Although both BAF and DEVD are able to rescue MNs from normal PCD on E8.5 in vivo (Li et al., 1997), they fail to prevent MN death after LBR. Furthermore, neither inhibitor reduced MN death on the side contralateral to LBR on E6.5. Because none of the ICE inhibitors we have tested prevent normal or LBR-induced MN death on E6.5, this suggests that the intracellular pathway of cell death is somehow different in this situation. This is somewhat surprising because cultured MNs are also target-deprived, yet they show a survival response to ICE inhibitors (Milligan et al., 1995; Li et al., 1997). One notable difference in the two situations is that after LBR, MNs never have an opportunity to contact their normal targets, whereas cultured MNs are removed from the embryo on E5.5 when they have already begun to innervate the limb. Limb-derived signals may be necessary for the development of intracellular death pathways mediated by ICE family proteases. Alternatively, because these agents were also ineffective in preventing the normal death of MNs contralateral to LBR on E6.5, all early dying MNs may use non-ICE pathways of cell death. Recently, similar examples of distinct cell death pathways have been described (Smith and Osborne, 1997). For example, sympathetic neurons and PC12 cells require a specific caspase (Nedd2) to undergo cell death after NGF withdrawal but not after downregulation of superoxide dismutase (Troy et al., 1997); the NGF-dependent survival of sympathetic but not sensory neurons involves a phosphoinostide-3 kinase signaling pathway (Bartlett et al., 1997); and the induction of apoptosis in Jurkat cells by the pro-apoptotic bcl-2 family member Bax also appears to occur independent of ICE-like proteases (Xiang et al., 1996). Finally, another alternative we cannot exclude entirely is the possibility that significantly higher doses of, or combinations of different, ICE inhibitors are required to block early MN death. Because the morphological features of dying MNs (e.g., apoptosis) after LBR are indistinguishable from normal PCD (Oppenheim et al., 1978), however, and because trophic agents and inhibitors of RNA and protein synthesis rescue MNs in both situations (Oppenheim et al., 1990; present data) even if intracellular mechanisms differ for early versus later PCD (i.e., NAC, ICE data), the two death pathways share many common features as well.
Footnotes
This work was supported by National Institutes of Health Grants NS31380 and NS20402 (R.W.O.), a grant from the Muscular Dystrophy Association (R.A.O.), and a postdoctoral grant (BE 92–31) from Commissio Interdepartmental de Recerca i Technologia (CRIT) to J.C.
Correspondence should be addressed to R. W. Oppenheim, Department of Neurobiology and Anatomy, Neuroscience Program, Wake Forest University School of Medicine, Winston–Salem, NC 27157.
REFERENCES
- 1.Acheson A, Connover JC, Fandi JP, Dechiara TM, Russell M, Thadani A, Squinto SP, Yancopoulas GD, Lindsay RM. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature. 1995;374:450–453. doi: 10.1038/374450a0. [DOI] [PubMed] [Google Scholar]
- 2.Bartlett SE, Reynolds AJ, Weible M, Haydon K, Hendry IA. In sympathetic but not sensory neurons PI-3 kinase is important for NGF-dependent survival and the retrograde transport of 125I-βNGF. Brain Res. 1997;761:257–262. doi: 10.1016/s0006-8993(97)00329-6. [DOI] [PubMed] [Google Scholar]
- 3.Bueker ED. Intracentral and peripheral factors in the differentiation of motor neurons in transplanted lumbo-sacral spinal cords of chick embryos. J Exp Zool. 1943;93:99–129. [Google Scholar]
- 4.Bueker ED. Limb ablation experiments on the embryonic chick and its effect as observed on the mature nervous system. Anat Rec. 1947;97:157–174. doi: 10.1002/ar.1090970204. [DOI] [PubMed] [Google Scholar]
