

Abstract
Marijuana consumption elicits diverse physiological and psychological effects in humans, including memory loss. Here we report that Δ9-tetrahydrocannabinol (THC), the major psychoactive component of marijuana, is toxic for hippocampal neurons. Treatment of cultured neurons or hippocampal slices with THC caused shrinkage of neuronal cell bodies and nuclei as well as genomic DNA strand breaks, hallmarks of neuronal apoptosis. Neuron death induced by THC was inhibited by nonsteroidal anti-inflammatory drugs, including indomethacin and aspirin, as well as vitamin E and other antioxidants. Furthermore, treatment of neurons with THC stimulated a significant increase in the release of arachidonic acid. We hypothesize that THC neurotoxicity is attributable to activation of the prostanoid synthesis pathway and generation of free radicals by cyclooxygenase. These data suggest that some of the memory deficits caused by cannabinoids may be caused by THC neurotoxicity.
Keywords: THC, cannabinoid receptors, CB1, cell death, hippocampal neurons, arachidonic acid, reactive oxygen species, SR141716A
Marijuana has a long history of consumption in human societies and is the most commonly used illicit drug today. Although marijuana is legalized in some states of the United States for therapeutic usage, its clinical use is controversial. One established therapeutic application of Δ-9-tetrahydrocannabinol (THC), the major psychoactive component of marijuana, is the treatment of nausea and vomiting associated with chemotherapy (Jaffe, 1990). Other uses of marijuana include appetite stimulation in cancer patients and abatement of pain (Abood and Martin, 1992; Voth and Schwartz, 1997). The effects of the drug in humans include hallucination, compromised cognition, memory loss, and immunosuppression (Abel, 1970;Reisine and Brownstein, 1994).
Human marijuana research is limited, and the retrospective nature of these studies compromise their interpretation. Nevertheless, marijuana usage disrupts short-term memory, working memory, and attentional skills (Fletcher et al., 1996), and induces deficits in mathematical skills, verbal expression, and memory retrieval (Block and Ghoneim, 1993). Long-lasting cannabis-dependent short-term memory deficits (Schwartz et al., 1989) and residual neuropsychological effects (Pope and Yurgelun-Todd, 1996) persist even after abstinence.
Administration of cannabis extracts to rodents also causes long-lasting memory defects. For example, a 6 month oral administration of THC reduces maze learning with a residual learning defect that persists (Fehr et al., 1976; Stiglick and Kalant, 1982b, 1983; Stiglick et al., 1984). Radial arm learning paradigm showed similar impairments (Stiglick and Kalant, 1982a). Collectively, studies indicate that chronic exposure to THC impairs cognition and memory (Carlini et al., 1970; Abel, 1971).
The effects of THC are mediated primarily through cannabinoid CB1 receptor in the brain (Matsuda et al., 1990) or CB2 receptor in peripheral tissues (Munro et al., 1993). Cannabinoid receptors couple to several regulatory pathways, including the cAMP signal transduction system (Howlett, 1995). Because cannabinoid receptors inhibit adenylyl cyclases (Howlett and Fleming, 1984) and cAMP regulates synaptic plasticity (Frey et al., 1993; Chavez Noriega and Stevens, 1994;Weisskopf et al., 1994; Impey et al., 1996), cannabinoids could inhibit synaptic function. For example, Schaffer collateral long-term potentiation in the hippocampus is depressed by THC (Nowicky et al., 1987; Collins et al., 1994). Alternatively, memory loss may be attributable to cannabinoid neurotoxicity.
Chronic cannabinoid administration to rats causes distinct morphological changes in the hippocampus indicative of neurotoxicity (Scallet, 1991). THC exposure for 3 months decreases the mean volume of neurons and their nuclei, the synaptic density, and the dendritic length of CA3 pyramidal neurons (Scallet et al., 1987). Exposure to THC also decreases neuronal density in rat hippocampus (Landfield et al., 1988). Although animal studies suggest that chronic exposure to THC may be toxic for hippocampal neurons, mechanisms for THC neurotoxicity have not been defined.
To determine whether THC is toxic for hippocampal neurons and to explore possible mechanisms, we examined its influence on the viability and morphology of cultured rat hippocampal neurons and hippocampal slices. Our data indicate that THC induces cell death in neurons that may be attributable to activation of the phospholipase A2–cyclooxygenase pathway.
MATERIALS AND METHODS
Primary neuron cultures and measurement of cell death. Primary hippocampal neurons were established from postnatal day one Sprague Dawley rat pups and maintained in defined medium with minimal supplements. Pups were killed by decapitation and the hippocampi were digested in 5 ml of 10 U/ml papain at 37°C for 20 min. After two rinses, the tissue was triturated (two rounds, 10–15 pipetting each) in dissociation medium (27 mmK2S04, 1 mm kynurenic acid, 15 mm MgCl2, 74 mmNa2SO4, 18 mm glucose, 225 μm CaCl2, 0.0012% phenol red, and 2 mm HEPES, pH 7.4) using a 5 ml disposable plastic pipette. Ninety-six-well plates were coated with poly-d-lysine (66 μg/ml). Cells were plated into the inner 60 wells of the 96-well plate (the peripheral wells were filled with water to act as humidity barrier) at 5 × 104cells/cm2 and maintained in 200–250 μl of Neurobasal medium (Life Technologies, Gaithersburg, MD) in the presence of N-2 supplement (Bottenstein and Sato, 1979) with 10 U/ml penicillin, 10 μg/ml streptomycin, and 0.5 μg/ml glutamine for 10–16 d before use. Because Neurobasal medium and N-2 did not maintain neuron viability for extended periods, the media was modified. These modifications included adding 0.1% chicken ovalbumin (Sigma, St. Louis, MO) and increasing sodium selenite to at least 300 nm. The amount of selenite added (up to micromolar) was determined empirically for each batch of N-2. Primary neurons cultured under these conditions survived 4–5 weeks. Serum-free Neurobasal medium with supplements has been used extensively for sustaining the survival of cultured CNS neurons (Brewer et al., 1993; Otsuki et al., 1994; Takeshima et al., 1994; Morrison et al., 1996; Xiang et al., 1996). However, supplements with very high contents of antioxidants, such as B27, were not suitable for our studies (see Results). Neurons maintained in the presence of serum experience serum withdrawal-induced cell death at the time of THC treatment. All cannabinoid dilutions were performed in siliconized tubes. Absolute ethanol was used as carrier or vehicle for THC. Controls and vehicle-treated controls were obtained from cells treated with comparable concentration of ethanol as THC-treated samples. MTT assays were performed as previously described (Hansen et al., 1989). At the end of the incubation, 25 μl of MTT (5 mg/ml) was added to the cells in 100 μl of growth media. The plates were incubated for 2 hr at 37°C in a CO2 incubator and solubilized overnight in 9% SDS and 22% DMF before determination of absorbance at 570 nm. SR141716A was provided by Research Biochemicals (Natick, MA) as part of the Chemical Synthesis Program of the National Institutes of Mental Health, Contract N01MH30003.
Inhibitor experiments were performed with 10–16 d cultured neurons. Unless otherwise stated, neurons in 96-well plates were pretreated with the inhibitors for 1 hr before treatment with THC. Various concentrations of inhibitors were tested for their effect on neuron viability in the absence of THC to define nontoxic concentrations for each inhibitor. The inhibitors and concentrations examined included quinacrine, 0.2–5.0 μm; indomethacin, 1.0–5.0 μm; nordihydroguaiaretic acid (NDGA), 1.0–5.0 μm; EGTA, 1.5 mm; vitamin E, 20–200 μm; Trolox, 100 μm; aspirin, 1.0–5.0 μm; dexamethasone, 100 nm; and actinomycin D, 2.0 μg/ml.
