

Abstract
Induction of the mitochondrial permeability transition (MPT) has been implicated in cellular apoptosis and in ischemia–reperfusion injury. During MPT, a channel in the inner mitochondrial membrane, the mitochondrial megachannel, opens and causes isolated mitochondria to swell. MPT and mitochondrial swelling is inhibited by cyclosporin A (CsA), which may also inhibit apoptosis in some cells.
Treatment with CsA (50 mg/kg, i.v.) showed a robust reduction of brain damage when administered 30 min before insulin-induced hypoglycemic isoelectricity of 30 min duration. Ultrastructural examination of the dentate gyrus revealed a marked swelling of dendrites and mitochondria during the hypoglycemic insult. In CsA-treated animals, mitochondria resumed a normal and contracted appearance during and after the hypoglycemic insult. Treatment with FK 506 (2 mg/kg, i.v.), a compound with immunosuppressive action similar to that of CsA, was not protective. Studies on the swelling kinetics of isolated mitochondria from the hippocampus showed that CsA, but not FK 506, inhibits calcium ion-induced MPT.
We conclude that CsA treatment during hypoglycemic coma inhibits the MPT and reduces damage and that mitochondria and the MPT are likely to be involved in the development of hypoglycemic brain damage in the rat.
Keywords: Cyclosporin A, hippocampal mitochondria, hypoglycemia, mitochondrial permeability transition, brain damage, cell death
When isolated mitochondria are exposed to high concentrations of calcium ions, a mitochondrial megachannel (MMC) opens in the inner mitochondrial membrane (Haworth and Hunter, 1979). This event, also named the mitochondrial permeability transition (MPT), is considered to be an early event in apoptosis in some cells (Marchetti et al., 1996a; Kroemer, 1997) and possibly a trigger of cell death in ischemia–reperfusion damage (Griffiths and Halestrap, 1993a; Uchino et al., 1995).
Cyclosporin A (CsA) and FK 506 are immunosuppressive compounds, believed to exert their action through binding to small intracellular regulatory proteins, the cyclophilins (CyPs) or FK binding proteins (FKBPs), also known as immunophilins (Galat and Metcalfe, 1995). When complexed with CsA and FK 506, the properties of the immunophilins change, leading for example to the inhibition of the phosphatase calcineurin (Liu et al., 1991) and subsequent inhibition of T-cell activation (Clipstone and Crabtree, 1992; O’Keefe et al., 1992).
Cyclosporin A, but not FK 506, prevents MPT by blocking translocation of the mitochondrial matrix-specific cyclophilin-D (CyP-D) to the inner membrane of liver mitochondria, thereby decreasing MMC sensitivity to calcium ions (Connern and Halestrap, 1994). Several other modulators of the MMC have been characterized in liver and heart mitochondria, among them free radicals, adenine nucleotide depletion, a low mitochondrial membrane potential, and increased matrix pH, which all stimulate the calcium-induced MPT, whereas decreasing the matrix pH below 7 is inhibitory (Halestrap, 1991; Bernardi et al., 1992; Zoratti and Szabo, 1995).
Although MPT is clearly involved in apoptosis in thymocytes (Marchetti et al., 1996b), its role in neuronal death attributable to energy shortage is still elusive. Early work by Shiga et al. (1992) showed brain protection by CsA in a model of focal ischemia. In other studies, FK 506, which readily penetrates the blood–brain barrier (BBB), and high doses of CsA showed a significant reduction of infarct volume in a model of endothelin-induced focal ischemia (Sharkey and Butcher, 1994;Butcher et al., 1997). In a rat model of global brain ischemia, FK 506 is neuroprotective when administered before the insult (Drake et al., 1996). Protection by FK 506 was also attained when administered after global ischemia in the gerbil, which may have been caused partially by hypothermia induced by the drug (Ide et al., 1996). Cyclosporin A, on the other hand, which has a limited ability to penetrate an undamaged BBB (Begley et al., 1990), has been shown to protect from damage after global ischemia only if combined with a needle lesion into the brain parenchyma (Uchino et al., 1995).
Severe insulin-induced hypoglycemia causes selective neuronal damage to certain brain regions, such as the outer layers of the cortex, the striatum, the medial CA1 region, and the crest of the dentate gyrus (Auer and Siesjo, 1993). Hypoglycemia is associated with severe energy failure and loss of ion homeostasis, which includes a massive influx of calcium ions into brain cells (Siesjo and Bengtsson, 1989). In contrast to ischemia, hypoglycemia is not accompanied by acidosis but rather by a mild alkalosis, because of reduced glycolytic flux and combustion of glycolytic metabolites in the tricarboxylic cycle. Early ultrastructural changes associated with the hypoglycemic insult include axon-sparing dendritic lesions with swollen dendrites and mitochondria (Auer et al., 1985). During hypoglycemic coma, conditions apparently favor the activation of MPT.
This study was performed to determine whether there are ultrastructural signs of MPT during hypoglycemia. Also, we aimed to assess whether CsA prevents calcium-induced swelling of isolated hippocampal mitochondria and whether treatment with CsA protects against hypoglycemic brain damage in vivo.
MATERIALS AND METHODS
Animal surgery. All animal experiments were approved by the ethical committee at the University of Lund. Male Wistar rats from Moellegard avlslaboratorium (Copenhagen, Denmark), weighing 300–340 gm, were fasted overnight with access to water. One hour before surgery, the rat received an intraperitoneal injection of 2 IU/kg of insulin (Actrapid, Novo Nordisk A/S, Copenhagen, Denmark). Induction of anesthesia was performed with 3% halothane in a 70:30 mixture of nitrous oxide and oxygen. The animal was intubated with polyethylene tubing and connected to a small animal respirator. Halothane concentration was reduced and kept at 1.5% during surgery. For glucose infusion, a venous line from a tail vein was used, and for blood sampling and recording of the mean arterial blood pressure (MABP), an arterial line from a tail artery was used. The jugular vein was exposed, and a soft SILASTIC catheter was placed into the inferior vena cava to control blood pressure by exsanguination or reinfusion of blood. After the surgical procedure, the halothane concentration was lowered to 0.5%, and the animals were allowed a steady-state period of ∼30 min before the induction of hypoglycemia. Heparin (50 IU) was given as an anticoagulant, temperature and MABP were measured continuously, and blood gases were controlled. An infusion of vecuronium (Norcuron, Organon Teknika, Amsterdam, The Netherlands) in Krebs’ solution was given. The arterial PCO2was adjusted to 35–40 mmHg, and the rectal temperature was kept at 37°C by external heating. Two subcutaneous biparietal needle electrodes were used to record a bipolar EEG, which was monitored continuously. At the onset of cerebral isoelectricity, blood pressure rose, and blood was therefore drawn from the central venous catheter to keep the blood pressure between 140 and 160 mmHg during the entire isoelectric period. Isoelectricity was terminated as 0.2 ml of 50% glucose was given at a rate of 1.5 ml/hr, followed by the infusion of 1:1 solution of 50% glucose and Krebs’ solution given at 0.5–1.0 ml/hr to maintain plasma glucose levels between 5 and 10 mmol/l. The animal was extubated by the time it recovered spontaneous respiration and made active movements to remove the endotracheal tube. Before the tail catheters were removed, blood glucose and blood gases were checked in the awake, conscious animal. The animals were allowed access to water and food pellets in the cage after surgery. Rectal temperature was monitored daily.
