

Abstract
The present study sought to determine whether doses of methamphetamine in the range of those used recreationally by humans produce brain dopamine (DA) neurotoxicity in baboons and to ascertain whether positron emission tomography (PET) imaging with the DA transporter (DAT) ligand [11C]WIN-35,428 ([11C]2β-carbomethoxy-3β-(4-fluorophenyl)-tropane) could be used to detect methamphetamine-induced DAT loss in living primates. Baboons were treated with saline (n = 3) or one of three doses of methamphetamine [0.5 mg/kg (n = 2); 1 mg/kg (n = 2); and 2 mg/kg (n = 3)], each of which was given intramuscularly four times at 2 hr intervals. PET studies were performed before and 2–3 weeks after methamphetamine treatment. After the final PET studies, animals were killed for direct neurochemical determination of brain DA axonal markers. PET-derived binding potential values, used to index striatal DAT density, were significantly decreased after methamphetamine, with larger decreases occurring after higher methamphetamine doses. Reductions in striatal DAT documented by PET were associated with decreases in DA, dihydroxyphenylacetic acid, and specific [3H]WIN-35,428 and [3H]DTBZ binding determined in vitro. Decreases in DAT detected with PET were highly correlated with decreases in specific [3H]WIN-35,428 binding determined in vitro in the caudate of the same animal (r= 0.77; p = 0.042). These results indicate that methamphetamine, at doses used by some humans, produces long-term reductions in brain DA axonal markers in baboons, and that it is possible to detect methamphetamine-induced DAT loss in living nonhuman primates by means of PET.
Keywords: methamphetamine; dopamine; neurotoxicity; PET; primates; WIN-35,428
Methamphetamine, a potent psychomotor stimulant drug, is known on the street as “speed,” “crystal meth,” or “crank.” Over the last 50 years, epidemics of methamphetamine abuse have occurred in many parts of the world, including Sweden (Inghe, 1969), the United Kingdom (Kiloh and Brandon, 1962), Japan (Brill and Hirose, 1969), and the United States (Kalant, 1966; Kramer et al., 1967; Miller and Hughes, 1994). To this day, abuse of methamphetamine continues, with recent surveys indicating that methamphetamine abuse is on the rise (Kozel, 1997; Lukas, 1997).
Methamphetamine is known to have the potential to damage brain dopamine (DA) and serotonin (5-HT) neurons. Laboratory animals given repeated high doses of methamphetamine show large, long-lasting depletions of brain DA and 5-HT (Kogan et al., 1976; Seiden et al., 1976; Hotchkiss and Gibb, 1980; Ricaurte et al., 1980; Wagner et al., 1980a,b; Slikker et al., 1988), as well as marked, long-term reductions in the activity of tyrosine hydroxylase and tryptophan hydroxylase (Hotchkiss et al., 1979; Hotchkiss and Gibb, 1980; Ricaurte et al., 1983; Schmidt et al., 1985), the concentration of DA and 5-HT metabolites (Seiden et al., 1976; Hotchkiss and Gibb, 1980; Ricaurte et al., 1980), and the number of DA and 5-HT transporters (5-HTT) (Ricaurte et al., 1980;Wagner et al., 1980 a,b, 1982). Anatomic studies indicate that loss of these presynaptic DA and 5-HT axonal markers is related to damage of distal DA and 5-HT axon projections (Ellison, 1978; Lorez, 1981; Nwanze and Jonsson, 1981; Ricaurte et al., 1982, 1984a,b; Fukui et al., 1989; Axt et al., 1991). Methamphetamine neurotoxicity has been demonstrated in mice, rats, guinea pigs, cats, and rhesus monkeys (Seiden and Ricaurte, 1987). Moreover, the toxic effects of methamphetamine on brain DA and 5-HT neurons in nonhuman primates persist for up to 4 years (Woolverton et al., 1989), suggesting that they may be permanent, at least in rhesus monkeys given repeated high doses.
In all animal species tested, the neurotoxicity of methamphetamine is dose-dependent, generally occurring only after the administration of repeated high doses (Seiden and Ricaurte, 1987; Gibb et al., 1994). However, lower doses of methamphetamine or amphetamine can also produce neurotoxic effects, if administered continuously (via osmotic minipumps) (Ricaurte et al., 1984a,b), with iprindole (Fuller and Hemrick-Luecke, 1980; Peat et al., 1983), or at short time intervals (e.g., every 2 hr) (Sonsalla et al., 1989; Melega et al., 1993). Whether repeated doses of methamphetamine in the range of those used recreationally by humans [20–40 mg (Jaffe, 1985)] produce long-term effects on brain DA neurons in baboons is unknown.
Although methamphetamine is a documented DA neurotoxin in animals, little is presently known regarding the DA neurotoxic potential of methamphetamine in humans, largely because of difficulties inherent in assessing DA neuronal integrity in the living human brain. Preclinical observations that amphetamines, if given repeatedly at short intervals (similar to the use pattern of “binge” methamphetamine users), can produce dopamine neurotoxicity (Sonsalla et al., 1989) suggest that human methamphetamine users may be at risk for methamphetamine-induced DA neural injury. The purpose of the present study was to determine whether methamphetamine, when given to baboons at a dose and in a pattern similar to that used by some humans [20–40 mg every 2–3 hr (Jaffe, 1985; Konuma, 1994)], produced long-term toxic effects on brain DA neurons. We also sought to determine whether PET imaging with [11C]WIN-35,428 could be used to detect methamphetamine-induced DAT loss in the brain of living nonhuman primates.
MATERIALS AND METHODS
Animals. Ten baboons (Papio anubis) were used in this study. Body weights ranged from 19 to 27 kg. Ages of the animals could not be determined with certainty because the baboons were feral-reared. Animals were housed in a colony room maintained at 22 ± 1°C, with free access to food and water. The experimental protocol was approved by the Animal Care and Use Committee of the Johns Hopkins Medical Institutions.
Drug treatment. Animals were treated with three different doses [0.5 mg/kg (n = 2); 1 mg/kg (n = 2); and 2 mg/kg (n = 3)] ofd-methamphetamine hydrochloride, with dose expressed as the salt. Methamphetamine was dissolved in a sterile 0.9% sodium chloride solution and injected intramuscularly. Each dose of methamphetamine was given four times, at 2 hr intervals, such that the total dose for each set of animals was 2, 4, and 8 mg · kg−1 · d−1, respectively. This particular regimen of methamphetamine administration was used because it is known to produce long-term effects on brain DA neurons in rodents (Sonsalla et al., 1989), and 2 mg/kg doses of amphetamine given according to a similar regimen produce long-term DA deficits in green vervet monkeys (Melega et al., 1993). Also, such a dose regimen closely approximates the binge use of methamphetamine by some humans [20–40 mg every 2–3 hr (Jaffe, 1985; Konuma, 1994)].
Study design. Ten baboons were studied, using a design that permitted both between- and within-subject comparisons. Methamphetamine-treated baboons were studied with PET before, and 2–3 weeks after, drug treatment. Control baboons also underwent repeat PET scans before and after saline treatment to establish test–retest reproducibility. On completion of the final PET studies, animals were killed by an overdose of pentobarbital (60 mg/kg), and the brains were removed for neurochemical studies. Tissues for neurochemical studies were kept frozen in liquid nitrogen; control and experimental samples were assayed in parallel.
Preparation of [11C]WIN-35,428.[11C]WIN-35,428 was synthesized as described previously (Dannals et al., 1993). The average specific activity of the final product calculated at the end of synthesis was >2000 mCi/μmol.