- 5.Burek MJ, Prevette D, Oppenheim RW. A temporal relationship between motoneuron birth date and onset of programmed cell death. Soc Neurosci Abstr. 1996;22:566. [Google Scholar]
- 6.Buttke TM, Sanstrom PA. Oxidative stress as a mediator of apoptosis. Immunol Today. 1994;15:7–10. doi: 10.1016/0167-5699(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 7.Campagna JA, Prevette D, Oppenheim RW, Bixby JL. Target contact regulates expression of synaptotagmin genes in spinal motor neurons in vivo. Mol Cell Neurosci. 1997;8:377–388. doi: 10.1006/mcne.1997.0596. [DOI] [PubMed] [Google Scholar]
- 8.Carr VM, Simpson SB. Proliferative and degenerative events in the early development of chick dorsal root ganglia: normal development. J Comp Neurol. 1978a;182:727–740. doi: 10.1002/cne.901820410. [DOI] [PubMed] [Google Scholar]
- 9.Carr VM, Simpson SB. Proliferation and degenerative events in the early development of chick dorsal root ganglia: responses to altered peripheral fields. J Comp Neurol. 1978b;182:741–756. doi: 10.1002/cne.901820411. [DOI] [PubMed] [Google Scholar]
- 10.Chu-Wang IW, Oppenheim RW. Cell death of motoneurons in the chick embryo spinal cord. J Comp Neurol. 1978;177:33–86. doi: 10.1002/cne.901770105. [DOI] [PubMed] [Google Scholar]
- 11.Clarke PGH, Oppenheim RW. Neuron death in vertebrate development: in vivo methods. In: Schwartz LM, Osborne BA, editors. Methods in cell biology, Vol 46, cell death. Academic; New York: 1995. pp. 227–321. [PubMed] [Google Scholar]
- 12.Dent JA, Polson AG, Klymkowsky MW. Whole mount immunocytochemical analysis of the expression of the intermediate filament protein vimentin in Xenopus. Development. 1989;105:61–74. doi: 10.1242/dev.105.1.61. [DOI] [PubMed] [Google Scholar]
- 13.Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM. Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Easter SS, Ross LS, Frankfurter A. Initial tract formation in the mouse brain. J Neurosci. 1993;13:285–299. doi: 10.1523/JNEUROSCI.13-01-00285.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrari G, Yan CY, Greene LA. N-acetylcysteine prevents apoptotic death of neuronal cells. J Neurosci. 1995;15:2857–2866. doi: 10.1523/JNEUROSCI.15-04-02857.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graybill TL, Dolle RE, Helaszek CT, Miller RE, Ator ME. Preparation and evaluation of peptidic aspartyl hemiacetals as reversible inhibitors of interleukin 1β converting enzyme (ICE). Int J Pept Protein Res. 1994;44:173–182. doi: 10.1111/j.1399-3011.1994.tb00573.x. [DOI] [PubMed] [Google Scholar]
- 17.Greenlund LJ, Deckwerth TL, Johnson EM. Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 18.Hamburger V. The effects of wing bud extirpation on the development of the central nervous system in chick embryos. J Exp Zool. 1934;68:449–494. [Google Scholar]
- 19.Hamburger V. Regression versus peripheral control of differentiation in motor hypoplasia. Am J Anat. 1958;102:365–402. doi: 10.1002/aja.1001020303. [DOI] [PubMed] [Google Scholar]
- 20.Hamburger V, Levi-Montalcini R. Proliferation, differentiation and degeneration in the spinal ganglia of the chick embryo under normal and experimental conditions. J Exp Zool. 1949;111:457–501. doi: 10.1002/jez.1401110308. [DOI] [PubMed] [Google Scholar]
- 21.Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J Morphol. 1951;88:49–92. [PubMed] [Google Scholar]
- 22.Hamburger V, Yip J. Reduction of experimentally induced neuronal death in spinal ganglia of the chick by nerve growth factor. J Neurosci. 1984;4:767–774. doi: 10.1523/JNEUROSCI.04-03-00767.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamburger V, Brunso-Bechtold JK, Yip J. Neuronal death in the spinal ganglia of the chick embryo and its reduction by nerve growth factor. J Neurosci. 1981;1:60–71. doi: 10.1523/JNEUROSCI.01-01-00060.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hollyday MA, Hamburger V. An autoradiographic study of the formation of the lateral motor column in the chick embryo. Brain Res. 1977;132:197–208. doi: 10.1016/0006-8993(77)90416-4. [DOI] [PubMed] [Google Scholar]
