

Abstract
Mutations in the gene encoding the K+ channel KCNQ2 cause neonatal epilepsy and myokymia, indicating that KCNQ2 regulates the excitability of CNS neurons and motor axons, respectively. We show here that KCNQ2 channels are functional components of axon initial segments and nodes of Ranvier, colocalizing with ankyrin-G and voltage-dependent Na+ channels throughout the CNS and PNS. Retigabine, which opens KCNQ channels, diminishes axonal excitability. Linopirdine, which blocks KCNQ channels, prolongs the repolarization of the action potential in neonatal nerves. The clustering of KCNQ2 at nodes and initial segments lags that of ankyrin-G during development, and both ankyrin-G and KCNQ2 can be coimmunoprecipitated in the brain. KCNQ3 is also a component of some initial segments and nodes in the brain. The diminished activity of mutant KCNQ2 channels accounts for neonatal epilepsy and myokymia; the cellular locus of these effects may be axonal initial segments and nodes.
Keywords: Kv, epilepsy, myelin, potassium channel, repolarization, neuromyotonia, M-current
Introduction
Voltage-gated sodium channels (Nav) mediate saltatory conduction of action potentials (APs) between the nodes of Ranvier. At nodes, these channels are clustered at high densities to the spectrin cytoskeleton by the adaptor protein, ankyrin-G (Arroyo and Scherer, 2000; Peles and Salzer, 2000; Rasband and Trimmer, 2001). The same molecular interactions are also found at axon initial segments, where neuronal action potentials are generated. Nodes also contain K+ channels. Three distinct K+ conductances have been recorded by patch clamping of mammalian nodes, the most prominent of which is a slowly activating current, Ks (Roper and Schwarz, 1989; Safronov et al., 1993). In mammals, Kv3.1b may generate the fast nodal current, Kf2 (Corrette et al., 1991; Devaux et al., 2003), but the channel responsible for Ks had not been identified.
We were intrigued by the report that a KCNQ2 mutation (R207W) causes myokymia (Dedek et al., 2001). Myokymia (Greek “myo,” muscle, plus “kyma,” wave) is an involuntary, repetitive muscle contraction that gives the appearance of rippling waves moving below the surface of the skin (Newsom-Davis, 1997; Vincent, 2000). Repetitive APs generated at ectopic locations in myelinated motor axons result in the repetitive activity of motor units, thereby causing myokymia and neuromyotonia, a highly related disorder (Gutmann et al., 2001). Myokymia–neuromyotonia occurs in various disorders, including episodic ataxia type-1, which is caused by dominant mutations in KCNA1, the gene that encodes Kv1.1 (Browne et al., 1994; Eunson et al., 2000). In addition, autoantibodies against Kv1.1 and Kv1.2 have been found in sera of many individuals with acquired neuromyotonia (Shillito et al., 1995). Thus, the abnormal function of Kv1.1 and Kv1.2 channels of myelinated motor axons causes inherited and acquired myokymia–neuromytonia.
KCNQ2 belongs to a family with five members, all of which have a membrane topology characteristic of the voltage-dependent K+ channel superfamily but possess large novel intracellular N and C termini (Robbins, 2001). Of these members, only KCNQ3 is known to coassemble with KCNQ2 to form heteromeric channels (Jentsch, 2000). KCNQ2 and KCNQ3 are colocalized in several neuronal populations (Wang et al., 1998; Cooper et al., 2000, 2001) and underlie the M-current (IM) of sympathetic ganglia and hippocampal pyramidal cells (Wang et al., 1998; Shah et al., 2002). The IM is modulated by metabotropic receptors via G-proteins, a classic example of vertebrate neuromodulation (Wang et al., 1998). Mutations in either KCNQ2 or KCNQ3 cause benign familial neonatal convulsions (BFNCs), suggesting that disruption of the IM causes excessive neuronal excitability (Castaldo et al., 2002).
Why does the R207W KCNQ2 mutation cause myokymia? This mutation eliminates a charged residue in the S4 voltage sensor, and in Xenopus oocytes, the voltage dependence of activation is shifted toward depolarized potentials and the activation kinetics are markedly slowed (Dedek et al., 2001). These changes are qualitatively similar to those resulting from some Kv1.1 mutants that cause myotonia (Zerr et al., 1998; Eunson et al., 2000). Kv1.1 is localized in the juxtaparanodal axonal membrane (Wang et al., 1993; Mi et al., 1995), a specialized region of myelinated axons (Scherer and Arroyo, 2002). Juxtaparanodal Kv1.1 channels form tetramers with the highly related Kv1.2 (Hopkins et al., 1994; Mi et al., 1995). Although M-channels have not been described in myelinated axons, the myokymic phenotype strongly suggested that KCNQ2 (and potentially KCNQ3) might be present. We have investigated this issue and herein report our results.
Materials and Methods
Generation of anti-KCNQ3N antisera. A 22 amino acid peptide sequence (AGDEERKVGLAPGDVEQVTLAL), corresponding to amino acids 36–57 from the N-terminal region of KCNQ3 (KCNQ3N), was selected for its high immunogenicity using MacVector software. BLAST database searches showed that the sequence was unique to KCNQ3 and fully conserved between mouse, rat, and human (Schroeder et al., 1998) (GenBank AAC36723, NP690887). The peptide was conjugated to keyhole limpet hemocyanin via a cysteine residue added at the C terminus of the peptide during synthesis. Two guinea pigs were immunized, and sera were collected (Animal Pharmacology Services, Healdsburg, CA). After screening experiments identified immunopositive sera, the antisera were purified against the peptide immunogen immobilized on 1 ml columns prepared using SulfoLink (Pierce, Rockford, IL). We used one of these affinity-purified antisera in all of the experiments reported here. The specificity of this antiserum was demonstrated by immunoblotting of cell lysates from human embryonic kidney cells transfected with KCNQ2 or KCNQ3 cDNA. No cross-reactivity with KCNQ2 was observed (data not shown).
Immunohistochemistry. Rats and mice were killed according to University of Pennsylvania Institutional Care and Use Committee guidelines. The sciatic nerves, spinal cords, and brains were embedded in OCT and frozen in acetone cooled with dry ice. Some animals were fixed by transcardial perfusion with 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4, and the dissected tissues were fixed for 30 min, cryoprotected with infiltration in 20% sucrose PB overnight, and embedded in OCT. Cryostat sections (5–10 μm thick) were thaw-mounted on Super-Frost Plus glass slides (Fisher Scientific, Pittsburgh, PA) and stored at –20°C. Teased nerve fibers were prepared from both fixed and unfixed adult rat sciatic nerves, dried on glass slides overnight at room temperature, and stored at –20°C. Sections and teased fibers were permeabilized by immersion in –20°C acetone for 10 min, blocked at room temperature for 1 hr with 5% fish skin gelatin (0.5% Triton X-100) in PBS, and incubated overnight at 4°C with various combinations of primary antibodies: a rabbit antisera against a peptide [KCNQ2N, diluted 1:200 (Cooper et al., 2001)] or a fusion protein [n-Q2, 1:1000 (Roche et al., 2002)] to the N-terminal region of KCNQ2, a guinea pig antiserum against the N-terminal region of KCNQ3 [KCNQ3N, 1:100 (Cooper et al., 2000, 2001)]; mouse monoclonal antibodies against NMDAR1 (1: 1000; Chemicon, Temecula, CA), Kv1.2 (1:50; Alomone Laboratories, Jerusalem, Israel), ankyrin-G [1:500 (Jenkins and Bennett, 2001; Jenkins et al., 2001)], or PanNav channels (1:50; Sigma, St. Louis, MO); rat monoclonal antibody against E-cadherin (1:50; Zymed, San Francisco, CA), or the heavy chain of neurofilament [NF-H, 1:10 (Lee et al., 1982, 1987)]. The slides were then washed several times and incubated with the appropriate fluorescein- and rhodamine-conjugated donkey cross-affinity-purified secondary antibodies (1:100; Jackson ImmunoResearch Laboratories, West Grove, PA). Slides were mounted with Vectashield (Vector Laboratories, Burlingame, CA), examined by epifluorescence with tetramethylrhodamine isothiocyanate and FITC optics on a Leica DMR light microscope, and photographed with a cooled Hamamatsu camera. Confocal microscopy was performed with a Leica TCS laser scanning confocal microscope.
