

Abstract
The inositol 1,4,5-trisphosphate receptor (InsP3R) is an intracellular Ca2+ channel that releases Ca2+ from internal Ca2+ stores in response to InsP3. Although InsP3R is highly expressed in various regions of the mammalian brain, the functional role of this receptor has not been clarified. We show here that cerebellar slices prepared from mice with a disrupted InsP3R type 1 gene, which is predominantly expressed in Purkinje cells, completely lack long-term depression (LTD), a model of synaptic plasticity in the cerebellum. Moreover, a specific antibody against InsP3R1, introduced into wild-type Purkinje cells through patch pipettes, blocked the induction of LTD. These data indicate that, in addition to Ca2+ influx through Ca2+ channels on the plasma membrane, Ca2+ release from InsP3R plays an essential role in the induction of LTD, suggesting a physiological importance for InsP3R in Purkinje cells.
Keywords: type 1 inositol 1,4,5-trisphosphate receptor; long-term depression; cerebellar Purkinje neuron; synaptic plasticity; brain slice; patch recording; caged-InsP3
Type 1 inositol 1,4,5-trisphosphate receptor (InsP3R1) is highly expressed in cerebellar Purkinje cells (Furuichi et al., 1989, 1993). Ca2+ release by this receptor has been detected in situ (Llano et al., 1991;Vranesic et al., 1991; Khodakhah and Ogden, 1993). In the Purkinje cell, the intracellular Ca2+ concentration ([Ca2+]i) is dynamically increased by excitatory synaptic stimulation or by artificial depolarization. However, the source of the transient Ca2+ has been assigned to voltage-gated Ca2+ channels (VGCCs) that are enriched on the plasma membrane of Purkinje cells (Tank et al., 1988;Lev-Ram et al., 1992; Miyakawa et al., 1992). Despite high levels of expression, there is little direct evidence for a physiological role for InsP3R1 in Purkinje cells.
Long-term depression (LTD) at the parallel fiber (PF)–Purkinje cell synapse is a candidate mechanism for the cellular basis of motor learning and motor coordination (Ito, 1989). LTD is induced by a conjunctive stimulation of PF and climbing fiber (CF) synapses. The initial step in the induction is the temporal overlap of the large elevation of [Ca2+]i caused by depolarization evoked by CF input and the activation of postsynaptic glutamate receptors at the PF synapse, including metabotropic glutamate receptors (mGluRs). Activation of mGluR results in the production of InsP3 and diacylglycerol. The former opens the InsP3R channel, and the latter activates protein kinase C (PKC) (Berridge, 1993). This signal transduction cascade is necessary for the induction of LTD, because inhibition of mGluR (Aiba et al., 1994; Conquet et al., 1994; Hartell, 1994b) or PKC (Crepel and Krupa, 1988; Hartell, 1994a; Chen et al., 1995) results in blockade of LTD. This feature is also shared by another form of LTD expressed in cultured Purkinje cells (culture-LTD) (Linden and Connor, 1991; Shigemoto et al., 1994). According to these reports, blockade of the mGluR response is caused by inhibition of PKC activation. However, it remains unclear whether InsP3R plays a role in this LTD scheme, mainly because of the lack of specific antagonists to this receptor.
LTD is blocked by the InsP3R inhibitor heparin and induced by an increase in InsP3 using caged-InsP3 in slices (Khodakhah and Armstrong, 1997) and culture-LTD (Kasono and Hirano, 1995). These experiments suggest that InsP3 is important in LTD. However, heparin may bind numerous other sites inside the cell, resulting in various nonspecific effects, including inhibition of PKC (Herbert and Maffrand, 1991). In addition, the caged-InsP3 experiments do not necessarily imply a role for InsP3R in the LTD mechanism. Thus, the necessity of Ca2+ release by InsP3R-sensitive intracellular stores to induce LTD is an unresolved question.
To examine this issue, we developed two strategies to eliminate the functional expression of InsP3R1. In one, we created a mouse strain lacking the InsP3R1 gene. In the other, we blocked the function of wild-type receptors with a specific antibody. The results of these experiments show that Ca2+ release from intracellular stores by the InsP3R1 channel is required for the induction of LTD.
Preliminary observations have been published previously (Inoue and Mikoshiba, 1997).
MATERIALS AND METHODS
Animals and preparation of slices. In experiments with mutant mice, 18- to 23-d-old InsP3R1+/+ and InsP3R1−/− animals (Matsumoto et al., 1996) were used, because InsP3R1−/− mice do not survive beyond postnatal day 23. In the antibody experiment, 25- to 50-d-old ddY mice were used, because we observed no LTD in mice younger than 25 d with the induction protocol of combined PF and CF stimulation (T. Inoue and K. Mikoshiba, unpublished observations). Transverse or sagittal cerebellar slices, 250 μm thick, were prepared according to standard procedures using a Vibratome tissue slicer (DSK-1000, Dosaka EM, Kyoto, Japan). Transverse slices were used in LTD induction experiments because PFs are not cut in this plane, enabling more stable recording of PF-mediated EPSPs (PF-EPSP) than in sagittally cut slices. However, sagittally cut slices, in the plane of Purkinje cell dendrites, were preferred for imaging experiments. Two types of superfusing saline were used: artificial CSF-A (ACSF-A) composed of (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 20 glucose; and ACSF-B composed of (in mm): 124 NaCl, 5 KCl, 2 CaCl2, 1 MgSO4, 1.25 NaH2PO4, 22 NaHCO3, and 10 glucose. Both solutions were bubbled with a mixture of 95% O2 and 5% CO2, and kept at 32–34°C. ACSF-A was used in the mutant mice experiments and ACSF-B was used in the antibody experiments. Bicuculline (10 μm) was always added to the external solution in LTD experiments. In either external solution, LTD was successfully observed in control experiments using the same protocol (Inoue and Miloshiba, unpublished observations).