PET imaging. On the day of the PET study, two intravenous catheters and an arterial catheter were placed for infusion of anesthesia, injection of radiotracer, and arterial blood sampling, respectively. Animals were initially anesthetized intramuscularly with 8–10 mg/kg alfadolone and alfaxolone acetate (Saffan) and intubated. Anesthesia was maintained throughout the study by a continuous intravenous infusion drip of 6–9 mg · kg−1 · hr[minus[1]alfadolone and alfaxolone acetate. The baboon was secured to the PET bed using an individually fitted thermoplastic mask, which allowed reproducible positioning between studies. Pulse, blood pressure, and oxygen saturation were monitored continuously during the studies. Blood oxygen saturation was always maintained above 85%. After baboons were positioned in the PET scanner, a transmission scan was performed with a 10 mCi 68Ga source to allow for attenuation correction. PET scanning was started immediately after intravenous injection of 20 mCi of high specific activity [11C]WIN-35,428 (corresponding to 0.12 nmol/kg). Fifteen simultaneous (eight direct planes, seven cross planes, z axis = ∼10 cm) sequential quantitative tomographic slices of the brain were obtained with the GE 4096+ PET tomograph in the high-resolution mode (∼6.5 mm full width at half maximum), over a 90 min period. Baboons were positioned so that the lowest plane was located approximately 5 cm below the canthomeatal line. Plasma radioactivity was corrected for decay and metabolites. In all cases, ∼30 arterial samples/PET study (for radioassay and protein binding) were obtained over 90 min. To correct the input function for unmetabolized [11C]WIN-35,428, arterial samples were also obtained at 10, 20, 30, 60, and 75 min after the injection of the radiotracer for analysis by means of high performance liquid chromatography (HPLC). PET images were reconstructed from the raw data with a standard filtered back-projection algorithm and a Hann filter (6.0 mm). Images were corrected for attenuation and decay. Regions of interest were placed manually over the striatum and the cerebellum, and time-activity curves were generated.
Postmortem monoamine and metabolite level determinations.Levels of monoamines and their major metabolites were determined by means of HPLC coupled with electrochemical detection (HPLC-EC), as described previously (Ricaurte et al., 1992).
In vitro [3H]WIN-35,428 binding studies. [3H]WIN-35,428-labeled DATs were measured using the method of Madras and colleagues (1989), with minor modification. Briefly, frozen striatal tissue was weighed, homogenized in 20 vol (w/v) of a 0.32 m sucrose phosphate buffer, pH 7.4, at 0–4°C, and centrifuged at ∼45,000 × g for 15 min at 0–4°C. The supernatant was discarded, and the pellet was resuspended in 20 vol of sucrose PO4 buffer and then centrifuged once again at ∼45,000 × g for 15 min at 0–4°C. The resulting pellet was suspended in buffer for a final tissue concentration of 10 mg/ml wet weight. [3H]WIN-35,428 was used at a predetermined saturating concentration of 30 nm. Cocaine, at a final concentration of 30 μm, was used to displace specific [3H]WIN-35,428 binding and estimate nonspecific binding. Tubes were incubated in sextuplicate for 60 min, at 0–4°C. Membranes were harvested by filtration through Whatman GFB filters soaked in 0.05% polyethyleneimine (PEI). Filters were washed three times using ice-cold sucrose PO4 buffer. Radioactivity was measured with a Packard-1500 Tricarb Liquid Scintillation Analyzer. Specific [3H]WIN35,428 binding was calculated by subtracting the average value of the six tubes containing excess cocaine from the average of the six tubes without cocaine. Specific [3H]WIN-35,428 binding was expressed in dpm/mg original wet weight tissue. Nonspecific binding represented ∼7–10% of the total binding.
In vitro [3H]dihydrotetrabenazine (TBNZ) binding studies. [3H]DTBZ binding, used to label type 2 vesicular monoamine transporter (VMAT) sites (Naudon et al., 1994; Vander Borght et al., 1996), was measured using the method of Wilson et al. (1996b), with minor modifications. Briefly, tissue samples were homogenized for 15 sec in 20 vol (w/v) of sodium phosphate buffer (25 mm, pH 7.7) and then centrifuged in a Sorvall RC2B at ∼45,000 × g for 15 min at 0–4°C. The resulting pellet was resuspended in 20 vol (w/v) of sodium phosphate buffer, homogenized again for 15 sec, and recentrifuged at ∼45,000 × g for 15 min at 0–4°C. The supernatant was discarded, and the resulting pellet was resuspended in buffer at a final concentration of 10 mg of original wet weight tissue per milliliter. Membrane preparations were incubated with a predetermined saturating concentration of [3H]DTBZ (15 nm) in 25 mm sodium phosphate buffer, pH 7.7, for 90 min at 30°C in a shaking water bath. Each sample was run in sextuplicate, such that six tubes were used to define total binding and six tubes were used to determine nonspecific binding. Nonspecific binding was determined in the presence of 1 μmtetrabenazine and represented ∼8–10% of total binding. The incubation was terminated by rapid filtration, using a 48-well cell harvester (Brandell, Gaithersburg, MD) and Whatman GFB filters soaked with 0.05% PEI. Filters were washed three times with 10 ml sodium phosphate buffer, and residual radioactivity was measured using a Packard-1500 Tricarb Liquid Scintillation Analyzer. Specific binding, calculated by subtracting nonspecific binding from total binding, was expressed as dpm/mg original wet weight tissue.
In vitro [3H]paroxetine binding studies. [3H]paroxetine-labeled 5-HTT were measured using the method of Habert et al. (1985), with the previously described minor modifications (Ricaurte et al., 1992).
In vitro [3H]nisoxetine binding studies. [3H]nisoxetine-labeled norepinephrine transporters (NETs) in tissue homogenates were measured using the method of Tejani-Butt (1992).
Autoradiographic studies. Autoradiograms with [3H]WIN-35,428 ([3H]-2β-carbomethoxy-3β-(4-fluorophenyl)-tropane) were prepared according to the method of Kaufman et al.(1991). [3H]WIN-35,428 was used at a concentration of 3 nm to measure total binding and along with 30 μm cocaine to determine nonspecific binding. Autoradiograms with [3H]mazindol were prepared using the method of Javitch et al. (1985). [3H]mazindol was used at a concentration of 4 nm to measure total binding and along with 1 μm mazindol to determine nonspecific binding.
Data analysis. PET data were analyzed by determining the binding potential (BP) (i.e.,Bmax/Kd), which represents the k3/k4 ratio of a three-compartment model described previously (Wong et al., 1993). In this approach, K1/k2 obtained in the cerebellum is used to constrain K1/k2 in the striatum, thereby reducing the number of parameters to 3. For each animal, the DAT BP change induced by methamphetamine was determined by calculating the difference between the BP value obtained in the baseline study and the BP value obtained in the PET study after methamphetamine. The significance of differences observed was determined using a paired two-tailed t test. Results from neurochemical studies were evaluated by ANOVA. When statistical differences were observed,post hoc comparisons were performed with Duncan’s multiple range tests, compensating for multiple comparisons. Pearson product moment and Spearman’s correlations were used to calculate the relation among the various DA axonal markers and imaging results, again compensating for multiple comparisons. All tests were two-tailed, and significance was set at p ≤ 0.05. Data analysis was performed using the Statistical Program for the Social Sciences (SPSS for Windows, Release 6).
Drugs and chemicals. Dopamine (DA) hydrochloride, 5-hydroxytryptamine (5-HT, serotonin) creatinine sulfate complex, and 5-hydroxyindole-3-acetic acid (dicyclohexylammonium salt) were purchased from Sigma (St. Louis, MO). Perchloric acid was purchased from J. T. Baker (Phillipsburg, NJ). [3H]WIN35,428 (specific activity, 83.5 Ci/mmol), [3H]paroxetine (specific activity, 17.1 Ci/mmol), [3H]mazindol (specific activity, 24.0 Ci/mmol), and [3H]nisoxetine (specific activity 74 Ci/mmol) were purchased from New England Nuclear (Boston, MA). [3H]TBNZ (specific activity, 150 Ci/mmol) was obtained from Amersham Life Science (Buckinghamshire, England).d-methamphetamine hydrochloride was obtained from the National Institute on Drug Abuse (Rockville, MD).