Immunostainings obtained with the rabbit antisera against a peptide (KCNQ2N) (Cooper et al., 2001) or a fusion protein (n-Q2, 1:1000) (Roche et al., 2002) were identical in sciatic nerve, spinal cord, and brain. Only the results obtained with the rabbit antisera against the peptide (KCNQ2N) are shown here.
Immunoblots and immunoprecipitation. Cell lysates were prepared from HeLa cells that stably expressed mouse KCNQ2 or rat KCNQ3, as described previously (Wen and Levitan, 2002). Membranes were prepared from frozen brain, spinal cord, sciatic nerve, and muscle from Wistar rats (10–12 weeks old) following a modified procedure of Hartshorne and Catterall (1984). Tissues were homogenized in ice-cold 0.32 m sucrose, 5 mm Tris-Cl, pH 7.4, containing protease inhibitors [2 mm EDTA, 1 μg/ml leupeptin and aprotinin, and 0.5 mm phenylmethylsulfonyl fluoride (Sigma)], and the homogenates were centrifuged for 10 min at 750 × g. The supernatants were sedimented for 60 min at 17,000 × g, and the resulting pellets were resuspended in 1 mm EDTA, 5 mm TRIS, pH 8.2, plus protease inhibitors, homogenized, and placed on ice for 30 min. The lysate membranes were then centrifuged 40 min at 27,000 × g, and the pellet (P3) was resuspended in 150 mm NaCl, 25 mm Tris, pH 7.4, plus protease inhibitors and stored at –80°C. Protein concentration was determined using the Bio-Rad kit (Bio-Rad, Hercules, CA). From each sample 100 μg of protein were loaded on a 5% SDS-PAGE gel and then transferred onto a polyvinylidene difluoride membrane. Membranes were blocked for 1 hr with 5% powdered skim milk–0.5% Tween 20 in PBS and incubated with KCNQ2N (1:500) or the guinea pig affinity-purified antiserum against KCNQ3 (1:500). After several washes the blots were incubated in peroxidase-coupled secondary antibodies against rabbit or guinea pig (1:2000; Jackson ImmunoResearch) for 1 hr at room temperature, washed several times, and revealed using ECL plus (Amersham Biosciences, Arlington Heights, IL).
The P3 pellet (described above) was solubilized in 1.25% Triton X-100, 0.1 mm KCl, 10 mm Tris-HCl, pH 7.4, plus protein inhibitors, stored on ice for 15 min, and then centrifuged at 27,000 × g for 60 min. The protein concentration of the supernatant (S4) was determined, and 200 μg of protein was incubated overnight with the primary antibody in the presence of 0.1% BSA. The following antisera were used: rabbit antiserum against ankyrin-G (Jenkins and Bennett, 2001; Jenkins et al., 2001) or KCNQ2 (KCNQ2N) and the guinea pig antiserum against KCNQ3 (KCNQ3N). Each sample was then incubated for 30 min at 4°C with 30 μl of protein-G–agarose beads (Invitrogen, Carlsbad, CA). After washing three times with 0.1% Triton X-100, 1% BSA plus protease inhibitors, bound proteins were released by boiling in 20 μl of SDS sample buffer for 2 min at 90°C. The released proteins were transferred to a 5% SDS-PAGE gel and processed as described above for immunoblotting.
Electrophysiology. The sciatic nerve or optic nerves were quickly dissected out from Wistar rats and transferred into artificial CSF (ACSF) that contained (in mm): 126 NaCl, 3 KCl, 2 CaCl2, 2 MgSO4, 1.25 NaH2PO4, 26 NaHCO3, and 10 dextrose, pH 7.4–7.5. Sciatic nerves were cut into 2 cm segments and desheathed to maximize the access to drugs. During the experiment, the nerves were kept in ACSF at 37°C in a 95% O2–5% CO2 atmosphere perfused at a flow rate of 1–2 ml/min. For optic nerves, suction electrodes were used for recording of compound action potentials (CAPs) (Devaux et al., 2002). For sciatic nerves, a three-compartment recording chamber was used; the distal end was stimulated supramaximally (40 μsec duration) through two electrodes isolated with Vaseline, and recordings were performed at the proximal end. Signals were amplified, digitized at 500 Hz, and stored on a hard disk. The effects of linopirdine (Sigma; solubilized in 50% ethanol) and retigabine (Wyeth, Philadelphia, PA; solubilized in DMSO) were measured 30–45 min after application once they had reached a steady state. For pretreatment experiments, nerves were incubated in linopirdine (20 μm) for 40 min and then retigabine was applied (in addition to linopirdine). For recruitment analysis, the CAP amplitude was measured and plotted as a function of the stimulation intensity. For the refractory period analysis, two successive stimuli were applied at different intervals. The amplitude of the highest peak of the second CAP was measured and plotted as a function of the delay between the two stimuli.
Results
KCNQZ is a functional nodal K+ channel in peripheral nerve
Using isoform-specific antibodies against N-terminal sequences of KCNQ2 and KCNQ3, we immunolabeled fixed and unfixed teased fibers and sections of adult rat sciatic nerve (Fig. 1). There was intense KCNQ2 and KCNQ3 staining in unfixed nerves; staining was weaker or even absent in fixed nerves. Two different antisera localized KCNQ2 to narrow (∼1 μm) bands that were regularly spaced along the length of myelinated axons (Fig. 1A–C). We directly demonstrated that these bands were nodes of Ranvier; KCNQ2 staining showed complete concordance with other known nodal makers: Nav channel α subunits (PanNav) (Fig. 1C) and ankyrin-G (data not shown). To demonstrate that KCNQ2 was localized to the axonal membrane, we double-labeled teased fibers for KCNQ2 and neurofilament-heavy, a marker of the cytosolic compartment, and used laser scanning confocal microscopy to obtain 0.5-μm-thick optical sections. We observed that KCNQ2 labeling closely apposed the neurofilament labeling in the nodal region, demonstrating that the labeling was membrane associated (Fig. 1B). All PNS nodes appeared to contain KCNQ2, not only in the sciatic nerve but also in the dorsal and ventral roots and intramuscular nerves (data not shown). Finally, nodal KCNQ2 and juxtaparanodal Kv1.2 staining did not overlap (Fig. 1D), demonstrating that these two kinds of K+ channels have distinct localizations in myelinated axons.
Figure 1.
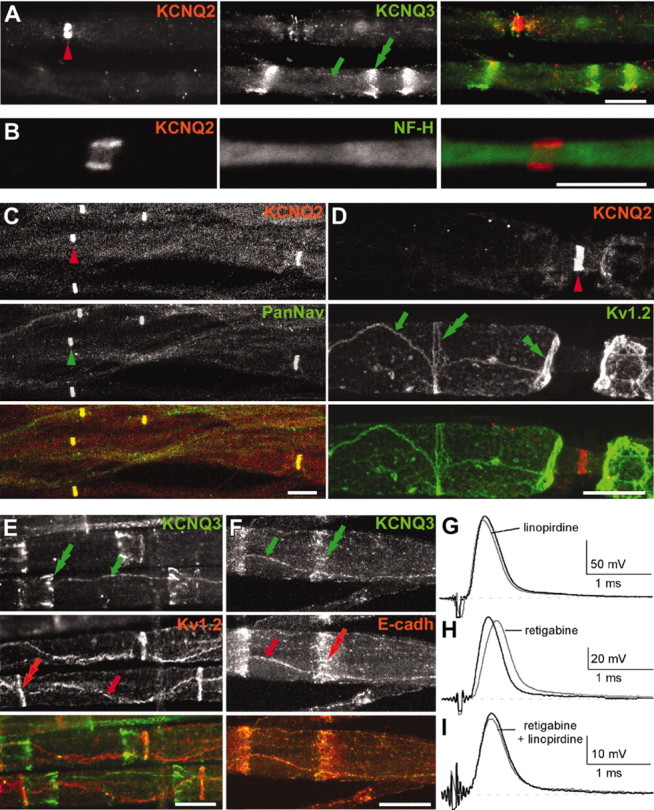
KCNQ2 is localized to PNS nodes of Ranvier. These are images of unfixed teased fibers from adult rat sciatic nerves immunolabeled for KCNQ2 (A–D) or KCNQ3 (A, E, F) and Nav channels α subunits (PanNav) (C), Kv1.2 (D, E), or E-cadherin (F). KCNQ2 and KCNQ3 are distinctly localized (A). A single optical section obtained by confocal microscopy shows that KNCQ2 (red) is localized to the axonal membrane, as shown by comparison to NF-H staining (B, green). KCNQ2 is colocalized with Nav channels at nodes of Ranvier (C), whereas KCNQ3 (A, E, F) is localized at incisures (green double arrows) and outer mesaxons (green arrows), where it is colocalized with E-cadherin (F). Kv1.2 is localized to the axonal membrane at juxtaparanodes (D, green double arrowheads), “juxta-mesaxons” (aligned with the glial inner mesaxon) (E, red arrows), and the “juxta-incisures” (aligned with the inner aspect of incisures) (E, red double arrows), locations that are distinct from those of KCNQ2 and KCNQ3. G–I, Compound action potential recorded from 3-month-old rat sciatic nerves. Linopirdine (100 μm) did not change the shape of the CAP recorded from adults (G). By contrast, retigabine (20 μm) delayed the onset of the CAP (H); these effects were blocked by pretreating the nerves with linopirdine (20 μm) (I). Scale bars: B, 5 μm; A, C–F, 10 μm.