LTD experiments in mutant Purkinje cells. All experiments were performed using whole-cell patch recording under direct visualization using a fixed-stage upright microscope (BX50WI; Olympus, Tokyo, Japan) and an objective lens (40× water immersion, NA 0.80; Olympus). Borosilicate pipettes (4–5 MΩ) were used and were filled with a solution containing (in mm): 70 KCl, 60 K-D-gluconate, 0.5 EGTA, 4 MgCl2, 4 Na-ATP, 0.4 Na-GTP, 30 HEPES, pH 7.3, and 280 mOsm. ACSF-A with 10 μmbicuculline was used as an external solution. Recordings were made with an AxoClamp 2A amplifier (Axon Instruments, Foster City, CA) in the current-clamp mode. For stimulation of PFs, monopolar square pulses (200 μsec) were applied through a glass pipette filled with the superfusing saline. The stimulation electrode was placed on the molecular layer, 100–200 μm from the Purkinje cell. The peak amplitude of the PF-EPSP was monitored every 5 sec. Although the stimulus artifact was relatively large, it did not affect the result. Membrane potentials were held between −65 and −68 mV manually. To monitor changes in Rm andRs, a hyperpolarizing square pulse (ranging from −100 to −150 pA, 60 msec duration, beginning 20 msec before the PF stimulus) was applied through the patch pipette. Changes in Rs were compensated for with a bridge balance circuit in the amplifier. Experiments in which the EPSP amplitude was not stable during the 10 min period before pairing were discarded. Instability was determined if the average in any 2 min period during the 10 min period exceeded ±5% range of the baseline value (baseline value was calculated as an average of the 6 min period just before the pairing). In addition, experiments in which the holding current exceeded −650 pA were discarded. In accepted experiments,Rm remained constant (based on the shape of the hyperpolarizing phase; see Fig. 1A,B, insets). During the LTD induction periods, the amplifier was switched to voltage-clamp mode (holding potential: −60 mV). LTD was induced by pairing depolarization of Purkinje cells (200 msec, −60 to 0 mV) with PF stimulation 240 times at 1 Hz (PF stimulus was delivered 50 msec after the onset of the depolarization). This protocol was always started 15–20 min after formation of the whole-cell patch. Electrophysiological data were filtered at 2 kHz, monitored, and stored on-line at a sampling rate of 10 kHz with an MS-DOS-based computer (PC-9801VX; NEC, Tokyo, Japan). The data were analyzed on a Macintosh computer with homemade software (TI WorkBench).
Fig. 1.
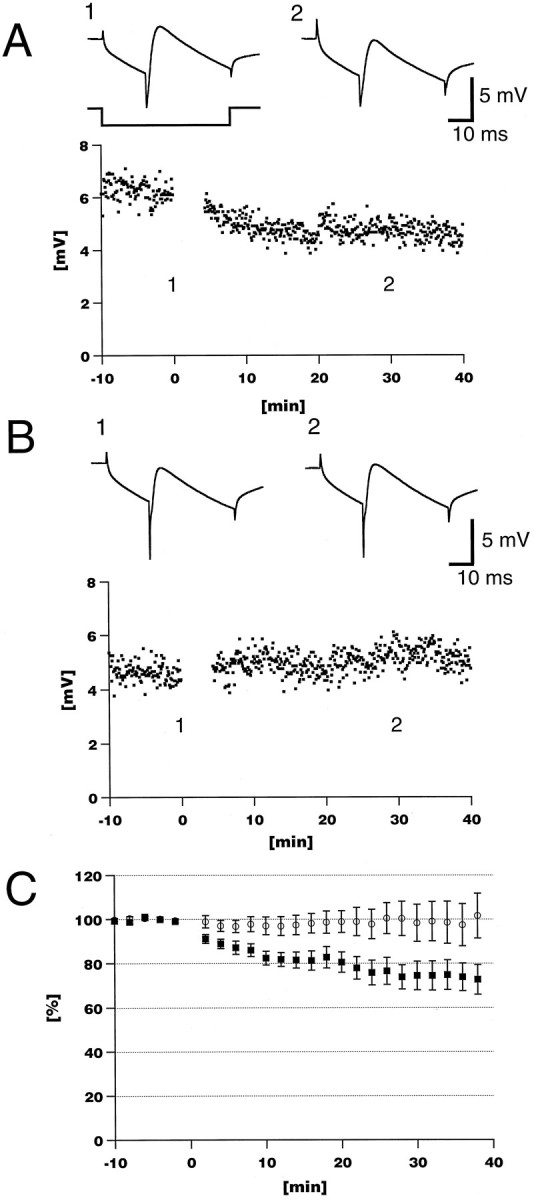
Loss of LTD in InsP3R1−/− Purkinje cells.A, Pairing depolarization and PF stimulation (1 Hz, 240 times) induced long-lasting depression of the PF-EPSP amplitude in control experiments using an InsP3R1+/+ cerebellar slice.B, LTD was lost in an InsP3R1−/− cerebellar slice.A, B, Insets show an average of 10 consecutive sweeps at time points indicated. Time course of hyperpolarizing current is also indicated at the bottom of the left insetin A. C, Averaged time course of normalized EPSP amplitude. LTD was not induced in InsP3R1−/− mice (○), whereas LTD was observed in control InsP3R+/+ mice (▪). Results are presented as mean ± SEM.
Calcium imaging in mutant cerebellar slices. Sagittally cut slices were used in ACSF-A. A patch pipette was filled with 400 μm Oregon Green 488 BAPTA-2 (Molecular Probes, Eugene, OR) and 200 μm 1-(2-nitrophenyl)ethyl (NPE)-caged inositol 1,4,5-trisphosphate (caged-InsP3; Molecular Probes) in internal solution. Fluorescence images (excitation at 470–490 nm; emission at 515–550 nm) were recorded with a cooled-CCD camera (PXL-37; Photometrics, Tucson, AZ) through a 60× water-immersion objective lens (NA 0.90; Olympus). The uncaging illumination for caged-InsP3 was provided by a pulsed laser source (λ = 337 nm, 20 Hz, 10 times) (VSL-337ND Nitrogen Laser; Laser Science, Newton, MA) through a quartz fiber light guide and the epi-fluorescence port of the upright microscope. The electrophysiological apparatus, the nitrogen pulse laser source, and the cooled-CCD camera were all controlled by and the data were recorded with TI WorkBench software running on a Power Macintosh 8500 (Apple Computer, Cupertino, CA). Ca2+transients were recorded by binned-pixel images (binning 10 × 10) at 12.5 frames/sec. To obtain the fluorescence amplitude (F) from each region of interest (ROI), pixel values in each region were averaged and a background level was subtracted from it. The background value was measured as an averaged value from a similar ROI in which the measured neuron was not included. The time course of the fluorescence change was plotted as a ratio,F/F0. The F value of each frame was divided by the value of the first frame (F0). The exposure time and neutral density filter were chosen to ensure that all pixel values were not saturated.
Ca imaging with antibodies. Experiments were performed as described in the previous section with the following differences. Transverse cerebellar slices were used in ACSF-B. mAb18A10 (160 μg/ml) or control rat IgG was added to the patch pipette solution.
Visualization of IgG penetration. Sagittally cut slices were used in ACSF-B. A patch pipette was filled with 2 mg/ml FITC-conjugated goat IgG in the patch pipette solution. Purkinje cells were voltage-clamped at −60 mV. Fluorescence images of FITC were taken with a cooled-CCD camera (PXL-37), with or without a confocal laser scanning unit (CSU10; Yokogawa Electric Corporation, Tokyo, Japan).