Treatment paradigms. Cyclosporin A, 50 mg/ml (Sandimmune), was diluted six times in NaCl. FK 506 (10 mg) was dissolved in 0.5 ml of a 9:1 mixture of ethanol/Tween 80 and further diluted once in water, yielding a 10 mg/ml stock solution. The vehicle for CsA and FK 506 was the same 9:1 mixture of ethanol/Tween 80, diluted once in H2O, and further diluted six times in NaCl, yielding a 7.5% ethanol, 0.8% Tween 80 solution.
Animals prepared for histopathological investigation were divided into six groups. Cyclosporin A [20 mg/kg (n = 6)], cyclosporin A [50 mg/kg (n = 8)], and vehicle (n = 8) were given intravenously as a single dose ∼30 min before the onset of isoelectric EEG. FK 506 [2 mg/kg (n = 4)] and vehicle (n = 4) were given intravenously as a single dose in the same manner. Finally, sham-operated control rats were given glucose after insulin administration to maintain a plasma glucose level between 5 and 10 mmol/l (n = 4).
Histopathology. After 1 week, animals were again anesthetized and perfused with a 4% formaldehyde solution. Brains were allowed to post-fix and later were sectioned into 2.8-mm-thick slices and processed in graded ethanol. They were embedded in paraffin, sectioned at 6 μm, and finally stained with 1% acid fuchsin and 0.05% toluidine blue. Damage in the dorsal dentate gyrus was assessed by a graded damage score: 0 = no damage, 1 = damage to the tip of the dentate gyrus, 2 = damage extended to half the ventral or dorsal blades of the dentate gyrus, and 3 = damage to the entire dentate gyrus. Damage in the CA1 region was determined at the same level as the dentate gyrus in a visual field of 400 μm diameter, using the following damage score scale: 0 = no damage, 1 = <30% damage, 2 = 30–60% damage, and 3 = 60–100% damage. Damage in the parietal cortex was assessed by neuronal counting in an area 400 μm wide, on the same brain slice where damage to the dentate gyrus was assessed. Striatal damage was assessed using the following damage score: 0 = no damage, 1 = scattered damage to the dorsal crest of the striatum, 2 = damage to the dorsal and ventral crest of the striatum, and 3 = dense damage to the ventral, dorsal, and medial striatum. Damage was assessed in two consecutive sections and is the mean of the two hemispheres. All histopathological assessments were performed by an observer blinded to treatment conditions.
Electron microscopy. Animals prepared for the electron microscopy (EM) study were divided into five groups. Cyclosporin A (50 mg/kg), its vehicle, or FK 506 (2 mg/kg) was given intravenously ∼30 min before the onset of isoelectric EEG, in the same manner as for animals prepared for the histopathological investigation. Animals were killed at either 30 min of isoelectric hypoglycemic coma [CsA (n = 4), vehicle (n = 4), FK 506 (n = 3)] or after 30 min of recovery after 30 min of hypoglycemic coma [CsA (n = 4), vehicle (n = 4)]. Animals were fixed with glutaraldehyde (2%) by intra-aortic perfusion, and brains were immediately removed and placed in fixation solution. The dentate gyrus was dissected and allowed to post-fix in glutaraldehyde solution for 1 hr, rinsed in Soerensen buffer overnight, and then further processed for EM.
To assess mitochondrial swelling at 30 min of isoelectric hypoglycemic coma, EM pictures of the distal stratum moleculare, superjacent to the crest of the dentate gyrus, were analyzed at 3800× magnification. The number of swollen mitochondria in an area measuring 26 μm × 26 μm was counted, and the mean from three areas calculated. We defined a swollen mitochondrion as being rounded, with a diameter >0.8 μm. This definition is based on the size of mitochondria in control animals and on data from the literature (Auer et al., 1985).
Measurements of cyclosporin A concentrations in vivo. Rats were given CsA [20 mg/kg (n = 4) or 50 mg/kg (n = 3)] intravenously, ∼30 min before the induction of hypoglycemic coma. Control animals (n = 6) that were not undergoing insulin-induced hypoglycemia were also given CsA (50 mg/kg, i.v.). The brains of three of the control animals were perfused with isotonic saline for 1 min through an intra-aortic catheter before decapitation. Blood samples were taken 45 min after the intravenous CsA injection, and the animals were then decapitated. The hippocampus (180 mg) was dissected out and homogenized in 350 μl of water. Samples were stored on ice and further analyzed for CsA content using a commercial EMIT kit (Behring Diagnostics, Cupertino, CA) on a Cobas Mira S equipment (Roche Products, Hertforshire, UK). Negative controls were rat blood and brain homogenate from animals not given CsA that showed CsA values of <50 ng/ml and 20 ng/ml, respectively. Addition of known amounts of CsA to brain homogenate yielded the expected concentrations. All samples were diluted with methanol to obtain values within the 100–500 ng/ml range.
Preparation of isolated mitochondria. Male Wistar rats (300–350 gm) were decapitated, and the brain were transferred to ice-cold isolation buffer (0.32 m sucrose, 2 mmEGTA, 10 mm Tris-base, pH 7.4). The hippocampus was dissected out from each hemisphere in ice-cold buffer in a Petri dish. Isolated mitochondria were prepared according to Sims, method B (Sims, 1990). Approximately 180 mg of hippocampal tissue was dissected out and homogenized in 12% Percoll, using a 2 ml Kontes Teflon homogenizer. The homogenate was added to a Percoll gradient, 40% and 26%, using a Pasteur pipette, and centrifuged according to published procedures (Sims, 1990).
Measurements of respiratory control ratio and MPT pore opening. Respiratory activity of mitochondrial preparations was measured by determining oxygen consumption in an airtight chamber in the presence of 5 mm malate and 5 mm glutamate and after the addition of ADP (Sims, 1990). Respiratory control ratio (RCR) was calculated as the ratio of state 3/state 4 respiration.