Double labeling for KCNQ3 and KCNQ2 directly demonstrated that these two subunits were not colocalized (Fig. 1A). KCNQ3 immunoreactivity was found in paranodes, in Schmidt-Lanterman incisures, and in the outer mesaxon of myelin sheaths, colocalizing with E-cadherin (Fig. 1F), an adhesion molecule previously shown in these same structures (Fannon et al., 1995). Thus, in contrast to KCNQ2, KCNQ3 is expressed by Schwann cells and localized to noncompact myelin. Double-labeling for KCNQ3 and Kv1.2 showed that the “juxtaincisural” staining of axonal Kv1.2 (Arroyo et al., 1999) complemented the incisural staining of KCNQ3 in myelin sheaths (Fig. 1E).
The above data suggested that KCNQ2 forms a nodal K+ channel. To investigate the role of KCNQ channels in modulating the excitability of myelinated axons, we studied the effects of compounds that affect KCNQ channels on 3-month-old rat sciatic nerves. Linopirdine (100 μm), a selective KCNQ channel blocker (Wang et al., 1998), had no significant effects on either the kinetics or the amplitude of the CAP (Fig. 1G) or on the refractory period of the nerves (data not shown). Retigabine (20 μm), a compound that shifts the threshold of activation of KCNQ2–5 channels to more negative potentials (Rundfeldt and Netzer, 2000), delayed the time-to-peak of the CAP from 0.27 ± 0.02 to 0.34 ± 0.01 msec (n = 4; p < 0.05) and increased its duration from 3.2 ± 0.3 to 4.8 ± 0.4 msec (n = 4; p < 0.01) (Fig. 1H). Both of these effects were abolished by linopirdine (20 μm) (Fig. 1I) or TEA (10 mm; data not shown). These data, taken together, indicate that functional KCNQ channels are present at PNS nodes.
KCNQ2 is localized to CNS nodes and initial segments
Although spinal cord neurons, including motoneurons, express KCNQ2 mRNA (Dedek et al., 2001), KCNQ2 has not been reported in CNS nodes (Wang et al., 1998; Cooper et al., 2000, 2001). We therefore immunolabeled sections of unfixed rat spinal cord for KCNQ2, using Nav and ankyrin-G antibodies to mark the locations of nodes. As in peripheral nerve, nodes were KCNQ2 positive (Fig. 2B,C), and KCNQ2 nodal staining did not overlap with Kv1.2 juxtaparanodal staining (Fig. 2D). Finally, we detected KCNQ2 in initial segments throughout the gray matter, colocalized with Nav or ankyrin-G (Fig. 2B, insets). Motoneuron cell bodies themselves, the largest neurons (10–30 μm in diameter) in the ventral horn, were diffusely KCNQ2 positive, but their initial segments were intensely labeled (Fig. 2A).
Figure 2.
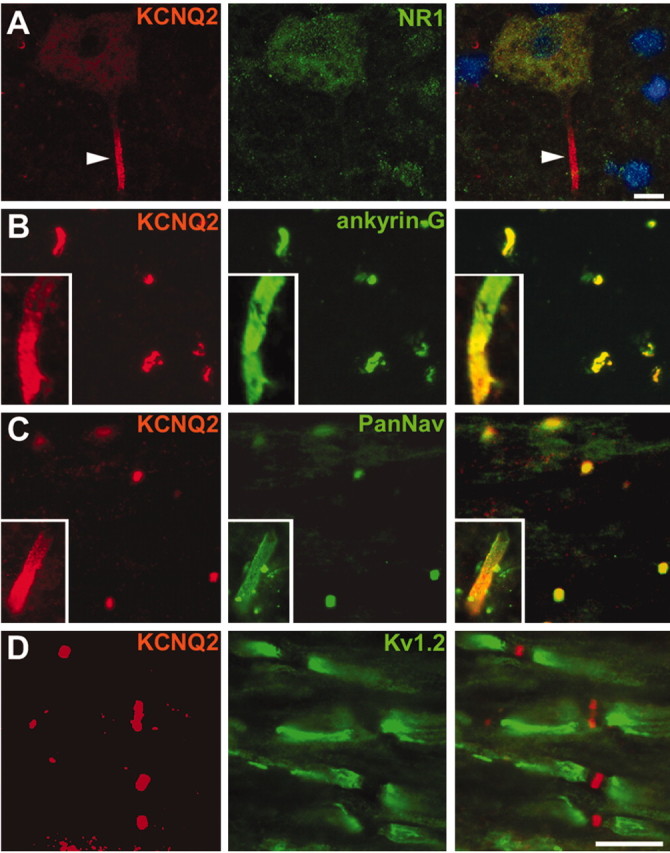
KCNQ2 is localized to CNS nodes and initial segments. These are images of sections of unfixed rat spinal cord, immunolabeled for KCNQ2 and NMDAR1 (A), ankyrin-G (B), Nav (C), or Kv1.2 (D); the merged image in A was also stained with the nuclear counterstain 4′,6′-diamidino-2-phenylindole. KCNQ2 labeling is concentrated in an initial segment (arrowhead) of an NMDAR1-positive motoneuron found in the ventral horn of spinal cord (A). KCNQ2 is colocalized with both ankyrin-G (B) and Nav (C) at nodes in the white matter and also at initial segments (B, C, insets are from motoneurons). By contrast, Kv1.2 is confined to the juxtaparanodes and does not colocalize with KCNQ2 (D). Scale bars: (including insets) 10 μm.
Distribution of KCNQ3 in spinal cord
To determine whether KCNQ3 colocalized with KCNQ2, we double- or triple-labeled unfixed spinal cord sections for KCNQ3 and KCNQ2, as well as for Kv1.2, Kv3.1b, and ankyrin-G. The entire gray matter had robust but diffuse KCNQ3 staining, but it was distinct from that of Kv1.2, Kv3.1b (data not shown), and KCNQ2; KCNQ3 and KCNQ2 staining showed little overlap. No KCNQ3 labeling was seen in neuronal cell bodies or initial segments, including those of motoneurons (Fig. 3A). White matter contained KCNQ3-positive strands that were in GFAP-positive cells (Fig. 3D), indicating that astrocytes express KCNQ3. In white matter, we could detect KCNQ3 staining in many but not all nodes (Fig. 3B). Nodal KCNQ3 staining was not apparent in gray matter, which contains an even higher density of nodes (Arroyo et al., 2001). Both the smaller sizes of nodes and the higher level of KCNQ3 staining in gray matter may contribute to the difficulty in detecting KCNQ3-positive nodes. In white matter, some KCNQ3 node-like clusters appeared to overlap with tenascin-R (Fig. 3C), a marker of nodal astrocytic processes (ffrench-Constant et al., 1986; Bartsch et al., 1992; Arroyo et al., 2001). Whether the KCNQ3 immunoreactivity detected at spinal cord nodes is neuronal or associated with the apposed membranes of the astrocytic end feet is beyond the resolution afforded by light microscopy. Finally, we also noted KNCQ3 labeling of the pial membrane, capillaries (see Fig. 5B, asterisk), and ependymal cells; in each of these cell types, KCNQ3 immunoreactivity was highly clustered at cell borders (data not shown).
Figure 3.