Arachidonic acid release assay. Hippocampal neurons (plated at 0.6 × 106/well in six-well plates) were grown in culture for 7–11 d before loading with 0.5 μCi/ml [5,6,8,9,11,12,14,15-3H(N)]arachidonic acid (223 Ci/mmol; DuPont NEN, Boston, MA) in a CO2incubator for 3.5–4.5 hr. The cells were rinsed twice with Neurobasal medium and treated in 2 ml conditioned media in the presence or absence of THC. At various time points after treatment with THC, 100 μl of supernatant was withdrawn and centrifuged at 14,000 ×g at 25°C for 5 min. The supernatant was transferred to a fresh tube for liquid scintillation counting. Data reported are averages from three independent experiments.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) and bisbenzimide staining of cultured hippocampal neurons. Neurons were cultured on 18 mm circular coverglass slips (Fisher Scientific, Houston, TX) coated with poly-d-lysine in 1 ml of growth medium in 12-well plates. TUNEL labeling was performed as recommended by the manufacturer (in situ cell death detection kit, POD; Boehringer Mannheim, Indianapolis, IN). Horseradish peroxidase reactions were performed as recommended using 3,3′-diaminobenzidine (DAB substrate kit for peroxidase; Vector Laboratories, Burlingame, CA). Counterstaining was done using 1% toluidine blue in 1% sodium borate. For bisbenzimide staining, neurons were fixed with 4% formaldehyde, rinsed with PBS, and stained with 2.5 μg/ml Hoechst 33342 (Sigma) at 25°C for 5 min. After rinsing with PBS and mounting on slides with GelMount, the slices were examined by fluorescence microscopy.
Calcium imaging. Primary hippocampal neurons were maintained for 8–16 d in four-chambered Lab-Tek coverglass (Nalge Nunc, Roskilde, Denmark) coated with poly-d-lysine. On the day of Ca2+ imaging, the conditioned media from the neurons were removed and saved. Cells were rinsed with 1 ml of imaging buffer and loaded with 1.5 μm fura-2 AM (Molecular Probes, Eugene, OR) in the same buffer in the dark for 20 min at 25°C. After rinsing with 1 ml imaging buffer, the conditioned media were added back to the cells, and the coverglass was incubated in the CO2incubator until use (within 2–4 hr). Calcium imaging was performed and interpreted as described (Wayman et al., 1995). Unless otherwise stated, stock THC and other reagents were first added to the buffer and then to hippocampal neurons incubating in the same volume of the identical buffer. Final concentrations were 10 μm THC, 5 μm SR141716A, and 4 mm LaCl3.
THC treatment of rat hippocampal slices. Brain transverse sections (400 μm) were cut from female adult (∼250 gm) Long–Evans rats that were killed by decapitation. Hippocampal slices were perfused in continuously oxygenated artificial CSF (ACSF; in mm: 120 NaCl, 3.5 KCl, 1.3 MgCl2, 2.5 CaCl2, 1.25 NaH2PO4, 25.6 NaHCO3, and 10 glucose) at 30°C for 1 hr before adding THC (Research Biochemicals). Stock solutions of THC were dissolved in 100% ethanol, and the final ethanol concentration after dilution was 0.01%. After 2 hr of perfusion with THC (flow rate, 75 ml/hr) and 3 additional hr with ACSF alone, the sections were fixed overnight in 4% formaldehyde and then cryostatically protected in 30% sucrose. Forty μm (for propidium iodide staining) or 25 μm (for TUNEL assay) sections were then made using a microtome. For propidium iodide staining, sections were treated with 5 ml of 10 μg/ml RNase A at 25°C for 30 min and stained with 1–2 ml of 0.5 μg/ml propidium iodide at 25°C for 5 min. For TUNEL assays, 25 μm sections were adhered to charged slides (Superfrost Plus, VWR Scientific) at 25°C and allowed to dry. Sections were treated with 10 μg/ml proteinase K in 10 mm Tris, pH 7.4, at 25°C for 30 min, rinsed four times in 10 ml PBS, permeabilized in 10 ml 1× PBS, 0.1% Triton X-100 at 25°C for 15 min, and then incubated in 250 μl terminal deoxytransferase (TdT) buffer (Life Technologies) and 4.0 μm biotin-14-dATP (Life Technologies) at 25°C for 10 min. Two hundred fifty microliters (100 U/ml) of a recombinant TdT (Life Technologies) were added in the same buffer and incubated at 37°C for 90 min. Reactions in small volume were performed in covered dishes in the CO2 incubator to minimize evaporation. The reaction was terminated by rinsing slides twice for 10 min with 10 ml of PBS at 25°C, followed by adding 10 ml of 300 mm NaCl with 30 mm sodium citrate and incubating at 25°C for 15 min. After rinsing with PBS, the sections were incubated in PBS containing 2% BSA and 4.0 μg/ml lissamine rhodamine-conjugated streptavidin (Jackson ImmunoResearch, West Grove, PA) at 25°C for 60 min. After rinsing several times with PBS, the slices were subjected to one more round of amplification with biotinylated anti-streptavidin antibodies from goat (1 μg/ml; Vector Laboratories) and lissamine rhodamine-conjugated streptavidin (8.0 μg/ml) before mounting with GelMount (Biomeda) for confocal microscopy.
RESULTS
THC is toxic for cultured hippocampal neurons
To determine whether THC is neurotoxic and to elucidate mechanisms for toxicity, we examined its effect on the viability of cultured primary hippocampal neurons from neonatal rats (Fig.1). Survival of hippocampal neurons was monitored as a function of THC concentration and time after treatment using the MTT tetrazolium salt [3,(4,5-dimethylthiazol-2-yl)2,5-diphenyl-tetrazolium bromide] assay as a measure of cell viability (Hansen et al., 1989). Concentrations of THC as low as 0.5 μm were toxic to hippocampal neurons, and the rate of neuron death increased with THC concentration (Fig. 1A,B). For example, 50% of the neurons were killed within 3 hr of treatment with 10 μm THC or 6 d after exposure to 1.0 μm THC. The lower doses used are comparable to THC concentrations measured in human plasma after consumption of a single marijuana cigarette (Chiang and Barnett, 1984). THC concentrations in the brain may be even higher after smoking marijuana, because THC is lipophilic (Thomas et al., 1990) and readily crosses the blood–brain barrier. Blood levels of THC reach a maximum shortly after smoking marijuana and decline rapidly thereafter. However, even a 15 min exposure to THC was toxic for cultured hippocampal neurons (Fig.1C). Cultured cortical neurons were much less sensitive to THC than hippocampal neurons (data not shown). Because THC is lipophilic, some of its effects may be attributable to interactions with membrane lipids rather than receptor activation. However, SR141716A, an antagonist of CB1 receptors (Rinaldi-Carmona et al., 1994), completely inhibited THC-induced neuron death (Fig.2). These data indicate that THC toxicity is mediated through CB1 receptors and not caused by nonspecific interactions with the membrane lipid phase.
Fig. 1.