In subsequent preparations for mitochondrial swelling experiments, the final pellet was resuspended in isolation buffer and kept on ice. Measurements of the calcium-induced mitochondrial swelling under de-energized conditions were performed in a Perkin-Elmer (Emeryville, CA) fluorometer by measuring the decrease in light-scattering at 520 nm. A fixed volume (10 μl) of mitochondrial suspension was added to 1.1 ml of isotonic buffer, pH 7.0, containing 150 mm KCl, 20 mm Mops, 10 mm Tris, 2 mmnitrilotriacetic acid (NTA), 0.5 μm rotenone, 0.5 μm antimycin, and 2 μm A23187 calcium ionophore, added to ensure complete equilibration of calcium ions across the mitochondrial membrane under de-energized conditions (Halestrap, 1991). Calcium ion concentrations were calculated from the NTA buffering (Connern and Halestrap, 1994). Protein concentrations were determined using a Bio-Rad assay (Bio-Rad, Richmond, CA). The protein concentration in the cuvette was ∼25 μg/ml, and the experiments were run at a temperature of 26°C.
Calcium dose–response measurements were performed, and the initial swelling rate, excluding the first 10 sec attributable to mixing artifacts, was calculated as the decrease in light scattering per time unit and per microgram of protein. When the effect of CsA or FK 506 on mitochondrial swelling was studied, the compound was added at 200 nm final concentration, before the addition of calcium chloride (see Fig. 3).
Fig. 3.

Left, Calcium ion-induced de-energized mitochondrial swelling measured as a decrease in light scattering. Mitochondria were isolated from the rat hippocampus and added to isotonic KCl buffer. The addition of 100 μm Ca (arrow) initiated rapid swelling, whereas swelling decreased significantly when 200 nm CsA was present.Right, The effect of 20 and 100 μm calcium chloride on de-energized mitochondrial swelling, in the absence and presence of 200 nm CsA or 200 nm FK 506. Swelling was measured as the decrease in light scattering at 520 nm initiated by the addition of calcium ions. A numerical value was calculated from the initial swelling (absorbency/time unit) divided by protein concentration. ** denotes significant differences from untreated samples with p < 0.01; Dunnett’s test.
All chemicals were from Sigma (St. Louis, MO), and the Percoll solution was from Pharmacia and Upjohn (Bedminster, NJ). Cyclosporin A (Sandimmune) was from Novartis, and FK 506 (tacrolimus) was a gift from Fujisawa (Tokyo, Japan).
RESULTS
The effect of cyclosporin A and FK 506 on hypoglycemic brain damage
Plasma glucose levels before, during, and after the hypoglycemic insult were not significantly different between the groups (Table1). Neither was there any significant difference in the average time span between injection of drugs or vehicle and the initiation of isoelectric EEG (data not shown). Body temperature in the recovery phase was not significantly different between the experimental groups, although in the CsA-treated rats a tendency to a slightly lower temperature was seen (Table2).
Table 1.
Plasma glucose levels at different time points after the induction of insulin-induced hypoglycemic coma in animals given either cyclosporin A or its vehicle
| n | Isoelectricity (30 min) | Reperfusion (15 min) | Reperfusion (2 hr) | |
|---|---|---|---|---|
| Cyclosporin A (20 mg/kg) | 6 | 0.4 ± 0.2 | 8.5 ± 1.2 | 6.9 ± 2.0 |
| Cyclosporin A (50 mg/kg) | 8 | 0.4 ± 0.2 | 7.5 ± 1.2 | 6.9 ± 1.6 |
| Vehicle | 8 | 0.4 ± 0.3 | 6.8 ± 1.8 | 6.9 ± 1.4 |
Plasma glucose levels (mmol/l) are shown as mean ± SD. No significant differences were found between the groups at any time point.
Table 2.
Rat rectal temperature at different time points after insulin-induced hypoglycemic coma
| n | 1 hr | 1 d | 3 d | 5 d | |
|---|---|---|---|---|---|
| Cyclosporin A (20 mg/kg) | 6 | 37.0 ± 0.4 | 37.7 ± 0.5 | 37.7 ± 0.3 | 37.7 ± 0.2 |
| Cyclosporin A (50 mg/kg) | 6 | 37.1 ± 0.4 | 37.5 ± 0.5 | 37.4 ± 0.5 | 37.5 ± 0.3 |
| Vehicle | 6 | 36.9 ± 0.3 | 37.9 ± 0.4 | 37.9 ± 0.2 | 38.0 ± 0.9 |
Rats were given either cyclosporin A or its vehicle. No significant difference in temperature (°C) was seen between the treated groups at any time point.
There was a dose-dependent neuroprotective effect of CsA in the dentate gyrus and in the CA1 area of the hippocampus (Fig.1). In vehicle-treated rats, neuronal damage was seen in the outer layers of the cortex, the striatum, the medial CA1 region, and the dentate gyrus granule cells at 1 week after 30 min of hypoglycemic isoelectricity. In all eight rats treated with vehicle, damage to the crest and blades of the dentate gyrus was seen. Also, ∼50% of neurons in the CA1 region had succumbed. In the cortex, a mean of eight damaged neurons were counted, and in the striatum, damage to the ventral and dorsal part was quite extensive (damage score = 3.0). In all rats treated with 50 mg/kg CsA, no damage to the dentate gyrus or the cortex was observed. In the CA1 pyramidal neurons, <30% damage was noted, whereas in the striatum damage was less effectively diminished (damage score = 2.4). When rats were treated with 20 mg/kg CsA, a variable degree of neuroprotection was seen, whereas treatment with 2 mg/kg FK 506 did not yield protection in any brain region (Fig. 1).
Fig. 1.

Damage score in the dentate gyrus and in the hippocampus CA1 region in rats, assessed at 7 d of recovery after 30 min of hypoglycemic coma. Rats were treated with a single dose of CsA (CsA20; 20 mg/kg, i.v.), CsA (CsA50; 50 mg/kg, i.v.), FK 506 (FK506; 2 mg/kg, i.v.), or vehicle (Vh). There was a significant (p < 0.05) decrease in damage in both regions in animals treated intravenously with 50 mg/kg, when compared with the control group. Kruskal–Wallis test.
Figure 2 shows a photomicrograph of the dentate gyrus of a sham-operated rat (Fig. 2A), a rat treated with vehicle (Fig. 2B), and a rat treated with 50 mg/kg CsA (Fig. 2C). No damage is seen in the CsA-treated rat, whereas dense neuronal damage is seen in the tip of the dentate gyrus of the vehicle-treated animal.
Fig. 2.

