

Abstract
We report that dipyridamole is neuroprotective for a variety of rat embryonic CNS neurons cultured in serum-free basal medium lacking trophic factors or other additives. We also describe the mechanism underlying this action. Neurons died rapidly in basal medium but were rescued in large measure by 10 μm dipyridamole. The protective action of dipyridamole seems to be attributable to its antioxidant property. Vitamin E and N-acetylcysteine provided comparable neuroprotection in basal medium, whereas an array of compounds that mimic other actions of dipyridamole (inhibition of phosphodiesterases, blockade of nucleoside and chloride transport, interference with the multidrug resistance protein, and enhancement of prostacyclin synthesis) failed to promote survival. Thus, a major cause of neuronal death in this system seems to be oxidative stress that is relieved by dipyridamole. Iron plays a significant role in generation of such stress, as indicated by the observations that addition of apotransferrin or iron chelators to basal medium or use of iron-free medium also afforded protection. Although oxidative stress was a major determinant of neuronal death, it was not the only factor. Dipyridamole or other antioxidant measures did not provide sustained neuroprotection. However, provision of insulin, which was not protective alone in basal medium, along with dipyridamole significantly enhanced long-term neuronal survival. Hence, optimal protection requires both trophic support and relief from oxidative stress. These findings lend credence to the potential use of dipyridamole or its derivatives in prevention and/or treatment of CNS injuries and degenerative disorders in which oxidative stress is a significant component.
Keywords: dipyridamole, transferrin, iron, oxidative stress, neuronal death, trophic factors
Survival of neurons is affected by multiple variables including the presence of appropriate neurotrophic agents, the supply of required nutrients, and protection from potentially toxic conditions such as oxidative stress. One strategy to identify both natural and synthetic products that are neuroprotective is to culture neurons in a nonsupportive basal medium and to supplement this with potential survival-promoting agents (Skaper et al., 1979,1982; Bottenstein et al., 1980; Huck, 1983). This approach has led to the development of a widely used defined medium for culturing and maintaining CNS neurons that consists of basal medium (often a 1:1 mixture of Ham’s F12 medium and Minimal Essential Medium) supplemented with insulin, transferrin, progesterone, selenium, and putrescine (Bottenstein and Sato, 1979; Bottenstein et al., 1980; di Porzio et al., 1980). The use of this and other defined basal media has permitted the identification and study of a variety of neurotrophic and neuroprotective agents (Barbin et al., 1984; Varon et al., 1984;Friedman et al., 1993).
In addition to primary neurons, cell lines such as the rat pheochromocytoma line PC12 have proved useful for evaluation of potential neuroprotective agents. Among the substances that have been found to prevent death of PC12 cells in serum-free RPMI 1640 medium are permeant derivatives of cAMP and cGMP (Rukenstein et al., 1991;Farinelli et al., 1996a). Moreover, nitric oxide (NO) generators and natriuretic peptides promote PC12 cell survival under such conditions by raising intracellular levels of cGMP (Farinelli et al., 1996a,b). We noted that coaddition of the drug dipyridamole markedly potentiated and prolonged the neuroprotective activity of NO and natriuretic peptides on PC12 cells. Although dipyridamole has diverse pharmacological targets, its actions in this case presumably were caused by its capacity to inhibit phosphodiesterase-mediated hydrolysis of cGMP (Thompson, 1991).
The potentiating actions of dipyridamole in serum-free PC12 cell cultures led us to explore the potential survival-promoting actions of this drug on CNS neurons. To do so, we used a system in which neurons from various regions of embryonic rat brain were plated in unsupplemented basal medium. In this report, we describe the neuroprotective effects of dipyridamole on CNS neurons and delineate the mechanism by which these occur.
MATERIALS AND METHODS
Materials. Minimal essential medium (MEM), catalog #11095–080, and Ham’s F12 nutrient mixture, catalog #11765–054, were obtained from Gibco, and all tissue culture plastic ware was from Becton Dickinson (Rutherford, NJ). All pharmacological and common tissue culture reagents were obtained from Sigma (St. Louis, MO) with the following exceptions.S-Nitroso-N-acetylpenicillamine (SNAP), diethylenetriamine nitric oxide adduct (DETA-NO), 7-nitroindazole, and MK-801 were from Research Biochemicals (Natick, MA); milrinone and trequinsin were from Calbiochem (La Jolla, CA); 8-(4-chlorophenylthio)-cAMP (8-CPT-cAMP) was from Boehringer Mannheim (Indianapolis, IN); iloprost was generously provided by Dr. Stephen Feinmark, Columbia University; and flavopiridol was graciously provided by Dr. David Park, Columbia University. Timed-pregnant Sprague Dawley rats were obtained from Zivic-Miller Labs or Taconic (Germantown, NY). Dipyridamole was made up in dimethylsulfoxide as a 20 mmstock, protected from light, and stored frozen at −20°C until use for up to 1 month. Cyanogen bromide-activated Sepharose was purchased from Sigma and was coupled to deferoxamine by the method of Feng et al. (1992). After coupling, the beads were washed extensively to remove unbound deferoxamine.
Preparation of neuronal cultures. Primary neuronal cultures from embryonic day 16 (E16) and E18 rats were prepared as described previously (Friedman et al., 1993) and maintained in serum-free conditions throughout the course of the investigations. After dissection, brain tissue was dissociated by mechanical trituration, and the cells were resuspended in MEM. The cell suspension was diluted to 200,000 cells/ml and then plated in 24-well poly-l-lysine-coated tissue culture dishes at a density of 100,000 cells per well in a final volume of 1 ml. The final experimental culture conditions were arrived at by adding 0.5 ml of the cell suspension in MEM to each well that contained 0.5 ml of tissue culture medium (Ham’s F12 or MEM) plus a twofold concentration of serum-free supplements and/or pharmacological agents when indicated. Thus, this 1:2 dilution resulted in the concentrations of agents reported in the Results. The standard culture conditions that served as the experimental control consisted of MEM/Ham’s F12 (1:1) containing insulin (25 μg/ml), glucose (6 mg/ml), transferrin (100 μg/ml), progesterone (20 nm), putrescine (60 μm), and selenium (30 nm) and is referred to as complete serum-free medium (SFM) (di Porzio et al., 1980). MEM/Ham’s F12 that did not include these supplements is referred to as basal medium. Cultures were maintained at 37°C in a humidified atmosphere of 95% air/5% CO2, and the medium was not changed during the course of the experiment unless otherwise indicated.
FeSO4 and H2O2toxicity. Neurons were plated and maintained in 0.5 ml of complete SFM for 3 d. On the third day, 0.5 ml of complete SFM containing twice the stated concentrations of FeSO4, H2O2, and any indicated pharmacological agents was added to experimental wells. Cultures were not pretreated with the protective agents before the addition of FeSO4 or H2O2. One day later, the cells were assessed for survival. In this paradigm, the control cultures received an additional 0.5 ml of complete SFM. In the experiments in which the cultures were plated in MEM alone, the medium was supplemented with 0.5 mm pyruvate, a nutrient that has been shown to be critical for neuronal survival (Selak et al., 1985). The mixture of MEM/F12 contains 0.5 mm pyruvate.
Thiobarbituric acid reactive substances assay. Neurons were dissected from E18 fetuses and plated in complete SFM, basal medium, or basal medium with 10 μm dipyridamole. After 5–6 hr, cells were washed twice in PBS and harvested in 300 μl of PBS. A volume of 0.5 ml of thiobarbituric acid reactive substances (TBARS) stock reagent was then added to the harvested material. The stock reagent consisted of 15% trichloroacetic acid, 0.375% 2-thiobarbituric acid, and 0.25N HCl (Buege and Aust, 1978). The cell suspensions were incubated in boiling water for 15 min and centrifuged at 14,000 rpm for 10 min, and fluorescence was measured at 553 nm with excitation at 515 nm (Keller et al., 1998).