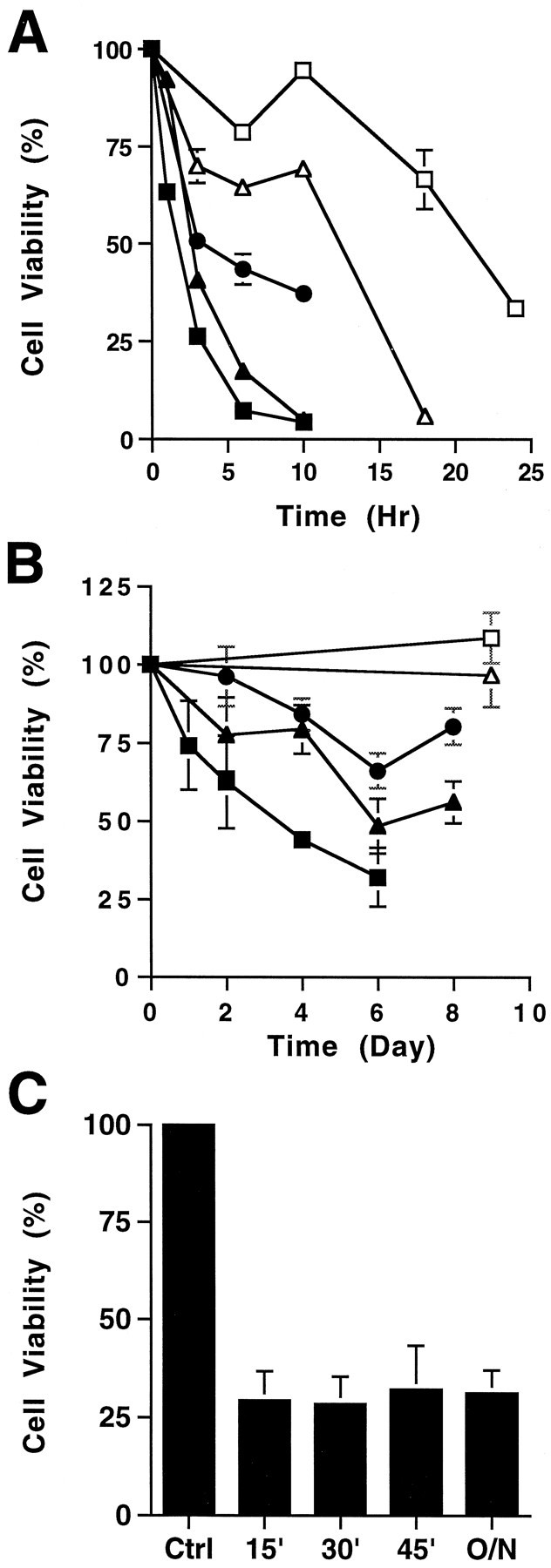
THC is toxic for cultured hippocampal neurons. Primary hippocampal neurons were treated with various concentrations of THC at high doses (A) (in μm):open squares, 3.5; open triangles, 5.0;filled circles, 7.5; filled triangles, 10; and filled squares, 20; or low doses (B) (in μm): open squares, 0.20; open triangles, 0.38;filled circles, 0.50; filled triangles, 1.0; and filled squares, 2.0. C, Primary hippocampal neurons were treated with 3.5 μm THC for the indicated periods (15, 30, or 45 min or 20 hr), followed by removal of the drug and reincubation in conditioned media overnight. Cell viability was measured by the MIT assay. One hundred percent viability is defined as the MTT conversion by cells treated with the carrier for the duration of the experiment.
Fig. 2.

THC toxicity requires activation of the CB1 receptor. Neurons were treated with 10 μm THC in the presence or absence of the CB1 antagonist SR141716A (SR). Cell viability was analyzed 5 hr after treatment by the MTT assay. Neurons were preincubated with SR for 20 min before treating with THC.
THC-induced neurotoxicity is not attributable to inhibition of adenylyl cyclases
THC neurotoxicity might be attributable to suppression of adenylyl cyclase activity, because cannabinoid receptors couple to inhibition of adenylyl cyclases through Gi (Howlett and Fleming, 1984;Howlett et al., 1986; Howlett, 1995), and neuron survival depends on optimal cAMP (Deckwerth and Johnson, 1993; Holcomb et al., 1995; Nobes and Tolkovsky, 1995; Xia et al., 1995). To evaluate this possibility, cultured neurons were treated with pertussis toxin (Ptx) to inactivate Gi and monitored for THC toxicity. Neither Ptx pretreatment nor forskolin, an adenylyl cyclase activator, protected neurons from THC-induced death (Fig. 3). This suggests that THC neurotoxicity is not caused by adenylyl cyclase inhibition. Interestingly, actinomycin D, an inhibitor of transcription, blocked THC-induced toxicity (Fig. 3). This suggests that THC may activate a transcription-dependent cell death program.
Fig. 3.

THC toxicity is not attributable to changes in cAMP. Hippocampal neuron viability was measured after treatment with 10 μm THC alone (THC), THC with 200 ng/ml pertussis toxin (Ptx; pretreated for 16 hr), with THC in the presence of 10 μm forskolin (Fsk), or with THC in the presence of 2 μg/ml actinomycin D (Act D). With the exception of Ptx, neurons were treated with reagents for 5 hr. p values are from Student’st test: NS, not significant; **p < 0.005. Data are triplicates from one representative experiment. Comparable results were obtained in four independent experiments. Ctrl, Control treatment with 0.01% ethanol, the carrier for THC. Error bars indicate ±SD.
THC increases intracellular Ca2+ in hippocampal neurons
Intracellular free Ca2+([Ca2+]i) is modulated by activation of cannabinoid receptors (Mackie and Hille, 1992; Mackie et al., 1993; Shen et al., 1996; Sugiura et al., 1996) and prolonged increases in [Ca2+]i are generally toxic (Trump and Berezesky, 1995; McConkey and Orrenius, 1996). Consequently, the effects of THC on [Ca2+]i were analyzed by fluorescent imaging using the Ca2+ fluorophore fura-2 AM (Fig.4). Addition of 10 μm THC to hippocampal neurons caused a delayed increase in [Ca2+]i that reached a maximum at 90 min (Fig. 4A). Treatment of neurons with 2 μm THC also increased [Ca2+]i but with slower kinetics (data not shown). When cells were treated with THC without extracellular Ca2+ and in the presence of EGTA, there was no significant increase in [Ca2+]i (Fig.4A). Addition of extracellular Ca2+ at the end of this incubation led to an instantaneous rise in [Ca2+]i, suggesting that a plasma membrane channel is activated by THC. Lanthanum, a potent blocker of Ca2+ channels, completely inhibited this rise in [Ca2+]i (Fig. 4B). THC-induced Ca2+ increases were also inhibited by the cannabinoid receptor antagonist SR141716A (Fig. 4C). Because stimulation of CB1 receptors increased [Ca2+]i, we examined the effect of chelating Ca2+ on THC neurotoxicity. Chelation of extracellular Ca2+ with EGTA did not rescue hippocampal neurons from THC toxicity (Fig.5). This suggests that THC neurotoxicity is not dependent on increases in intracellular Ca2+, because THC-stimulated increases in [Ca2+]i required extracellular Ca2+. This distinguishes THC neurotoxicity from other forms of toxicity that are triggered by increased [Ca2+]i.
Fig. 4.