LTD experiments with antibodies. Experiments were performed as described in “LTD experiments in mutant Purkinje cells,” with the following differences. ACSF-B with 10 μm bicuculline was used as an external saline. During pairing periods, the CF was stimulated with an electrode in the granule cell layer in conjunction with PF stimulation. The Purkinje cell was held in current-clamp mode. Nonspecific rat IgG was purchased from Sigma (St. Louis, MO).
RESULTS
LTD was induced by pairing the depolarization of Purkinje cells with PF stimulation (240 times at 1 Hz) in the InsP3R1-deficient mouse experiments. LTD induction by pairing CF and PF stimulations was not used with the mutant mice for two reasons. First, we wanted to avoid any developmental changes at presynaptic and postsynaptic sites at the CF synapse; there was a difference in paired pulse depression of CF-mediated EPSCs in the InsP3R1−/− Purkinje cell (Matsumoto et al., 1996). Second, in young wild-type Purkinje cells (younger than 25 d old), LTD was not induced by pairing CF and PF stimuli (4 Hz, 480 times), which is one of the typical LTD-induction protocols (Inoue and Mikoshiba, unpublished observations). The InsP3R1-deficient mice do not survive beyond postnatal day 23 (Matsumoto et al., 1996). In wild-type (InsP3R1+/+) Purkinje cells, the amplitude of the PF-EPSP was reduced to 73.8 ± 16.5% of the control response 30 min after pairing (mean ± SD; n = 9 from eight animals) (Fig.1A,C). The initial slope of PF-EPSPs was also decreased without any significant variation in latency, in time to peak, and in the input resistance of Purkinje cells (Fig. 1A, insets). In contrast, in InsP3R1−/− Purkinje cells, the amplitude of the PF response was 100.2 ± 25.9% after the pairing (n = 11 from seven animals) (Fig. 1B,C) and was significantly different from InsP3R1+/+ animals (p < 0.05;t test) between 4–40 min after the pairing except at the 36 and 38 min time points. Thus, LTD was not induced in InsP3R1−/− Purkinje cells.
We have shown previously that InsP3R1 is functionally knocked out in the InsP3R1−/− cerebellum using an InsP3-binding assay and an InsP3-induced Ca2+ release (IICR) assay of microsome fractions from cerebellum (Matsumoto et al., 1996). To confirm these observations in living Purkinje cells, we performed the IICR assay in Purkinje cells in slices. Figure2C shows that an InsP3R1−/− Purkinje cell completely lacked IICR activity induced by release of caged-InsP3 (n = 5 for InsP3R1−/−; n= 4 for InsP3R1+/+). In contrast, there was no apparent difference in the time course of depolarization-induced Ca2+transients in the soma and dendritic regions between InsP3R1−/− and InsP3R1+/+ Purkinje cells (Fig. 2B). This result indicates that there was no apparent alteration in the plasma membrane Ca2+ channels and Ca2+ buffering mechanisms in the mutant mice, at least at a qualitative level. Ca2+ transients evoked by depolarizations were stronger at the soma than dendritic regions in both types of Purkinje cells. Because Ca2+ transients are stronger in dendritic regions than at somata when Ca spikes occur (Tank et al., 1988; Lev-Ram et al., 1992) (Inoue and Mikoshiba, unpublished observations), and because Ca spikes occur much less frequently in mouse Purkinje cells at ages when InsP3R−/− and control mice were used (18–23 d old) (Inoue and Mikoshiba, unpublished observations), we infer that the Ca2+ transients observed in Figure2B were not caused by Ca spikes. The fluorescence was attenuated in the distal dendrites of InsP3R1+/+ Purkinje cells and at the soma and dendrites of the InsP3R1−/− Purkinje cells (Fig.2C) after the UV pulses. In the proximal dendrite and soma of the InsP3R1+/+, this attenuation appeared to be hidden by IICR. Photo bleaching of the dye by the UV laser pulses, and not cell damage, was probably the cause of this attenuation, because caged-InsP3-induced Ca2+ release was observed repeatedly in InsP3R1+/+ Purkinje cells. In addition, depolarization-evoked Ca2+ transients (Fig. 2B) did not change after several UV pulses in both types of Purkinje cells. The attenuation by photo bleaching was not constant in different parts of a neuron, presumably because of the uneven efficacy of the UV flash. In particular, the fine dendrites in focus were effectively illuminated, but the soma was not because not all parts of the thick soma were in focus. This experiment shows that the InsP3R1−/− Purkinje cells were functionally unable to release Ca2+, although there was no obvious abnormality in the plasma membrane Ca2+ channels and the Ca2+buffering mechanisms.