Medium replacement experiment. Neurons were plated in 1 ml of complete SFM or basal medium containing 10 μmdipyridamole. One day later, 0.8 ml of the medium was carefully removed from the well and replaced with 0.8 ml of the fresh medium containing the indicated additives. This partial removal and the readdition were necessary because it was determined that complete removal of the medium resulted in decreased viability of the neurons. The residual 20% of dipyridamole or serum-free supplements had no impact on the outcome of this experiment because these levels were unable to promote survival. The following day, the cells were assessed for survival.
Assay for neuronal survival. Neuronal survival was assayed by a method routinely used to assess PC12 cell viability and described previously (Batistatou and Greene, 1991; Rukenstein et al., 1991). After removal of the culture medium, the neurons were lysed at daily intervals, and intact nuclei were counted using a hemacytometer (Soto and Sonnenschein, 1985). In this assay, nuclei of dead cells generally disintegrate or, if in the process of dying, appear pyknotic and irregularly shaped. In contrast, nuclei of living cells are phase bright and have clearly defined limiting membranes. Cell counts were performed on triplicate wells. The data are expressed as a percentage of the number of neurons alive in complete SFM 1 d after plating. All data shown are representative experiments of at least three replicate experiments.
RESULTS
Dipyridamole rescues multiple neuronal populations from death in basal medium
One of our primary aims was to use a simple model system for testing potential agents that are neuroprotective for CNS neurons. To do so, we used a system in which freshly dissociated embryonic rat CNS neurons were plated in medium with no trophic factors or other additives. We predicted that neuronal survival in this minimal medium would be poor and, therefore, that the effect of exogenously supplied protective agents would be readily apparent. When E18 hippocampal neurons were plated in complete serum-free medium with a high level of insulin and other serum-free additives (complete SFM) (di Porzio et al., 1980), neuronal survival at 24 hr was excellent, as judged by morphological criteria, and the cells attached to the substrate and began to elaborate neuritic processes (Fig.1A). When the same neurons were plated in wells containing unsupplemented, basal medium, the cells failed to survive beyond 16 hr, and virtually all were dead by 24 hr. There were very few intact cell bodies and no cells with visible neuritic processes (Fig. 1B). When 10 μm dipyridamole was added at the time of plating, there was substantial rescue of the hippocampal neurons with 70–80% survival after 24 hr. The neurons surviving in basal medium with dipyridamole were nearly indistinguishable from the neurons grown in complete SFM (Fig. 1C).
Fig. 1.

Phase contrast micrographs of E18 hippocampal neurons maintained in complete SFM for 24 hr (A), no additives (B), 10 μmdipyridamole (C), or 100 μg/ml (230 μm) vitamin E (D).
Dipyridamole prevented neuronal death in a concentration-dependent manner with the maximal effect in the range of 10–20 μm(Fig. 2A). The dose–response curve is very steep as concentrations ≤1 μm had no effect on neuronal survival. Concentrations of dipyridamole that were >20 μm seemed toxic to neurons because viability was significantly less than that in cultures treated with 10 μm dipyridamole. To confirm the viability of the neurons maintained by dipyridamole, we plated cultures in basal medium with dipyridamole and then 1 d later switched a portion to either complete SFM or basal medium, in each case without dipyridamole. The cultures were scored for viability the next day. Under these conditions, the cells that had been treated for 1 d with dipyridamole and then exposed to complete SFM were still alive on day 2, whereas those cultures that were switched to basal medium had almost no surviving cells (Fig. 2B). These observations confirm that the neurons judged to be maintained by dipyridamole were alive and not irreversibly committed to death.
Fig. 2.

Dipyridamole promotes survival of primary CNS neurons in basal medium. A, Dose–response relationship for the effect of dipyridamole on survival of E18 hippocampal neurons in basal medium (1 d). Survival data are expressed relative to the number of neurons alive in complete SFM 1 d after plating (arbitrarily set at 100). B, Viability of dipyridamole-supported cells confirmed by responsiveness to complete SFM. Cells were cultured for 24 hr in basal medium with or without 10 μm dipyridamole or complete SFM, and the culture medium was then replaced as indicated. Cells maintained continuously in complete SFM or in basal medium containing 10 μmdipyridamole are shown for reference. Cell numbers were quantified 2 d after plating and expressed relative to the number of neurons in replicate cultures maintained continuously in complete SFM.C, Protection of neurons from several different brain regions by dipyridamole. Survival data are expressed relative to the number of neurons from the indicated brain region alive in complete SFM 1 d after plating. D, Time course. E18 hippocampal neurons were plated in the indicated media, and replicate cultures were assessed for surviving neurons at various times. No subsequent addition of dipyridamole was given. Data are the mean ± SEM of three samples. In this and subsequent figures, the apparent absence of error bars indicates that the error was smaller than thesymbol used. DP, Dipyridamole.
We extended our model system to test whether dipyridamole could promote survival of neurons from regions of the CNS in addition to the hippocampus. We assessed several brain regions including the cerebral cortex, basal forebrain, ventral mesencephalon, spinal cord, and striatum. In every brain region tested, dipyridamole significantly increased neuronal survival in comparison with that in cultures in basal medium. Dipyridamole was most effective on cultures taken from E18 hippocampus and cortex, rescuing 70–80% of the neurons relative to cultures in complete SFM. For neurons taken from the other brain regions on E16, the optimal age for culturing basal forebrain and ventral mesencephalon (Friedman et al., 1993), dipyridamole rescued 50–60% of the neurons (Fig. 2C).
Figure 2D shows a time course for dipyridamole-promoted survival of hippocampal neurons in basal medium. Dipyridamole rescued ∼50% of the neurons after 3 d in culture, 25% after 5 d, and <10% by day 7. Thus dipyridamole, unlike complete SFM, does not confer long-term survival. When dipyridamole was added to complete SFM, there was no increase in neuronal viability compared with that in cultures maintained in complete SFM alone (Fig.2D). Readdition of dipyridamole on days 1–5 did not prolong neuronal survival beyond that observed when the drug was added just once at the time of plating (data not shown). The time course of dipyridamole-mediated survival in basal medium was similar regardless of the brain region from which the neurons were cultured (data not shown).
Mechanism of the neuroprotective action of dipyridamole
We next studied the mechanism by which dipyridamole rescues neurons from death. Because dipyridamole is a compound with multiple pharmacological actions, we tested whether we could protect CNS neurons by mimicking any of these known activities by other means. We initially focused on the well established ability of dipyridamole to inhibit phosphodiesterases (Thompson, 1991) because it was this property that led to the potentiation of NO and natriuretic peptides on the survival of PC12 cells (Farinelli et al., 1996a,b) and also because elevated cAMP can rescue neurons from trophic factor deprivation (Rydel and Greene, 1988; Chang et al., 1996; Miller et al., 1997). However a number of different phosphodiesterase inhibitors as well as permeant cAMP and cGMP analogs failed to prevent neuronal death (Table1). Nitric oxide generators and nitric oxide synthase (NOS) inhibitors were also without effect. In addition to its actions on phosphodiesterases, dipyridamole has been shown to block nucleoside uptake (Zhang and Fredholm, 1994; Thorn and Jarvis, 1996), inhibit the multidrug resistance protein (Ayesh et al., 1996), block chloride transport (Garcia and Lodish, 1989), and increase prostacyclin synthesis (Jackson et al., 1982). None of the variety of agents tested that mimic these as well as several other activities promoted survival of CNS neurons in basal medium (Table 1). Transfer to a basal, serum-free medium lacking growth factors has also been used to probe survival and death mechanisms of PC12 cells. In the latter system, rescue from death is conferred by cyclin-dependent kinase inhibitors (Park et al., 1996) and blockers of RNA synthesis (Mesner et al., 1992). In contrast, these were ineffective on CNS neurons in basal medium. Conversely, dipyridamole did not rescue PC12 cells or sympathetic neurons deprived of trophic support (Farinelli et al., 1996a).
Table 1.