RESULTS
PET studies
Assessment of test–retest variability in three untreated baboons, each of which was imaged twice with [11C]WIN-35,428 in a manner identical to that of methamphetamine-treated animals, showed the mean (±SD) percent variability to be 9.3 ± 4.7%.
The difference between pre- and postmethamphetamine PET-derived [11C]WIN-35,428 BP values, used to index DAT density in vivo (Wong et al., 1993), was highly significant (mean ± SEM; paired difference: 0.82 ± 0.21,t = 3.84, p = 0.009). Figure1 shows the average PET images (between 70–90 min after injection of the radiotracer) normalized to injected activity from studies in three different baboons before and after treatment with methamphetamine at three different doses of methamphetamine (0.5, 1, and 2 mg/kg, respectively). BP reductions ranged from 35 to 44% at the 0.5 mg/kg dose, 40 to 48% at the 1 mg/kg dose, and 50 to 69% at the 2 mg/kg dose of methamphetamine (Table1).
Fig. 1.
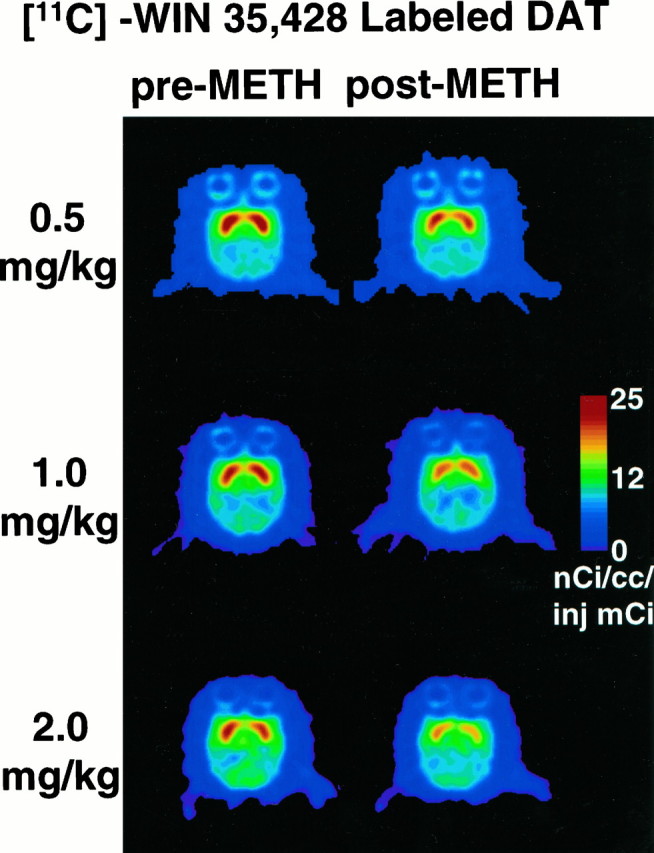
[11C]WIN-35,428 images in baboons before (left) and 2–3 weeks after (right) treatment with three different doses of methamphetamine. Each dose was given four times, at 2 hr intervals. Images shown are at midstriatal level from representative baboons. Images shown represent average PET images (between 70 and 90 min after injection of the radiotracer) normalized to injected activity.
Table 1.
PET-derived BP values (Bmax/kd) in the striatum of control and methamphetamine-treated baboons
| Before | After | |
|---|---|---|
| Control 1.54 ± 0.22; n = 101-a | ||
| Methamphetamine | ||
| 0.5 mg/kg | 1.6 | 0.9 |
| 2.6 | 1.7 | |
| 1.0 mg/kg | 1.2 | 0.7 |
| 1.1 | 0.6 | |
| 2.0 mg/kg | 1.1 | 0.5 |
| 1.0 | 0.5 | |
| 2.9 | 0.9 | |
Based on PET imaging results of 10 untreated baboons, including the animals that subsequently received methamphetamine.
The linear correlation coefficient between BP reduction and methamphetamine dose was r = 0.75 (p = 0.05), with a Spearman correlation coefficient of 0.87 (p = 0.01) (Fig.2).
Fig. 2.

Relation between dose of methamphetamine and reduction in the DAT density, as measured by means of PET imaging with [11C]WIN-35,428. Methamphetamine was given intramuscularly at three different doses. Each dose was given four times, at 2 hr intervals. Animals underwent PET studies 2–3 weeks after methamphetamine treatment.
Postmortem neurochemical studies
Tables 2 and3 show the results of postmortem neurochemical studies performed using tissue samples from the caudate and putamen of the same baboons that had undergone PET studies. Methamphetamine significantly reduced DA axonal markers in the caudate in a dose-dependent manner (Table 1), with larger dopaminergic deficits resulting from higher methamphetamine doses (DA:F(3,6) = 4.91, p = 0.047; DOPAC:F(3,6) = 5.95, p = 0.031; [3H]WIN-35,428 binding:F(3,6) = 36.3, p < 0.001; [3H]DTBZ: F(3,6) = 16.7,p = 0.002). Similar results were obtained in the putamen (Table 3), again with larger dopaminergic deficits resulting from higher methamphetamine doses (DA: F(3,6) = 9.15, p = 0.012; DOPAC: F(3,6) = 4.93, p = 0.046; [3H]WIN-35,428 binding: F(3,6) = 10.0, p = 0.009; [3H]DTBZ: F(3,6) = 5.3, p = 0.01).
Table 2.
Monoaminergic neuronal markers in control and methamphetamine-treated baboons
| Caudate | ||||||
|---|---|---|---|---|---|---|
| DA | DOPAC | [3H]WIN | [3H]DTBZ | 5-HT | 5-HIAA | |
| Control (n = 3) | 7.34 ± 0.80 | 0.52 ± 0.05 | 1695 ± 173 | 2541 ± 108 | 0.53 ± 0.07 | 0.44 ± 0.04 |
| Methamphetamine | ||||||
| 0.5 mg/kg | 5.23 (−29) | 0.38 (−27) | 830 (−51) | 2237 (−12) | 0.25 (−53) | 0.34 (−23) |
| 5.36 (−27) | 0.36 (−31) | 1150 (−32) | 2037 (−20) | 0.49 (−05) | 0.28 (−36) | |
| 1.0 mg/kg | 2.99 (−59) | 0.27 (−48) | 819 (−52) | 1091 (−57) | 0.30 (−43) | 0.40 (−09) |
| 5.12 (−30) | 0.40 (−23) | 674 (−60) | 1289 (−49) | 0.26 (−51) | 0.29 (−34) | |
| 2.0 mg/kg | 2.73 (−63) | 0.28 (−46) | 543 (−68) | 836 (−67) | 0.05 (−91) | 0.11 (−75) |
| 1.93 (−74) | 0.26 (−50) | 512 (−70) | 1097 (−57) | 0.13 (−75) | 0.21 (−48) | |
| 3.10 (−58) | 0.31 (−40) | 546 (−68) | 1638 (−35) | 0.26 (−51) | 0.21 (−48) | |
Methamphetamine was given intramuscularly at a dose of 0.5, 1, and 2 mg/kg. Each dose was given four times, at 2 hr intervals. Animals were killed for neurochemical studies 2–3 weeks after methamphetamine treatment. Control values represent the mean ± SEM (n = 3) and are provided in the following units: dopamine (DA), dihydroxyphenylacetic acid (DOPAC), serotonin (5-HT), and 5-hydroxyindoleacetic acid (5-HIAA) in μg/gm tissue; [3H]WIN-35,428 and [3H]dihydrotetrabenazine (DTBZ) in dpm/mg tissue. Values for each methamphetamine-treated baboon are provided, with the percentage change shown in parentheses.
Table 3.