Fig. 2.
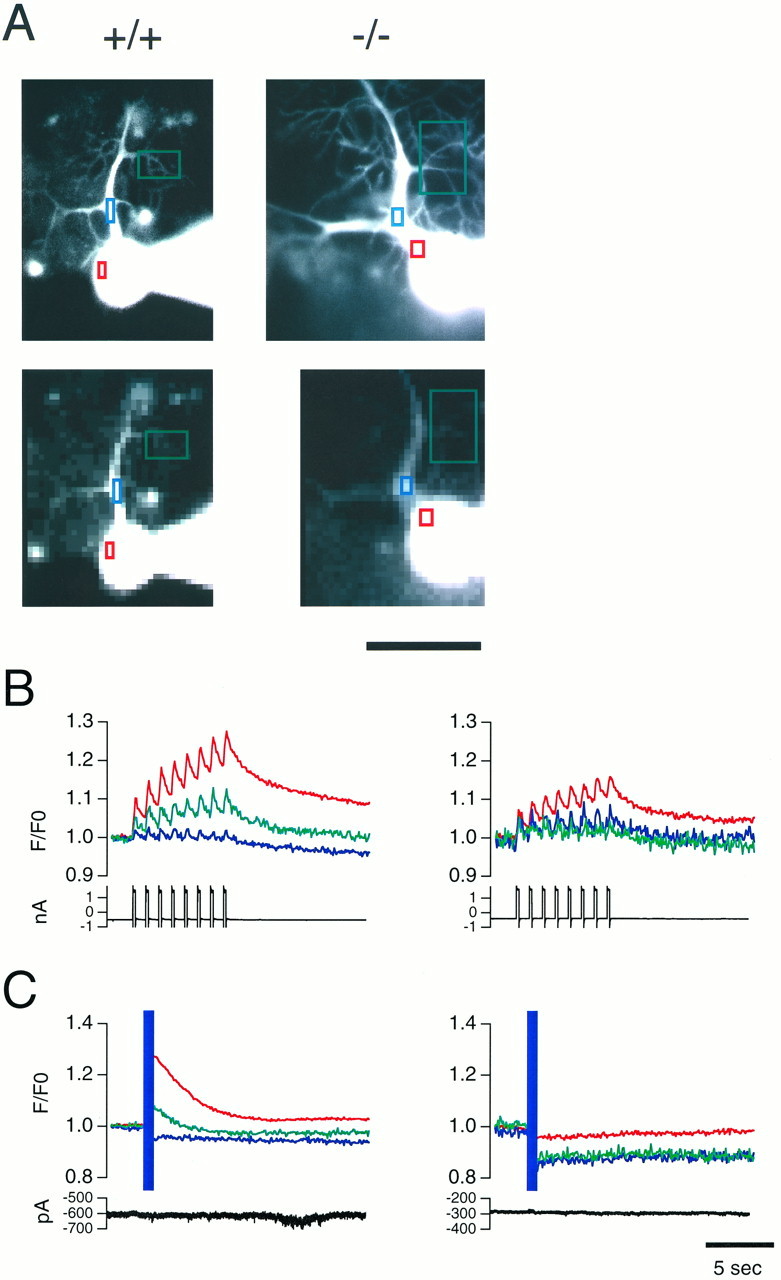
Calcium imaging shows normal Ca2+ transients evoked by depolarization and lack of InsP3-induced Ca2+ release in InsP3R−/− Purkinje cells. A, Morphology of InsP3R1+/+ and InsP3R1−/− Purkinje cells. Colored rectangles indicate regions where time courses of fluorescent changes were plotted. Top panels show fine structures of dendrites with high spatial resolution images. Bottom panels show actual images in which resolution changes in Ca2+ were measured (binning 10 × 10). Scale bar, 50 μm. B, Ca2+ transients were evoked by depolarization pulses. Purkinje cells from InsP3R1+/+ (left) and InsP3R1−/− (right) cerebellum were voltage-clamped at −70 mV and depolarizing pulses (−70–0 mV, 200 msec, 1 Hz, 8 times) were applied to the soma. Fluorescence intensities of indicatedrectangles were averaged, corrected for background, normalized to those from the first frame (resting level), and plotted in the same color as in A. Current traces are also shown at the bottom of the plots. Ca2+transients were observed at proximal (blue) and distal (green) dendritic regions as well as at the soma (red). C, Ca2+ release was induced by photolysis of caged-InsP3 in the InsP3R1+/+ Purkinje cell (left) by UV laser pulses (purple band), whereas no increase in [Ca2+]i was observed in the InsP3R1−/− Purkinje cell (right). Purkinje cells were voltage-clamped at −70 mV; current traces are also shown at the bottom of the plots. Data from the same cells are shown in A–C. Calibration bar, 5 sec.
Several lines of evidence suggest that the observed lack of LTD in InsP3R1−/− Purkinje cells is a direct consequence of the gene knockout rather than an indirect developmental effect. There was no difference in the input resistance between the two types of Purkinje cells (InsP3R1+/+, 180 ± 71 MΩ, n = 17; InsP3R1−/−, 180 ± 57 MΩ, n = 12). In addition, other electrophysiological characteristics of InsP3R−/− Purkinje cells, such as the complex of Na and Ca spikes, paired-pulse facilitation of PF-EPSC, and pharmacological profiles of the PF and CF synapses were indistinguishable from wild-type Purkinje cells (Matsumoto et al., 1996). There were no abnormalities in the morphology of Purkinje cells in InsP3R1−/− mice at the light microscopic level (Matsumoto et al., 1996). The expression levels of mGluR1 and mGluR5, both of which are linked to InsP3 production, were not altered in the InsP3R1−/− cerebellum (E. Nagata, personal communication). These observations strongly suggest that the lack of LTD in InsP3R1−/− Purkinje cells is a direct result of the lack of the InsP3R1 function.
To further rule out the possibility of an indirect effect of the gene knockout, we conducted a second set of experiments using the monoclonal antibody 18A10 (mAb18A10), which is a potent and selective blocker of InsP3R1 in vitro (Nakade et al., 1991), in hamster oocytes (Miyazaki et al., 1992) and in a gastric epithelial cell line (Hamada et al., 1993). We confirmed this activity of mAb18A10 in Purkinje cells in slices using caged-InsP3. The amplitude of Ca2+transients induced by releasing caged-InsP3 declined in parallel with the diffusion of 160 μg/ml mAb18A10 from the patch pipette, whereas they remained constant in a Purkinje cell loaded with 160 μg/ml of nonspecific rat IgG (Fig. 3A). Figure 3B shows the mean normalized changes in fluorescence at the soma. Five minutes after break-in, the change in fluorescence in Purkinje cells filled with mAb18A10 was 60% smaller than that with control IgG. The difference became larger and more significant at 25 and 35 min. Although we could detect Ca2+ transients in dendritic regions, the signals were too small to be analyzed quantitatively. Because the inhibitory potency of mAb18A10 depends on the amount of InsP3 (Nakade et al., 1991; Miyazaki et al., 1992), and because the amount of InsP3 produced during synaptic transmission is not known, the actual extent of inhibition of InsP3R1 by mAb18A10 in synaptic transmission could vary from the value obtained in this experiment.
Fig. 3.