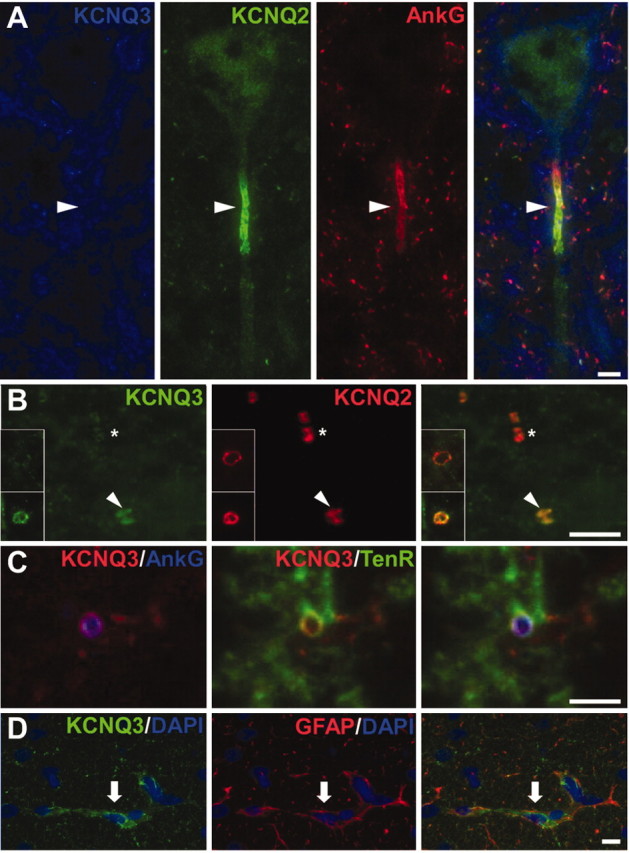
Distribution of KCNQ3 in the spinal cord. These are images of sections of unfixed rat spinal cord, immunostained as indicated. A, KCNQ3 staining surrounds the soma of a motoneuron in the ventral horn of the spinal cord but does not label the soma itself or the initial segment (arrowhead). B, Longitudinal section of the white matter stained for KCNQ3 (green) and KCNQ2 (red). Some KCNQ2-positive nodes are also KCNQ3 positive (arrowheads), whereas others are KCNQ3 negative (asterisks). Insets show transverse sections of both KCNQ3-positive (bottom) and KCNQ3-negative (top) nodes. C, Transverse section of the white matter stained for KCNQ3, ankyrin-G, and tenascin-R. KCNQ3 and tenascin-R appear to surround ankyrin-G-positive nodes. D, Transverse section of the white matter stained for KCNQ3 and GFAP, showing partially overlapping immunoreactivity in astrocytes (arrows). Scale bars: B, C, 5 μm; A, D, 10 μm.
Figure 5.

Colocalization of KCNQ2 and KCNQ3 in initial segments of cortex. These are images of horizontal sections of unfixed mouse brain immunolabeled for KCNQ2, KCNQ3, and ankyrin-G; DAPI was used as a nuclear counterstain in A and B. In the CA3 region of the hippocampus, many initial segments in the stratum pyramidale (sp), as well as the mossy fibers of the stratum lucidum (sl), are strongly KCNQ2 positive (A). The stratum radiatum (sr) and stratum oriens (so) are indicated. KCNQ3 was found with KCNQ2 in the initial segments of some pyramidal cells in CA3 but also in the mossy fibers (B). KCNQ3 colocalized with both ankyrin-G and KCNQ2 in the initial segment of neurons from the CA1 (C) and temporal neocortex (D). The asterisk in B marks a KCNQ3-positive blood vessel. Scale bars, 20 μm.
Ankyrin-G clustering precedes that of KCNQ2
Ankyrin-G is the first component that becomes clustered at nodes and initial segments during development (Jenkins and Bennett, 2001, 2002). To evaluate KCNQ2 in this context, we examined immunolabeled sections of unfixed rat spinal cord at intervals of postnatal development. At postnatal day (P) 4, 26% of ankyrin-G nodal clusters in presumptive white matter tracts were KCNQ2 positive (n = 84) (Fig. 4A); none of the ankyrin-G-positive initial segments in gray matter were KCNQ2 positive (Fig. 4A, insets). At P8, both the number and the density of ankyrin-G-positive nodes were higher, and 68% were KCNQ2 positive (n = 393) (Fig. 4B); many initial segments were also KCNQ2 positive (Fig. 4B, insets). At P12 and P15, the percentage of KCNQ2-positive nodes increased to 81% (n = 382) and 91% (n = 263) (Fig. 4C), respectively. Thus, KCNQ2 clustering at nodes and initial segments, like Nav, occurs after that of ankyrin-G. To determine whether KCNQ2 is retained at nodes after demyelination, we double-labeled sections of P21 myelin-deficient rat spinal cord. In these mutants, clusters of ankyrin-G and Nav channels initially form adjacent to ensheathed axonal segments and persist even after oligodendrocyte death (Arroyo et al., 2002). Similarly, KCNQ2 colocalized with Nav in node-like clusters (Fig. 4D). Thus, the axoglial contact is not essential to maintain node-like KCNQ2 clusters.
Figure 4.
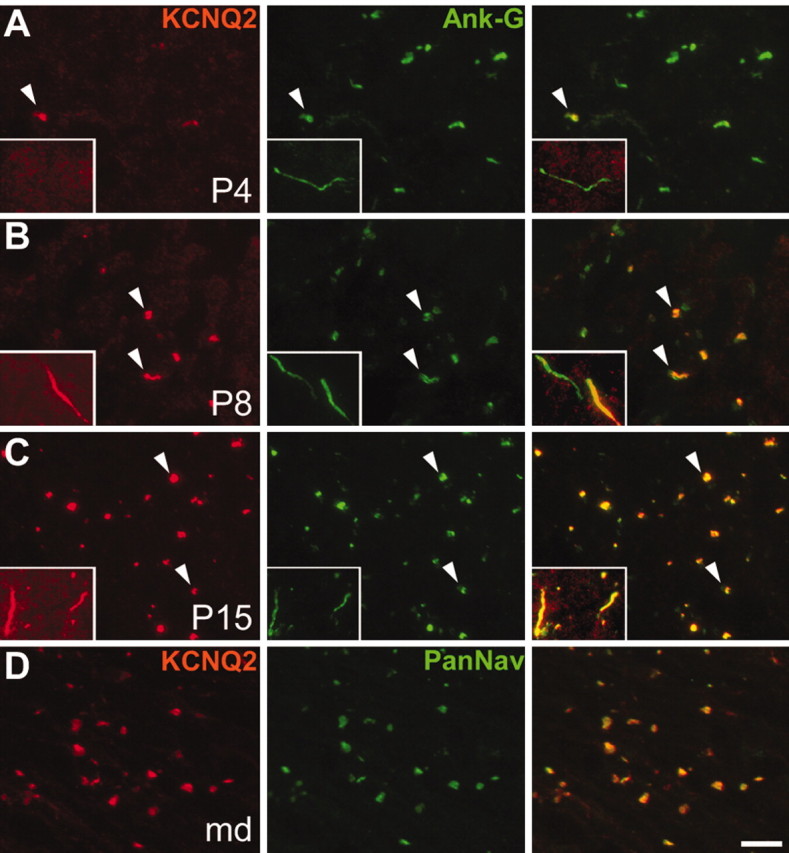
Localization of KCNQ2 in the developing rat spinal cord. These are images of unfixed longitudinal sections of spinal cord from P4 (A), P8 (B), and P15 (C) rats, as well as P21 myelin-deficient rats (D), double labeled for KCNQ2 (red) and ankyrin-G or Nav (green). At P4 (A), few nodes (arrowheads) and no initial segments (insets) are KCNQ2 positive. At P8 (B), many nodes (arrowheads) and initial segments (insets) are KCNQ2 positive. At P15 (C), nearly all nodes and initial segments are KCNQ2 positive. In the spinal cord white matter of P21 myelin-deficient rats (D), where axons are undergoing demyelination because of oligodendrocytes cell death, KCNQ2 remained colocalized with Nav in node-like clusters. Scale bar: (including insets) 10 μm.