Monoaminergic neuronal markers in control and methamphetamine-treated baboons
| Putamen | ||||||
|---|---|---|---|---|---|---|
| DA | DOPAC | [3H]WIN | [3H]DTBZ | 5-HT | 5-HIAA | |
| Control (n = 3) | 8.13 ± 0.79 | 0.85 ± 0.11 | 2451 ± 362 | 3700 ± 575 | 0.32 ± 0.05 | 0.60 ± 0.07 |
| Methamphetamine | ||||||
| 0.5 mg/kg | 5.43 (−33) | 0.68 (−20) | 1224 (−50) | 2705 (−27) | 0.23 (−28) | 0.49 (−18) |
| 4.67 (−42) | 0.47 (−45) | 1471 (−40) | 2704 (−27) | 0.30 (−06) | 0.53 (−12) | |
| 1.0 mg/kg | 4.10 (−50) | 0.40 (−53) | 986 (−60) | 2040 (−45) | 0.24 (−25) | 0.42 (−30) |
| 4.43 (−45) | 0.47 (−45) | 799 (−67) | 1957 (−47) | 0.18 (−44) | 0.53 (−12) | |
| 2.0 mg/kg | 4.60 (−43) | 0.28 (−67) | 483 (−80) | 1126 (−70) | 0.04 (−88) | 0.22 (−63) |
| 1.79 (−78) | 0.56 (−34) | 651 (−73) | 1516 (−59) | 0.10 (−69) | 0.24 (−60) | |
| 3.74 (−54) | 0.31 (−64) | 1082 (−56) | 2797 (−24) | 0.21 (−34) | 0.35 (−42) | |
Methamphetamine was given intramuscularly at a dose of 0.5, 1, and 2 mg/kg. Each dose was given four times, at 2 hr intervals. Animals were killed for neurochemical studies 2–3 weeks after methamphetamine treatment. Control values represent the mean ± SEM (n = 3) and are provided in the following units: dopamine (DA), dihydroxyphenylacetic acid (DOPAC), serotonin (5-HT), and 5-hydroxyindoleacetic acid (5-HIAA) in μg/gm tissue; [3H]WIN-35,428 and [3H]dihydrotetrabenazine (DTBZ) in dpm/mg tissue. Values for each methamphetamine-treated baboon are provided, with the percentage change shown in parentheses.
The linear correlation coefficient between the reductions in striatal [11C]WIN-35,428-labeled DAT detected by means of PET in vivo and the decreases in [3H]WIN-35,428 binding determined in vitro in caudate homogenates of the same animals wasr = 0.77 (p = 0.042), with a Spearman correlation coefficient of 0.90 (p = 0.006) (Fig. 3). In the putamen, the correlation did not achieve significance (r = 0.54,p = 0.21). Figure 3 illustrates the reduction in striatal DAT density, as shown by coronal PET images of [11C]WIN-35,428 binding in living animals (top) and by autoradiography using [3H]WIN-35,428 (middle) and [3H]mazindol in postmortem tissue of the same animal (bottom).
Fig. 3.
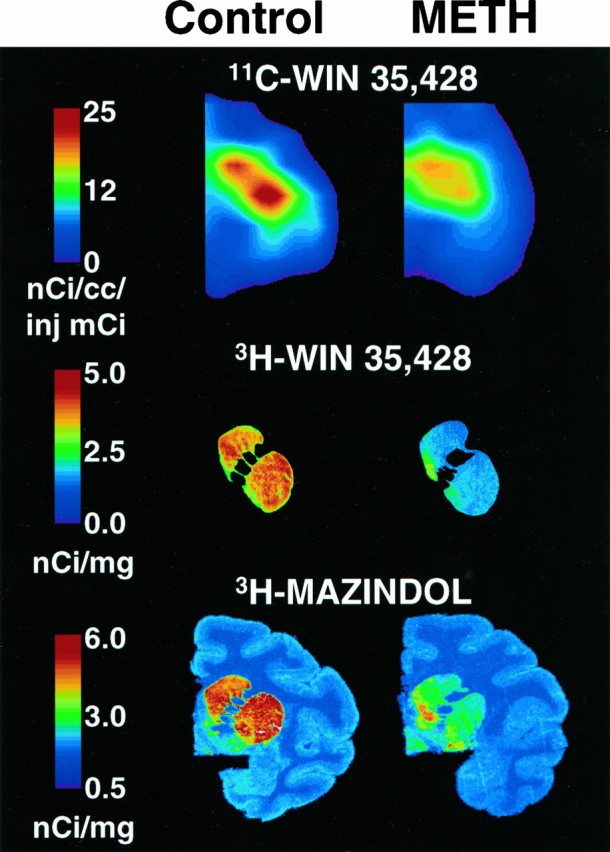
Reductions in striatal DAT density, as shown by coronal PET images of [11C]WIN-35,428 binding in living animal (top) and autoradiography using [3H]WIN-35,428 (middle) and [3H]mazindol (bottom) in postmortem tissue of the same animal. Methamphetamine (2 mg/kg) was given four times, at 2 hr intervals. [11C]WIN-35,428 binding was reduced by 69% (control BP value: 2.99; methamphetamine: 0.93), [3H]WIN-35,428 was reduced by 75% (control: 2.7 nCi/mg; methamphetamine: 0.7 nCi/mg), and [3H]mazindol was reduced by 68% (control: 4.3 nCi/mg; methamphetamine: 1.4 nCi/mg). PET studies were performed 2–3 weeks after methamphetamine treatment. Tissue for autoradiographic studies was also obtained 2–3 weeks after methamphetamine treatment, shortly after PET studies were completed.
Despite the relatively small sample size, correlational analyses were conducted to assess the relation among different DA axonal markers (DA, DOPAC, [3H]WIN-35,428, and [3H]DTBZ) in both the caudate and putamen of control and methamphetamine-treated baboons. In control caudate, statistically significant Pearson correlations (p < 0.05) were limited to DA versus DOPAC (r = 0.99); in control putamen, no significant correlations were observed. After methamphetamine, all DA axonal markers in the caudate were significantly intercorrelated [r = 0.87 to r = 0.95 except DA and [3H]DTBZ (r = 0.74,p = 0.056), DOPAC and [3H]WIN-35,428 (r = 0.72,p = 0.065), and [3H]WIN-35,428 and [3H]DTBZ (r = 0.68,p = 0.094)]. In the putamen of methamphetamine-treated animals, only [3H]WIN-35,428 and [3H]DTBZ were significantly correlated (r = 0.92, p = 0.003).
Methamphetamine also significantly reduced 5-HT axonal markers in the caudate in a dose-dependent manner (Table 2), with higher doses producing larger serotonergic deficits (5-HT:F(3,6) = 4.78, p = 0.049; 5-HIAA: F(3,6) = 6.78, p = 0.023). In the putamen, methamphetamine-induced 5-HT deficits did not achieve statistical significance (p = 0.12) (Table 3). By contrast, methamphetamine-induced 5-HIAA deficits in the putamen were significant, with higher doses producing larger serotonergic deficits (F(3,6) = 5.42,p = 0.038).
Methamphetamine also produced long-term reductions in regional cortical 5-HT, but not NE, axonal markers (Tables4 and5).
Table 4.
Regional cortical 5-HT and NE concentrations in control and methamphetamine-treated baboons
| Frontal cortex | Parietal cortex | Temporal cortex | ||||
|---|---|---|---|---|---|---|
| 5-HT | NE | 5-HT | NE | 5-HT | NE | |
| Control (n = 3) | 101.1 ± 16.8 | 36.7 ± 2.8 | 95.1 ± 4.6 | 39.1 ± 5.7 | 175.0 ± 5.5 | 29.4 ± 3.5 |
| Methamphetamine (n = 3) | 52.3 ± 2.04-a | 31.2 ± 2.5 | 57.0 ± 2.34-a | 42.6 ± 6.1 | 119.6 ± 5.14-a | 22.3 ± 3.1 |
Methamphetamine was given intramuscularly at a dose of 2 mg/kg. Each dose was given four times, at 2 hr intervals. Animals were killed for neurochemical studies 2–3 weeks after methamphetamine treatment. Control values represent the mean ± SEM (n = 3). Values are in μg/mg tissue.
Designates significant difference from control (p < 0.05).
Table 5.