- 25.Honig MG, Hume RI. Fluorescent carbocyanine dyes allow living neurons of identified origin to be studies in long term cultures. J Cell Biol. 1986;103:171–187. doi: 10.1083/jcb.103.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hory-Lee F, Frank E. The nicotinic blocking agents d-tubocurare and α-bungarotoxin save motoneurons from naturally occurring death in the absence of neuromuscular blockade. J Neurosci. 1995;15:6453–6460. doi: 10.1523/JNEUROSCI.15-10-06453.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson JE, Yin QW, Prevette D, Oppenheim RW. Brain-derived proteins that rescue spinal motoneurons from cell death in the chick embryo. J Neurobiol. 1995;27:573–589. doi: 10.1002/neu.480270411. [DOI] [PubMed] [Google Scholar]
- 28.Kalchiem C, Carmeli C, Rosenthal A. Neurotrophin-3 is a mitogen for cultured neural crest cells. Proc Natl Acad Sci USA. 1992;89:1661–1665. doi: 10.1073/pnas.89.5.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kane DJ, Sarafian TA, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE. Bcl-2 inhibition of neuronal death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 30.Lanser ME, Fallon JF. Development of the lateral motor column in the limbless mutant chick embryo. J Neurosci. 1984;4:2043–2050. doi: 10.1523/JNEUROSCI.04-08-02043.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lanser ME, Fallon JF. Development of the brachial lateral motor column in the wingless mutant chick embryo. J Comp Neurol. 1987;261:423–434. doi: 10.1002/cne.902610307. [DOI] [PubMed] [Google Scholar]
- 32.Lanser ME, Carrington JL, Fallon JF. Survival of motoneurons in the brachial lateral motor column of limbless mutant chick embryos depends on the periphery. J Neurosci. 1986;6:2551–2557. doi: 10.1523/JNEUROSCI.06-09-02551.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee MK, Tuttle JB, Rebhun LI, Cleveland DW, Frankfurter A. The expression and post-translational modification of a neuron-specific beta tubulin isoform during chick embryogenesis. Cell Motil Cytoskeleton. 1990;17:118–132. doi: 10.1002/cm.970170207. [DOI] [PubMed] [Google Scholar]
- 34.Lemke G. Neuregulins in development. Mol Cell Neurosci. 1996;7:247–262. doi: 10.1006/mcne.1996.0019. [DOI] [PubMed] [Google Scholar]
- 35.Levi-Montalcini R, Levi G. Les consequences de la destruction d’un territoire d’innervation peripherigue sur le developpment des centres nerveux correspondants dans l’embryon de Poulet. Arch Biol. 1942;53:537–545. [Google Scholar]
- 36.Li L, Prevette D, Milligan CE, Oppenheim RW. Motoneuron cell death following target deletion is not prevented by the inhibition of ICE family, cysteine proteases. Soc Neurosci Abstr. 1997;23:1157. [Google Scholar]
- 37.Linden R. The survival of developing neurons: a review of afferent control. Neuroscience. 1996;58:671–682. doi: 10.1016/0306-4522(94)90447-2. [DOI] [PubMed] [Google Scholar]
- 38.Mayer M, Noble M. N-acetyl-l-cysteine is a pluripotent protector against cell death and enhancer of trophic factor mediated cell survival in vitro. Proc Natl Acad Sci USA. 1994;91:7496–7500. doi: 10.1073/pnas.91.16.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKay SE, Garner AS, Caldero J, Tucker RP, Large T, Oppenheim RW. The expression of trkB and p75 and the role of BDNF in the developing neuromuscular system of the chick embryo. Development. 1996;122:715–724. doi: 10.1242/dev.122.2.715. [DOI] [PubMed] [Google Scholar]