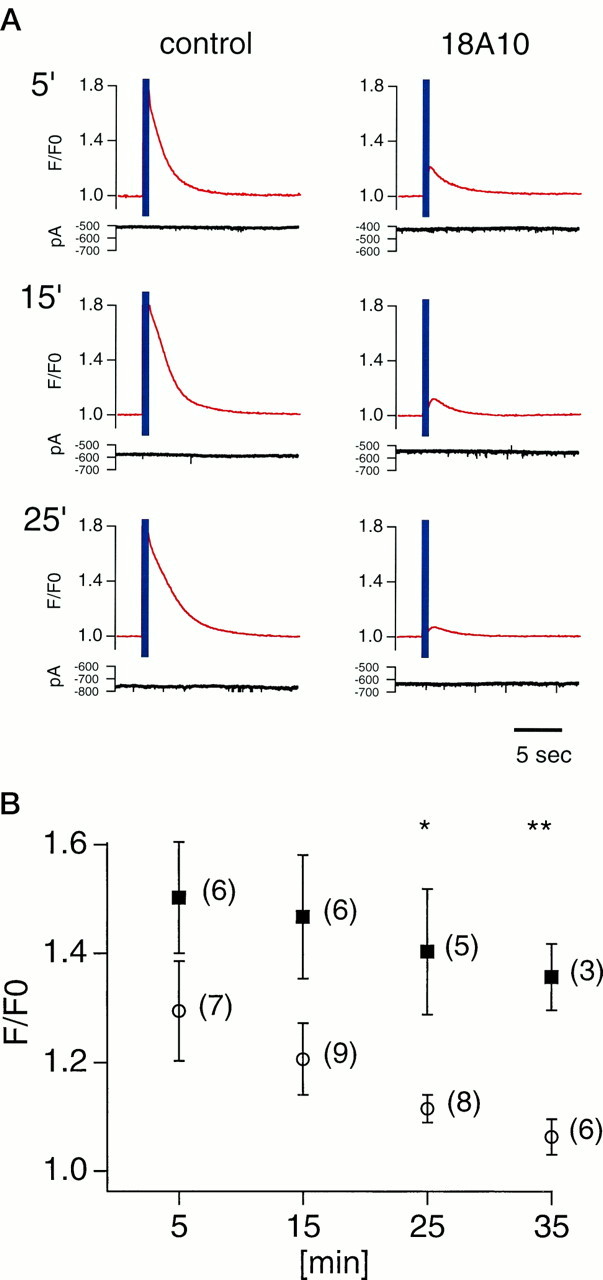
An InsP3R1-specific monoclonal antibody inhibited InsP3-induced Ca2+ release in Purkinje cells. A, UV illumination (purple band) evoked InsP3-induced Ca2+ release at the soma. The amplitude of caged-InsP3-induced Ca2+transients decreased in a mAb18A10-injected Purkinje cell, whereas it did not change in a Purkinje cell loaded with control IgG. Traces were recorded at time points indicated after whole-cell patch recording was started. Fluorescence intensities were normalized to those from the first frame after subtraction of background. MAb18A10 and control IgG diffused into the cells from patch pipettes. Purkinje cells were voltage-clamped at −70 mV; current traces are also shown at thebottom of the plots. B, Averaged result of caged-InsP3-induced Ca2+ release in Purkinje cells loaded with mAb18A10 (○) and control IgG (▪). Normalized changes in fluorescence at the soma were averaged and plotted against time after whole-cell recording was started. A single asterisk indicates p < 0.05, and adouble asterisk indicates p < 0.01 (t test). Numbers beside plot marks indicate number of cells tested.
To estimate the diffusion time of these IgGs in Purkinje cell dendrites, we investigated the migration of FITC-conjugated IgG delivered by patch pipette. Figure4A shows images obtained with a confocal unit and a CCD camera, and Figure4B shows images obtained without a confocal unit. Confocal imaging removed interference by high background fluorescence from the surface of the slice attributable to leakage of FITC-labeled IgG from the patch pipette while approaching the Purkinje cell, especially at early time points (Fig. 4, compareA,B). However, longer recordings were preferentially performed with a conventional CCD imaging system because of lower photobleaching of the fluorescence. The fluorescence intensity at secondary and tertiary dendrites (arrowheads) was detected as early as 3.5 min (Fig. 4A) and did not increase much further beyond 15 min after breaking into the cell (Fig. 4B). These results suggest that IgGs loaded from patch pipettes can reach dendritic regions within 10–20 min.
Fig. 4.

Penetration of IgG into Purkinje cells from patch pipette. Purkinje cells were labeled with FITC-labeled goat IgG. Theabscissa indicates the time after break-in. The IgG reached the secondary and tertiary dendritic regions (arrowheads) of the Purkinje cell within 3.5 min after patch formation (A), and the fluorescence intensity did not increase much more after the 15 min time point. Each set of images was taken and displayed with the same exposure and display conditions in A and B. A confocal laser scanning unit was used in A but was not used inB. Scale bar, 50 μm.
Figure 5 shows LTD experiments using mAb18A10. In this set of experiments, we used pairing of PF and CF stimuli as the LTD-inducing protocol to more closely approach in vivo conditions. In control experiments using nonspecific rat IgG or no IgG, LTD was induced by pairing PF and CF stimulation 480 times at 4 Hz (Fig. 5A,C). The average normalized amplitude of the PF-EPSPs was 78.3 ± 16.3% (n = 11 from eight animals) and 76.6 ± 18.7% (n = 10 from seven animals) 30 min after the pairing in the presence of 160 μg/ml nonspecific rat IgG and no IgG, respectively. In contrast, when 160 μg/ml mAb18A10 was used, no LTD was induced by the same pairing protocol (98.1 ± 21.8%, n = 11 from nine animals) (Fig. 5B,C). The difference between mAb18A10 and nonspecific rat IgG was significant (p < 0.05,t test) at all times between 4 and 40 min after the pairing except at the 10 min time point. The paired stimulation was started 15–20 min after break-in to ensure that the IgG reached the dendritic region of the Purkinje cell. Neither nonspecific rat IgG nor mAb18A10 altered PF-EPSPs without paired CF stimulation (Fig. 5D). Thus, 18A10 specifically blocked the induction of LTD, confirming that functional InsP3R1 is necessary for the induction of LTD.
Fig. 5.
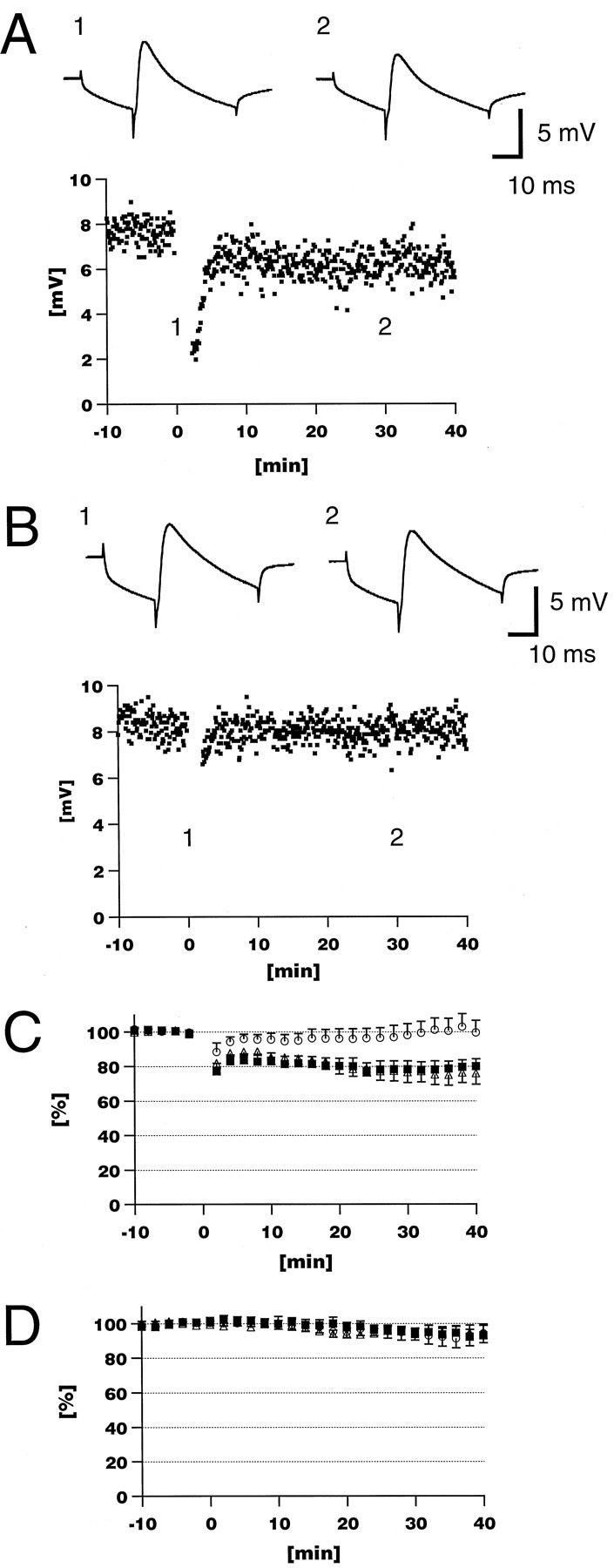
An InsP3R1-specific monoclonal antibody blocked induction of LTD. A, Paired CF and PF stimulation (4 Hz, 480 times) induced long-lasting depression of the PF-EPSP amplitude in a control experiment in which nonspecific rat IgG was included in the patch pipette (160 μg/ml). B, mAb18A10 blocked the induction of LTD. In insets, the averages of 10 consecutive sweeps obtained at time points indicated are shown.C, The averaged time course of the normalized EPSP amplitude, indicating that mAb18A10 (160 μg/ml) blocked induction of LTD (○), whereas LTD was induced in the presence of nonspecific rat IgG (160 μg/ml; ▪) and without IgG (▵). D, Without the pairing protocol, the EPSP was stable with mAb18A10 (160 μg/ml; ○; n = 8 from 5 animals) and nonspecific rat IgG (160 μg/ml; ▪; n = 5 from 4 animals) as well as without IgG (▵; n = 9 from 9 animals).