Regional cortical 5-HT and NET densities in control and methamphetamine-treated baboons
| Frontal cortex | Parietal cortex | Temporal cortex | ||||
|---|---|---|---|---|---|---|
| 5-HT transporter | NE transporter | 5-HT transporter | NE transporter | 5-HT transporter | NE transporter | |
| Control (n = 3) | 772.2 ± 74.4 | 362.7 ± 79.2 | 396.1 ± 68.3 | 446.9 ± 57.7 | 921.0 ± 75.8 | 263.6 ± 24.7 |
| Methamphetamine (n = 3) | 537.7 ± 81.55-a | 345.9 ± 54.3 | 268.5 ± 55.3 | 464.5 ± 37.0 | 575.6 ± 84.55-a | 268.6 ± 30.3 |
5-HTT and NET densities were measured with [3H]paroxetine and [3H]nisoxetine, respectively. Methamphetamine was given intramuscularly at a dose of 2 mg/kg. Each dose was given four times, at 2 hr intervals. Animals were killed for neurochemical studies 2–3 weeks after methamphetamine treatment. Control values represent the mean ± SEM (n = 3). Values are in μg/mg tissue.
Designates significant difference from control (p < 0.05).
DISCUSSION
The results of the present study indicate that doses of methamphetamine on the order of those used recreationally by some humans (see below) produce lasting effects on brain DA axonal markers in baboons. Furthermore, the present results indicate that PET imaging with [11C]WIN-35,428 is suitable for detecting partial methamphetamine-induced reductions in brain DAT in living nonhuman primates. Together, these findings suggest that PET imaging with [11C]WIN-35,428 will be useful for evaluating humans for possible methamphetamine-induced DA neurotoxicity, and they raise further concerns about the neurotoxic potential of methamphetamine in humans.
Surprisingly, methamphetamine produced long-term decreases on brain DA axonal markers in baboons at all doses tested, including the 0.5 mg/kg dose (Fig. 1, Tables 1–3). These results confirm and extend those of Melega and colleagues (1993) who observed decreases in 6-[18F]fluoro-DOPA uptake in vervet monkeys injected with 4 mg/kg amphetamine (two injections of 2 mg/kg, i.m.) 1 week and 1 month previously. Using interspecies scaling methods (Harwood, 1963; Mordenti and Chappell, 1989; Chappell and Mordenti, 1991), it is possible to estimate equivalent doses in humans by taking into account known relationships between body mass and surface area. When this is done for the two lower doses of methamphetamine used in the present study (0.5 and 1 mg/kg), the equivalent human doses (for an individual weighing 70 kg) are 26 and 52 mg, respectively. These doses are well within the range of those reported to be used by human methamphetamine users on a repeated basis (Jaffe, 1985; Konuma, 1994), particularly after tolerance has developed (Kalant, 1966; Kramer et al., 1967; Lukas, 1997). The notion that only extraordinarily high doses of methamphetamine can produce DA neurotoxicity needs to be reconsidered, at least in primates exposed to repeated doses of methamphetamine.
As mentioned above, if methamphetamine-induced DAT loss occurs in humans, PET imaging with [11C]WIN-35,428 should prove useful for its detection. Baboons with known methamphetamine-induced DAT loss show clear evidence of decreased striatal DAT density (Fig. 1, Table 1). Moreover, there is an excellent correlation between decreased striatal DAT density, as measured by PET, and decreases in specific [3H]WIN-35,428 binding determined in vitro in the caudate of the same animal (r = 0.77, p = 0.042). The fact that reductions in striatal DAT in methamphetamine-treated baboons are associated with comparable decreases in other markers of DA axon terminals (Tables 2, 3) suggests that PET imaging with [11C]WIN-35,428 will be useful for detecting methamphetamine-induced DA neurotoxicity in humans, if it occurs.
Recently, Wilson and colleagues (1996a) examined striatal DA nerve terminal markers in postmortem tissue from humans with a history of chronic methamphetamine abuse and found reduced levels of DA, tyrosine hydroxylase (TH), and DAT, but normal levels of DOPAC, aromatic amino acid decarboxylase (AADC), and the VMAT. On the basis of these findings, Wilson et al. (1996a) concluded that chronic exposure to methamphetamine did not cause permanent degeneration of striatal dopamine nerve terminals in the subjects in their study. In large part, these investigators based their conclusion on the fact that in patients with Parkinson’s disease, in whom striatal dopamine nerve terminal degeneration is well documented (Hornykiewicz, 1966), not only are the levels of DA, TH, and DAT reduced but so are the levels of DOPAC, AADC, and VMAT (Wilson et al., 1996b). The basis for apparent differences between methamphetamine-treated baboons in the present study and chronic methamphetamine users in the study by Wilson and colleagues (1996a) is not certain but could possibly be related to differences in drug use patterns (e.g., it is possible that none of the methamphetamine users engaged in binge methamphetamine use, a pattern that is more likely to produce neurotoxicity). It is also possible that nondopaminergic sources of VMAT and AADC (e.g., other monoaminergic neurons) in the chronic methamphetamine users masked DA-specific decreases of these enzymes. A third possible explanation for this apparent discrepancy could be that the neuropathology of Parkinson’s disease is not identical to that of methamphetamine neurotoxicity, although the reductions in DOPAC and VMAT observed in methamphetamine-treated baboons (Tables 1, 2) suggest that there are similarities. Finally, the presence of residual drug (methamphetamine and amphetamine) in the human brain tissue samples analyzed by Wilson et al. (1996a) may complicate the interpretation of alterations of DA axonal markers, as could neuroadaptive processes involving the DA transporter. Clearly, additional studies are needed to determine the DA neurotoxic potential of methamphetamine in humans.
The present findings are in good agreement with those of previous PET studies using other presynaptic DA neuroligands to assess the integrity of brain DA neurons after toxic drug exposure. For instance, using PET with 6-[18F]fluoro-DOPA, Melega and colleagues (1996) have shown lasting decrements in striatal DA synthesis capacity in vervet monkeys treated with escalating doses of amphetamine. Similarly, using PET with 6-[18F]fluoro-DOPA, Calne and colleagues (1985) successfully detected dopaminergic deficits in young drug addicts who unwittingly experimented with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), a potent dopaminergic neurotoxin (Langston, 1985). More recently, brain DA neural injury has also been detected with PET using [11C]- or [18F]-labeled radioligands that bind to DA axonal markers, including [11C]nomifensine (Aquilonius et al., 1987,Leenders et al., 1988, Tedroff et al., 1988), [18F]GBR 13119 (Kilbourn et al., 1988, 1989a,b), [11C]DTBZ (Frey et al., 1996), [11C]cocaine (Fowler et al., 1989), and [11C]WIN-35,428 (Frost et al., 1993; Wong et al., 1993, 1996; Brownell et al., 1996; Morris et al., 1996), which is also known as 2β-carbomethoxy-3β-(4-fluorophenyl)-[N-[11C]methyl]tropane or [11C]CFT (Madras et al., 1989; Canfield et al., 1990; Kaufman et al., 1991). Compared to 6-[18F]fluoro-DOPA, DAT and VMAT ligands (Frey et al., 1996), may prove preferable for detecting neurotoxic injury (Nyhan and Wong, 1996), because they label “structural” macromolecular elements of the neuron rather than its transmitter precursor pool, which may be more amenable to metabolic perturbation.
As indicated in the introductory remarks, methamphetamine is toxic to both DA and 5-HT axon terminals, and 5-HT neuronal markers were found to be significantly decreased in methamphetamine-treated baboons in the present study. Hence, the question arises as to whether any of the long-term reductions in [11C]WIN-35,428 binding observed in methamphetamine-treated baboons might be attributable to loss of the 5-HTT. Previous in vitro studies indicate that although [11C]WIN-35,428 has high affinity for the DAT, it has much lower affinity for the 5-HTT (Madras et al., 1989; Canfield et al., 1990; Kaufman et al., 1991). Furthermore, in vivo studies have failed to reveal binding of [11C]WIN-35,428 to the 5-HTT (Scheffel et al., 1991). Thus, it seems unlikely that any of the observed reductions in [11C]WIN-35,428 binding after methamphetamine treatment are caused by methamphetamine-induced loss of the 5-HTT.