Distribution of KCNQ2 and KCNQ3 in brain
Previous studies did not note KCNQ2 in nodes or initial segments of hippocampal and cortical neurons (Wang et al., 1998; Cooper et al., 2000, 2001), although Nav and ankyrin-G are known to be present there (Komada and Soriano, 2002). To investigate this issue, we double- and triple-labeled horizontal sections of unfixed mouse brain for KCNQ2, KCNQ3, and ankyrin-G. In CA3 (Fig. 5A), CA1 (Fig. 5C), and the polymorphic layer of the dentate gyrus (data not shown), most ankyrin-G-positive initial segments were KCNQ2 positive. As reported previously (Wang et al., 1998; Cooper et al., 2001), KCNQ2 labeling was seen in the somatodendritic compartment of pyramidal cells but was more intense in the axons of the mossy fibers layer (Fig. 5A, B). In our material, however, the most pronounced KCNQ2 staining was in initial segments, although this differed strikingly between brain regions. Many initial segments were strongly KCNQ2 positive in the brainstem, striatum, and neocortex (Fig. 5D), particularly layer 3, whereas those in the cerebellum (Purkinje cells) and olfactory bulb typically had little or no KCNQ2 staining (data not shown). This variation was reliably and reproducibly observed, even in the same section. The proportion of nodal KCNQ2 also appeared to vary in different brain regions, raising the possibility that the neurons that have KCNQ2 in their initial segments also have KCNQ2 in their nodes.
Unlike spinal cord, many initial segments of pyramidal neurons in CA3 (Fig. 5B), CA1 (Fig. 5C), and temporal neocortex (Fig. 5D), as well as striatum (data not shown), were both KCNQ2 and KCNQ3 positive. KCNQ3 labeling was typically weaker than that of KCNQ2, and some KCNQ2-positive initial segments were KCNQ3 negative (even in regions with the highest proportion of KCNQ2 labeling). All KCNQ3-positive initial segments were also KCNQ2 positive. These results, taken together, suggest that a subset of neurons express KCNQ3 in their initial segments, always in association with KCNQ2, which in turn is present in most but not all initial segments.
KCNQ2 and KCNQ3 are present in the myelinated tracts
To confirm the presence of KCNQ2 and KCNQ3 in myelinated tracts, we prepared immunoblots of membranes from rat sciatic nerve, spinal cord, and brain. HeLa cells that expressed mouse KCNQ2 or rat KCNQ3 were used as positive controls and molecular weight indicators for KCNQ2 and KCNQ3; skeletal muscle membranes were used as a negative control. Bands corresponding to KCNQ2 (Fig. 6A) and KCNQ3 (Fig. 6B) were detected in transfected HeLa cells as well as in brain and spinal cord membranes. KCNQ3 was also detected in sciatic nerve (Fig. 6B). The failure to detect KCNQ2 was expected because the nodal membrane is a tiny fraction of this membrane preparation, and we have not been able to detect other nodal components in immunoblots of peripheral nerve (data not shown). Bands corresponding to KCNQ2 and KCNQ3 could be immunoprecipitated from optic nerve membranes (a purely central myelinated tract), confirming that KCNQ2 and KCNQ3 are expressed in myelinated tracts (Fig. 6C). Hippocampal membranes were used as positive control (Wang et al., 1998; Cooper et al., 2000, 2001).
Figure 6.

Immunoblots and immunoprecipitations. A, B, Immunoblot analysis. Membrane proteins (100 μg) from rat muscle, sciatic nerve, spinal cord, and brain, and HeLa celllysates (10 μg) were separated by electrophoresis and immunoblotted for KCNQ2 (A) or KCNQ3 (B). Bands corresponding to the molecular mass of KCNQ2 and KCNQ3 expressed in HeLa cells (∼97 kDa) were detected in both spinal cord and brain. KCNQ3, but not KCNQ2, was detected in sciatic nerve membrane. C, Immunoprecipitations of KCNQ2 and KCNQ3. Rat optic nerve and hippocampal membranes (200 μg) were immunoprecipitated for KCNQ2 and KCNQ3 and then immunoblotted with KCNQ2 or KCNQ3 antisera. KCNQ2 and KCNQ3 were detected in both samples. MW markers are shown on the left (in kilodaltons). D, E, Coimmunoprecipitations of KCNQ2 and ankyrin-G. Rat spinal cord membranes (200 μg) were immunoprecipitated for KCNQ2 or ankyrin-G and then immunoblotted for ankyrin-G (D) and KCNQ2 (E). A ∼97 kDa isoform of ankyrin-G was pulled down by the KCNQ2. The ankyrin-G antiserum pulled down multiple ankyrin-G isoforms, including the ∼97 kDa isoform. KCNQ2 (asterisk) was immunoprecipitated by both the ankyrin-G and the KCNQ2 antisera. MW markers are shown on the left (in kilodaltons). In E, the immunoblot for KCNQ2 is shown for two film exposure times: 3 min (3′) and 10 min (10′).
Nodal Nav channels are linked to the spectrin cytoskeleton by ankyrin-G (Jenkins and Bennett, 2001, 2002). To determine whether KCNQ2 interacts with ankyrin-G, we performed coimmunoprecipitation experiments of solubilized rat brain membranes. An ankyrin-G antiserum immunoprecipitated numerous ankyrin-G isoforms (Fig. 6D) and also KCNQ2 (Fig. 6E, asterisk). The KCNQ2 antiserum immunoprecipitated KCNQ2 (Fig. 6E) and also a ∼97 kDa ankyrin-G isoform; this band was also seen in the ankyrin-G-immunoprecipitated lane (Fig. 6D, arrowhead). Ankyrin-G isoforms were not immunoprecipitated by antisera against KCNQ3 (Fig. 6D) or Kv1.2 (data not shown). These results suggest that KCNQ2 interacts with ankyrin-G, either directly or indirectly.
Differential effects of KCNQ modulators on CNS axons during development
To determine whether CNS nodes have functional KCNQ channels, we tested the effects of linopirdine and retigabine on 3-month-old rat optic nerve in which nearly every (ankyrin-G positive) node was strongly KCNQ2 positive (Fig. 7A), whereas a few were weakly KCNQ3 positive (Fig. 7B). The CAP has a triphasic shape because of the presence of three populations of myelinated axons. Linopirdine (100 μm) did not affect the shape of the CAP (Fig. 7C) or its refractory period (Fig. 7G). Retigabine (20 μm) delayed the onset of all three phases of the CAP (Fig. 7D) and significantly increased its duration, from 4.9 ± 0.5 to 11.7 ± 2.9 msec (n = 5; p < 0.01). These effects were maximal at 20 μm and were abolished by linopirdine (20 μm) (Fig. 7E). Thus, CNS nodes appear to have functional KCNQ channels.
Figure 7.