- 40.Milligan CE, Prevette D, Yaginuma H, Homma S, Cardwell C, Fritz LC, Tomaselli KJ, Oppenheim RW, Schwartz LM. Peptide inhibitors of the ICE protease family arrest programmed cell death of motoneurons in vivo and in vitro. Neuron. 1995;15:385–393. doi: 10.1016/0896-6273(95)90042-x. [DOI] [PubMed] [Google Scholar]
- 41.Morrissey TK, Levi AD, Nujens A, Sliwkowski MX, Bunge RP. Axon-induced mitogenesis of human Schwann cells involves heregulin and p185 erbβ2. Proc Natl Acad Sci USA. 1995;92:1431–1435. doi: 10.1073/pnas.92.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neff NT, Prevette D, Houenou LJ, Lewis ME, Glicksman MA, Yin MA, Yin QW, Oppenheim RW. Insulin-like growth factors: putative muscle-derived trophic agents that promote motoneuron survival. J Neurobiol. 1993;24:1578–1588. doi: 10.1002/neu.480241203. [DOI] [PubMed] [Google Scholar]
- 43.Oakley RA, Lefcourt FB, Clary DO, Reichardt LF, Prevette D, Oppenheim RW, Frank E. Neurotrophin-3 promotes the differentiation of muscle spindle afferents in the absence of peripheral targets. J Neurosci. 1997;17:4262–4274. doi: 10.1523/JNEUROSCI.17-11-04262.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ockel M, Lavin GR, Barde YA. In vivo effects of neurotrophin-3 during sensory neurogenesis. Development. 1996;122:301–307. doi: 10.1242/dev.122.1.301. [DOI] [PubMed] [Google Scholar]
- 45.Oppenheim RW. Neuronal cell death and some related regressive phenomena during neurogenesis: a selective historical review and progress report. In: Cowan WM, editor. Studies in developmental neurobiology: essays in honor of Viktor Hamburger. Oxford UP; Oxford: 1981. pp. 74–133. [Google Scholar]
- 46.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 47.Oppenheim RW. The concept of uptake and retrograde transport of neurotrophic molecules during development: history and present status. Neurochem Res. 1996a;21:769–777. doi: 10.1007/BF02532299. [DOI] [PubMed] [Google Scholar]
- 48.Oppenheim RW. Neurotrophic survival molecules for motoneurons: an embarrassment of riches. Neuron. 1996b;17:195–197. doi: 10.1016/s0896-6273(00)80151-8. [DOI] [PubMed] [Google Scholar]
- 49.Oppenheim RW, Chu-Wang IW, Maderdrut JL. Cell death of motoneurons in the chick embryo spinal cord. III. The differentiation of motoneurons prior to their induced degeneration following limb-bud removal. J Comp Neurol. 1978;177:87–112. doi: 10.1002/cne.901770107. [DOI] [PubMed] [Google Scholar]
- 50.Oppenheim RW, Haverkamp LJ, Prevette D, McManaman JL, Appel SH. Reduction of naturally occurring motoneuron death in vivo by a target-derived neurotrophic factor. Science. 1988;240:919–922. doi: 10.1126/science.3363373. [DOI] [PubMed] [Google Scholar]
- 51.Oppenheim RW, Prevette D, Tytell M, Homma S. Naturally occurring and induced neuronal death in the chick embryo in vivo requires protein and RNA synthesis. Dev Biol. 1990;138:104–113. doi: 10.1016/0012-1606(90)90180-q. [DOI] [PubMed] [Google Scholar]
- 52.Oppenheim RW, Prevette D, Yin QW, Collins F, MacDonald J. Control of embryonic motoneuron survival in vivo by ciliary neurotrophic factor. Science. 1991;251:1616–1618. doi: 10.1126/science.2011743. [DOI] [PubMed] [Google Scholar]
- 53.Oppenheim RW, Prevette D, Fuller F. The lack of effect of basic and acidic fibroblast growth factors on the naturally occurring death of neurons in the chick embryo. J Neurosci. 1992;12:2726–2734. doi: 10.1523/JNEUROSCI.12-07-02726.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oppenheim RW, Prevette D, Haverkamp LJ, Houenou L, Yin Q-W, McManaman J. Biological studies of a putative avian muscle-derived neurotrophic factor that prevents naturally occurring motoneuron death in vivo. J Neurobiol. 1993;24:1065–1079. doi: 10.1002/neu.480240806. [DOI] [PubMed] [Google Scholar]