Effects of various agents on survival of cultured CNS neurons
| Treatment | Mechanism tested | Concentration range tested | Brain region tested1-a | CNS neuron survival1-b | PC12 cell survival1-c | Referenced |
|---|---|---|---|---|---|---|
| Zaprinast | Type V PDE inhibitor | 1–300 μm | hpc | n | n | Maurice and Haslam, 1990 |
| IBMX | General PDE inhibitor | 10–100 μm | hpc | n | y | Hartell, 1996 |
| Milrinone | Type III PDE inhibitor | 10–100 μm | hpc | n | y | Marcoz et al., 1993 |
| Trequinsin | General PDE inhibitor | 10 nm–10 μm | hpc | n | y | Sparwasser et al., 1994 |
| 8-CPT-cAMP | cAMP analog | 1–100 μm | str, hpc | n | y | Rydel and Greene, 1988 |
| 8-Br-cGMP | cGMP analog | 10 μm–10 mm | hpc | n | y | Farinelli et al., 1996a |
| Adenosine | Ado receptor agonist | 1–300 μm | hpc | n | y | Zhang and Fredholm, 1994 |
| Nitrobenzylthioinosine | Ado uptake blocker | 300 nm–100 μm | hpc | n | ? | Thorn and Jarvis, 1996 |
| Propentofylline | Ado uptake blocker | 1–300 μm | hpc | n | ? | Zhang and Fredholm, 1994 |
| Dilazep | Ado uptake blocker | 10 nm–100 μm | hpc | n | ? | Thorn and Jarvis, 1996 |
| Verapamil | MDR inhibitor, Ca2+ channel blocker | 30 nm–300 μm | hpc | n | n | Ayesh et al., 1996 |
| Cyclosporin A | MDR inhibitor, immunophilin | 3 nm–30 μm | ctx, hpc | n | n | Ayesh et al., 1996 |
| Iloprost | Prostacyclin analog | 100 nm–10 μm | hpc | n | ? | Jackson et al., 1982 |
| Indomethacin | Cyclooxygenase inhibitor | 3–300 μm | hpc | n | ? | Jackson et al., 1982 |
| DIDS | Chloride transport inhibitor | 300 nm–100 μm | hpc | n | ? | Garcia and Lodish, 1989 |
| Dideoxyforskolin | Chloride transport inhibitor | 30 nm–20 μm | hpc | n | ? | Sanchez-Olea et al., 1996 |
| Flavopiridol | Cell cycle inhibitor, cdk inhibitor | 100–3 μm | hpc | n | y | Park et al., 1996 |
| Ciclopirox olamine | Cell cycle inhibitor | 10 nm–3 μm | str, hpc | n | y | Farinelli and Greene, 1996 |
| Actinomycin D | RNA synthesis inhibitor | 1–10 μm | hpc | n | y | Mesner et al., 1992 |
| Cycloheximide | Protein synthesis inhibitor | 1 nm–10 μm | hpc | n | y/n | Furukawa et al., 1997 |
| SNAP | Nitric oxide generator | 1–100 μm | ctx, hpc, str | n | y | Farinelli et al., 1996a |
| DETA-NO | Nitric oxide generator | 1–100 μm | ctx, hpc, str | n | y | Farinelli et al., 1996a |
| l-NAME | General NOS inhibitor | 10 μm–10 mm | hpc | n | n | Dawson et al., 1991 |
| 7-Nitroindazole | Neuronal NOS inhibitor | 100 nm–200 μm | hpc | n | n | O’Neill et al., 1996 |
| Kynurenic acid | Glutamate receptor antagonist | 1 μm–10 mm | hpc | n | Zeevalk and Nicklas, 1989 | |
| MK-801 | NMDA receptor antagonist | 100 nm–10 μm | hpc | n | n | Zeevalk and Nicklas, 1989 |
| Insulin | Component of complete SFM | 25 μg/ml | hpc | n | y | di Porzio et al., 1980 |
| Progesterone | Component of complete SFM | 20 nm | hpc | n | ? | di Porzio et al., 1980 |
| Putrecine | Component of complete SFM | 60 μm | hpc | n | ? | di Porzio et al., 1980 |
| Selenium | Component of complete SFM | 30 nm | hpc | n | ? | di Porzio et al., 1980 |
| Transferrin | Component of complete SFM | 0.01–1 mg/ml | hpc | y | n | di Porzio et al., 1980 |
| Dipyridamole | all | y | n |
8-Br-cGMP, 8-Bromo-cGMP; cdk, cyclin-dependent kinase; ctx, cortex; DIDS, 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid; hpc, hippocampus; IBMX, 3-isobutyl-l-methylxanthine; n, no;l-NAME, Nω-nitro-l-arginine methyl ester; str, striatum; y, yes.
Brain regions from which the neurons were cultured and tested for this report.
Survival in MEM/F12 basal medium with indicated drug observed this study.
Survival of trophic factor-deprived PC12 cells as observed or reported by our laboratory; ? indicates not tested by us in PC12 cells.
Deontes either action of drug or action on PC12 cells.
The failure of all these different agents to mimic the protective actions of dipyridamole on CNS neurons led us to examine one of its lesser-studied properties, namely its role as an antioxidant. Dipyridamole has been shown to inhibit lipid peroxidation and to scavenge superoxide and hydroxyl radicals (Iuliano et al., 1989, 1995). We therefore tested whether the antioxidants vitamin E andN-acetylcysteine (NAC) might also prevent neuronal death in basal medium. Both vitamin E, a free-radical scavenger, and NAC, an antioxidant that increases intracellular glutathione levels, prevented neuronal death in a concentration-dependent manner (Fig.3A,B). A vitamin E-treated culture is shown in Figure 1D. Vitamin E was maximally effective at a concentration of 100 μg/ml (230 μm), and NAC was maximally effective at 100–300 μm; both saved ∼80% of the hippocampal neurons after 24 hr relative to cultures maintained in complete SFM. The time course of survival in the presence of vitamin E more closely resembled that observed with dipyridamole treatment than it did that after NAC treatment (Fig. 3C). After 3 d, survival in the presence of NAC was no different than that in the cultures maintained in basal medium alone. In contrast, vitamin E afforded significant protection after 5 d, similar to dipyridamole. Readdition of NAC or vitamin E did not prolong survival beyond that observed when either was added only once at the time of plating (data not shown). As observed with dipyridamole, addition of vitamin E or NAC to complete SFM did not increase neuronal survival compared with that seen in cultures maintained in complete SFM alone (Fig. 3D) The protective effects of the three antioxidants were not additive. When dipyridamole, vitamin E, and NAC were combined, the combination was no more effective than each of them alone, suggesting a shared mechanism of action (Fig. 3E).
Fig. 3.