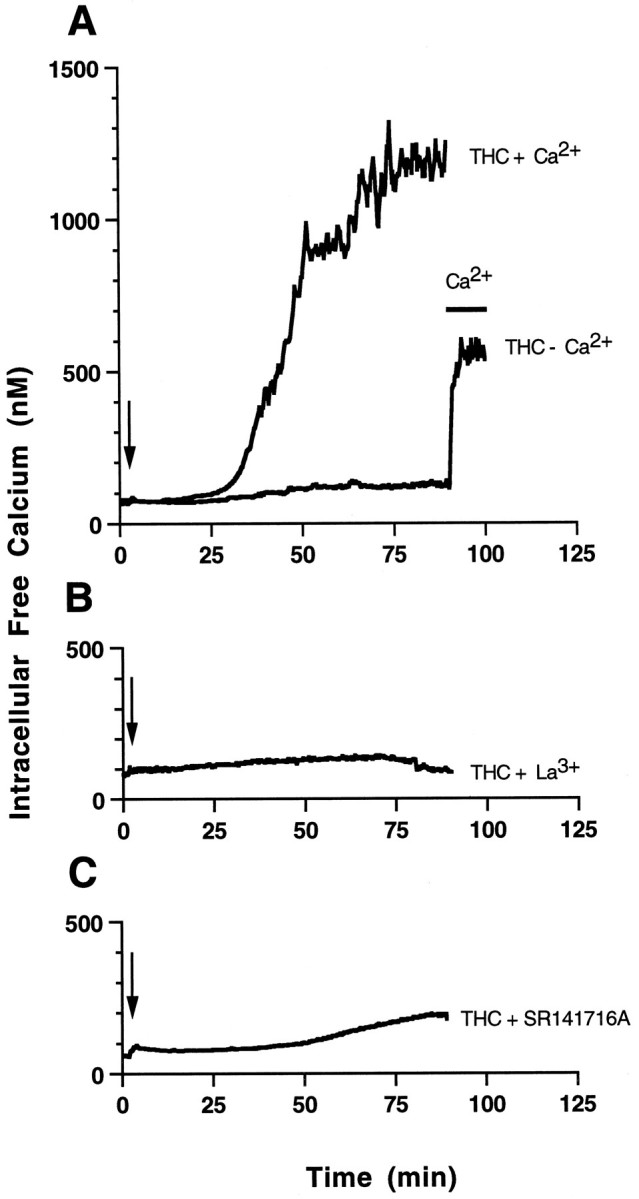
THC induces a delayed increase in intracellular Ca2+ that is dependent on extracellular Ca2+. [Ca2+]i of THC-treated primary hippocampal neurons was monitored by fura-2 fluorescent imaging. The calibrated [Ca2+]i (nm) was plotted against time (minutes). A, THC was added to neurons in the presence of 1.5 mm CaCl2 (THC + Ca2+) at the indicated time (arrow). No increase in intracellular Ca2+ was seen when the ethanol carrier (0.01%) was added alone (data not shown) or when THC was added to cells incubated without Ca2+ in the presence of 5 mm EGTA (THC − Ca2+). Four mm LaCl3(B) or 5 μm SR141716A (C) blocked increases in [Ca2+]i caused by 10 μmTHC.
Fig. 5.

THC neurotoxicity is blocked by inhibitors of the PLA2 cyclooxygenase pathway and antioxidants, but not by Ca2+ chelation. Neurons were treated with 0.2% ethanol (Ctrl), 3.5 μm THC (THC), or THC in the presence of 0.2 μmquinacrine (Quin), 1 μm indomethacin (Indo), 5 μm aspirin (Asp), 5 μm NDGA, 1.5 mm EGTA, or 20 μm α-tocopherol (Vit E). After 20 hr, neurons were assayed for viability as described in Materials and Methods. Neurons were preincubated with inhibitors for 60 min before adding THC. Numbers in parentheses represent the number of independent experiments, each performed in triplicate. Error bars indicate SD.p values are from Student’s t test. *p < 0.05; **p < 0.0005; NS, not significant.
THC neurotoxicity is abrogated by vitamin E as well as inhibitors of phospholipase A2 and cyclooxygenase
Activation of cannabinoid receptors can stimulate phospholipase A2 (PLA2), which catalyzes release of arachidonic acid (Reichman et al., 1988; Audette et al., 1991;Shivachar et al., 1996), the substrate for several major pathways. One branch, catalyzed by cyclooxygenases (COX), generates prostanoids, prostaglandins, and thromboxanes. Another pathway catalyzed by lipoxygenases produces leukotrienes. Both pathways generate free radicals (Wei et al., 1981; Pourcyrous et al., 1990; Yamamoto, 1991;Lafon-Cazal et al., 1993) that can lead to lipid peroxidation and cell death. Preincubation with quinacrine, an inhibitor of PLA2, partially protected neurons from THC-induced cell death (Fig. 5). Neurons were also protected by dexamethasone (data not shown), which induces the expression of annexin I (Flowers, 1988). Annexin I inhibits PLA2 (Croxtall et al., 1995) and the expression of COX (Fu et al., 1990). Indomethacin and aspirin, inhibitors of COX, completely protected neurons from THC, whereas NDGA, a lipoxygenase inhibitor, was not protective (Fig. 5). Because COX generates reactive oxygen species (ROS), which can cause cell death by stimulating the oxidation of lipids, proteins, and nucleic acid (Yamamoto, 1991; Troy and Shelanski, 1994; Greenlund et al., 1995;Simonian and Coyle, 1996), we examined the effect of antioxidants on THC-induced neuron death. Vitamin E completely protected neurons from THC (Fig. 5). These data suggest that THC may kill neurons by stimulation of the cyclooxygenase pathway with generation of ROS.
THC stimulates the release of arachidonic acid from primary hippocampal neurons
If THC neurotoxicity is attributable to activation of the PLA2–COX pathway in neurons, THC should stimulate arachidonic acid release. Treatment of cultured hippocampal neurons with THC induced the release of arachidonic acid from cultured hippocampal neurons, which was completely blocked by SR141716A, the CB1 receptor antagonist (Fig. 6). The kinetics for arachidonic acid release were dependent on THC concentration. For example, the rate of arachidonic acid release was three times faster in the presence of 10 μm THC (Fig.6B) compared with 3.5 μm THC (Fig.6A). Interestingly, pertussis toxin did not affect THC-induced increases in arachidonic acid release, suggesting that the enhancement of arachidonic acid release by THC is not mediated through Gi coupling. To determine whether exogenously added arachidonic acid affects neuron viability, primary hippocampal neurons were treated with increasing concentrations of arachidonic acid and monitored for cell viability. Exogenous arachidonic acid at 10 μm killed 100% of the cultured hippocampal neurons 24 hr after exposure (data not shown). However, it is unlikely that free arachidonic acid added to the outside surface of a cell is comparable to arachidonic acid release by activation of PLA2 caused by receptor stimulation.
Fig. 6.

THC induces arachidonic acid release from hippocampal neurons. Arachidonic acid release was monitored in cultured hippocampal neurons after treatment with THC. Hippocampal neurons were maintained in culture for 7–8 d and loaded with [3H]arachidonic acid for 3–4 hr and treated as described in Materials and Methods with 3.5 μm(A) or 10 μm(B) THC. SR141716A (SR) pretreatment (5 μm) was done 5 min before THC treatment. Pertussis toxin (Ptx) was added at 300 ng/ml, 12 hr before the addition of THC. Error bars indicate SD.
THC induces cell body shrinkage, nuclear condensation, and genomic DNA strand breakage in cultured hippocampal neurons
To determine whether THC induces morphological changes in cultured hippocampal neurons, we examined its effects on cell morphology and genomic DNA integrity using TUNEL for in situ DNA labeling (Gavrieli et al., 1992). Neuron cell bodies were lightly counterstained (Fig. 7A), whereas DNA strand breaks were detectable as small dark bodies in culture (Fig.7B,C). THC increased DNA strand breaks that were evident within days after treatment with 2 μm THC (Fig. 7B) or within 6 hr after treatment with 10 μm THC (Fig. 7C). In addition, THC caused a marked decrease in cell body size relative to control cells.