Photomicrographs of the dentate gyrus at 7 d of recovery after 30 min of hypoglycemic coma. A control section (toluidine blue stain) is shown (A) with normal neurons with owl eye-appearing nuclei. Widespread neuronal death is seen in the vehicle-treated rat (B) with dark pyknotic neurons. In rats treated with a single dose of CsA (50 mg/kg, i.v.), no neuronal damage is seen (C). Scale bar, 10 μm.
Cyclosporin A concentrations in blood and brain
Concentrations of CsA in blood and hippocampal parenchyma, 45 min after a 20 mg/kg or 50 mg/kg intravenous dose, are shown in Table3. Hypoglycemic animals and control animals, not subjected to hypoglycemia, showed similar CsA concentrations and blood/brain parenchyma ratios at the given dose. At a dose of 20 mg/kg, the CsA concentration in the brain was 2.5 μg/g (2 μm) and at 50 mg/kg the concentration increased multifold (10–12 μm). The ratios between blood and brain CsA concentrations at 45 min after the injected dose of 20 mg/kg or 50 mg/kg were 0.36 and 0.26, respectively. There was no significant difference in the levels of CsA in brains that were perfused with saline and those that were not (data not shown), indicating that the measured CsA concentrations in brain were not attributed to blood remaining in the intracerebral vessels.
Table 3.
Concentrations of cyclosporin A in blood and hippocampal parenchyma, 45 min after an intravenous injection
| n | Blood (μg/ml) | Brain (μg/mg) | |
|---|---|---|---|
| Cyclosporin A (20 mg/kg)3-a | 4 | 6.9 ± 1.3 | 2.5 ± 0.9 |
| Cyclosporin A (50 mg/kg)3-a | 3 | 56 ± 4.2 | 14.6 ± 2.5 |
| Cyclosporin A (50 mg/kg)3-b | 3 | 45 ± 7.3 | 12.0 ± 0.5 |
Concentrations are shown as mean ± SD.
Samples were taken during hypoglycemic coma, as described in Materials and Methods.
Samples were taken from control animals, as described in Materials and Methods.
Respiratory rate and mitochondrial swelling
Mitochondria isolated from the hippocampus were well-coupled with an RCR of 5.5 ± 0.4 (n = 3).
When the mitochondrial suspension was added to the isotonic KCl buffer, a low basal mitochondrial swelling was seen (Fig.3, left). On addition of calcium ions the swelling rate increased in a dose-dependent manner, hence the initial swelling rate at 100 μm calcium chloride was ∼10 times faster than at 20 μm. Addition of 200 nm CsA effectively inhibited swelling at 20 μm calcium chloride and decreased significantly the swelling rate induced by 100 μm calcium chloride (Fig.3). Addition of 200 nm FK 506 did not affect the calcium-induced mitochondrial swelling (Fig. 3).
Ultrastructural changes during hypoglycemic coma
Ultrastructural morphology of the stratum moleculare superjacent to the crest of the dentate gyrus in a sham-operated animal is shown in Figure 4A. The morphology displays a compact neuropil. At higher magnification, dendrites containing neurofilaments and axonal endings with synaptic vesicles are seen embutting on dendrites with clear postsynaptic densities (Fig. 4B). The mitochondria are compact and contracted. In vehicle-treated animals exposed to 30 min of isoelectric EEG induced by hypoglycemia, dendrites are markedly swollen (Fig.4C). Dendritic mitochondria are also swollen, and some have disrupted cristae (Fig. 4C,D). In animals treated with 50 mg/kg CsA intravenously before 30 min of hypoglycemic isoelectricity, dendrites appear similarly swollen as in the vehicle-treated rats (Fig.4E). However, dendritic mitochondria are contracted and appear to be morphologically normal (Fig.4F) and similar to those in the sham-operated animals (Fig. 4B). In untreated and vehicle-treated animals, the presynaptic structures seem unaltered, with mitochondria appearing morphologically intact (Fig.5). At 30 min of recovery after 30 min of isoelectric EEG, the neuropil becomes loose and structurally disordered (Fig. 6A). The dendrites collapse, shrink, and become dark, whereas mitochondria are swollen with disrupted or electron-dense matrix (Fig.6A). In the CsA-treated animal, dendrites are still swollen, but the mitochondria appear normal and compact (Fig.6B). The neuropil is also remarkably compact. The ultrastructural changes in FK 506-treated animals were not different from vehicle-treated ones (data not shown).
Fig. 4.
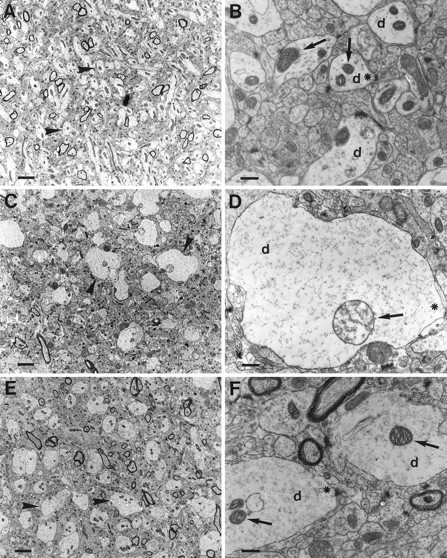
Ultrastructural changes in the stratum moleculare superjacent to the crest of the dentate gyrus in a sham-operated animal (A, B), showing a compact structure with normal dendrites (arrowhead or d), normal axons, and compact and contracted mitochondria (arrow). At higher magnification (B), the postsynaptic densities are evident as dark electrondense structures on the dendritic membrane (*). In the vehicle-treated animals (C, D), dendrites (arrowhead or d) are markedly swollen at 30 min of isoelectric EEG, whereas axonal endings (*) impinging on dendrites are unremarkable. Mitochondria (arrow) in the dendrites of the vehicle-treated animals are swollen, and some have disrupted cristae. In animals treated intravenously with 50 mg/kg CsA (E, F), dendritic mitochondria (arrow) are contracted and appear to be morphologically normal and similar to those in the sham-operated animals (A, B). Still, dendrites (arrowhead or d) appear similarly swollen as in the vehicle-treated rats. Scale bars; A, C, E, 2.7 μm; B, D, F, 0.54 μm.
Fig. 5.