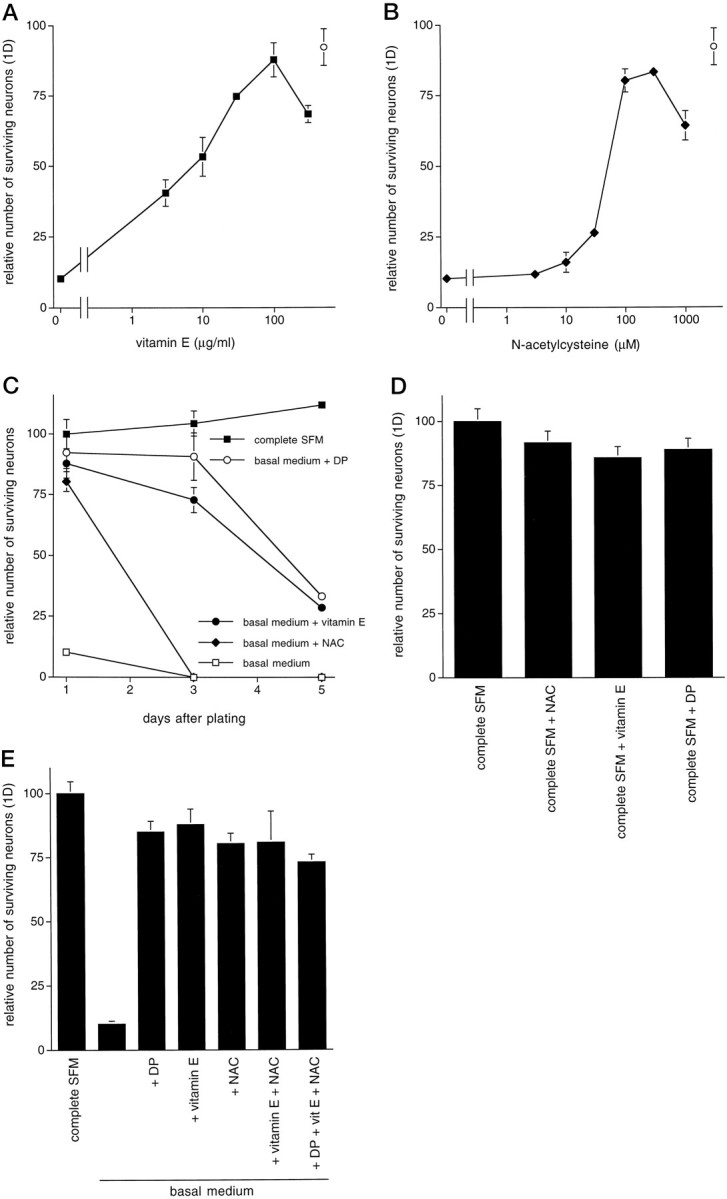
The antioxidants vitamin E and NAC promote survival of primary CNS neurons in basal medium. A,B, Dose–response relationships for the effect of vitamin E (A) and NAC (B) on promotion of survival of E18 hippocampal neurons in basal medium (1 d) are shown. The open circle represents survival mediated by 10 μm dipyridamole. Survival data are expressed relative to the number of neurons alive in complete SFM 1 d after plating (arbitrarily set at 100). C, Time course is shown. E18 hippocampal neurons were plated in either complete SFM or basal medium in the presence of the indicated additives. No subsequent addition of these agents was given. D, Antioxidants do not afford additional protection beyond that provided by complete SFM alone. E, Antioxidant effects on survival in basal medium are not additive and fail to increase dipyridamole-promoted survival. All survival data are expressed relative to the number of neurons alive in complete SFM 1 d after plating.
Dipyridamole protects neurons from oxidative stress
In view of the above data, we had reason to suspect that death in our neuronal cultures in basal medium was primarily attributable to oxidative stress rather than to loss of trophic support, at least during the first several days in vitro. We tested this hypothesis by placing healthy neurons cultured in complete SFM under conditions known to cause oxidative stress, namely, exposure to FeSO4 or H2O2 (Zhang et al., 1993), and then we assessed whether dipyridamole could mimic the effects of other antioxidants and be protective under these circumstances. For example, Chow et al. (1994) reported that Trolox, a vitamin E analog, protects cultured cortical neurons from exposure to iron salts, and thus the iron toxicity model seemed to be a good test system in which to determine whether the antioxidant property of dipyridamole was sufficient to confer protection. One day after treatment with either 10 μm FeSO4 or 30 μm H2O2 in complete SFM, ∼80% of the neurons died compared with the effects seen in untreated controls (Fig.4A,B). When added at the time of exposure to FeSO4, dipyridamole, vitamin E, or NAC afforded very good protection from the oxidative insult (Figs. 4A,5). Pretreatment of the cultures with the antioxidants before addition of FeSO4 was not required for protection. Dipyridamole provided complete protection, whereas NAC and vitamin E protected 75–80% of the neurons. These agents were less effective against H2O2 toxicity, but all provided protection compared with untreated cultures (Fig.4B). NAC was the most effective at protecting against H2O2 toxicity, rescuing ∼70% of the neurons at 1 d.
Fig. 4.
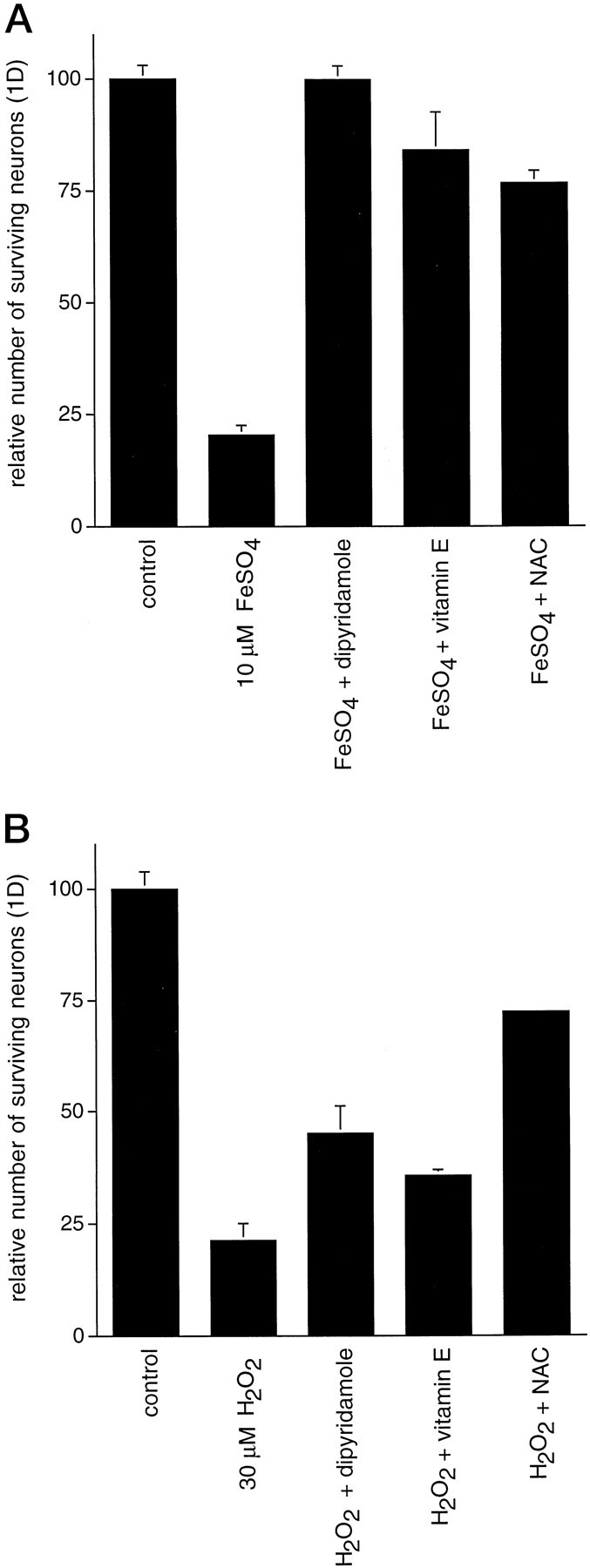
Dipyridamole, vitamin E, and NAC provide protection to E18 hippocampal neurons under conditions of oxidative stress. Neurons were plated in complete SFM and allowed to become established for 3 d. On the third day, cultures were treated with 10 μm FeSO4 (A) or 30 μm H2O2 (B) in complete SFM with the indicated additions. Cultures were not pretreated with the agents before addition of FeSO4 or H2O2. Cultures were assessed for survival 24 hr later. All survival data are expressed relative to the number of neurons in untreated control cultures.
Fig. 5.

Phase contrast micrographs of E18 hippocampal neurons maintained in complete SFM for 3 d followed by a 1 d treatment with no additives (complete SFM control) (A), 10 μm FeSO4(B), or 10 μm FeSO4plus 10 μm dipyridamole (C).