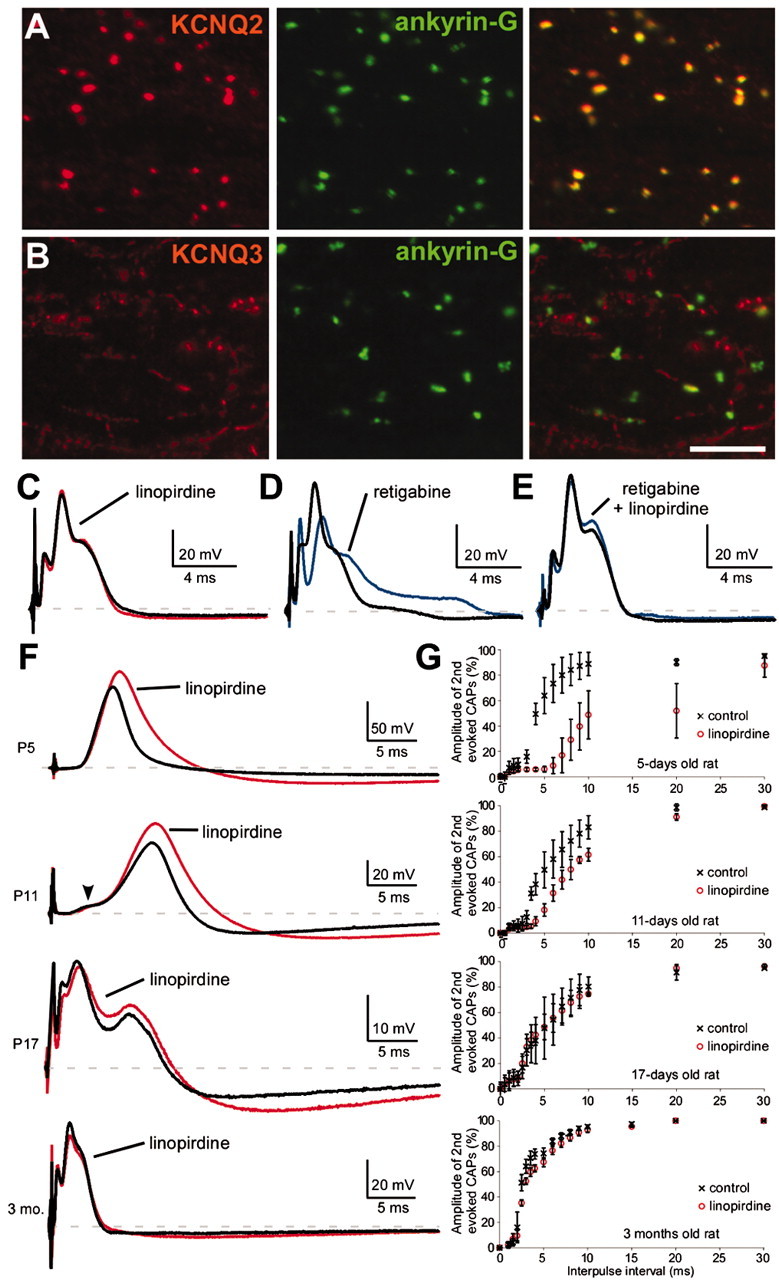
KCNQ2 modulates the excitability of premyelinated fibers. A, B, Images of horizontal sections of unfixed rat optic nerve immunolabeled for ankyrin-G and KCNQ2 (A) or KCNQ3 (B). Virtually all ankyrin-G nodes are strongly KCNQ2 positive, but a few are weakly positive for KCNQ3. Scale bar, 10 μm. C, E, CAPs recorded from 3-month-old rat optic nerves. Linopirdine (100 μm) did not affect the CAP (n = 5) (C), but retigabine (20 μm) delayed the onset of the CAP (n = 5) (D). The effects of retigabine were blocked by pretreating the nerves with linopirdine (20 μm; n = 4) (E). F, G, CAPs recorded from P5, P11, P17, and 3-month-old rat optic nerves. Linopirdine (100 μm) increased the duration (F) and refractory period (G) of the CAP at both P5 (n = 2) and P11 (n = 4) but not at P17 (n = 2) or 3 months (n = 5). For the refractory period, two stimuli are applied, and the amplitude of the highest peak of the second evoked CAP is measured.
Mutations in KCNQ2 and KCNQ3 cause seizures during the first months of life, a period of robust CNS myelination (Rorke and Riggs, 1969). To investigate the possibility that the effects of KCNQ channels might be more robust before myelination, we examined the effects of linopirdine on the rat optic nerve at several developmental stages (P5, P11, P17, and 3 months). In this preparation, myelination starts at approximately P8 and is complete by P40 (Devaux et al., 2002). As shown previously (Devaux et al., 2002), the CAP recorded at P5 is slow and monophasic; an additional faster phase first appears around P11, corresponding to the first myelinated fibers (Fig. 7F, arrowhead); the CAP becomes triphasic and faster as myelination proceeds (Fig. 7F). At P5 and P11, linopirdine (100 μm) increased the duration and amplitude of the CAP (Fig. 7F), resulting in an increased refractory period (Fig. 7G). At P17 and later, linopirdine had little effect on the CAP amplitude (except for increasing the late hyperpolarization) and no effect on the refractory period (Fig. 7F,G). Similar results have been observed previously with TEA (Devaux et al., 2002), suggesting that TEA and linopirdine may block the same channels. In accord with this possibility, pretreating neonatal optic nerves with linopirdine completely occluded the effects of TEA (10 mm; data not shown). These results suggest that KCNQ channels have a more prominent role in axons before myelination.
Discussion
KCNQ2 is localized at nodes and initial segments
Unlike previous studies (Wang et al., 1998; Cooper et al., 2000, 2001), we found KCNQ2 and KCNQ3 in nodes and initial segments. We used similar methods and even some of the same antisera against KCNQ2 and KCNQ3, but we did not fix the tissue with aldehydes. Previous reports typically fixed specimens for many hours. Furthermore, Triton improved the detection of KCNQ2 and KCNQ3 at initial segments and nodes (data not shown), suggesting that they may be relatively Triton insoluble and tethered to the cytoskeleton, as described for L1 (Winckler et al., 1999). Given our findings, the cellular localization of KCNQ2 and KCNQ3 in other brain regions should be reinvestigated on unfixed sections, as should the localization of KCNQ5, which is widely expressed in the CNS and can coassemble with KCNQ3 (Shah et al., 2002; Yus-Najera et al., 2003). KCNQ2 and KCNQ3 may have an important role in initial segments, because they can be modulated by metabotropic receptors via G-proteins (Selyanko et al., 2000; Shapiro et al., 2000). Thus, the activity generated in initial segments could be regulated by modulatory neurotransmitters secreted at axo-axonic synapses (Conradi, 1969).
Most nodal proteins (Na+ channel α and β subunits, βIV spectrin, neurofascin, and Nr-CAM) interact with ankyrin-G (Bennett and Chen, 2001; Bouzidi et al., 2002; Malhotra et al., 2002) and are recruited after ankyrin-G in nodes and initial segments during development (Jenkins and Bennett, 2001, 2002), as observed for KCNQ2. These proteins fail to cluster in Purkinje cell initial segments in ankyrin-G-null cerebella (Zhou et al., 1998). Because Purkinje cell initial segments express little KCNQ2, ankyrin-G-null Purkinje cells were not informative in this regard (data not shown). The isoform of ankyrin-G that interacts with KCNQ2 appears identical to the one that interacts with Kv3.1b, a CNS nodal K+ channel (Devaux et al., 2003). This ∼97 kDa isoform is much smaller than the established nodal isoforms (270 and 480 kDa) (Kordeli et al., 1995). In addition to the possibility that it is a novel nodal isoform, it could be a nonnodal isoform or a protease degradation product. Thus, it remains to be determined whether KCNQ2 and ankyrin-G interact directly or indirectly. Because our KCNQ2 antisera recognize all of the described KCNQ2 splice variants (Cooper et al., 2001; Pan et al., 2001; Smith et al., 2001), we cannot evaluate the possibility that different isoforms interact with distinct anchoring proteins that dictate their localization in axonal versus somatodendritic compartments.
KCNQ2 and Ks
A slowly activating, voltage-dependent K+ current, Ks, has long been described in vertebrate nodes (Dubois, 1981; Roper and Schwarz, 1989; Corrette et al., 1991; Safronov et al., 1993). The characteristics of its activation and pharmacology are similar to those of KCNQ2: slow kinetics of activation and deactivation, no inactivation, and blockage by TEA but not by 4-aminopyridine (4-AP). The similarities between Ks and IM were noted soon after these currents were first recorded (Dubois, 1983), and KCNQ channels are the only known K+ channels with these characteristics (for review, see Coetzee et al., 1999).
The effects of linopirdine and retigabine on axonal conduction favor the idea that axons have functional KCNQ channels. The slowing in conduction velocity observed after retigabine may be accounted for by a decrease in nodal membrane resistance. Indeed, at the concentration used here, retigabine shifts the threshold of activation of the IM recorded in rat sympathetic neurons and in cells expressing KCNQ2, KCNQ3, or KCNQ4 (Tatulian et al., 2001) but not of Kv1, Kv2, Kv3, or ether-a-go go (eag) channels (Rundfeldt, 1999; Rundfeldt and Netzer, 2000). Linopirdine antagonized these effects at a concentration that completely blocks KCNQ channels but only partially affects other K+ channels; the IC50 on Kv1.2, Kv4.3, and eag channels is three- to fourfold higher than the concentration that we used (Wang et al., 1998). Furthermore, linopirdine but not dendrotoxin-I or 4-AP (Devaux et al., 2002) mimics the effects of TEA on neonatal nerves. The above data, taken together, suggest that KCNQ2 mediates the Ks. Although Ks activates at more hyperpolarized voltages and with a less steep voltage dependence than that of KCNQ2, a similar discrepancy exists between the hair cell current, Kn, and KCNQ4, the subunit that is thought to underlie it (Kharkovets et al., 2000).
How do KCNQ2 mutations cause BFNC and myokymia?