The results of the present studies have basic and clinical relevance. Scientifically, it is important to determine whether studies of methamphetamine neurotoxicity in animals accurately predict possible long-term effects of methamphetamine in humans. In addition, if methamphetamine neurotoxicity is found to occur in humans, a better understanding of underlying mechanisms may provide insight into the basis for dopamine neuronal degeneration in Parkinson’s disease and related neurodegenerative conditions (Fuller, 1992). Clinically, detection of possible DA neurotoxicity in humans previously exposed to methamphetamine is important, because such individuals may have an increased risk for developing Parkinson’s disease as they age (Calne and Langston, 1983; Calne et al., 1985), and they could be candidates for putative prophylactic pharmacotherapies such as deprenyl (Tetrud and Langston, 1989; The Parkinson Study Group, 1989, 1993).
In summary, the results of the present study indicate that doses of methamphetamine in the range of those used recreationally by some humans produce significant decrements in brain DA axonal markers in baboons, and that it is possible to detect lasting DAT loss in living nonhuman primates by means of PET. The fact that PET imaging with [11C]WIN-35,428 can detect partial decrements in striatal DAT, decrements not associated with clinical signs of overt parkinsonism, is important because methamphetamine-induced reductions in DA axonal markers are typically in the range of 30–70% (depending on dose, species, brain region, and survival time) (Seiden and Ricaurte, 1987) and are not detectable clinically. Thus, if subclinical methamphetamine-induced DA neural injury in humans occurs, PET imaging with [11C]WIN-35,428 should facilitate its detection and help define the health risks associated with the use and abuse of methamphetamine.
Footnotes
This work was supported by Public Health Service Grants DA05707, DA06275 (G.A.R.), and DA09482 (D.F.W.). The technical support of Robert C. Smoot, David J. Clough, Karen Edmonds, and Paige Finley is gratefully acknowledged.
Correspondence should be addressed to Dr. George A. Ricaurte, Department of Neurology, The Johns Hopkins Medical Institutions, 5501 Bayview Drive, Room 5B71E, Baltimore, MD 21224.
REFERENCES
- 1.Aquilonius SM, Bergstrom K, Eckernas SA, Hartvig P, Leenders KL, Lundquist H, Antoni G, Gee A, Rimoand A, Uhlin J, Langstrom B. In vivo evaluation of striatal dopamine reuptake sites using [11C]nomifensine and positron emission tomography. Acta Neurol Scand. 1987;76:283–287. doi: 10.1111/j.1600-0404.1987.tb03582.x. [DOI] [PubMed] [Google Scholar]
- 2.Axt KJ, Molliver ME. Immunocytochemical evidence for methamphetamine-induced serotonergic axon loss in the rat brain. Synapse. 1991;9:302–313. doi: 10.1002/syn.890090405. [DOI] [PubMed] [Google Scholar]
- 3.Brill H, Hirose T. The rise and fall of a methamphetamine epidemic: Japan 1945–1955. Semin Psychiatry. 1969;1:179–213. [Google Scholar]
- 4.Brownell AL, Elmaleh DR, Meltzer PC, Shoup TM, Brownell GL, Fischman AJ, Madras BK. Cocaine congeners as PET imaging probes for dopamine terminals. J Nucl Med. 1996;37:1186–1192. [PubMed] [Google Scholar]
- 5.Calne DB, Langston JW. On the etiology of Parkinson’s disease. Lancet. 1983;2:1457–1459. doi: 10.1016/s0140-6736(83)90802-4. [DOI] [PubMed] [Google Scholar]
- 6.Calne DB, Langston JW, Martin W, Stoessel A, Ruth T, Adam M, Schulzer M. Positron emission tomography after MPTP: observations relating to the cause of Parkinson’s disease. Nature. 1985;317:246–248. doi: 10.1038/317246a0. [DOI] [PubMed] [Google Scholar]
- 7.Canfield DR, Spealman RD, Kaufman MJ, Madras BK. Autoradiographic localization of cocaine binding sites by ([3H]WIN-35,428) in the monkey brain. Synapse. 1990;6:189–195. doi: 10.1002/syn.890060211. [DOI] [PubMed] [Google Scholar]
- 8.Chappell W, Mordenti J. Extrapolation of toxicological and pharmacological data from animals to humans. In: Testa B, editor. Advances in drug research. Academic; San Diego: 1991. pp. 1–116. [Google Scholar]
- 9.Dannals RF, Neumeyer JL, Milius RA, Ravert HT, Wilson AA, Wagner HN., Jr Synthesis of a radiotracer for studying dopamine uptake sites in vivo using PET: 2b-carbomethoxy-3b-(4-fluorophenyl)-[N-11C-methyl]tropane ([11C]CFT or [11C]WIN-35,428). J Label Comp Radiopharmacol. 1993;33:147–152. [Google Scholar]
- 10.Ellison G, Elson MS, Huberman HS, Daniel F. Long-term changes in dopaminergic innervation of caudate nucleus after continuous amphetamine administration. Science. 1978;201:276–278. doi: 10.1126/science.26975. [DOI] [PubMed] [Google Scholar]
- 11.Fowler JS, Volkow ND, Wolf AP, Dewey SL, Schyler DJ, MacGregor RR, Hitzemann R, Logan J, Bendrienn B, Gately SJ, Christman D. Mapping cocaine binding sites in human and baboon brain in vivo. Synapse. 1989;4:371–377. doi: 10.1002/syn.890040412. [DOI] [PubMed] [Google Scholar]
- 12.Frey K, Koeppe R, Kilbourn M, Vander Borght T, Albin R, Gilman S, Kuhl D. Presynaptic monoaminergic vesicles in Parkinson’s disease and normal aging. Ann Neurol. 1996;40:873–884. doi: 10.1002/ana.410400609. [DOI] [PubMed] [Google Scholar]
- 13.Frost JJ, Rosier AJ, Reich SG, Smith JS, Ehlers MD, Snyder SH, Ravert HT, Dannals RF. Positron emission tomographic imaging of the dopamine transporter with 11C-WIN-35,428 reveals marked declines in mild Parkinson’s disease. Ann Neurol. 1993;34:423–431. doi: 10.1002/ana.410340331. [DOI] [PubMed] [Google Scholar]
- 14.Fuller R, Hemrick-Luecke S. Long-lasting depletion of striatal dopamine by a single injection of amphetamine in iprindole-treated rats. Science. 1980;209:305–307. doi: 10.1126/science.7384808. [DOI] [PubMed] [Google Scholar]
- 15.Fuller RW. Comparison of MPTP and amphetamines as dopaminergic neurotoxins. Ann NY Acad Sci. 1992;648:87–95. doi: 10.1111/j.1749-6632.1992.tb24526.x. [DOI] [PubMed] [Google Scholar]
- 16.Fukui K, Nakajima T, Kariyama H, Kashiba A, Kato N, Tohyama I, Kimura H. Selective reduction of serotonin immunoreactivity in some forebrain regions of rats induced by acute methamphetamine treatment: quantitative morphometric analysis by serotonin immunocytochemistry. Brain Res. 1989;482:198–203. doi: 10.1016/0006-8993(89)90562-3. [DOI] [PubMed] [Google Scholar]
- 17.Gibb JW, Hanson GR, Johnson M. Neurochemical mechanisms of toxicity. In: Cho AK, Segal DS, editors. Amphetamine and its analogs. Academic; San Diego: 1994. pp. 269–295. [Google Scholar]