Neurons cultured in basal medium are under oxidative stress, and this is reduced by dipyridamole
The above findings suggested that dipyridamole protects neurons not by mimicking or replacing required neurotrophic support but rather by protecting them from oxidative stress. To determine whether the neurons cultured in basal medium were, in fact, under oxidative stress, we used the TBARS assay to measure membrane lipid peroxidation in cultures maintained under various conditions for 5–6 hr after plating. At this time, the cultures do not yet show overt signs of neuronal degeneration. The levels of TBARS in neurons cultured in basal medium were considerably higher than that for neurons cultured in SFM (176 ± 6% of the levels in SFM controls; n = 4), indicating a greater level of oxidative stress. When present in basal medium, dipyridamole substantially decreased the level of lipid peroxidation measured by the TBARS assay (119 ± 6% of the levels in SFM controls; n = 4). Similar results were obtained in two independent experiments. Comparable findings were achieved with cultures grown in complete SFM for 4–5 d and then challenged with 3–10 μm FeSO4 in the presence or absence of dipyridamole.
Iron as a major contributor to oxidative stress in basal medium
We next proceeded to investigate the potential source of oxidative stress in neurons cultured in basal medium. An important clue in this direction was furnished by testing the individual components of the normal supplements that are included in complete medium. None of these agents was able to protect hippocampal neurons in basal medium except apotransferrin (Table 1; Fig. 6). Addition of 100 μg/ml apotransferrin, the concentration that is present in complete SFM, protected ∼70% of the neurons after 1 d (Fig. 6). Based on the protective actions of the transferrin and the antioxidants, it seemed possible that such agents and dipyridamole protect the neurons from components of the MEM/F12 mixture that constitute basal medium and, in particular, from the pro-oxidant actions of iron. Examination of the components of basal medium revealed the presence of 1.5 μm FeSO4. We therefore assessed the hypothesis that, in the absence of supplements to the basal medium, iron-catalyzed production of free radicals is toxic to the neurons and that dipyridamole is protective because it inhibits this process. If this hypothesis were correct, one would predict that iron chelators should protect neurons in basal medium and that medium formulated without FeSO4 would not be as toxic to neurons as basal medium.
Fig. 6.

Transferrin promotes survival of primary hippocampal neurons in basal medium. Survival data are expressed relative to the number of neurons alive in complete SFM 1 d after plating (arbitrarily set at 100). Tf, Transferrin.
We tested the first prediction by plating neurons in basal medium supplemented with the iron chelators deferoxamine or mimosine (Hashiguchi and Takahashi, 1977; Ganeshaguru et al., 1980). Both deferoxamine and mimosine partially blocked neuronal death in a concentration-dependent manner. Both agents were maximally effective at a concentration of 30 μm, rescuing ∼70% of the neurons after 24 hr relative to cultures plated in complete SFM (Fig.7). This level of survival, although not complete, is comparable with that promoted by dipyridamole, vitamin E, or NAC. To exclude the possibility that the chelators might rescue neurons by a mechanism unrelated to their chelating properties as well as to ensure that the 30% death (relative to SFM) that occurs with dipyridamole and the chelators was not caused by iron bound to the chelators but still present in the medium, we incubated basal MEM/F12 with deferoxamine coupled to Sepharose beads. The medium was then centrifuged to remove the beads and bound iron. Neuronal survival in this iron-depleted medium was similar to survival in medium supplemented with the iron chelators (Figs. 7,8B). To assess the second prediction, we tested whether neurons plated in unsupplemented MEM (without F12), a formulation that does not contain iron, had any survival advantage over neurons plated in basal medium. Compared with neurons plated in complete SFM, there was ∼65–75% survival after 24 hr when neurons were plated in unsupplemented MEM, whereas in contrast, nearly all the neurons plated in unsupplemented MEM/F12 were dead (Fig.8A). There was little, if any, further enhancement of survival with addition of dipyridamole. Thus, removal of iron by chelators or culture in iron-free medium confers a significant degree of neuronal protection.
Fig. 7.

The iron chelators deferoxamine and mimosine promote survival of primary CNS neurons in basal medium. Dose–response relationships for the effects of deferoxamine and mimosine on survival of E18 hippocampal neurons in basal medium (1 d) are shown. Theclosed circle represents survival mediated by 10 μm dipyridamole. The open circle indicates survival in basal medium depleted of iron by immobilized deferoxamine (DF). All survival data are expressed relative to the number of neurons in complete SFM 1 d after plating.
Fig. 8.

Death of neurons is enhanced in basal medium (MEM/F12), compared with MEM, a culture medium lacking FeSO4. E18 hippocampal neurons were plated in the indicated medium and assessed for survival at daily intervals. A, Survival is enhanced in iron-free MEM compared with that in iron-containing basal medium and is diminished by the addition of
We next determined whether the presence of iron is sufficient to cause neuronal death. Various concentrations of FeSO4 were added to neurons plated in iron-free MEM medium. At 1.5 μm, the same concentration that is in basal medium, there was a significant decrease in viability relative to neurons cultured in MEM alone (Fig.8A). However, even when FeSO4 was added to reach concentrations as high as 10 μm, the degree of neuronal death was not as great as that observed in cultures plated in basal MEM/F12 (Fig. 8A). This suggests that although iron is a major factor in generation of oxidative stress in the cultures, there may be other contributing factors as well. To assess this possibility, we maintained neuronal cultures for 3 d in iron-free MEM or in MEM/F12 depleted of iron by the deferoxamine-coupled beads and with or without dipyridamole. Additionally, to ensure that the MEM did not contain trace amounts of iron that contributed to neuronal death over time, we also incubated this medium with deferoxamine-coupled beads. Under these conditions, neuronal death at 3 d of culture continued in MEM alone, deferoxamine-treated MEM, and deferoxamine-treated MEM/F12 but was significantly arrested in the presence of dipyridamole in all three media (Fig. 8B). Moreover, the presence of vitamin E in deferoxamine-treated MEM or deferoxamine-treated MEM/F12 also rescued ∼50% of the neurons at 3 d (Fig. 8B). Thus, even in the absence of iron, there seems to be a source of oxidative stress that is alleviated by dipyridamole or vitamin E.
Contribution of trophic factors to acute and longer-term neuronal survival
Although dipyridamole conferred protection from death, the level of rescue achieved with this drug never reached >75–80% of that attained with complete SFM. This raised the possibility that neurotrophic support, which is present in complete SFM in the form of high concentrations of insulin, may be an additional factor in survival. We therefore assessed the effects of supplying insulin (but not other additives to complete medium) with or without dipyridamole. As shown in Figure 8C, insulin did not substantially enhance survival when added alone to basal medium. However, when added together with dipyridamole, the rescue was comparable with that in complete SFM. This finding suggests that although insulin alone cannot protect the neurons from oxidative stress, it can rescue an additional population when oxidative stress is effectively suppressed. This is corroborated by the observation that addition of insulin to iron-free MEM also brings the relative level of survival to 100% (Fig.8C).
As noted above, in contrast to complete SFM, dipyridamole provided neuroprotection for only a limited length of time in basal medium. One possible explanation for this is that neurons require both protection from oxidative stress and neurotrophic support and that provision of either alone is insufficient for long-term survival. To test this, we plated hippocampal neurons in basal medium supplemented with either dipyridamole, insulin, or dipyridamole plus insulin and assessed the survival of these neurons at 1 week. As anticipated, there was no survival with insulin alone and poor long-term survival with dipyridamole alone. However, a substantial proportion of the cells remained viable at 1 week in the presence of both dipyridamole and insulin (Fig. 9).
Fig. 9.

Trophic factors contribute to longer-term neuronal survival. E16 hippocampal neurons were plated in basal medium supplemented with either dipyridamole, insulin, or dipyridamole plus insulin and assessed for survival at 1 week. The survival data are expressed relative to the number of neurons alive in complete SFM 1 d after plating.
DISCUSSION
The reductionist approach has proved to be a powerful strategy for defining the variables that affect neuronal survival as well as for uncovering and characterizing drugs with neuroprotective activity. This has highlighted the importance of nutrients, antioxidants, and trophic factors. In the present study we used a serum-free basal medium to test the potential neuroprotective role of the drug dipyridamole. We report that although a wide variety of CNS neurons cannot survive in a standard basal medium, they are acutely supported under such conditions by dipyridamole. Moreover, dipyridamole permits prolonged survival in basal medium when supplied in conjunction with insulin.