Fig. 7.

THC induces nuclear shrinkage and genomic DNA breakage in primary hippocampal neurons. Neurons were maintained for 10 d before treatment with 0.01% ethanol for 3 d (A, D), 2.0 μm THC for 3 d (B, E), or 10 μmTHC for 6 hr (C, F). Cells were analyzed using the TUNEL assay (A–C) coupled to horseradish peroxidase assay or stained with 2.5 μg/ml of Hoechst 33342 (D–F) as described in Materials and Methods. Light microscopic images of horseradish peroxidase staining and counter staining with 0.1% toluidine blue (A–C) and fluorescent images (D–F) are shown. Thin arrows indicate representative examples of condensed chromatin;thick arrows indicate normal chromatin. Scale bars, 50 μm.
Neurons were also stained with Hoechst 33342, a cell-permeant bisbenzimide DNA dye, to examine the effects of THC on nuclear morphology. Treatment of neurons with 2 μm (Fig.7E) or 10 μm THC (Fig. 7F) increased the percentage of cells exhibiting nuclear condensation (50.4%, n = 241; and 56.5%, n = 250, respectively) compared with neurons treated with carrier alone (Fig.7D; 13.6%, n = 301).
THC treatment causes nuclear contraction and genomic DNA breakage in hippocampal slices
Rat hippocampal slices were exposed to THC, fixed, and treated with RNase A before staining with propidium iodide to stain DNA or with terminal deoxytransferase for TUNEL assay. The CA1 pyramidal cell layer of hippocampal slices was then examined by confocal microscopy (Fig.8). In control slices, nuclear propidium iodide staining in the CA1 pyramidal cell layer was diffuse and relatively uniform (Fig. 8A, m.). In contrast, nuclei in slices treated with 2.5 μm THC were condensed and contracted compared with controls (Fig.8B, m.). An analysis at higher magnification indicated that the nuclei of THC-treated neurons were ∼50% smaller (Fig. 8D) than untreated nuclei (Fig.8C). However, the distance between neighboring nuclei and the size of nucleoli were unaffected.
Fig. 8.

THC induces nuclear shrinkage and DNA cleavage in neurons of hippocampal slices. Hippocampal slices perfused with 0.01% ethanol, the carrier for THC, (A, C,E, G) or with 2.5 μm THC (B, D, F, H) for 2 hr followed by 3 hr of perfusion in buffer alone. Slices were stained with propidium iodide (A–D) or subjected to TUNEL assays (E–H) as described in Materials and Methods. Confocal microscopic images of the CA1 pyramidal cell layer are shown. s.o., Stratum oriens; m., CA1 pyramidal cell layer;s.r., stratum radiatum of the hippocampus. Scale bars: A, B, 50 μm;C, D, 10 μm; E,F, 100 μm; and G, H, 25 μm.
TUNEL staining of CA1 pyramidal neurons in hippocampal slices was markedly increased by THC (Figs.8F,H) compared with control slices (Figs. 8E,G). Although other areas of the hippocampus, including area CA3 and the dentate gyrus, were also affected by THC, the increase in TUNEL staining was most evident in the CA1 pyramidal cell layer. These data indicate that CA1 pyramidal neurons are sensitive to THC in tissue slices where many normal cellular interactions are preserved.
DISCUSSION
Although marijuana is known to cause memory deficits in humans and laboratory animals, the mechanism(s) for these effects have not been elucidated. Because administration of marijuana and cannabinoids to rodents causes a reduction in neuron density in the hippocampus (Landfield et al., 1988), deficiencies in memory might be attributable to neurotoxicity. The objectives of this study were to determine whether THC, a major psychoactive component of marijuana, is toxic for hippocampal neurons and to characterize mechanisms for neurotoxicity. We examined THC neurotoxicity in vitro using cultured hippocampal neurons and hippocampal slices to identify signal transduction pathways contributing to toxicity.
On the basis of the data presented in this study, we conclude that binding of THC to cannabinoid CB1 receptors in hippocampal neurons leads to neuronal death. THC is neurotoxic at concentrations as low as 0.5–1.0 μm, which are comparable to THC levels measured in human plasma after consumption of marijuana cigarettes. Although THC neurotoxicity was unaffected by Ca2+ chelators and activators of adenylyl cyclase, it was abated by inhibitors of PLA2 and completely blocked by aspirin and indomethacin, inhibitors of COX. This suggests that activation of PLA2 by THC may contribute to increases in arachidonic acid. Because protection by quinacrine was only partial, other mechanisms may contribute to the arachidonic acid release. For example, THC may also increase intracellular arachidonic acid by inhibition of arachidonic acid acylation (Reichman et al., 1991). We hypothesize that THC-induced neuron death is triggered by a signal transduction cascade that increases arachidonic acid and activates COX with the formation of ROS (Fig. 9). THC enhancement of arachidonic acid release from cultured neurons and protection of neurons from THC by vitamin E supports this hypothesis.
Fig. 9.

Signaling pathways contributing to THC-induced neuronal death. It is hypothesized that excessive stimulation of cannabinoid receptors (CB1) by THC stimulates the production of arachidonic acid (AA) by several pathways. Cyclooxygenase (COX) catalyzes the formation of prostaglandins (PGs), thromboxanes (TXs), and reactive oxygen species (ROS), which stimulates peroxidation of lipids, proteins, and DNA. In addition, cannabinoid-induced transcriptional events may also contribute to THC induction of neuron death.
Our data implicating increases in arachidonic acid and activation of COX in THC neurotoxicity are consistent with other reports in the literature that cannabinoids stimulate arachidonic acid release from cultured astrocytes (Shivachar et al., 1996), cortical slices (Reichman et al., 1987), and lymphocytes (Audette et al., 1991). Furthermore, cannabinoids have been reported to stimulate cyclooxygenases (Reichman et al., 1987). The release of arachidonic acid and the subsequent generation of ROS in other systems are both implicated in cell damage (Kuehl and Egan, 1980) and cell death (Tang et al., 1996; Chen et al., 1997; Li et al., 1997; Wissing et al., 1997). ROS oxidizes membrane lipids, nucleic acids, and proteins, all of which can lead to necrotic or apoptotic cell death. Interestingly, THC causes DNA strand breaks, nuclear contraction, and cell body shrinkage, which are characteristic of cells undergoing apoptosis (Hockenbery, 1995; Majno and Joris, 1995).
Data in this paper suggest that stimulation of transcription may be important for THC neurotoxicity. Although the transcriptional events necessary for THC neurotoxicity were not defined, there are several possibilities. For example, activation of cannabinoid receptors induces the expression of immediate early genes Krox24, Krox20, and Jun B (Bouaboula et al., 1995). It also increases nuclear factor-κB activity (Daaka et al., 1997), which activates the expression of several genes including tumor necrosis factor α (TNFα) and other cytokines (for review, see Baeuerle and Baltimore, 1996). Interestingly, TNFα induces apoptotic cell death. Coactivation of these pathways with THC stimulation of the arachidonic acid release and generation of ROS may be required for THC neurotoxicity. Because inhibitors of protein synthesis induce the expression of Bcl-2 and activate antioxidant pathways (Furukawa et al., 1997), inhibitors of transcription may have similar effects. Consequently, the generation of ROS by THC may be antagonized by induction of Bcl-2, superoxide dismutases, and catalase.