DISCUSSION
The initial step in the induction of LTD is considered to be the temporal overlap of the large elevation of [Ca2+]i caused by the CF input with activation of postsynaptic glutamate receptors at the PF synapse. Although the large [Ca2+]i increase has been thought to be mediated through VGCCs opened by the CF-induced depolarization (Tank et al., 1988; Sakurai, 1990; Crepel and Jaillard, 1991; Konnerth et al., 1992;Lev-Ram et al., 1992; Miyakawa et al., 1992), our results indicate that the increase in [Ca2+]i caused by the release of intracellular Ca2+ by InsP3R plays a specific role in the induction of LTD. Because Ca2+ influx through VGCCs occurs at the plasma membrane, and because Ca2+ is an intracellular signal with a short-acting range attributable to intracellular Ca2+ buffers (Allbritton et al., 1992; Kasai and Petersen, 1994), Ca2+ released from intracellular pools through InsP3R may reach different regions than those affected by the Ca2+ influx mediated through VGCCs. In addition to spatial differences, the two types of [Ca2+]i regulatory mechanisms may differ temporally. Thus, InsP3R1 may mediate spatiotemporal-specific Ca2+ signals that are essential for the induction of LTD.
IICR could be modulated by the high [Ca2+]i resulting from Ca2+ influx. The activity of InsP3R1 is sensitive to changes in [Ca2+]i in a biphasic manner (Bezprozvanny et al., 1991). The InsP3R1 channel could be activated by elevated [Ca2+]i even at the resting InsP3 concentration, which would boost [Ca2+]i elevation further. On the other hand, Khodakhah and Ogden (1995)reported that IICR was inhibited by high [Ca2+]i in the Purkinje cell, suggesting that there could be a negative interaction between [Ca2+]i and the IICR activity. The details of the [Ca2+]i dynamics during the pairing stimulation, especially in fine dendritic regions including the spine, remain to be elucidated.
We reported previously that the activation of ryanodine receptors, another type of Ca2+ channel located on intracellular Ca2+ stores, is necessary for the induction of culture-LTD (Kohda et al., 1995). Because ryanodine receptors are also functionally expressed in Purkinje cells (Ellisman et al., 1990; Kuwajima et al., 1992; Llano et al., 1994), they could play a role in the LTD mechanism in slices, simply being triggered by high [Ca2+]i coming through VGCCs independent of InsP3R. Alternatively, they could be functioning in concert with InsP3R; Ca2+ released from InsP3R could stimulate the ryanodine receptor.
The role of InsP3R in culture LTD remains unclear. Although Kasono and Hirano (1995) reported that heparin blocked the induction of culture-LTD, the concentration of heparin in their study (2.5 mg/ml) was sufficient to inhibit other cellular components, including PKC and cAMP-dependent protein kinase (Herbert and Maffrand, 1991). In addition, Narasimhan and Linden observed that xestospongin C, a potent antagonist of InsP3R (Gafni et al., 1997), did not block culture-LTD, whereas heparin did (Narasimhan and Linden, 1997; K. Narasimhan and D. Linden, personal communication). These data suggest that InsP3R may not be needed in the induction of culture-LTD, whereas it is needed in LTD in slices. In the culture-LTD protocol, Ca2+ influx through VGCCs, and possibly Ca2+ release from the ryanodine-sensitive stores, might be sufficient to induce LTD. However, in the present study in slices, Ca2+ from the InsP3-operated intracellular store was also necessary for LTD. Other differences between the culture and slice LTD systems are known (e.g., a requirement for nitric oxide) (for review, see Linden, 1994). Relevant to these experiments are differences in the anatomical and electrical geometry of the dendrites, density and distribution pattern of VGCCs, and properties of InsP3R. The most important difference may be whether the phenomenon occurs at a synapse. LTD in slices takes place at real PF-Purkinje cell synapses, whereas in culture-LTD, PF stimulation is replaced by artificial glutamate application. Our results in slices may be more relevant and imply that InsP3R1 may play an important role in Purkinje cells in vivo.
Hemart et al. (1995) reported that thapsigargin, which inhibits intracellular Ca2+ release by blocking intracellular Ca-ATPases, did not block the induction of LTD in slices. In their LTD induction protocol, PF stimulation (1 Hz) was paired with Ca spike firing evoked by continuous depolarization for 1 min. The discrepancy between their observations and the data presented here may be explained by differences in experimental conditions. During their pairing protocol, continuous Ca spike firing might keep [Ca2+]i at a higher level than the protocols used in this study. Periodic depolarizations for 200 msec at 1 Hz (Fig. 1; in experiments using mutant mice) would cause less Ca2+ influx than the continuous depolarization protocol. In addition, as described previously, the less frequent occurrence of Ca spikes in young mouse Purkinje cells might lead to less Ca2+ influx than continuous Ca spike firing. CF stimuli at 4 Hz (Fig. 5; in experiments using antibodies) would also load less Ca2+ than the Ca spike-firing protocol, because the frequency of Ca spike firing induced by current injection (range, 6–15 Hz) (Llinas and Sugimori, 1980; Lev-Ram et al., 1992) (Inoue and Mikoshiba, unpublished observations) is considerably higher. A single CF stimulus would cause a [Ca2+]i increase similar to that of a single Ca spike (Lev-Ram et al., 1992). Thus, the Ca2+ released by InsP3R1, which was required in this study, could be supplemented by such a high level of [Ca2+)i. The induction conditions for LTD used in the present study appear to be less artificial than the conditions thatHemart et al. (1995) adopted, because continuous Ca spike bursting for 1 min is unlikely to occur in vivo (Armstrong and Rawson, 1979).