Dipyridamole has multiple pharmacological actions that could potentially account for its neuroprotective effects. It was especially important to establish whether the survival-promoting actions of dipyridamole were attributable to mimicry of neurotrophic activity. For instance, the capacity of dipyridamole to inhibit phosphodiesterase activity was of particular interest in that elevated levels of cAMP or cGMP protect certain neurons from loss of trophic support. The ineffectiveness of other phosphodiesterase inhibitors in our system both eliminates a mechanism involving cyclic nucleotides and suggests an action other than exertion of trophic factor-like effects. Another means by which dipyridamole might have protected neurons was its inhibition of nucleoside transport that could have resulted in enhanced levels of extracellular adenosine that in turn might have promoted survival by promoting receptor-mediated elevation of intracellular cAMP and thereby providing trophic support. However, here again other inhibitors of this action were not neuroprotective. Additional evidence against a mechanism whereby dipyridamole exerts trophic factor-like actions includes the inability of high levels of insulin alone to inhibit death in basal medium and the ineffectiveness of dipyridamole as well as of vitamin E or 100 μm NAC to block neuronal death in other systems caused by withdrawal of trophic support (Ferrari et al., 1995). NAC has been found to protect neurons from loss of trophic support, but in these cases, much higher concentrations (20–60 mm) were required, and protection did not correlate with antioxidant activity (Ferrari et al., 1995; Yan et al., 1995). An array of additional agents that provide protection from withdrawal of neurotrophic factors also failed to rescue neurons in the present paradigm.
Positive evidence regarding the mechanism of dipyridamole was provided by the observations that its neuroprotective actions on neurons cultured in basal medium were effectively mimicked by the antioxidants vitamin E and NAC (100 μm) and that dipyridamole, like vitamin E and NAC, protected neurons from oxidative stress evoked by exposure to ferrous iron or peroxide. It has been demonstrated that dipyridamole, like vitamin E, is an effective free radical-scavenging agent (Iuliano et al., 1995) and that it is effective at inhibiting FeSO4-induced lipid peroxidation in experimental systems including rat lens and lung (De la Cruz et al., 1994, 1996). We show here that dipyridamole also substantially reduces the formation of TBARS in cultures grown in basal medium or in complete medium supplemented with 3–10 μm iron. Taken together, these findings reveal that a major reason neurons rapidly die in basal medium is that they are under oxidative stress and that this is alleviated by dipyridamole.
Our studies indicate that a major generator of oxidative stress for neurons in basal medium is iron. It has been well established that iron can cause oxidative stress by promoting lipid peroxidation as well as DNA and protein damage by catalyzing the formation of hydroxyl radicals (Halliwell and Gutteridge, 1986a,b). Moreover, a number of studies have shown that ferrous iron is toxic to cultured neurons (Brewer et al., 1993; Zhang et al., 1993; Chow et al., 1994). Our findings reinforce the potentially harmful effects of iron salts on cultured neurons, even when present at relatively low levels (1.5 μm in this case). The most likely mechanism by which dipyridamole protects neurons from iron seems to be by virtue of its capacity to scavenge free radicals. There is no evidence that dipyridamole directly interacts with iron, and the structure of dipyridamole does not suggest iron-chelating activity.
Among the evidence pointing to the possibility that FeSO4in basal medium might be responsible for oxidative stress and cell death was the observation that addition of apotransferrin alone provides protection similar to that conferred by dipyridamole. It was originally conceived that the role of transferrin in defined medium would be to provide a source of iron transport into cells (Bottenstein and Sato, 1979; Aizenman et al., 1986). However, transferrin binds iron and as such prevents its participation in formation of the hydroxyl radical and lipid peroxidation (Gutteridge et al., 1979, 1981). Thus, apotransferrin also seems to play an important neuroprotective role in defined medium by preventing iron-mediated oxidative stress.
Another piece of evidence implicating FeSO4 as a source of oxidative stress in our cultures was the observation that the iron chelators deferoxamine and mimosine are neuroprotective in basal medium. It was established previously that these agents protect PC12 cells and sympathetic neurons from trophic factor withdrawal (Farinelli and Greene, 1996). However, this effect occurs only at 10–100-fold higher concentrations than that required here to protect CNS neurons. This suggests differences in the mechanisms of death as well as in the actions of the iron chelators in the two systems. In the trophic factor deprivation system, iron was not present in the medium, and it was postulated that the neuroprotective effects of mimosine and deferoxamine were attributable to their known actions as cell cycle blockers at the G1/S transition rather than to protection from oxidative stress. In contrast, for the current studies, the low concentrations of the chelators were likely protective because they bound iron present in the basal medium. This is corroborated by our observation that incubating basal medium with immobilized deferoxamine was equally effective in preventing death.
Our observations indicate that iron alone is not the sole source of the oxidative stress on cultured neurons. Although addition of FeSO4 to MEM produced substantial levels of death, the effect was not as great as that observed in basal medium. Moreover, neurons did not show longer-term survival in iron-free MEM unless dipyridamole was present. The source of such stress is presently unclear but may reflect both the composition of the culture medium as well as the status of intracellular antioxidant mechanisms.
Even though oxidative stress was a major factor in the death of neurons, it was not the only contributory element. Exposure to dipyridamole or other antioxidants or the use of iron-free medium did not support the same level of acute survival as that obtained with complete SFM. Moreover, suppression of oxidative stress by vitamin E or dipyridamole was not in itself sufficient to maintain long-term support of neurons. Survival of embryonic neurons both in vivo andin vitro requires an adequate supply of trophic factors. We observed that provision of insulin in addition to dipyridamole resulted in undiminished acute survival (relative to complete SFM) as well as longer-term maintenance. Insulin and insulin-like growth factors (whose receptors are activated by the high concentrations of insulin used here) have been shown to possess widespread trophic actions on neurons (Snyder and Kim, 1979; Skaper et al., 1982; Huck, 1983). It therefore seems likely that the incomplete survival of dipyridamole-treated neurons on day 1 in culture as well as their subsequent loss over time reflects the absence of required trophic support and that this is provided, at least in part, by insulin. Optimal neuronal survival in culture was thus achieved when the neurons received both protection from oxidative stress and a source of trophic activity. Such findings substantiate the points that oxidative stress and trophic factor deprivation evoke death by means of distinct pathways and that trophic factors and antioxidants promote survival by different mechanisms.
The present studies support the potential use of dipyridamole or its analogs as effective agents for protection of neurons from oxidative stress. In contrast to vitamin E, which acts only at the cell membrane, dipyridamole is an effective scavenger in both the aqueous and lipid phases (Iuliano et al., 1995). It is also notable that dipyridamole was optimally active in our experiments at 10 μm, whereas comparable efficacy of vitamin E and NAC required concentrations of approximately an order of magnitude higher. In an animal model of induced stroke, an analog of dipyridamole that passes the blood–brain barrier provided significant levels of protection (De la Cruz, 1992). Given the suggested role of free radicals in neuronal death after ischemia or trauma (Halliwell, 1992) or in neurodegenerative disorders (Gerlach et al., 1994) as well as the recent report that vitamin E administration significantly delays progression of Alzheimer’s disease (Sano et al., 1997), our in vitro findings lend credence to the potential use of dipyridamole or its derivatives in prevention and/or treatment of certain injuries and diseases of the nervous system.
Footnotes
This work was supported in part by National Institutes of Health-National Institute of Neurological Disorders and Stroke (NINDS) Grants 31357 (W.J.F.) and 33689 (L.A.G.) and by grants from the Amyotrophic Lateral Sclerosis Foundation and the Blanchette Rockefeller Foundation. S.E.F. was supported in part by a National Research Service Award from the NINDS. We thank Dania Alarcon-Vargas for her excellent technical assistance.