- 55.Oppenheim RW, Houenou LJ, Johnson JE, Lin LFH, Li L, Lo AC, Newsome AL, Prevette DM, Wang SW. Developing motor neurons rescued from programmed and axotomy-induced cell death by GDNF. Nature. 1995;373:344–346. doi: 10.1038/373344a0. [DOI] [PubMed] [Google Scholar]
- 56.Oppenheim RW, Prevette D, Wang SW. The rescue of avian motoneurons by activity blockade at the neuromuscular junction. Soc Neurosci Abstr. 1996;22:44. [Google Scholar]
- 57.Oppenheim RW, Prevette D, Houenou LJ, Pinçon-Raymond M, Dimitriadou V, Donevan A, O’Donovan M, Wenner P, McKemy DD, Allen PD. Neuromuscular development in the avian paralytic mutant crooked neck dwarf (cn/cn): further evidence for the role of neuromuscular activity in motoneuron survival. J Comp Neurol. 1997;381:353–372. doi: 10.1002/(sici)1096-9861(19970512)381:3<353::aid-cne7>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 58.Phelan KA, Hollyday M. Embryonic development and survival of brachial motoneurons projecting to muscle-less chick wings. J Comp Neurol. 1991;311:313–320. doi: 10.1002/cne.903110302. [DOI] [PubMed] [Google Scholar]
- 59.Pittman R, Oppenheim RW. Neuromuscular blockade increases motoneuron survival during normal cell death in the chick embryo. Nature. 1978;271:364–366. doi: 10.1038/271364a0. [DOI] [PubMed] [Google Scholar]
- 60.Pittman R, Oppenheim RW. Cell death of motoneurons in the chick embryo spinal cord. J Comp Neurol. 1979;187:425–466. doi: 10.1002/cne.901870210. [DOI] [PubMed] [Google Scholar]
- 61.Schwartz LM, Milligan CA. Cold thoughts of death: the role of ICE proteases in neuronal death. Trends Neurosci. 1996;19:555–562. doi: 10.1016/s0166-2236(96)10067-9. [DOI] [PubMed] [Google Scholar]
- 62.Shorey MC. the effect of the destruction of peripheral areas on the differentiation of the neuroblasts. J Exp Zool. 1909;7:25–63. [Google Scholar]
- 63.Smith SW, Osborne BA. Private pathways to a common death. J NIH Res. 1997;9:33–37. [Google Scholar]
- 64.Smith SW, Osborne BA. Private pathways to a common death. J NIH Res. 1997;9:33–37. [Google Scholar]
- 65.Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 66.Thornberry NA, Molineaux SM. Interleukin-1β converting enzyme: a novel cysteine protease required for IL-1β production and implications in programmed cell death. Protein Sci. 1995;4:3–12. doi: 10.1002/pro.5560040102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tosney KW, Landmesser L. Pattern and specificity of axonal outgrowth following varying degrees of chick limb bud ablation. J Neurosci. 1984;4:2518–2527. doi: 10.1523/JNEUROSCI.04-10-02518.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Troy CM, Stefanis L, Greene LA, Shelanski ML. Nedd2 is required for apoptosis after trophic factor withdrawal, but not superoxide dismutase (SOD1) downregulation, in sympathetic neurons and PC12 cells. J Neurosci. 1997;17:1911–1918. doi: 10.1523/JNEUROSCI.17-06-01911.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tuschida T, Ensini M, Morton SB, Baldassare M, Edlund T, Jessell TM, Pfaff SL. Topographic organization of embryonic motor neurons defined by expression of LIM homeobox genes. Cell. 1994;79:957–970. doi: 10.1016/0092-8674(94)90027-2. [DOI] [PubMed] [Google Scholar]
- 70.Xiang J, Chao DT, Korsmeyer SJ. Bax-induced cell death may not require interleukin 1β-converting enzyme-like proteases. Proc Natl Acad Sci USA. 1996;93:14553–14563. doi: 10.1073/pnas.93.25.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yaginuma H, Tomita M, Takashita N, McKay SE, Cardwell C, Yin QW, Oppenheim RW. A novel type of programmed neuronal death in the cervical spinal cord of the chick embryo. J Neurosci. 1996;16:3685–3703. doi: 10.1523/JNEUROSCI.16-11-03685.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]