The pharmacology of cannabinoids is characterized by a multiplicity of effects in humans and rodents. Treatment of rats and other laboratory animals with THC elicits several responses, including decreases in memory, antinociception, hypothermia, catalepsy, and diminished spontaneous motion (for review, see Dewey, 1986). The hippocampal complex is pivotal for memory in humans and laboratory animals. Hippocampal lesions in humans (Scoville and Milner, 1957; Damasio et al., 1985) or rodents (Olton, 1987) decrease the ability to acquire new information or recover previously learned knowledge. Intrahippocampal injection of cannabinoids in rats impairs spatial memory, suggesting that memory defects are mediated primarily by cannabinoid receptors in the hippocampus (Lichtman et al., 1995). The data reported in our study are consistent with this hypothesis and suggest that marijuana-induced memory defects in rodents may be attributable to THC neurotoxicity. Also consistent with this hypothesis is the expression of high levels of cannabinoid receptors in the hippocampus (Herkenham et al., 1991).
Marijuana is used therapeutically for several conditions, including treatment of pain and inflammation, nausea caused by cancer chemotherapy, and muscle spasms associated with multiple sclerosis (for review, see Hollister, 1986). However, clinical use of cannabinoids may be compromised by their disruptive effects on information processing and storage. Although there is no direct evidence that marijuana is neurotoxic for human brain, some of the memory deficits associated with its use may be caused by neuronal death in the hippocampus. Consequently, memory defects caused by marijuana might be reduced by aspirin, indomethacin, or vitamin E. Interestingly, clinical studies indicate that elevation of prostaglandins caused by marijuana smoking can be suppressed by indomethacin (Perez Reyes et al., 1991). This indicates that marijuana use in humans activates the PLA2–COX pathway and suggests that indomethacin or aspirin might be appropriate for protection against cannabinoids. Although speculative, this hypothesis is based on an analysis of the molecular mechanisms underlying THC neurotoxicity. The identification of pharmacological methods to prevent marijuana neurotoxicity may be an important consideration when using marijuana medicinally.
In summary, treatment of hippocampal neurons with THC induces transcriptionally dependent cell death. This suggests that memory loss associated with marijuana treatment of rodents, and perhaps humans, may be attributable to THC neurotoxicity. The identification of a specific signal transduction system responsible for THC toxicity and the discovery that aspirin and vitamin E inhibit this neurotoxicity suggest pharmacological tools to block THC-induced cell death. In addition, endogenous ligands for cannabinoid receptors have been identified (Devane et al., 1992; Priller et al., 1995). Cannabinoid receptors may play an important role during neural development by mediating cell death in specific populations of neurons in response to the local release of endogenous cannabinoids such as anandamide.
Footnotes
This research was supported by National Institutes of Health Grant NS20598 and the Alcohol and Drug Abuse Institute, University of Washington. Dr. G. C. Chan was supported by postdoctoral training Grants DE07023-21 and DA07278-01A1. We thank Lisa Prichard, Scott Wong, Mark Nielsen, Jia Wei, Dr. Christoff Deloulme, Dr. Lauren Baker, and Dr. Enrique Villacres for critical reading of this manuscript.
Correspondence should be addressed to Daniel R. Storm, Department of Pharmacology, Box 357280, University of Washington, Seattle, WA 98195.
REFERENCES
- 1.Abel EL. Marijuana and memory. Nature. 1970;227:1151–1152. doi: 10.1038/2271151b0. [DOI] [PubMed] [Google Scholar]
- 2.Abel EL. Retrieval of information after use of marijuana. Nature. 1971;231:58. doi: 10.1038/231058a0. [DOI] [PubMed] [Google Scholar]
- 3.Abood ME, Martin BR. Neurobiology of marijuana abuse. Trends Pharmacol Sci. 1992;13:201–206. doi: 10.1016/0165-6147(92)90064-d. [DOI] [PubMed] [Google Scholar]
- 4.Audette CA, Burstein SH, Doyle SA, Hunter SA. G-protein mediation of cannabinoid-induced phospholipase activation. Pharmacol Biochem Behav. 1991;40:559–563. doi: 10.1016/0091-3057(91)90363-7. [DOI] [PubMed] [Google Scholar]
- 5.Baeuerle PA, Baltimore D. NF-κB: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 6.Block RI, Ghoneim MM. Effects of chronic marijuana use on human cognition. Psychopharmacology. 1993;110:219–228. doi: 10.1007/BF02246977. [DOI] [PubMed] [Google Scholar]
- 7.Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci USA. 1979;70:514–519. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bouaboula M, Bourrie B, Rinaldi-Carmona M, Shire D, Le Fur G, Casellas P. Stimulation of cannabinoid receptor CB1 induces krox-24 expression in human astrocytoma cells. J Biol Chem. 1995;270:13973–13980. doi: 10.1074/jbc.270.23.13973. [DOI] [PubMed] [Google Scholar]
- 9.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 10.Carlini EA, Hamaoui A, Bieniek D, Korte F. Effects of (–) delta-9-trans-tetrahydrocannabinol and a synthetic derivative on maze performance of rats. Pharmacology. 1970;4:359–368. doi: 10.1159/000136165. [DOI] [PubMed] [Google Scholar]
- 11.Chavez Noriega LE, Stevens CF. Increased transmitter release at excitatory synapses produced by direct activation of adenylate cyclase in rat hippocampal slices. J Neurosci. 1994;14:310–317. doi: 10.1523/JNEUROSCI.14-01-00310.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Q, Galleano M, Cederbaum AI. Cytotoxicity and apoptosis produced by arachidonic acid in Hep G2 cells overexpressing human cytochrome P4502E1. J Biol Chem. 1997;272:14532–14541. doi: 10.1074/jbc.272.23.14532. [DOI] [PubMed] [Google Scholar]
- 13.Chiang CWN, Barnett G. Marijuana effect and delta-9-tetrahydrocannabinol plasma level. Clin Pharmacol Ther. 1984;36:234–238. doi: 10.1038/clpt.1984.168. [DOI] [PubMed] [Google Scholar]
- 14.Collins DR, Pertwee RG, Davies SN. The action of synthetic cannabinoids on the induction of long-term potentiation in the rat hippocampal slice. Eur J Pharmacol. 1994;259:R7–R8. doi: 10.1016/0014-2999(94)90666-1. [DOI] [PubMed] [Google Scholar]
- 15.Croxtall JD, Choudhury Q, Tokumoto H, Flower RJ. Lipocortin-1 and the control of arachidonic acid release in cell signal—glucocorticoids inhibit G-protein-dependent activation of cPLA2 activity. Biochem Pharmacol. 1995;50:465–474. doi: 10.1016/0006-2952(95)00156-t. [DOI] [PubMed] [Google Scholar]
- 16.Daaka Y, Zhu W, Friedman H, Klein TW. Induction of interleukin-2 receptor α gene by Δ9-tetrahydrocannabinol is mediated by nuclear factor κB and CB1 cannabinoid receptor. DNA Cell Biol. 1997;16:301–309. doi: 10.1089/dna.1997.16.301. [DOI] [PubMed] [Google Scholar]