The R207W mutation has markedly slowed and shifted activation kinetics and has a dominant-negative effect on wild-type KCNQ2 (Dedek et al., 2001). This mutation may decrease the K+ conductance at nodes and thereby depolarize the membrane potential and cause spontaneous firing in myelinated axons. Although neither TEA nor linopirdine induces spontaneous firing in nerve fibers, clinical evidence demonstrates that myokymia is likely generated in the intramuscular aspect of motor axons (Auger et al., 1984; Dedek et al., 2001). In KCNA1-null mice, the last node of Ranvier (at the neuromuscular junction) appears more prone to produce spontaneous APs and underlie myokymia (Zhou et al., 1999). Kv1.1 and Kv1.2 may also compensate for the blockade of KCNQ2, because spontaneous APs have been observed in mammalian PNS myelinated fibers only after treatment with both TEA (which blocks KCNQ channels) and 4-AP (which blocks Kv1 channels) (Bergmann et al., 1968; Eng et al., 1988; Zhou et al., 1999).
The effects of linopirdine before myelination raise the possibility that unmyelinated or premyelinating axons play a key role in the origin of seizures in BFNC. If unmyelinated axons express KCNQ2 and KCNQ3, then KCNQ2 and KCNQ3 mutants with reduced channel activity might cause excessive firing that leads to seizures. Kv1.1 and Kv1.2 channels provide an important precedent for this suggestion, because these channels prevent the generation of repetitive firing after the AP in neonatal PNS myelinated nerve (Vabnick et al., 1999). Seizures also occur in patients with mutations in KCNA1 (Browne et al., 1994; Zuberi et al., 1999) and KCNA1-null mice (Smart et al., 1998). These similarities imply that axonal defects may be the cause of some kinds of epilepsy. Just as spontaneous firing in motor axons can lead to myokymia in the PNS, in the CNS it could lead to seizures.
Footnotes
This work was supported by a Charcot-Marie-Tooth Association Fellowship to J.J.D., by a National Multiple Sclerosis Society Fellowship to K.A.K., by Academic Dean Funds of the University of Pennsylvania Medical Center to E.C., and by National Institutes of Health Grant RO1 NS42878 to S.S.S. We thank Drs. Steve Lambert and Ken Mackie for antibodies, Dr. Irwin Levitan for stably transfected cells expressing KCNQ2/KCNQ3, Dr. Rita Balice-Gordon for the use of an electrophysiology set-up, and Dr. Philip Haydon for critical reading of this manuscript.
DOI:10.1523/JNEUROSCI.4512-03.2004
Correspondence should be addressed to Dr. Jérôme J. Devaux, University of Pennsylvania Medical Center, Room 464, Stemmler Hall, 36th Street and Hamilton Walk, Philadelphia, PA 19104-6077. E-mail: jdevaux@mail.med.upenn.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/241236-09$15.00/0
E.C.C. and S.S.S. contributed equally to this work.
References
- Arroyo EJ, Scherer SS (2000) On the molecular architecture of myelinated fibers. Histochem Cell Biol 113: 1–18. [DOI] [PubMed] [Google Scholar]
- Arroyo EJ, Xu Y-T, Zhou L, Messing A, Peles E, Chiu SY, Scherer SS (1999) Myelinating Schwann cells determine the internodal localization of Kv1.1, Kv1.2, Kvβ2, and Caspr. J Neurocytol 28: 333–347. [DOI] [PubMed] [Google Scholar]
- Arroyo EJ, Xu T, Poliak S, Watson M, Peles E, Scherer SS (2001) Internodal specializations of myelinated axons in the CNS. Cell Tissue Res 305: 53–66. [DOI] [PubMed] [Google Scholar]
- Arroyo EJ, Xu T, Grinspan J, Lambert S, Levinson SR, Brophy PJ, Peles E, Scherer SS (2002) Genetic dysmyelination alters the molecular architecture of the nodal region. J Neurosci 22: 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger RG, Daube JR, Gomez MR, Lambert EH (1984) Hereditary form of sustained muscle activity of peripheral nerve origin causing generalized myokymia and muscle stiffness. Ann Neurol 15: 13–21. [DOI] [PubMed] [Google Scholar]
- Bartsch S, Bartsch U, Dorries U, Faissner A, Weller A, Ekblom P, Schachner M (1992) Expression of tenascin in the developing and adult cerebellar cortex. J Neurosci 12: 736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett V, Chen L (2001) Ankyrins and cellular targeting of diverse membrane proteins to physiological sites. Curr Opin Cell Biol 13: 61–67. [DOI] [PubMed] [Google Scholar]
- Bergmann C, Nonner W, Stampfli R (1968) Sustained spontaneous activity of Ranvier nodes induced by the combined actions of TEA and lack of calcium. Pflügers Arch 302: 24–37. [DOI] [PubMed] [Google Scholar]
- Bouzidi M, Tricaud N, Giraud P, Kordeli E, Caillol G, Deleuze C, Couraud F, Alcaraz G (2002) Interaction of the Nav1.2a subunit of the voltage-dependent sodium channel with nodal AnkyrinG: in vitro mapping of the interacting domains and association in synaptosomes. J Biol Chem 277: 28996–29004. [DOI] [PubMed] [Google Scholar]
- Browne DL, Gancher ST, Nutt JG, Brunt ERP, Smith EA, Kramer P, Litt M (1994) Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 8: 136–140. [DOI] [PubMed] [Google Scholar]
- Castaldo P, del Giudice EM, Coppola G, Pascotto A, Annunziato L, Taglialatela M (2002) Benign familial neonatal convulsions caused by altered gating of KCNQ2/KCNQ3 potassium channels. J Neurosci 22: RC199(1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega-Saenz de Miera E, Rudy B (1999) Molecular diversity of K+ channels. Ann NY Acad Sci 868: 233–285. [DOI] [PubMed] [Google Scholar]
- Conradi S (1969) Observations on the ultrastructure of the axon hillock and initial segment of lumbosacral motoneurons in the cat. Acta Physiol Scand [Suppl] 332: 65–92. [PubMed] [Google Scholar]
- Cooper EC, Aldape KD, Abosch A, Barbaro NM, Berger MS, Peacock WS, Jan YN, Jan LY (2000) Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy. Proc Natl Acad Sci USA 97: 4914–4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EC, Harrington E, Jan YN, Jan LY (2001) M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci 21: 9529–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrette B, Repp H, Dreyer F, Schwarz J (1991) Two types of fast K+ channels in rat myelinated fibres and their sensitivity to dendrotoxin. Pfügers Arch 418: 408–416. [DOI] [PubMed] [Google Scholar]
- Dedek K, Kunath B, Kananura C, Reuner U, Jentsch TJ, Steinlein OK (2001) Myokymia and neonatal epilepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel. Proc Natl Acad Sci USA 98: 12272–12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaux J, Gola M, Jacquet G, Crest M (2002) Effects of K+ channel blockers on developing rat myelinated CNS axons: identification of four types of K+ channels. J Neurophysiol 87: 1376–1385. [DOI] [PubMed] [Google Scholar]
- Devaux J, Alcaraz G, Grinspan J, Bennett V, Joho R, Crest M, Scherer SS (2003) Kv3.1b is a novel component of CNS nodes. J Neurosci 23: 4509–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois JM (1981) Evidence for the existence of three types of potassium channels in the frog Ranvier node membrane. J Physiol (Lond) 318: 297–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois JM (1983) Potassium currents in the frog node of Ranvier. Prog Biophys Mol Biol 42: 1–20. [DOI] [PubMed] [Google Scholar]
- Eng DL, Gordon TR, Kocsis JD, Waxman SG (1988) Development of 4-AP and TEA sensitivities in mammalian myelinated nerve fibers. J Neurophysiol 60: 2168–2179. [DOI] [PubMed] [Google Scholar]
- Eunson LH, Rea R, Zuberi SM, Youroukos S, Panayiotopoulos CP, Liguori R, Avoni P, McWilliam RC, Stephenson JBP, Hanna MG, Kullmann DM, Spauschus A (2000) Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol 48: 647–656. [PubMed] [Google Scholar]
- Fannon AM, Sherman DL, Ilyina-Gragerova G, Brophy PJ, Friedrich VL, Colman DR (1995) Novel E-cadherin mediated adhesion in peripheral nerve: Schwann cell architecture is stabilized by autotypic adherens junctions. J Cell Biol 129: 189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ffrench-Constant C, Miller RH, Kruse J, Schachner M, Raff MC (1986) Molecular specialization of astrocyte processes at nodes of Ranvier in rat optic nerve. J Cell Biol 102: 844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann L, Libell D, Gutmann L (2001) When is myokymia neuromyotonia? Muscle Nerve 24: 151–153. [DOI] [PubMed] [Google Scholar]
- Hartshorne RP, Catterall WA (1984) The sodium channel from rat brain. Purification and subunit composition. J Biol Chem 259: 1667–1675. [PubMed] [Google Scholar]