In conclusion, our findings clearly demonstrate that intracellular Ca2+ release through the InsP3R1 channel plays an essential role in the induction of LTD in Purkinje cells in slices.
Footnotes
This work was supported by grants from the Ministry of Education, Science and Culture of Japan (T.I., K.M.). We thank D. Linden and E. Nagata for valuable information; M. Yuzaki, W. N. Ross, F. Crepel, T. Furuichi, L. G. Sayers, M. Kessler, and A. Arai for critical reading of this manuscript; H. Miyakawa for valuable discussions; T. Michikawa and A. Takahashi for preparing mAb18A10; and A. Hoshino, W. Saikawa, and M. Saito for technical assistance.
Correspondence should be addressed to Dr. Takafumi Inoue, Department of Molecular Neurobiology, The Institute of Medical Science, The University of Tokyo, 4-6-1 Shirokane-dai, Minato-ku, Tokyo-108, Japan.
Dr. Kohda’s present address: Department of Developmental Neurobiology, St. Jude Children’s Research Hospital, Memphis, TN 38105.
REFERENCES
- 1.Aiba A, Kano M, Chen C, Stanton ME, Gregory DF, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- 2.Allbritton NL, Meyer T, Stryer L. Range of messenger action of calcium ion and inositol 1,4,5-trisphosphate. Science. 1992;258:1812–1815. doi: 10.1126/science.1465619. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong DM, Rawson JA. Activity patterns of cerebellar cortical neurones and climbing fibre afferents in the awake cat. J Physiol (Lond) 1979;289:425–448. doi: 10.1113/jphysiol.1979.sp012745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 5.Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- 6.Chen C, Kano M, Abeliovich A, Chen L, Bao S, Kim JJ, Hashimoto K, Thompson RF, Tonegawa S. Impaired motor coordination correlates with persistent multiple climbing fiber innervation in PKCγ mutant mice. Cell. 1995;83:1233–1242. doi: 10.1016/0092-8674(95)90148-5. [DOI] [PubMed] [Google Scholar]
- 7.Conquet F, Bashir ZI, Davies CH, Daniel H, Ferraguti F, Bordi F, Franz-Bacon K, Reggiani A, Matarese V, Conde F, Collingridge GL, Crepel F. Motor deficit and impairment of synaptic plasticity in mice lacking mGluR1. Nature. 1994;372:237–243. doi: 10.1038/372237a0. [DOI] [PubMed] [Google Scholar]
- 8.Crepel F, Jaillard D. Pairing of pre- and postsynaptic activities in cerebellar Purkinje cells induces long-term changes in synaptic efficacy in vitro. J Physiol (Lond) 1991;432:123–141. doi: 10.1113/jphysiol.1991.sp018380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crepel F, Krupa M. Activation of protein kinase C induces a long-term depression of glutamate sensitivity of cerebellar Purkinje cells. An in vitro study. Brain Res. 1988;458:397–401. doi: 10.1016/0006-8993(88)90486-6. [DOI] [PubMed] [Google Scholar]
- 10.Ellisman MH, Deerinck TJ, Ouyang Y, Beck CF, Tanksley SJ, Walton PD, Airey JA, Sutko JL. Identification and localization of ryanodine binding proteins in the avian central nervous system. Neuron. 1990;5:135–146. doi: 10.1016/0896-6273(90)90304-x. [DOI] [PubMed] [Google Scholar]
- 11.Furuichi T, Yoshikawa S, Miyawaki A, Wada K, Maeda N, Mikoshiba K. Primary structure and functional expression of the inositol 1,4,5-trisphosphate-binding protein P400. Nature. 1989;342:32–38. doi: 10.1038/342032a0. [DOI] [PubMed] [Google Scholar]
- 12.Furuichi T, Simon-Chazottes D, Fujino I, Yamada N, Hasegawa M, Miyawaki A, Yoshikawa S, Guenet J-L, Mikoshiba K. Widespread expression of inositol 1,4,5-trisphosphate receptor type 1 gene (Insp3r1) in the mouse central nervous system. Receptors Channels. 1993;1:11–24. [PubMed] [Google Scholar]
- 13.Gafni J, Munsch JA, Lam TH, Catlin MC, Costa LG, Molinski TF, Pessah IN. Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron. 1997;19:723–733. doi: 10.1016/s0896-6273(00)80384-0. [DOI] [PubMed] [Google Scholar]
- 14.Hamada E, Nakajima T, Ota S, Terano A, Omata M, Nakade S, Mikoshiba K, Kurachi Y. Activation of Ca2+-dependent K+ current by acetylcholine and histamine in a human gastric epithelial cell line. J Gen Physiol. 1993;102:667–692. doi: 10.1085/jgp.102.4.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartell NA. cGMP acts within cerebellar Purkinje cells to produce long term depression via mechanisms involving PKC and PKG. NeuroReport. 1994a;5:833–836. doi: 10.1097/00001756-199403000-00024. [DOI] [PubMed] [Google Scholar]
- 16.Hartell NA. Induction of cerebellar long-term depression requires activation of glutamate metabotropic receptors. NeuroReport. 1994b;5:913–916. doi: 10.1097/00001756-199404000-00015. [DOI] [PubMed] [Google Scholar]
- 17.Hemart N, Daniel H, Jaillard D, Crepel F. Receptors and second messengers involved in long-term depression in rat cerebellar slices in vitro: a reappraisal. Eur J Neurosci. 1995;7:45–53. doi: 10.1111/j.1460-9568.1995.tb01019.x. [DOI] [PubMed] [Google Scholar]
- 18.Herbert J-M, Maffrand J-P. Effect of pentosan polysulphate, standard heparin and related compounds on protein kinase C activity. Biochim Biophys Acta. 1991;1091:432–441. [PubMed] [Google Scholar]
- 19.Inoue T, Mikoshiba K. Type 1 inositol 1,4,5-trisphosphate receptor is essential in long-term depression in cerebellar Purkinje neuron. Soc Neurosci Abstr. 1997;23:1965. doi: 10.1523/JNEUROSCI.18-14-05366.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito M. Long-term depression. Annu Rev Neurosci. 1989;12:85–102. doi: 10.1146/annurev.ne.12.030189.000505. [DOI] [PubMed] [Google Scholar]