- 17.Damasio AR, Eslinger PJ, Damasio H, Van Hoesen GW, Cornell S. Multimodal amnesic syndrome following bilateral temporal and basal forebrain damage. Arch Neurol. 1985;42:252–259. doi: 10.1001/archneur.1985.04060030070012. [DOI] [PubMed] [Google Scholar]
- 18.Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 20.Dewey WL. Cannabinoid pharmacology. Pharmacol Rev. 1986;38:151–178. [PubMed] [Google Scholar]
- 21.Fehr KA, Kalant H, LeBlanc AE. Residual learning deficit after heavy exposure to cannabis or alcohol in rats. Science. 1976;192:1249–1251. doi: 10.1126/science.1273591. [DOI] [PubMed] [Google Scholar]
- 22.Fletcher JM, Page JB, Francis DJ, Copeland K, Naus MJ, Davis CM, Morris R, Krauskopf D, Satz P. Cognitive correlates of long-term cannabis use in Costa Rican men. Arch Gen Psychiatry. 1996;53:1051–1057. doi: 10.1001/archpsyc.1996.01830110089011. [DOI] [PubMed] [Google Scholar]
- 23.Flowers J. Lipocortin and the mechanism of action of the glucocorticoids. Br J Pharmacol. 1988;94:987–1015. doi: 10.1111/j.1476-5381.1988.tb11614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frey U, Huang YY, Kandel ER. Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons. Science. 1993;260:1661–1664. doi: 10.1126/science.8389057. [DOI] [PubMed] [Google Scholar]
- 25.Fu JY, Masferrer JL, Seibert K, Raz A, Needleman P. The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes. J Biol Chem. 1990;265:16737–16740. [PubMed] [Google Scholar]
- 26.Furukawa K, Estus S, Fu W, Mark RJ, Mattson MP. Neuroprotective action of cycloheximide involves induction of Bcl-2 and antioxidant pathways. J Cell Biol. 1997;136:1137–1149. doi: 10.1083/jcb.136.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenlund LJS, Deckwerth TL, Johnson EM., Jr Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 29.Jaffe JH. Drug addiction and drug abuse. In: Gilman AG, Rall TW, Nies AS, Taylor P, editors. Goodman and Gilman’s pharmacological basis of therapeutics, Eighth Edition. Pergamon; New York: 1990. pp. 522–573. [Google Scholar]
- 30.Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 31.Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hockenbery D. Defining apoptosis. Am J Pathol. 1995;146:16–19. [PMC free article] [PubMed] [Google Scholar]
- 33.Holcomb JD, Mumm JS, Calof AL. Apoptosis in the neuronal lineage of the mouse olfactory epithelium: regulation in vivo and in vitro. Dev Biol. 1995;172:307–323. doi: 10.1006/dbio.1995.0025. [DOI] [PubMed] [Google Scholar]
- 34.Hollister LE. Health aspects of cannabis. Pharmacol Rev. 1986;38:1–20. [PubMed] [Google Scholar]
- 35.Howlett AC. Pharmacology of cannabinoid receptors. Annu Rev Pharmacol Toxicol. 1995;35:607–634. doi: 10.1146/annurev.pa.35.040195.003135. [DOI] [PubMed] [Google Scholar]
- 36.Howlett AC, Fleming RM. Cannabinoid inhibition of adenylate cyclase. Mol Pharmacol. 1984;26:532–538. [PubMed] [Google Scholar]
- 37.Howlett AC, Qualy JM, Khachatrian L. Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Mol Pharmacol. 1986;29:307–313. [PubMed] [Google Scholar]
- 38.Impey S, Mark M, Villacres EC, Poser S, Chavkin C, Storm DR. Induction of CRE-mediated gene expression by stimuli that generate long-lasting LTP in area CA1 of the hippocampus. Neuron. 1996;16:973–982. doi: 10.1016/s0896-6273(00)80120-8. [DOI] [PubMed] [Google Scholar]
- 39.Kuehl FA, Egan RW. Prostaglandins, arachidonic acid, and inflammation. Science. 1980;210:978–984. doi: 10.1126/science.6254151. [DOI] [PubMed] [Google Scholar]
- 40.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 41.Landfield PW, Cadwallader LB, Vinsant S. Quantitative changes in hippocampal structure following long-term exposure to THC: possible mediation by glucocorticoid systems. Brain Res. 1988;443:47–62. doi: 10.1016/0006-8993(88)91597-1. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- 43.Lichtman AH, Dimen KR, Martin BR. Systemic or intrahippocampal cannabinoid administration impairs spatial memory in rats. Psychopharmacology. 1995;119:282–290. doi: 10.1007/BF02246292. [DOI] [PubMed] [Google Scholar]
- 44.Mackie K, Hille B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci USA. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mackie K, Devane WA, Hille B. Anandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Mol Pharmacol. 1993;44:498–503. [PubMed] [Google Scholar]
- 46.Majno G, Joris I. Apoptosis, oncosis, and necrosis: an overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 47.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 48.McConkey DJ, Orrenius S. The role of calcium in the regulation of apoptosis. J Leukoc Biol. 1996;59:775–783. [PubMed] [Google Scholar]
- 49.Morrison RS, Wenzel HJ, Kinoshita Y, Robbins CA, Donehower LA, Schwartzkroin PA. Loss of the p53 tumor suppressor gene protects neurons from kainate-induced cell death. J Neurosci. 1996;16:1337–1345. doi: 10.1523/JNEUROSCI.16-04-01337.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 51.Nobes CD, Tolkovsky AM. Neutralizing anti-p21 ras Fabs suppress rat sympathetic neuron survival induced by NGF, LIF, CNTF and cAMP. Eur J Neurosci. 1995;7:344–350. doi: 10.1111/j.1460-9568.1995.tb01069.x. [DOI] [PubMed] [Google Scholar]
- 52.Nowicky AV, Teyler TJ, Vardaris RM. The modulation of long-term potentiation by delta-9-tetrahydrocannabinol in the rat hippocampus, in vitro. Brain Res Bull. 1987;19:663–672. doi: 10.1016/0361-9230(87)90052-9. [DOI] [PubMed] [Google Scholar]
- 53.Olton DS. The radial arm maze as a tool in behavioral pharmacology. Physiol Behav. 1987;40:793–797. doi: 10.1016/0031-9384(87)90286-1. [DOI] [PubMed] [Google Scholar]
- 54.Otsuki H, Yamada K, Yuguchi T, Taneda M, Hayakawa T. Prostaglandin E1 induces c-Fos and Myc proteins and protects rat hippocampal cells against hypoxic injury. J Cereb Blood Flow Metab. 1994;14:150–155. doi: 10.1038/jcbfm.1994.20. [DOI] [PubMed] [Google Scholar]
- 55.Perez-Reyes M, Burstein SH, White WR, McDonald SA, Hicks RE. Antagonism of marijuana effects by indomethacin in humans. Life Sci. 1991;48:507–515. doi: 10.1016/0024-3205(91)90465-n. [DOI] [PubMed] [Google Scholar]
- 56.Pope HG, Yurgelun-Todd D. The residual cognitive effects of heavy marijuana use in college students. J Am Med Assoc. 1996;275:521–527. [PubMed] [Google Scholar]
- 57.Pourcyrous M, Leffler CW, Mirro R, Busija DW. Brain superoxide anion generation during asphyxia and reventilation in newborn pigs. Pediatr Res. 1990;28:618–621. doi: 10.1203/00006450-199012000-00015. [DOI] [PubMed] [Google Scholar]
- 58.Priller J, Briley EM, Mansouri J, Devane WA, Mackie K, Felder CC. Mead ethanolamide, a novel eicosanoid, is an agonist for the central (CB1) and peripheral (CB2) cannabinoid receptors. Mol Pharmacol. 1995;48:288–292. [PubMed] [Google Scholar]