- 18.Habert E, Graham G, Tahraoui L, Claustre Y, Langer SZ. Characterization of [3H]-paroxetine binding in rat cortical membranes. Eur J Pharmacol. 1985;118:107–114. doi: 10.1016/0014-2999(85)90668-5. [DOI] [PubMed] [Google Scholar]
- 19.Harwood PD. Therapeutic dosage in small and large mammals. Science. 1963;139:684–685. doi: 10.1126/science.139.3555.684. [DOI] [PubMed] [Google Scholar]
- 20.Hornykkiewicz O. Dopamine (3-hydroxytyramine) and brain function. Pharmacol Rev. 1966;18:925–965. [PubMed] [Google Scholar]
- 21.Hotchkiss AJ, Gibb JW. Long-term effects of multiple doses of methamphetamine on tryptophan hydroxylase and tyrosine hydroxylase activity in rat brain. J Pharmacol Exp Ther. 1980;214:257–262. [PubMed] [Google Scholar]
- 22.Hotchkiss AJ, Morgan ME, Gibb JW. The long-term effects of multiple doses of methamphetamine on neostriatal tryptophan hydroxylase, tyrosine hydroxylase, choline acetyltransferase and glutamate decarboxylase activities. Life Sci. 1979;25:1373–1378. doi: 10.1016/0024-3205(79)90414-4. [DOI] [PubMed] [Google Scholar]
- 23.Inghe G. The present state of abuse and addiction to stimulant drugs in Sweden. In: Sjoqvist F, Tottie M, editors. Abuse of central stimulants. Almqvist and Wiksell; Stockholm: 1969. pp. 19–27. [Google Scholar]
- 24.Jaffe J. Drug addiction and drug abuse (1985). In: Goodman L, Gilman S, editors. Pharmacological basis of therapeutics. McMillan; New York: 1985. pp. 284–324. [Google Scholar]
- 25.Javitch J, Strittmatter S, Snyder S. Differential visualization of dopamine and norepinephrine uptake sites in rat brain using [3H]mazindol autoradiography. J Neurosci. 1985;5:1513–1521. doi: 10.1523/JNEUROSCI.05-06-01513.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalant O. The amphetamines: toxicity and addiction (Thomas C, ed). University of Toronto; Toronto: 1966. [Google Scholar]
- 27.Kaufman MJ, Spealman RD, Madras BK. Distribution of cocaine recognition sites in monkey brain. I. In vitro autoradiography with [3H]CFT. Synapse. 1991;9:177–187. doi: 10.1002/syn.890090304. [DOI] [PubMed] [Google Scholar]
- 28.Kilbourn MR. In vivo binding of [18F]GBR 13119 to the brain dopamine uptake system. Life Sci. 1988;42:1347–1353. doi: 10.1016/0024-3205(88)90163-4. [DOI] [PubMed] [Google Scholar]
- 29.Kilbourn MR, Carey JE, Koeppe RA, Haka MS, Hutchins GD, Sherman PS, Kuhl DE. Biodistribution, dosimetry, metabolism and monkey PET studies of [18F]GBR 13119: imaging the dopamine uptake system in vivo. Int J Radiat Appl Instrum [Part B] 1989a;16:569–576. doi: 10.1016/0883-2897(89)90072-x. [DOI] [PubMed] [Google Scholar]
- 30.Kilbourn MR, Haka MS, Mulholland GK, Jewett DM, Kuhl DE. Synthesis of radiolabeled inhibitors of presynaptic monoamine uptake systems: fluorine-18 GBR-13119 DA, carbon-11 nisoxetine NE and carbon-11 fluoxetine 5-HT. J Label Comp Radiopharmacol. 1989b;26:412–414. [Google Scholar]
- 31.Kiloh LG, Brandon S. Habituation and addiction to amphetamines. Br Med J. 1962;40:3. doi: 10.1136/bmj.2.5296.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kogan FJ, Nichols WK, Gibb JW. Influence of methamphetamine on nigral and striatal tyrosine hydroxylase activity and on striatal dopamine levels. Eur J Pharmacol. 1976;36:363–371. doi: 10.1016/0014-2999(76)90090-x. [DOI] [PubMed] [Google Scholar]
- 33.Konuma K. Use and abuse of amphetamines in Japan. In: Cho AK, Segal DS, editors. Amphetamine and its analogs. Academic; San Diego: 1994. pp. 415–435. [Google Scholar]
- 34.Kozel N (1997) Epidemiologic trends in drug abuse, Vol 1. National Institute on Drug Abuse, NIH Publication No. 97–4204.SS.
- 35.Kramer JC, Fischman VS, Littlefield DC. Amphetamine abuse: pattern and effects of high doses taken intravenously. JAMA. 1967;210:305–309. doi: 10.1001/jama.201.5.305. [DOI] [PubMed] [Google Scholar]
- 36.Langston JW. MPTP and Parkinson’s disease. Trends Neurosci. 1985;8:79–83. [Google Scholar]
- 37.Leenders KL, Aquilonius SM, Bergstrom K, Bjurling P, Crossman AR, Eckernas SA, Gee AG, Hartvig P, Lundquist H, Langstrom B, Rimland A, Tedroff J. Unilateral MPTP lesion in a rhesus monkey: effects on the striatal dopaminergic system measured in vivo with PET using various novel tracers. Brain Res. 1988;445:61–67. doi: 10.1016/0006-8993(88)91074-8. [DOI] [PubMed] [Google Scholar]
- 38.Lorez H. Fluorescence histochemistry indicates damage of striatal dopamine nerve terminals in rats after multiple doses of methamphetamine. Life Sci. 1981;28:911–916. doi: 10.1016/0024-3205(81)90053-9. [DOI] [PubMed] [Google Scholar]
- 39.Lukas SE. Proceedings of the National Consensus Meeting on the use, abuse and sequelae of abuse of methamphetamine with implications for prevention, treatment and research. DHHS Publication SMA 96-8013. USDHHS; Rockville, MD: 1997. [Google Scholar]
- 40.Madras BK, Spealman RD, Fahey MA, Neumeyer JL, Saha JK, Milius RA. Cocaine receptors labeled by [3H]-b-carbomethoxy-3-β-(4-fluorophenyl)-tropan. Mol Pharmacol. 1989;36:518–524. [PubMed] [Google Scholar]
- 41.Melega WP, Yu DC, Raleigh MJ, Huang SC, Phelps ME. FDOPA-PET studies show neurotoxic effects of low-dose amphetamine in monkeys. Soc Neurosci Abstr. 1993;19:822. [Google Scholar]
- 42.Melega WP, Quintana J, Raleigh MJ, Stout DB, Yu DC, Lin KP, Huang SC, Phelps ME. 6-[18F]fluoro-[sca]l-DOPA-PET studies show partial reversibility of long-term effects of chronic amphetamine in monkeys. Synapse. 1996;22:63–69. doi: 10.1002/(SICI)1098-2396(199601)22:1<63::AID-SYN7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 43.Miller M, Hughes A. Epidemiology of amphetamine abuse in the United States. In: Cho AK, Segal DS, editors. Amphetamine and its analogs. Academic; San Diego: 1994. pp. 439–457. [Google Scholar]
- 44.Mordenti J, Chappell W. The use of interspecies scaling in toxicokinetics. In: Yacobi A, Kelly J, Batra V, editors. Toxicokinetics in new drug development. Pergamon; New York: 1989. pp. 42–96. [Google Scholar]
- 45.Morris ED, Babich JW, Alpert NM, Bonab AA, Livni E, Weise S, Hsu H, Christian BT, Madras BK, Fischman AJ. Quantification of dopamine transporter density in monkeys by dynamic PET imaging of multiple injections of 11C-CFT. Synapse. 1996;24:262–272. doi: 10.1002/(SICI)1098-2396(199611)24:3<262::AID-SYN9>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 46.Naudon L, Leroux-Nicollet I, Constentin J. Short-term treatments with haloperidol or bromocryptine do not alter the density of the vesicular monoamine transporter. Neurosci Lett. 1994;173:1–4. doi: 10.1016/0304-3940(94)90136-8. [DOI] [PubMed] [Google Scholar]