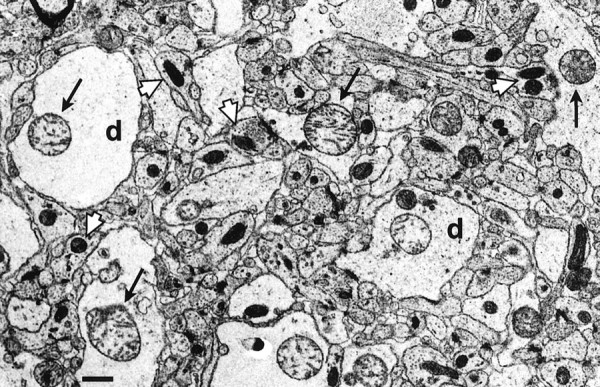
Ultrastructural changes in the stratum moleculare superjacent to the crest of the dentate gyrus in a vehicle-treated animal at 30 min of hypoglycemic coma. Presynaptic structures appear normal with normal mitochondria in nerve endings (white arrow). Dendrites (d), on the other hand, are markedly swollen, containing swollen mitochondria with disrupted cristae (arrow). Scale bar, 0.68 μm.
Fig. 6.

Ultrastructural changes in the stratum moleculare superjacent to the crest of the dentate gyrus at 30 min recovery after 30 min of isoelectric EEG. A, The dendrites are disrupted in the vehicle-treated animals, and the mitochondria (arrow) appear swollen with disrupted or electrondense matrix. Some cells (arrowhead) are condensed.B, In the animal treated intravenously with 50 mg/kg CsA, dendrites (d) are still swollen, whereas the mitochondria (arrow) appear normal and contracted. Scale bar, 0.54 μm.
The number of swollen mitochondria was significantly larger in the vehicle and in FK 506-treated animals compared with the CsA-treated ones (Fig. 7). In vehicle and FK 506-treated animals, the number of swollen mitochondria was 38.5 ± 6.6 and 36 ± 4.4, respectively, whereas in the CsA-treated group the number was 4.5 ± 3.8 (p < 0.01).
Fig. 7.

Mean number of swollen mitochondria (>0.8 μm in diameter) in an area measuring 26 × 26 μm in the distal stratum moleculare, superjacent to the crest of the dentate gyrus. The animals were subjected to 30 min of hypoglycemic isoelectricity and treated with 50 mg/kg CsA (n = 4), 2 mg/kg FK 506 (n = 3), or vehicle (n = 4). ** denotes significant difference in CsA-treated animals, compared with vehicle and FK 506-treated animals. p < 0.01; Scheffe’s test.
DISCUSSION
The main finding of this investigation is a marked neuroprotective effect of cyclosporin A in a model of severe insulin-induced hypoglycemia. Our data show that mitochondria swell noticeably during and after hypoglycemic coma and that this swelling is prevented by cyclosporin A. We also show that calcium-induced swelling in isolated mitochondria from the hippocampus is depressed by cyclosporin A but not by FK 506. Because calcium-induced pathophysiological swelling of mitochondria is believed to be caused by the activation of the MPT and because MPT may be involved in cell death, the following discussion will be focused on the possible involvement of MPT in the development of hypoglycemic brain damage.
Mitochondrial permeability transition
When mitochondria are exposed to high levels of calcium ions and oxidative stress, MPT is induced, caused by the opening of a large proteinaceous pore, the MMC. The opening of the MMC and subsequent collapse of the mitochondrial potential allows solutes of up to 1500 Da size to flow across the membrane, and when studied in vitro, swelling of the isolated mitochondria ensues (Haworth and Hunter, 1979;Crompton et al., 1987; Gunter and Pfeiffer, 1990). Reactive oxygen species (ROS), adenine nucleotide depletion, and a low mitochondrial membrane potential all facilitate MPT in vitro, seemingly by increasing pore sensitivity to calcium ions (Bernardi et al., 1994;Halestrap, 1994). In addition, calpain-like proteases (Aguilar et al., 1996) and ICE-like caspases (Susin et al., 1997) have been found to induce MPT. Low matrix pH, on the other hand, is a potent inhibitor of MPT (Halestrap, 1991; Bernardi et al., 1992). The Bcl-2 family of proteins also influences MPT through several possible mechanisms (Kroemer, 1997).
Recently, the matrix-specific rat CyP-D showing striking homology to the human hCyp-3, was cloned (Woodfield, 1997). Cyclophilin-D has been shown to be of importance in the regulation of the mitochondrial permeability transition, and it has been suggested that CyP-D interacts with an integral membrane protein, possibly the adenine nucleotide translocase, thereby facilitating MPT on calcium binding (Halestrap and Davidson, 1990; Connern and Halestrap, 1992, 1994). Nanomolar concentrations of CsA inhibit the MPT by blocking translocation of CyP-D to the mitochondrial inner membrane in isolated liver mitochondria, decreasing MMC sensitivity to calcium ions (Connern and Halestrap, 1994).
The activation of MPT by calcium ions and the regulation of the pore by different modulators has been studied extensively in isolated mitochondria from the liver and the heart (Crompton et al., 1987;Gunter and Pfeiffer, 1990; Halestrap and Davidson, 1990) and recently in isolated mitochondria from whole brain (Kristal and Dubinsky, 1997). Variations in sensitivity to calcium ion-induced MPT in different species and tissue, which have been attributed to differences in the content of MPT modulators (Griffiths and Halestrap, 1993b; Jurkowitz and Brierley, 1982), have been demonstrated (Zoratti and Szabo, 1995). The data presented here show that MPT is induced in isolated hippocampal mitochondria by calcium ions in a dose-dependent manner. We also demonstrate that cyclosporin A, but not FK 506, is a potent inhibitor of hippocampal mitochondrial swelling. We conclude that hippocampal mitochondria undergo MPT during exposure to supraphysiological levels of calcium ions. This process is inhibited by cyclosporin A, in a manner similar to that reported in other studies (Crompton et al., 1988; Griffiths and Halestrap, 1991; Kristal and Dubinsky, 1997).
MPT and hypoglycemic brain damage
The pathophysiological conditions prevailing during hypoglycemia favor MPT, and the effect of CsA on the ultrastructural changes of dentate gyrus granule cells during hypoglycemia supports this notion. The ultrastructural data presented here demonstrate that dendrites and mitochondria swell considerably during the hypoglycemic insult. This is presumably caused by the energy failure, loss of ion homeostasis, and excitotoxic stress with concomitant massive influx of sodium and calcium ions and water. During hypoglycemic coma, the tissue ATP levels drop to below 1 mm (Wieloch et al., 1984), which indicates that mitochondrial ATP production is limited, most probably because of a decrease in mitochondrial membrane potential secondary to the shortage in glucose supply. The mitochondria therefore can be considered to be at least partly de-energized. The extracellular calcium ion concentration in the brain decreases from ∼1 mm to below 100 μm (Harris et al., 1984). Because the plasma membrane is depolarized, an equilibration of the ionic gradients across the cell membrane occurs. Therefore, an intracellular calcium ion concentration of ∼100 μm can be expected, which is in the range where we observe extensive swelling of isolated de-energized mitochondria.