- 59.Reichman M, Nen W, Hokin LE. Effects of delta-9-tetrahydrocannabinol on prostaglandin formation in brain. Mol Pharmacol. 1987;32:686–690. [PubMed] [Google Scholar]
- 60.Reichman M, Nen W, Hokin LE. Delta-9-tetrahydrocannabinol increases arachidonate levels in guinea pig cerebral cortex slices. Mol Pharmacol. 1988;34:823–828. [PubMed] [Google Scholar]
- 61.Reichman M, Nen W, Hokin LE. Delta 9-tetrahydrocannabinol inhibits arachidonic acid acylation of phospholipids and triacylglycerols in guinea pig cerebral cortex slices. Mol Pharmacol. 1991;40:547–555. [PubMed] [Google Scholar]
- 62.Reisine T, Brownstein MJ. Opioid and cannabinoid receptors. Curr Opin Neurobiol. 1994;4:406–412. doi: 10.1016/0959-4388(94)90103-1. [DOI] [PubMed] [Google Scholar]
- 63.Rinaldi-Carmona M, Barth F, Heaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Neliat G, Caput D, Ferrara P, Soubrie P, Breliere JC, Le Fur G. SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- 64.Scallet AC. Neurotoxicology of cannabis and THC: a review of chronic exposure studies in animals. Pharmacol Biochem Behav. 1991;40:671–676. doi: 10.1016/0091-3057(91)90380-k. [DOI] [PubMed] [Google Scholar]
- 65.Scallet AC, Uemura E, Andrews A, Ali SF, McMillan DE, Paule MG, Brown RM, Slikker W., Jr Morphometric studies of the rat hippocampus following chronic delta-9-tetrahydrocannabinol (THC). Brain Res. 1987;436:193–198. doi: 10.1016/0006-8993(87)91576-9. [DOI] [PubMed] [Google Scholar]
- 66.Schwartz RH, Gruenewald PJ, Klitzner M, Fedio P. Short-term memory impairment in cannabis-dependent adolescents. Am J Dis Child. 1989;143:1214–1219. doi: 10.1001/archpedi.1989.02150220110030. [DOI] [PubMed] [Google Scholar]
- 67.Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. J Neurol Neurosurg Psychiatry. 1957;20:11–21. doi: 10.1136/jnnp.20.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen M, Piser TM, Seybold VS, Thayer SA. Cannabinoid receptor agonists inhibit glutamatergic synaptic transmission in rat hippocampal cultures. J Neurosci. 1996;16:4322–4334. doi: 10.1523/JNEUROSCI.16-14-04322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shivachar AC, Martin BR, Ellis EF. Anandamide- and delta-9-tetrahydrocannabinol-evoked arachidonic aid mobilization and blockade by SR141716A. Biochem Pharmacol. 1996;51:669–676. doi: 10.1016/s0006-2952(95)02248-1. [DOI] [PubMed] [Google Scholar]
- 70.Simonian NA, Coyle JT. Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol. 1996;36:83–106. doi: 10.1146/annurev.pa.36.040196.000503. [DOI] [PubMed] [Google Scholar]
- 71.Stiglick A, Kalant H. Learning impairment in the radial-arm maze following prolonged cannabis treatment in rats. Psychopharmacology. 1982a;77:117–123. doi: 10.1007/BF00431932. [DOI] [PubMed] [Google Scholar]
- 72.Stiglick A, Kalant H. Residual effects of prolonged cannabis administration on exploration and DRL performance in rats. Psychopharmacology. 1982b;77:124–128. doi: 10.1007/BF00431933. [DOI] [PubMed] [Google Scholar]
- 73.Stiglick A, Kalant H. Behavioral effects of prolonged administration of delta 9-tetrahydrocannabinol in the rat. Psychopharmacology. 1983;80:325–330. doi: 10.1007/BF00432114. [DOI] [PubMed] [Google Scholar]
- 74.Stiglick A, Llewellyn ME, Kalant H. Residual effects of prolonged cannabis treatment on shuttle-box avoidance in the rat. Psychopharmacology. 1984;84:476–479. doi: 10.1007/BF00431452. [DOI] [PubMed] [Google Scholar]
- 75.Sugiura T, Kodaka T, Kondo S, Tonegawa T, Nakane S, Kishimoto S, Yamashita A, Waku K. 2-Arachidonoylglycerol, a putative endogenous cannabinoid receptor ligand, induces rapid, transient elevation of intracellular free Ca2+ in neuroblastoma x glioma hybrid NG108–15 cells. Biochem Biophys Res Commun. 1996;229:58–64. doi: 10.1006/bbrc.1996.1757. [DOI] [PubMed] [Google Scholar]
- 76.Takeshima T, Johnston JM, Commissiong JW. Mesencephalic type 1 astrocytes rescue dopaminergic neurons from death induced by serum deprivation. J Neurosci. 1994;14:4769–4779. doi: 10.1523/JNEUROSCI.14-08-04769.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang DG, Chen YQ, Honn KV. Arachidonate lipoxygenases as essential regulators of cell survival and apoptosis. Proc Natl Acad Sci USA. 1996;93:5241–5246. doi: 10.1073/pnas.93.11.5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thomas BF, Compton DR, Martin BR. Characterization of the lipophilicity of natural and synthetic analogs of delta-9-THC and its relationship to pharmacological potency. J Pharmacol Exp Ther. 1990;255:624–630. [PubMed] [Google Scholar]
- 79.Troy CM, Shelanski ML. Down-regulation of copper/zinc superoxide dismutase causes apoptotic death in PC12 neuronal cells. Proc Natl Acad Sci USA. 1994;91:6384–6387. doi: 10.1073/pnas.91.14.6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Trump BF, Berezesky IK. Calcium-mediated cell injury and cell death. FASEB J. 1995;9:219–228. doi: 10.1096/fasebj.9.2.7781924. [DOI] [PubMed] [Google Scholar]
- 81.Voth EA, Schwartz RH. Medicinal applications of delta-9-tetrahydrocannabinol and marijuana. Ann Intern Med. 1997;126:791–798. doi: 10.7326/0003-4819-126-10-199705150-00008. [DOI] [PubMed] [Google Scholar]
- 82.Wayman GA, Hinds TR, Storm DR. Hormone Stimulation of type III adenylyl cyclase causes Ca2+ oscillations in HEK-293 cells. J Biol Chem. 1995;270:24108–24115. doi: 10.1074/jbc.270.41.24108. [DOI] [PubMed] [Google Scholar]
- 83.Wei EP, Kontos HA, Dietrich WD, Povlishock JT, Ellis EF. Inhibition by free radical scavengers and by cyclooxygenase inhibitors of pial arteriolar abnormalities from concussive brain injury in cats. Circ Res. 1981;48:95–103. doi: 10.1161/01.res.48.1.95. [DOI] [PubMed] [Google Scholar]
- 84.Weisskopf MG, Castillo PE, Zalutsky RA, Nicoll RA. Mediation of hippocampal mossy fiber long-term potentiation by cyclic AMP. Science. 1994;23:1878–1882. doi: 10.1126/science.7916482. [DOI] [PubMed] [Google Scholar]
- 85.Wissing D, Mouritzen H, Egeblad M, Poirier GG, Jaattela M. Involvement of caspase-dependent activation of cytosolic phospholipase A(2) in tumor necrosis factor-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:5073–5077. doi: 10.1073/pnas.94.10.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP Kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 87.Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS. Evidence for p53-mediated modulation of neuronal viability. J Neurosci. 1996;16:6753–6765. doi: 10.1523/JNEUROSCI.16-21-06753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yamamoto S. Enzymatic lipid peroxidation: reactions of mammalian lipoxygenases. Free Radic Biol Med. 1991;10:149–159. doi: 10.1016/0891-5849(91)90008-q. [DOI] [PubMed] [Google Scholar]