Correspondence should be addressed to Dr. Lloyd A. Greene, Department of Pathology and Center for Neurobiology and Behavior, Columbia University, College of Physicians and Surgeons, 630 West 168th Street, New York, NY 10032.
REFERENCES
- 1.Aizenman Y, Weichsel ME, Jr, de Vellis J. Changes in insulin and transferrin requirements of pure brain neuronal cultures during embryonic development. Proc Natl Acad Sci USA. 1986;83:2263–2266. doi: 10.1073/pnas.83.7.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ayesh S, Shao YM, Stein WD. Co-operative, competitive and non-competitive interactions between modulators of p-glycoprotein. Biochim Biophys Acta. 1996;1316:8–18. doi: 10.1016/0925-4439(96)00008-7. [DOI] [PubMed] [Google Scholar]
- 3.Barbin G, Selak I, Manthorpe M, Varon S. Use of central neuronal cultures for the detection of neuronotrophic agents. Neuroscience. 1984;12:33–43. doi: 10.1016/0306-4522(84)90135-0. [DOI] [PubMed] [Google Scholar]
- 4.Batistatou A, Greene LA. Aurintricarboxylic acid rescues PC12 cells and sympathetic neurons from cell death caused by nerve growth factor deprivation: correlation with suppression of endonuclease activity. J Cell Biol. 1991;115:461–471. doi: 10.1083/jcb.115.2.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci USA. 1979;76:514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bottenstein JE, Skaper SD, Varon SS, Sato GH. Selective survival of neurons from chick embryo sensory ganglionic dissociates utilizing serum-free supplemented medium. Exp Cell Res. 1980;125:183–190. doi: 10.1016/0014-4827(80)90202-5. [DOI] [PubMed] [Google Scholar]
- 7.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 8.Buege JA, Aust SD. Microsomal lipid peroxidation. Methods Enzymol. 1978;52:303–310. doi: 10.1016/s0076-6879(78)52032-6. [DOI] [PubMed] [Google Scholar]
- 9.Chang JY, Korolev VV, Wang JZ. Cyclic AMP and pituitary adenylate cyclase-activating polypeptide (PACAP) prevent programmed cell death of cultured rat cerebellar granule cells. Neurosci Lett. 1996;206:181–184. doi: 10.1016/s0304-3940(96)12468-x. [DOI] [PubMed] [Google Scholar]
- 10.Chow HS, Lynch JJ, III, Rose K, Choi DW. Trolox attenuates cortical neuronal injury induced by iron, ultraviolet light, glucose deprivation or AMPA. Brain Res. 1994;639:102–108. doi: 10.1016/0006-8993(94)91769-8. [DOI] [PubMed] [Google Scholar]
- 11.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci USA. 1991;88:6368–6371. doi: 10.1073/pnas.88.14.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De la Cruz JP, Villalobos MA, Carrasco T, Smith-Agreda JM, Sanchez de la Cuesta F. The pyrimido-pyrimidine derivative RA-642 protects from brain injury in a combined model of permanent focal ischemia and global ischemia reperfusion. Brain Res. 1992;597:250–256. doi: 10.1016/0006-8993(92)91481-s. [DOI] [PubMed] [Google Scholar]
- 13.De la Cruz JP, Moreno A, Merida F, Garcia-Campos J, Sanchez de la Cuesta F. The pyrimido-pyrimidine derivatives, dipyridamole and RA-642, reduce opacification of crystalline lens in diabetic rats. Pharmacol Toxicol. 1994;75:250–254. doi: 10.1111/j.1600-0773.1994.tb00356.x. [DOI] [PubMed] [Google Scholar]
- 14.De la Cruz JP, Olveira C, Gonzalez-Correa JA, Benitez A, Sanchez de la Cuesta F. Inhibition of ferrous-induced lipid peroxidation by dipyridamole, RA-642 and mopidamol in human lung tissue. Gen Pharmacol. 1996;27:855–859. doi: 10.1016/0306-3623(95)02098-5. [DOI] [PubMed] [Google Scholar]
- 15.di Porzio U, Daguet MC, Glowinski J, Prochiantz A. Effect of striatal cells on in vitro maturation of mesencephalic dopaminergic neurones grown in serum-free conditions. Nature. 1980;288:370–373. doi: 10.1038/288370a0. [DOI] [PubMed] [Google Scholar]
- 16.Farinelli SE, Greene LA. The cell cycle blockers mimosine, ciclopirox and deferoxamine prevent the death of PC12 cells and post-mitotic sympathetic neurons following removal of trophic support. J Neurosci. 1996;16:1150–1162. doi: 10.1523/JNEUROSCI.16-03-01150.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farinelli SE, Park DS, Greene LA. Nitric oxide delays the death of trophic factor-deprived PC12 cells and sympathetic neurons by a cGMP-mediated mechanism. J Neurosci. 1996a;16:2325–2334. doi: 10.1523/JNEUROSCI.16-07-02325.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farinelli SE, Friedman WJ, Greene LA. Natriuretic peptides prevent the death of trophic factor-deprived PC12 cells and promote the survival of basal forebrain cholinergic neurons. Soc Neurosci Abstr. 1996b;22:1485. [Google Scholar]
- 19.Feng M, van der Does L, Bantjes A. Iron III chelating resins–I. Preparation and properties of Sepharose-desferrioxamine gels. J Biomater Sci Polym Ed. 1992;4:99–105. [PubMed] [Google Scholar]
- 20.Ferrari G, Yan CYI, Greene LA. N-Acetylcysteine (d- and l-stereoisomers) prevents apoptotic death of neuronal cells. J Neurosci. 1995;15:2857–2866. doi: 10.1523/JNEUROSCI.15-04-02857.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Friedman WJ, Ibanez CF, Hallbook F, Persson H, Cain LD, Dreyfus CF, Black IB. Differential actions of neurotrophins in the locus coeruleus and basal forebrain. Exp Neurol. 1993;119:71–78. doi: 10.1006/exnr.1993.1007. [DOI] [PubMed] [Google Scholar]
- 22.Furukawa K, Estus S, Fu W, Mark RJ, Mattson MP. Neuroprotective action of cycloheximide involves induction of bcl-2 and antioxidant pathways. J Cell Biol. 1997;136:1137–1149. doi: 10.1083/jcb.136.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ganeshaguru K, Hoffbrand AV, Grady RW, Cerami A. Effects of various iron chelating agents on DNA synthesis in human cells. Biochem Pharmacol. 1980;29:1275–1279. doi: 10.1016/0006-2952(80)90285-3. [DOI] [PubMed] [Google Scholar]
- 24.Garcia AM, Lodish HF. Lysine 539 of human band 3 is not essential for ion transport or inhibition by stilbene disulfonates. J Biol Chem. 1989;264:19607–19613. [PubMed] [Google Scholar]
- 25.Gerlach M, Ben-Shachar D, Riederer P, Youdim MBH. Altered brain metabolism of iron as a cause of neurodegenerative diseases? J Neurochem. 1994;63:793–807. doi: 10.1046/j.1471-4159.1994.63030793.x. [DOI] [PubMed] [Google Scholar]
- 26.Gutteridge JMC, Richmond R, Halliwell B. Inhibition of the iron-catalyzed formation of hydroxyl radicals from superoxide and of lipid peroxidation by desferrioxamine. Biochem J. 1979;184:469–472. doi: 10.1042/bj1840469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gutteridge JMC, Paterson SK, Segal AW, Halliwell B. Inhibition of lipid peroxidation by the iron-binding protein lactoferrin. Biochem J. 1981;199:259–261. doi: 10.1042/bj1990259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 29.Halliwell B, Gutteridge JMC. Iron and free radical reactions: two aspects of antioxidant protection. Trends Biochem Sci. 1986a;11:372–375. [Google Scholar]
- 30.Halliwell B, Gutteridge JMC. Oxygen free radicals and iron in relation to biology and medicine: some problems and concepts. Arch Biochem Biophys. 1986b;246:501–514. doi: 10.1016/0003-9861(86)90305-x. [DOI] [PubMed] [Google Scholar]