Cyclosporin A dramatically prevents the mitochondrial swelling in granule cells but not the swelling of dendrites, which are still swollen 30 min after the hypoglycemic insult. Apparently, membrane depolarization seems to occur also in cyclosporin A-treated rats during the insult, leading to dendritic swelling. This ensues during early recovery after hypoglycemia because dendrites still may experience glutamate receptor overactivation (Sandberg et al., 1986). Mitochondria, on the other hand, are still compact during recovery in CsA-treated animals. Hence there is a clear correlation between preservation of mitochondrial ultrastructure and neuronal survival after CsA treatment, which suggests strongly that CsA-sensitive mitochondrial swelling is associated with the activation of a deleterious process.
The activation of the MMC has several consequences. First, loss of ion gradients and depolarization of the mitochondrial membrane potential uncouples oxidative phosphorylation. Second, a burst of ROS production is induced (Zamzami et al., 1995), leading to depletion of reduced glutathione and NADPH. Third, mitochondrial proteins activating the initial steps of apoptosis may be released (Susin et al., 1996). However, it is not clear how the release of these factors, which requires an increase in the permeability of the outer membrane to large molecules, is related to the MPT, which is associated with an increase in the permeability of the inner membrane to smaller molecules. Indeed, in some systems, the release of apoptogenic proteins occurs before the initiation of MPT (Kluck et al., 1997; Yang et al., 1997; Zou et al., 1997). In a yet unidentified manner, these proteins encompassing apoptosis-inducing factor, cytochrome c, and apoptosis protease-activating factor, in turn activate caspases, the executioners of apoptotic cell death (Nicholson and Thornberry, 1997). During hypoglycemia, the induction of MPT is followed by a series of events leading to DNA fragmentation (M. Ferrand-Drake, H. Friberg, T. Wieloch, unpublished observations).
Cyclosporin A, FK 506, and neuroprotection
A decrease in brain temperature to 33°C during or after ischemia has repeatedly been shown to markedly diminish brain damage (Ginsberg, 1997). Neuroprotection observed by some pharmacological interventions has been attributed to hypothermia that accompanies drug treatment, rather than to the drug effect (Buchan and Pulsinelli, 1990). In contrast, hypothermia during or after a hypoglycemic insult does not affect the neuronal damage incurred (Agardh et al., 1992). Therefore, the tendency to a slightly lower body temperature after hypoglycemia observed in CsA-treated rats compared with vehicle-treated ones in the present investigation could not have contributed to the observed dramatic neuroprotective effect.
Cyclosporin A has a restricted passage into the brain parenchyma (Begley et al., 1990; Sakata et al., 1994), but it accumulates to pharmacological concentrations in the brain at high blood levels (Bernareggi and Rowland, 1991). We also found a dose-dependent increase of CsA levels in brain parenchyma after single intravenous injections, clearly showing permeability of CsA across the BBB at high doses. Although it is highly probable that the accumulated CsA is found mainly in the parenchyma and therefore also in neurons, accumulation of CsA in the endothelium cannot be excluded. Cyclosporin A concentrations in the brain obtained in the present investigation were higher than those needed to inhibit MPT in isolated hippocampal mitochondria, but were comparable with those shown to delay and block the MPT in mixed neuronal cultures after NMDA exposure (Nieminen et al., 1996; Schinder et al., 1996; White and Reynolds, 1996). It is therefore reasonable to believe that the concentrations of CsA attained in the brain in this study are sufficient to inhibit MPT during hypoglycemia. In previous investigations in models of brain ischemia, CsA was neuroprotective at doses generally lower than those used in the present study (Shiga et al., 1992; Butcher et al., 1997). For example, in models of focal ischemia the CsA dose showing neuroprotection was in the range of 15–20 mg/kg, which is similar to the dose where neuroprotection is seen in the present study, whereas in global ischemia, a 10 mg/kg dose was not sufficient to provide protection (Uchino et al., 1995). Tissue CsA concentrations, however, were not measured in these studies, and the effective pharmacological doses can therefore not be compared with those reported here.
FK 506, on the other hand, has been shown to readily penetrate the BBB (Dawson et al., 1993; Butcher et al., 1997). The 2 mg/kg dose used in this study is in the same order of magnitude as published earlier, showing neuroprotection in a model of transient global ischemia (Drake et al., 1996) and in a model of focal ischemia (Sharkey and Butcher, 1994).
Several different mechanisms have been proposed to be responsible for the neuroprotective properties of immunophilin ligands, such as CsA, FK 506, or their analogues. In some models, the immunosuppressive action of CsA and FK 506 via calcineurin inhibition may be of importance (Butcher et al., 1997), especially if an inflammatory component is obvious in neuronal death. Other neuroprotective effects may involve the regulatory role of immunophilins at the ryanodine receptor or the IP3 receptor (Brillantes et al., 1994; Cameron et al., 1995). A direct neurotrophic effect by immunophilin ligands has been proposed as well, although the cellular mechanisms behind this remain unclear (Steiner et al., 1997). Patch-clamp studies in hippocampal neurons have shown that CsA action is not mediated through NMDA-receptor blockade (V. S. Vorobjev, personal communication).
On the basis of our experiments and earlier published data, we propose that the decreased flux in the electron transport chain caused by glucose deprivation and the ensuing loss of membrane potentials, as well as the calcium ion flow into neurons concomitant with mild alkalosis, enhance MPT during hypoglycemia. This is evident as mitochondrial swelling, which is blocked by CsA. In animals treated with CsA, MPT is therefore not initiated, mitochondrial integrity is preserved, and the cascade leading to cell death is prevented. Although the lack of a protective effect in FK 506-treated animals in this study implies that calcineurin is not involved in the process leading to hypoglycemic cell death, we cannot exclude a combinatorial protective effect by CsA through the blockade of MPT and calcineurin inhibition (Shibasaki et al., 1997).
Footnotes
This work was supported by the Swedish Medical Research Council (Grant 8644), the European Union BIOMED II (Grant BMH4-CT96-0851), and The Bergendahl Foundation. We thank Kerstin Beirup for excellent technical assistance and Chief Chemist Henrik Björk at the Department of Clinical Pharmacology for skillfully analyzing CsA.
Correspondence should be addressed to Hans Friberg, Laboratory for Experimental Brain Research, Wallenberg Neuroscience Center, University Hospital, S-221 85 Lund, Sweden.