- 21.Kasai H, Petersen OH. Spatial dynamics of second messengers: IP3 and cAMP as long-range and associative messengers. Trends Neurosci. 1994;17:95–101. doi: 10.1016/0166-2236(94)90112-0. [DOI] [PubMed] [Google Scholar]
- 22.Kasono K, Hirano T. Involvement of inositol trisphosphate in cerebellar long-term depression. NeuroReport. 1995;6:569–572. doi: 10.1097/00001756-199502000-00040. [DOI] [PubMed] [Google Scholar]
- 23.Kohda K, Inoue T, Mikoshiba K. Ca2+ release from Ca2+ stores, particularly from ryanodine-sensitive Ca2+ stores, is required for the induction of LTD in cultured cerebellar Purkinje cells. J Neurophysiol. 1995;74:2184–2188. doi: 10.1152/jn.1995.74.5.2184. [DOI] [PubMed] [Google Scholar]
- 24.Khodakhah K, Armstrong C. Induction of long-term depression and rebound potentiation by inositol trisphosphate in cerebellar Purkinje neurons. Proc Natl Acad Sci USA. 1997;94:14009–14014. doi: 10.1073/pnas.94.25.14009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khodakhah K, Ogden D. Functional heterogeneity of calcium release by inositol trisphosphate in single Purkinje neurones, cultured cerebellar astrocytes, and peripheral tissues. Proc Natl Acad Sci USA. 1993;90:4976–4980. doi: 10.1073/pnas.90.11.4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khodakhah K, Ogden D. Fast activation and inactivation of inositol trisphosphate-evoked Ca2+ release in rat cerebellar Purkinje neurones. J Physiol (Lond) 1995;487:343–358. doi: 10.1113/jphysiol.1995.sp020884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Konnerth A, Dreessen J, Augustine GJ. Brief dendritic calcium signals initiate long-lasting synaptic depression in cerebellar Purkinje cells. Proc Natl Acad Sci USA. 1992;89:7051–7055. doi: 10.1073/pnas.89.15.7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuwajima G, Futatsugi A, Niinobe M, Nakanishi S, Mikoshiba K. Two types of ryanodine receptors in mouse brain: skeletal muscle type exclusively in Purkinje cells and cardiac muscle type in various neuron. Neuron. 1992;9:1133–1142. doi: 10.1016/0896-6273(92)90071-k. [DOI] [PubMed] [Google Scholar]
- 29.Lev-Ram V, Miyakawa H, Lasser-Ross N, Ross WN. Calcium transients in cerebellar Purkinje neurons evoked by intracellular stimulation. J Neurophysiol. 1992;68:1167–1177. doi: 10.1152/jn.1992.68.4.1167. [DOI] [PubMed] [Google Scholar]
- 30.Linden DJ. Long-term synaptic depression in the mammalian brain. Neuron. 1994;12:457–472. doi: 10.1016/0896-6273(94)90205-4. [DOI] [PubMed] [Google Scholar]
- 31.Linden DJ, Connor JA. Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science. 1991;254:1656–1659. doi: 10.1126/science.1721243. [DOI] [PubMed] [Google Scholar]
- 32.Llano I, Dreessen J, Kano M, Konnerth A. Intradendritic release of calcium induced by glutamate in cerebellar Purkinje cells. Neuron. 1991;7:577–583. doi: 10.1016/0896-6273(91)90370-f. [DOI] [PubMed] [Google Scholar]
- 33.Llano I, DiPolo R, Marty A. Calcium-induced calcium release in cerebellar Purkinje cells. Neuron. 1994;12:663–673. doi: 10.1016/0896-6273(94)90221-6. [DOI] [PubMed] [Google Scholar]
- 34.Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell somata in mammalian cerebellar slices. J Physiol (Lond) 1980;305:171–195. doi: 10.1113/jphysiol.1980.sp013357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsumoto M, Nakagawa T, Inoue T, Nagata E, Tanaka K, Takano H, Minowa O, Kuno J, Sakakibara S, Yamada M, Yoneshima H, Miyawaki A, Fukuuchi Y, Furuichi T, Okano H, Mikoshiba K, Noda T. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature. 1996;379:168–171. doi: 10.1038/379168a0. [DOI] [PubMed] [Google Scholar]
- 36.Miyakawa H, Lev-Ram V, Lasser-Ross N, Ross WN. Calcium transients evoked by climbing fiber and parallel fiber synaptic inputs in guinea pig cerebellar Purkinje neurons. J Neurophysiol. 1992;68:1178–1189. doi: 10.1152/jn.1992.68.4.1178. [DOI] [PubMed] [Google Scholar]
- 37.Miyazaki S, Yuzaki M, Nakada K, Shirakawa H, Nakanishi S, Nakade S, Mikoshiba K. Block of Ca2+ wave and Ca2+ oscillation by antibody to the inositol 1,4,5-trisphosphate receptor in fertilized hamster eggs. Science. 1992;257:251–255. doi: 10.1126/science.1321497. [DOI] [PubMed] [Google Scholar]
- 38.Nakade S, Maeda N, Mikoshiba K. Involvement of the C-terminus of the inositol 1,4,5-trisphosphate receptor in Ca2+ release analyzed using region-specific monoclonal antibodies. Biochem J. 1991;277:125–131. doi: 10.1042/bj2770125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Narasimhan K, Linden DJ. A test of the calcium threshold hypothesis for cerebellar LTD induction. Soc Neurosci Abstr. 1997;23:1178. [Google Scholar]
- 40.Sakurai M. Calcium is an intracellular mediator of the climbing fiber in induction of cerebellar long-term depression. Proc Natl Acad Sci USA. 1990;87:3383–3385. doi: 10.1073/pnas.87.9.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shigemoto R, Abe T, Nomura S, Nakanishi S, Hirano T. Antibodies inactivating mGluR1 metabotropic glutamate receptor block long-term depression in cultured Purkinje cells. Neuron. 1994;12:1245–1255. doi: 10.1016/0896-6273(94)90441-3. [DOI] [PubMed] [Google Scholar]
- 42.Tank DW, Sugimori M, Connor JA, Llinas RR. Spatially resolved calcium dynamics of mammalian Purkinje cells in cerebellar slice. Science. 1988;242:773–777. doi: 10.1126/science.2847315. [DOI] [PubMed] [Google Scholar]
- 43.Vranesic I, Batchelor A, Gahwiler BH, Garthwaite J, Staub C, Knopfel T. Trans-ACPD-induced Ca2+ signals in cerebellar Purkinje cells. NeuroReport. 1991;2:759–762. doi: 10.1097/00001756-199112000-00007. [DOI] [PubMed] [Google Scholar]