- 31.Hartell NA. Inhibition of cGMP breakdown promotes the induction of cerebellar long-term potentiation. J Neurosci. 1996;16:2881–2890. doi: 10.1523/JNEUROSCI.16-09-02881.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hashiguchi H, Takahashi H. Inhibition of two copper-containing enzymes, tyrosine and dopamine β-hydroxylase, by l-mimosine. Mol Phamacol. 1977;13:362–367. [PubMed] [Google Scholar]
- 33.Huck S. Serum-free medium for cultures of the postnatal mouse cerebellum: only insulin is essential. Brain Res Bull. 1983;10:667–674. doi: 10.1016/0361-9230(83)90036-9. [DOI] [PubMed] [Google Scholar]
- 34.Iuliano L, Violi F, Ghiselli A, Alessandri C, Balsano F. Dipyridamole inhibits lipid peroxidation and scavenges oxygen radicals. Lipids. 1989;24:430–433. [Google Scholar]
- 35.Iuliano L, Pedersen JZ, Rotilio G, Ferro D, Violi F. A potent chain-breaking antioxidant activity of the cardiovascular drug dipyridamole. Free Radic Biol Med. 1995;18:239–247. doi: 10.1016/0891-5849(94)e0123-z. [DOI] [PubMed] [Google Scholar]
- 36.Jackson CA, Greaves M, Preston FE. A study of the stimulation of human venous prostacyclin synthesis by dipyridamole. Thrombosis Res. 1982;27:563–573. doi: 10.1016/0049-3848(82)90303-6. [DOI] [PubMed] [Google Scholar]
- 37.Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen H-C, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marcoz P, Prigent AF, Lagarde M, Nemoz G. Modulation of rat thymocyte proliferative response through the inhibition of different cyclic nucleotide phosphodiesterase isoforms by means of selective inhibitors and cGMP-elevating agents. Mol Pharmacol. 1993;44:1027–1035. [PubMed] [Google Scholar]
- 39.Maurice DH, Haslam RJ. Molecular basis of the synergistic inhibition of platelet function by nitrovasodilators and activators of adenylate cyclase: inhibition of cyclic AMP breakdown by cyclic GMP. Mol Pharmacol. 1990;37:671–681. [PubMed] [Google Scholar]
- 40.Mesner PW, Winters TR, Green SH. Nerve growth factor-withdrawal induced cell death in neuronal PC12 cells resembles that in sympathetic neurons. J Cell Biol. 1992;119:1669–1680. doi: 10.1083/jcb.119.6.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller TM, Tansey MG, Johnson EM., Jr Inhibition of phosphotidylinositol 3-kinase activity blocks depolarization- and insulin-like growth factor I-mediated survival of cerebellar granule cells. J Biol Chem. 1997;272:9847–9853. doi: 10.1074/jbc.272.15.9847. [DOI] [PubMed] [Google Scholar]
- 42.O’Neill MJ, Hicks C, Ward M. Neuroprotective effects of 7-nitroindazole in the gerbil model of global cerebral ischaemia. Eur J Pharmacol. 1996;310:115–122. doi: 10.1016/0014-2999(96)00387-1. [DOI] [PubMed] [Google Scholar]
- 43.Park D, Farinelli SE, Green LA. Inhibitors of cyclin dependent kinases promote survival of post-mitotic neuronally differentiated PC12 cells and sympathetic neurons. J Biol Chem. 1996;271:8161–8169. doi: 10.1074/jbc.271.14.8161. [DOI] [PubMed] [Google Scholar]
- 44.Rukenstein A, Rydel RE, Greene LA. Multiple agents rescue PC12 cells from serum-free cell death by translation- and transcription-independent mechanisms. J Neurosci. 1991;11:2552–2563. doi: 10.1523/JNEUROSCI.11-08-02552.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rydel RE, Greene LA. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci USA. 1988;85:1257–1261. doi: 10.1073/pnas.85.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanchez-Olea R, Morales M, Garcia O, Pasantes-Morales H. Cl channel blockers inhibit the volume-activated efflux of Cl and taurine in cultured neurons. Am J Physiol. 1996;270:C1703–C1708. doi: 10.1152/ajpcell.1996.270.6.C1703. [DOI] [PubMed] [Google Scholar]
- 47.Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, Schneider LS, Thal LJ. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. N Engl J Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 48.Selak I, Skaper SD, Varon S. Pyruvate participation in the low molecular weight trophic activity for central nervous system neurons in glia-conditioned media. J Neurosci. 1985;5:23–28. doi: 10.1523/JNEUROSCI.05-01-00023.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Skaper SD, Adler R, Varon S. A procedure for purifying neuron-like cells in cultures from central nervous tissue with a defined medium. Dev Neurosci. 1979;2:233–237. doi: 10.1159/000112485. [DOI] [PubMed] [Google Scholar]
- 50.Skaper SD, Selak I, Varon S. Molecular requirements for survival of cultured avian and rodent dorsal root ganglionic neurons responding to different trophic factors. J Neurosci Res. 1982;8:251–261. doi: 10.1002/jnr.490080215. [DOI] [PubMed] [Google Scholar]
- 51.Snyder EY, Kim SU. Hormonal requirements for neuronal survival in culture. Neurosci Lett. 1979;13:225–230. doi: 10.1016/0304-3940(79)91498-8. [DOI] [PubMed] [Google Scholar]
- 52.Soto AM, Sonnenschein C. The role of estrogens on the proliferation of human breast tumor cells (MCF-7). J Steroid Biochem. 1985;23:87–94. doi: 10.1016/0022-4731(85)90265-1. [DOI] [PubMed] [Google Scholar]
- 53.Sparwasser C, Drescher P, Will JA, Madsen PO. Smooth muscle tone regulation in rabbit cavernosal and spongiosal tissue by cyclic AMP- and cyclic GMP-dependent mechanisms. J Urology. 1994;152:2159–2163. doi: 10.1016/s0022-5347(17)32343-1. [DOI] [PubMed] [Google Scholar]
- 54.Thompson WJ. Cyclic nucleotide phosphodiesterases: pharmacology, biochemistry and function. Pharmacol Ther. 1991;51:13–33. doi: 10.1016/0163-7258(91)90039-o. [DOI] [PubMed] [Google Scholar]
- 55.Thorn JA, Jarvis SM. Adenosine transporters. Gen Pharmacol. 1996;27:613–620. doi: 10.1016/0306-3623(95)02053-5. [DOI] [PubMed] [Google Scholar]
- 56.Varon S, Skaper SD, Barbin G, Selak I, Manthorpe M. Low molecular weight agents support survival of cultured neurons for the central nervous system. J Neurosci. 1984;4:654–658. doi: 10.1523/JNEUROSCI.04-03-00654.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yan CYI, Ferrari G, Greene LA. N-Acetylcysteine-promoted survival of PC12 cells is glutathione-independent but transcription-dependent. J Biol Chem. 1995;45:26827–26832. doi: 10.1074/jbc.270.45.26827. [DOI] [PubMed] [Google Scholar]
- 58.Zeevalk GD, Nicklas WJ. Acute excitotoxicity in chick retina caused by the unusual amino acids BOAA and BMAA: effects of MK-801 and kynurenate. Neurosci Lett. 1989;102:284–290. doi: 10.1016/0304-3940(89)90093-1. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y, Fredholm BB. Propentofylline enhancement of the actions of adenosine on neutrophil leukocytes. Biochem Pharmacol. 1994;48:2025–2032. doi: 10.1016/0006-2952(94)90501-0. [DOI] [PubMed] [Google Scholar]
- 60.Zhang Y, Tatsuno T, Carney JM, Mattson MP. Basic FGF, NGF, and IGFs protect hippocampal and cortical neurons against iron-induced degeneration. J Cereb Blood Flow Metab. 1993;13:378–388. doi: 10.1038/jcbfm.1993.51. [DOI] [PubMed] [Google Scholar]