REFERENCES
- 1.Agardh CD, Smith ML, Siesjö BK. The influence of hypothermia on hypoglycemia-induced brain damage in the rat. Acta Neuropathol (Berl) 1992;83:379–385. doi: 10.1007/BF00713529. [DOI] [PubMed] [Google Scholar]
- 2.Aguilar HI, Botla R, Arora AS, Bronk SF, Gores GJ. Induction of the mitochondrial permeability transition by protease activity in rats: a mechanism of hepatocyte necrosis. Gastroenterology. 1996;110:558–566. doi: 10.1053/gast.1996.v110.pm8566604. [DOI] [PubMed] [Google Scholar]
- 3.Auer R, Kalimo H, Olsson Y, Wieloch T. The dentate gyrus in hypoglycemia: pathology implicating excitotoxin-mediated neuronal necrosis. Acta Neuropathol (Berl) 1985;67:279–288. doi: 10.1007/BF00687813. [DOI] [PubMed] [Google Scholar]
- 4.Auer RN, Siesjo BK. Hypoglycaemia: brain neurochemistry and neuropathology. Baillieres Clin Endocrinol Metab. 1993;7:611–625. doi: 10.1016/s0950-351x(05)80210-1. [DOI] [PubMed] [Google Scholar]
- 5.Begley DJ, Squires LK, Zlokovic BV, Mitrovic DM, Hughes CC, Revest PA, Greenwood J. Permeability of the blood-brain barrier to the immunosuppressive cyclic peptide cyclosporin A. J Neurochem. 1990;55:1222–1230. doi: 10.1111/j.1471-4159.1990.tb03128.x. [DOI] [PubMed] [Google Scholar]
- 6.Bernardi P, Vassanelli S, Veronese P, Colonna R, Szabo I, Zoratti M. Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J Biol Chem. 1992;267:2934–2939. [PubMed] [Google Scholar]
- 7.Bernardi P, Broekemeier KM, Pfeiffer DR. Recent progress on regulation of the mitochondrial permeability transition pore: a cyclosporin-sensitive pore in the inner mitochondrial membrane. J Bioenerg Biomembr. 1994;26:509–517. doi: 10.1007/BF00762735. [DOI] [PubMed] [Google Scholar]
- 8.Bernareggi A, Rowland M. Physiologic modeling of cyclosporin kinetics in rat and man. J Pharmacokinet Biopharm. 1991;19:21–50. doi: 10.1007/BF01062191. [DOI] [PubMed] [Google Scholar]
- 9.Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR. Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein. Cell. 1994;77:513–523. doi: 10.1016/0092-8674(94)90214-3. [DOI] [PubMed] [Google Scholar]
- 10.Buchan A, Pulsinelli WA. Hypothermia but not the N-methyl-d-aspartate antagonist, MK-801, attenuates neuronal damage in gerbils subjected to transient global ischemia. J Neurosci. 1990;10:311–316. doi: 10.1523/JNEUROSCI.10-01-00311.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butcher SP, Henshall DC, Teramura Y, Iwasaki K, Sharkey J. Neuroprotective actions of FK506 in experimental stroke: in vivo evidence against an antiexcitotoxic mechanism. J Neurosci. 1997;17:6939–6946. doi: 10.1523/JNEUROSCI.17-18-06939.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cameron AM, Steiner JP, Sabatini DM, Kaplin AI, Walensky LD, Snyder SH. Immunophilin FK506 binding protein associated with inositol 1,4,5-trisphosphate receptor modulates calcium flux. Proc Natl Acad Sci USA. 1995;92:1784–1788. doi: 10.1073/pnas.92.5.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clipstone NA, Crabtree GR. Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature. 1992;357:695–697. doi: 10.1038/357695a0. [DOI] [PubMed] [Google Scholar]
- 14.Connern CP, Halestrap AP. Purification and N-terminal sequencing of peptidyl-prolyl cis-trans-isomerase from rat liver mitochondrial matrix reveals the existence of a distinct mitochondrial cyclophilin. Biochem J. 1992;284:381–385. doi: 10.1042/bj2840381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Connern CP, Halestrap AP. Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem J. 1994;302:321–324. doi: 10.1042/bj3020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crompton M, Costi A, Hayat L. Evidence for the presence of a reversible Ca2+-dependent pore activated by oxidative stress in heart mitochondria. Biochem J. 1987;245:915–918. doi: 10.1042/bj2450915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crompton M, Ellinger H, Costi A. Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J. 1988;255:357–360. [PMC free article] [PubMed] [Google Scholar]
- 18.Dawson TM, Steiner JP, Dawson VL, Dinerman JL, Uhl GR, Snyder SH. Immunosuppressant FK506 enhances phosphorylation of nitric oxide synthase and protects against glutamate neurotoxicity. Proc Natl Acad Sci USA. 1993;90:9808–9812. doi: 10.1073/pnas.90.21.9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drake M, Friberg H, Boris-Moller F, Sakata K, Wieloch T. The immunosuppressant FK506 ameliorates ischaemic damage in the rat brain. Acta Physiol Scand. 1996;158:155–159. doi: 10.1046/j.1365-201X.1996.535298000.x. [DOI] [PubMed] [Google Scholar]
- 20.Galat A, Metcalfe SM. Peptidylproline cis/trans isomerases. Prog Biophys Mol Biol. 1995;63:67–118. doi: 10.1016/0079-6107(94)00009-x. [DOI] [PubMed] [Google Scholar]
- 21.Ginsberg MD. Hypothermic neuroprotection in cerebral ischemia. In: Welch KMA, Reis DJ, Siesjo BK, Weir B, editors. Primer on cerebrovascular disease. Academic Press; Los Angeles: 1997. pp. 272–275. [Google Scholar]
- 22.Griffiths EJ, Halestrap AP. Further evidence that cyclosporin A protects mitochondria from calcium overload by inhibiting a matrix peptidyl-prolyl cis-trans isomerase. Implications for the immunosuppressive and toxic effects of cyclosporin. Biochem J. 1991;274:611–614. doi: 10.1042/bj2740611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Griffiths EJ, Halestrap AP. Protection by cyclosporin A of ischemia/reperfusion-induced damage in isolated rat hearts. J Mol Cell Cardiol. 1993a;25:1461–1469. doi: 10.1006/jmcc.1993.1162. [DOI] [PubMed] [Google Scholar]
- 24.Griffiths EJ, Halestrap AP. Pyrophosphate metabolism in the perfused heart and isolated heart mitochondria and its role in regulation of mitochondrial function by calcium. Biochem J. 1993b;290:489–495. doi: 10.1042/bj2900489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- 26.Halestrap AP. Calcium-dependent opening of a non-specific pore in the mitochondrial inner membrane is inhibited at pH values below 7. Implications for the protective effect of low pH against chemical and hypoxic cell damage. Biochem J. 1991;278:715–719. doi: 10.1042/bj2780715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halestrap AP. Interaction between oxidative stress and calcium overload on mitochondrial function. In: Darley-Usmar V, Schapira AHV, editors. Mitochondria: DNA, proteins and disease. Portland; London: 1994. pp. 113–142. [Google Scholar]