- 47.Nyhan WL, Wong DF. New approaches to understanding Lesch-Nyhan disease. N Engl J Med. 1996;334:1602–1604. doi: 10.1056/NEJM199606133342411. [DOI] [PubMed] [Google Scholar]
- 48.Nwanze F, Jonsson G. Amphetamine neurotoxicity on dopamine nerve terminals in the caudate nucleus of mice. Neurosci Lett. 1981;26:163–168. doi: 10.1016/0304-3940(81)90343-8. [DOI] [PubMed] [Google Scholar]
- 49.Peat MA, Warren PF, Gibb JW. Effects of a single dose of methamphetamine and iprindole on the serotonergic and dopaminergic system of the rat brain. J Pharmacol Exp Ther. 1983;225:126–131. [PubMed] [Google Scholar]
- 50.Ricaurte GA, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on dopamine and serotonin neurons in the rat brain: a regional study. Brain Res. 1980;193:153–163. doi: 10.1016/0006-8993(80)90952-x. [DOI] [PubMed] [Google Scholar]
- 51.Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- 52.Ricaurte GA, Seiden LS, Schuster CR. Increased dopamine metabolism in the rat neostriatum after toxic doses of d-methylamphetamine. Neuropharmacology. 1983;22:1383–1388. doi: 10.1016/0028-3908(83)90228-9. [DOI] [PubMed] [Google Scholar]
- 53.Ricaurte GA, Guillery RW, Seiden LS, Schuster CR. Nerve terminal degeneration after a single injection of d-amphetamine in iprindole-treated rats: relation to selective long-lasting dopamine depletion. Brain Res. 1984a;291:378–382. doi: 10.1016/0006-8993(84)91273-3. [DOI] [PubMed] [Google Scholar]
- 54.Ricaurte GA, Seiden LS, Schuster CR. Further evidence that amphetamines produce long-lasting dopamine neurochemical deficits by destroying dopamine nerve fibers. Brain Res. 1984b;303:359–364. doi: 10.1016/0006-8993(84)91221-6. [DOI] [PubMed] [Google Scholar]
- 55.Ricaurte GA, Martello AL, Katz JL. Lasting effects of MDMA on central serotonergic neurons in non-human primates: neurochemical observations. J Pharmacol Exp Ther. 1992;261:616–622. [PubMed] [Google Scholar]
- 56.Scheffel U, Pogun S, Stathis M, Boja J, Kuhar M. In vivo labeling of cocaine binding sites on dopamine transporters with [3H]WIN 35,428. J Pharmacol Exp Ther. 1991;257:954–958. [PubMed] [Google Scholar]
- 57.Schmidt CJ, Ritter JK, Sonsalla KP, Hanson GR, Gibb JW. Role of dopamine in the neurotoxic effects of methamphetamine. J Pharmacol Exp Ther. 1985;233:539–543. [PubMed] [Google Scholar]
- 58.Seiden LS, Ricaurte GA. Neurotoxicity of methamphetamine and related drugs. In: Meltzer HY, editor. Psychopharmacology–a generation of progress. Raven; New York: 1987. pp. 359–366. [Google Scholar]
- 59.Seiden LS, Fischman MW, Schuster CR. Long-term methamphetamine induced changes in brain catecholamines in tolerant rhesus monkeys. Drug Alcohol Depend. 1976;1:215–219. doi: 10.1016/0376-8716(76)90030-2. [DOI] [PubMed] [Google Scholar]
- 60.Sonsalla P, Nicklas W, Heikkila R. Role for excitatory amino acids in methamphetamine-induced nigrostriatal dopaminergic toxicity. Science. 1989;243:398–400. doi: 10.1126/science.2563176. [DOI] [PubMed] [Google Scholar]
- 61.Tedroff J, Aquilonius SM, Hartvig P, Lundquist H, Gee AG, Uhlin J, Langstrom B. Monoamine re-uptake sites in the human brain evaluated in vivo by means of [11C]nomifensine and positron emission tomography: the effects of age and Parkinson’s disease. Acta Neurol Scand. 1988;77:192–201. doi: 10.1111/j.1600-0404.1988.tb05894.x. [DOI] [PubMed] [Google Scholar]
- 62.Tejani-Butt SM. [3H]nisoxetine: a radioligand for quantitation of norepinephrine uptake sites by autoradiography or by homogenate binding. J Pharmacol Exp Ther. 1992;260:427–436. [PubMed] [Google Scholar]
- 63.Tetrud JW, Langston JW. The effect of deprenyl selegiline on the natural history of Parkinson’s disease. Science. 1989;245:519–522. doi: 10.1126/science.2502843. [DOI] [PubMed] [Google Scholar]
- 64.The Parkinson Study Group. Effect of deprenyl on the progression of disability in early Parkinson’s disease. N Engl J Med. 1989;321:1364–1371. doi: 10.1056/NEJM198911163212004. [DOI] [PubMed] [Google Scholar]
- 65.The Parkinson Study Group. Effects of tocopherol and deprenyl on the progression in early Parkinson’s disease. N Engl J Med. 1993;328:176–183. doi: 10.1056/NEJM199301213280305. [DOI] [PubMed] [Google Scholar]
- 66.Vander Borght T, Sima A, Kilbourn M, Desmond T, Kuhl D, Frey K. The vesicular monoamine transporter is not regulated by dopaminergic drug treatments. Eur J Pharmacol. 1996;294:577–584. doi: 10.1016/0014-2999(95)00594-3. [DOI] [PubMed] [Google Scholar]
- 67.Wagner GC, Ricaurte GA, Seiden LS, Schuster CR, Miller RJ, Westley J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980a;181:151–160. doi: 10.1016/0006-8993(80)91265-2. [DOI] [PubMed] [Google Scholar]
- 68.Wagner GC, Ricaurte GA, Johansen C, Schuster CR, Seiden LS. Amphetamine induces caudate dopamine depletions in monkeys. Neurology. 1980b;30:547–550. doi: 10.1212/wnl.30.5.547. [DOI] [PubMed] [Google Scholar]
- 69.Wagner GC, Preston K, Ricaurte GA, Schuster CR, Seiden LS. Neurochemical similarities between d,l-cathinone and d-amphetamine. Drug Alcohol Depend. 1982;9:279–284. doi: 10.1016/0376-8716(82)90067-9. [DOI] [PubMed] [Google Scholar]
- 70.Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, Schmunk GA, Shannak K, Haycock JW, Kish SJ. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nature Med. 1996a;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]
- 71.Wilson JM, Levey AI, Rajput A, Ang L, Guttman M, Shannak K, Niznik H, Hornykiewicz O, Pifl C, Kish SJ. Differential changes in neurochemical markers of striatal dopamine nerve terminals in idiopathic Parkinson’s disease. Neurology. 1996b;47:718–728. doi: 10.1212/wnl.47.3.718. [DOI] [PubMed] [Google Scholar]
- 72.Wong DF, Yung BCK, Dannals RF, Shaya EK, Ravert HT, Chen CA, Chan B, Folio T, Scheffel U, Ricaurte GA, Neumeyer JL, Wagner HN, Jr, Kuhar MJ. In vivo imaging of baboon and human dopamine transporters by positron emission tomography using [11C]WIN-35,428. Synapse. 1993;15:130–142. doi: 10.1002/syn.890150205. [DOI] [PubMed] [Google Scholar]
- 73.Wong DF, Harris JC, Naidu S, Yokoi F, Marenco S, Dannals RF, Ravert HT, Yaster M, Evans A, Rousset O, Bryan RN, Gjedde A, Kuhar M, Breese GR. Dopamine transporters are markedly reduced in Lesch-Nyhan disease in vivo. Proc Natl Acad Sci USA. 1996;93:5539–5543. doi: 10.1073/pnas.93.11.5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Woolverton WL, Ricaurte GA, Forno LS, Seiden LS. Long-term effects of chronic methamphetamine administration in rhesus monkeys. Brain Res. 1989;486:73–78. doi: 10.1016/0006-8993(89)91279-1. [DOI] [PubMed] [Google Scholar]