- Hopkins WF, Allen ML, Mouamed KM, Tempel BL (1994) Properties of voltage-gated K+ currents expressed in Xenopus oocytes by mKv1.1, mKv1.2 and their heteromultimers as revealed by mutagenesis of the dendrotoxin-binding site in Kv1.1. Pflügers Arch 428: 382–390. [DOI] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V (2001) Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol 155: 739–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Bennett V (2002) Developing nodes of Ranvier are defined by ankyrin-G clustering and are independent of paranodal axoglial adhesion. Proc Natl Acad Sci USA 99: 2303–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins SM, Kizhatil K, Kramarcy NR, Sen A, Sealock R, Bennett V (2001) FIGQY phosphorylation defines discrete populations of L1 cell adhesion molecules at sites of cell-cell contact and in migrating neurons. J Cell Sci 114: 3823–3835. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ (2000) Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci 1: 21–30. [DOI] [PubMed] [Google Scholar]
- Kharkovets T, Hardelin JP, Safieddine S, Schweizer M, El-Amraoui A, Petit C, Jentsch TJ (2000) KCNQ4, a K+ channel mutated in a form of dominant deafness, is expressed in the inner ear and the central auditory pathway. Proc Natl Acad Sci USA 97: 4333–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komada M, Soriano P (2002) Beta IV-spectrin regulates sodium channel clustering through ankyrin-G at axon initial segments and nodes of Ranvier. J Cell Biol 156: 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordeli E, Lambert S, Bennett V (1995) AnkyrinG: a new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem 270: 2352–2359. [DOI] [PubMed] [Google Scholar]
- Lee V, Wu HL, Schlaepfer WW (1982) Monoclonal antibodies recognized individual neurofilament triplet proteins. Proc Natl Acad Sci USA 79: 6089–6092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM-Y, Carden MJ, Schlaepfer WW, Trojanowski JQ (1987) Monoclonal antibodies distinguish several differentially phosphorylated states of the two largest rat neurofilament subunits (NF-H and NF-M) and demonstrate their existence in the normal nervous system of adult rats. J Neurosci 7: 3474–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Koopmann MC, Kazen-Gillespie KA, Fettman N, Hortsch M, Isom LL (2002) Structural requirements for interaction of sodium channel beta 1 subunits with ankyrin. J Biol Chem 277: 26681–26688. [DOI] [PubMed] [Google Scholar]
- Mi HY, Deerinck TJ, Ellisman MH, Schwarz TL (1995) Differential distribution of closely related potassium channels in rat Schwann cells. J Neurosci 15: 3761–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newsom-Davis J (1997) Autoimmune neuromyotonia (Isaacs' syndrome): an antibody-mediated potassium channelopathy. Ann NY Acad Sci 835: 111–119. [DOI] [PubMed] [Google Scholar]
- Pan Z, Selyanko AA, Hadley JK, Brown DA, Dixon JE, McKinnon D (2001) Alternative splicing of KCNQ2 potassium channel transcripts contributes to the functional diversity of M-currents. J Physiol (Lond) 531: 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peles E, Salzer JL (2000) Molecular domains of myelinated fibers. Curr Opin Neurobiol 10: 558–565. [DOI] [PubMed] [Google Scholar]
- Rasband MN, Trimmer JS (2001) Developmental clustering of ion channels at and near the node of Ranvier. Dev Biol 236: 5–16. [DOI] [PubMed] [Google Scholar]
- Robbins J (2001) KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol Ther 90: 1–19. [DOI] [PubMed] [Google Scholar]
- Roche JP, Westenbroek R, Sorom AJ, Hille B, Mackie K, Shapiro MS (2002) Antibodies and a cysteine-modifying reagent show correspondence of M current in neurons to KCNQ2 and KCNQ3 K+ channels. Br J Pharmacol 137: 1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper J, Schwarz JR (1989) Heterogeneous distribution of fast and slow potassium channels in myelinated rat nerve fibres. J Physiol (Lond) 416: 93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorke LB, Riggs HE (1969) Myelination of the brain in the newborn. Philadelphia: Lippincott.
- Rundfeldt C (1999) Characterization of the K+ channel opening effect of the anticonvulsant retigabine in PC12 cells. Epilepsy Res 35: 99–107. [DOI] [PubMed] [Google Scholar]
- Rundfeldt C, Netzer R (2000) The novel anticonvulsant retigabine activates M-currents in Chinese hamster ovary cells tranfected with human KCNQ2/3 subunits. Neurosci Lett 282: 73–76. [DOI] [PubMed] [Google Scholar]
- Safronov BV, Kampe K, Vogel W (1993) Single voltage-dependent potassium channels in rat peripheral nerve membrane. J Physiol (Lond) 460: 675–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Arroyo EJ (2002) Recent progress on the molecular organization of myelinated axons. J Periph Nerv Syst 7: 1–12. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Kubisch C, Stein V, Jentsch TJ (1998) Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 396: 687–690. [DOI] [PubMed] [Google Scholar]
- Selyanko AA, Hadley JK, Wood IC, Abogadie FC, Jentsch TJ, Brown DA (2000) Inhibition of KCNQ1–4 potassium channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J Physiol (Lond) 522 3: 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Mistry M, Marsh SJ, Brown DA, Delmas P (2002) Molecular correlates of the M-current in cultured rat hippocampal neurons. J Physiol (Lond) 544: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B (2000) Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3(+) channels that underlie the neuronal M current. J Neurosci 20: 1710–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shillito P, Molenaar PC, Vincent A, Leys K, Zheng W, van den Berg RJ, Plomp JJ, Van Kempen GTH, Chauplannaz G, Wintzen AR, Vandijk JG, Newsom-Davis J (1995) Acquired neuromyotonia: evidence for autoantibodies directed against K+ channels of peripheral nerves. Ann Neurol 38: 714–722. [DOI] [PubMed] [Google Scholar]
- Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A, Tempel BL (1998) Deletion of the Kv1.1 potassium channel causes epilepsy in mice. Neuron 20: 809–819. [DOI] [PubMed] [Google Scholar]
- Smith JS, Iannotti CA, Dargis P, Christian EP, Aiyar J (2001) Differential expression of KCNQ2 splice variants: implications to M current function during neuronal development. J Neurosci 21: 1096–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA (2001) Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci 21: 5535–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabnick I, Trimmer JS, Schwarz TL, Levinson SR, Risal D, Shrager P (1999) Dynamic potassium channel distributions during axonal development prevent aberrant firing patterns. J Neurosci 19: 747–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent A (2000) Understanding neuromyotonia. Muscle Nerve 23: 655–657. [DOI] [PubMed] [Google Scholar]
- Wang H, Kunkel DD, Martin TM, Schwartkroin PA, Tempel BL (1993) Heteromultimeric K+ channels in terminal and juxtaparanodal regions of neurons. Nature 365: 75–79. [DOI] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282: 1890–1893. [DOI] [PubMed] [Google Scholar]
- Wen H, Levitan IB (2002) Calmodulin is an auxiliary subunit of KCNQ2/3 potassium channels. J Neurosci 22: 7991–8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winckler B, Forscher P, Melman I (1999) A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature 397: 698–701. [DOI] [PubMed] [Google Scholar]
- Yus-Najera A, Munoz A, Salvador N, Jensen BS, Rasmussen HB, Defelipe J, Villarroel A (2003) Localization of KCNQ5 in the normal and epileptic human temporal neocortex and hippocampal formation. Neuroscience 120: 353–364. [DOI] [PubMed] [Google Scholar]
- Zerr P, Adelman JP, Maylie J (1998) Episodic ataxia mutations in Kv1.1 alter potassium channel function by dominant negative effects or haploin sufficiency. J Neurosci 18: 2842–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou DX, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V (1998) AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol 143: 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Messing A, Chiu SY (1999) Determinants of excitability at transition zones in Kv1.1-deficient myelinated nerves. J Neurosci 19: 5768–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuberi SM, Eunson LH, Spauschus A, De Silva R, Tolmie J, Wood NW, McWilliam RC, Stephenson JP, Kullmann DM, Hanna MG (1999) A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain 122: 817–825. [DOI] [PubMed] [Google Scholar]