- 28.Halestrap AP, Davidson AM. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harris RJ, Wieloch T, Symon L, Siesjo BK. Cerebral extracellular calcium activity in severe hypoglycemia: relation to extracellular potassium and energy state. J Cereb Blood Flow Metab. 1984;4:187–193. doi: 10.1038/jcbfm.1984.27. [DOI] [PubMed] [Google Scholar]
- 30.Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys. 1979;195:460–467. doi: 10.1016/0003-9861(79)90372-2. [DOI] [PubMed] [Google Scholar]
- 31.Ide T, Morikawa E, Kirino T. An immunosuppressant, FK506, protects hippocampal neurons from forebrain ischemia in the mongolian gerbil. Neurosci Lett. 1996;204:157–160. doi: 10.1016/0304-3940(96)12352-1. [DOI] [PubMed] [Google Scholar]
- 32.Jurkowitz MS, Brierley GP. H+-dependent efflux of Ca2+ from heart mitochondria. J Bioenerg Biomembr. 1982;14:435–449. doi: 10.1007/BF00743069. [DOI] [PubMed] [Google Scholar]
- 33.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 34.Kristal BS, Dubinsky JM. Mitochondrial permeability transition in the central nervous system: induction by calcium cyclin-dependent and -independent pathways. J Neurochem. 1997;69:524–538. doi: 10.1046/j.1471-4159.1997.69020524.x. [DOI] [PubMed] [Google Scholar]
- 35.Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 37.Marchetti P, Castedo M, Susin SA, Zamzami N, Hirsch T, Macho A, Haeffner A, Hirsch F, Geuskens M, Kroemer G. Mitochondrial permeability transition is a central coordinating event of apoptosis. J Exp Med. 1996a;184:1155–1160. doi: 10.1084/jem.184.3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marchetti P, Hirsch T, Zamzami N, Castedo M, Decaudin D, Susin SA, Masse B, Kroemer G. Mitochondrial permeability transition triggers lymphocyte apoptosis. J Immunol. 1996b;157:4830–4836. [PubMed] [Google Scholar]
- 39.Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 40.Nieminen AL, Petrie TG, Lemasters JJ, Selman WR. Cyclosporin A delays mitochondrial depolarization induced by N-methyl-d-aspartate in cortical neurons: evidence of the mitochondrial permeability transition. Neuroscience. 1996;75:993–997. doi: 10.1016/0306-4522(96)00378-8. [DOI] [PubMed] [Google Scholar]
- 41.O’Keefe SJ, Tamura J, Kincaid RL, Tocci MJ, O’Neill EA. FK-506- and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature. 1992;357:692–694. doi: 10.1038/357692a0. [DOI] [PubMed] [Google Scholar]
- 42.Sakata A, Tamai I, Kawazu K, Deguchi Y, Ohnishi T, Saheki A, Tsuji A. In vivo evidence for ATP-dependent and P-glycoprotein-mediated transport of cyclosporin A at the blood-brain barrier. Biochem Pharmacol. 1994;48:1989–1992. doi: 10.1016/0006-2952(94)90601-7. [DOI] [PubMed] [Google Scholar]
- 43.Sandberg M, Butcher SP, Hagberg H. Extracellular overflow of neuroactive amino acids during severe insulin-induced hypoglycemia: in vivo dialysis of the rat hippocampus. J Neurochem. 1986;47:178–184. doi: 10.1111/j.1471-4159.1986.tb02847.x. [DOI] [PubMed] [Google Scholar]
- 44.Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharkey J, Butcher SP. Immunophilins mediate the neuroprotective effects of FK506 in focal cerebral ischaemia. Nature. 1994;371:336–339. doi: 10.1038/371336a0. [DOI] [PubMed] [Google Scholar]
- 46.Shibasaki F, Kondo E, Akagi T, McKeon F. Suppression of signalling through transcription factor NF-AT by interactions between calcineurin and Bcl-2. Nature. 1997;386:728–731. doi: 10.1038/386728a0. [DOI] [PubMed] [Google Scholar]
- 47.Shiga Y, Onodera H, Matsuo Y, Kogure K. Cyclosporin A protects against ischemia-reperfusion injury in the brain. Brain Res. 1992;595:145–148. doi: 10.1016/0006-8993(92)91465-q. [DOI] [PubMed] [Google Scholar]
- 48.Siesjo BK, Bengtsson F. Calcium fluxes, calcium antagonists, and calcium-related pathology in brain ischemia, hypoglycemia, and spreading depression: a unifying hypothesis. J Cereb Blood Flow Metab. 1989;9:127–140. doi: 10.1038/jcbfm.1989.20. [DOI] [PubMed] [Google Scholar]
- 49.Sims NR. Rapid isolation of metabolically active mitochondria from rat brain and subregions using Percoll density gradient centrifugation. J Neurochem. 1990;55:698–707. doi: 10.1111/j.1471-4159.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- 50.Steiner JP, Connolly MA, Valentine HL, Hamilton GS, Dawson TM, Hester L, Snyder SH. Neurotrophic actions of nonimmunosuppressive analogues of immunosuppressive drugs FK506, rapamycin and cyclosporin A. Nat Med. 1997;3:421–428. doi: 10.1038/nm0497-421. [DOI] [PubMed] [Google Scholar]
- 51.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Susin SA, Zamzami N, Castedo M, Daugas E, Wang HG, Geley S, Fassy F, Reed JC, Kroemer G. The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J Exp Med. 1997;186:25–37. doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uchino H, Elmer E, Uchino K, Lindvall O, Siesjo BK. Cyclosporin A dramatically ameliorates CA1 hippocampal damage following transient forebrain ischaemia in the rat. Acta Physiol Scand. 1995;155:469–471. doi: 10.1111/j.1748-1716.1995.tb09999.x. [DOI] [PubMed] [Google Scholar]
- 54.White RJ, Reynolds IJ. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wieloch T, Harris RJ, Symon L, Siesjo BK. Influence of severe hypoglycemia on brain extracellular calcium and potassium activities, energy, and phospholipid metabolism. J Neurochem. 1984;43:160–168. doi: 10.1111/j.1471-4159.1984.tb06692.x. [DOI] [PubMed] [Google Scholar]
- 56.Woodfield KY, Price NT, Halestrap AP. cDNA cloning of rat mitochondrial cyclophilin. Biochem Biophys Acta. 1997;1351:27–30. doi: 10.1016/s0167-4781(97)00017-1. [DOI] [PubMed] [Google Scholar]
- 57.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]
- 58.Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B, Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]
- 60.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4: participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]