

Abstract
Leukemia inhibitory factor (LIF) receptor β (LIFRβ) is a receptor for a variety of neurotrophic cytokines, including LIF, ciliary neurotrophic factor (CNTF), and cardiotrophin-1 (CT-1). These cytokines play an essential role for the survival and maintenance of developing and postnatal somatic motoneurons. CNTF may also serve the maintenance of autonomic, preganglionic sympathetic neurons (PSNs) in the spinal cord, as suggested by its capacity to prevent their death after destruction of one of their major targets, the adrenal medulla. Although somatic motoneurons and PSNs share a common embryonic origin, they are distinct in several respects, including responses to lesions. We have studied PSNs in mice with targeted deletions of the LIFRβ or CT-1 genes, respectively. We show that LIF, CNTF, and CT-1 are synthesized in embryonic adrenal gland and spinal cord and that PSNs express LIFRβ. In embryonic day 18.5 LIFRβ (−/−) and CT-1 (−/−) mice, PSNs were reduced by ∼20%. PSNs projecting to the adrenal medulla were more severely affected (−55%). Although LIFRβ (−/−) mice revealed normal numbers of adrenal chromaffin cells and axons terminating on chromaffin cells, levels of adrenaline and numbers of adrenaline-synthesizing cells were significantly reduced. We conclude that activation of LIFRβ is required for normal development of PSNs and one of their prominent targets, the adrenal medulla. Thus, both somatic motoneurons and PSNs in the spinal cord depend on LIFRβ signaling for their development and maintenance, although PSNs seem to be overall less affected than somatic motoneurons by LIFRβ deprivation.
Keywords: preganglionic sympathetic neurons, intermediolateral column, adrenal medulla, LIFRβ, ciliary neurotrophic factor, cardiotrophin-1
Introduction
The ciliary neurotrophic factor (CNTF) family of growth factors comprises a number of different cytokines with broad structural similarities (Bazan, 1991) that share very similar signaling pathways (for review, see Heinrich et al., 2003). These cytokines have different biological activities and include leukemia inhibitory factor (LIF) (Gearing et al., 1987), CNTF (Lin et al., 1989; Stöckli et al., 1989), oncostatin M (OsM) (for review, see Morikawa, 2005), growth promoting activity (Leung et al., 1992), cardiotrophin-1 (CT-1) (Pennica et al., 1995), cardiotrophin-2 (CT-2) (Derouet et al., 2004), and cardiotrophin-like cytokine (CLC)/cytokine-like factor (CLF) (Forger et al., 2003). CNTF, LIF, CT-1, and CLC/CLF promote survival of cultured spinal cord motoneurons (Arakawa et al., 1990; Martinou et al., 1992; Pennica et al., 1996; Forger et al., 2003) and lesioned motoneurons in vivo (Sendtner et al., 1990; Oppenheim, 1991; Cheema et al., 1994; Pennica et al., 1996). Gene targeting experiments have revealed that CNTF and LIF cooperate in maintaining postnatal survival and functional integrity of motoneurons. However, although cntf and lif single and double knock-out mice do not show any deficits in the survival of embryonic motoneurons, a significant loss of motoneurons has been reported for CT-1-deficient mice (Oppenheim et al., 2001). Analyses of the triple knock-out of cntf, lif, and ct-1 have revealed both cooperative and distinct roles of these factors for the maintenance and function of motoneurons (Holtmann et al., 2005).
CNTF, LIF, CT-1, CT-2, CLC/CLF, and OsM share one receptor component, the LIF receptor β (LIFRβ) (Gearing et al., 1991, 1992; Heinrich et al., 2003). Mice lacking LIFRβ, in contrast to mice lacking CNTF, LIF, and/or CT-1, die shortly after birth (Ware et al., 1995) and show severe losses of motoneurons (Li et al., 1995).
Spinal motoneurons innervating striated muscle (i.e., somatic motoneurons) share a common developmental origin with preganglionic sympathetic neurons (PSNs) located in the intermediolateral column of the spinal cord (for review, see Schober and Unsicker, 2001). PSNs serve essential roles in the regulation of cardiovascular and other autonomic functions; their targets are the peripheral autonomic ganglia, intra-adrenal and extra-adrenal chromaffin cells. Although a number of neurotrophic factors [FGF-2, CNTF, TGF-β, glial cell line-derived neurotrophic factor (GDNF), and neurotrophin-4 (NT-4)] have been shown to prevent PSN death after destruction of one of their major targets, the adrenal medulla (for review, see Schober and Unsicker, 2001), the physiological relevance of neurotrophic factors for PSN development and maintenance is less well understood than that of somatic motoneurons. The present study was therefore designed to analyze a putative requirement of LIFRβ signaling for the development and embryonic maintenance of PSNs.
Our analyses of LIFRβ- and CT-1-deficient mice support the notion that PSNs require LIFRβ signaling for their embryonic development and maintenance. Mutant mice lack an average of 20% PSNs. However, PSN dependence on LIFRβ signaling may quantitatively differ among PSNs innervating different targets, as suggested by a significantly greater loss of PSNs projecting to the adrenal gland (−55%). PSN losses are partially compensated, as shown by normal numbers of axon terminals in the LIFRβ (−/−) adrenal medulla.
Materials and Methods
Animals.
Thirty-two mice deficient for LIFRβ, eight CT-1-deficient mice, four CNTF-deficient mice, and 50 respective wild-type littermates were analyzed at embryonic day 13.5 (E13.5), E16.5, E18.5, and postnatally at 4 months. In addition, 20 Hanover–Wistar rats at ages 6 d and 3 months were used. Animals were kept at the Central Animal Facilities of Heidelberg University and Kopfklinikum of the Würzburg University, respectively, according to the regulations of animal welfare (Tierschutzgesetz).
RNA preparation and reverse transcription.
Total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA) according to the guidelines of the manufacturer. Two units of RNase inhibitor (Fermentas, St. Leon-Rot, Germany) and 1 U of RQ1 DNase (Stratagene, Heidelberg, Germany) were added to 3 μg of total RNA in a final volume of 12 μl. Reactions were incubated for 15 min at 37°C and for 5 min at 70°C. First-strand cDNA was synthesized in a final volume of 40 μl. Reactions consisted of 3 μg of total RNA and final concentrations of 1× first-strand buffer [5× first-strand buffer (in mm): 250 Tris-HCl, pH 8.3, 375 KCl, and 15 MgCl2; Invitrogen, Karlsruhe, Germany], 10 mm dithiothreitol (DTT), 1 mm each of deoxynucleotide triphosphates (dNTPs; Amersham Biosciences, Freiburg, Germany), 25 ng/μl random hexamer primers (Roche Diagnostics, Mannheim, Germany) or oligo-dT primers (Promega, Madison, WI), 1 U/μl RNase inhibitor (Fermentas), and 20 U/μl Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen). The reaction mixture was incubated at 37°C for 2 h, heated for 5 min at 90°C, cooled to 4°C for 5 min, and supplemented with 2 U of RNase H (Invitrogen). Samples were incubated for 20 min at 37°C. Total RNA (1 μg per reaction) was denatured for 5 min at 70°C and then reverse transcribed for 60 min at 42°C in 1× first-strand buffer (Invitrogen) containing 20 U of RNase inhibitor (Fermentas), 10 mm DTT, 1.5 μm oligo-dT, 0.3 mm dNTPs (Amersham Biosciences), and 200 U of M-MLV reverse transcriptase (Invitrogen). Reactions were stopped by incubation at 75°C for 10 min.
PCR analysis.
After reverse transcription (RT), cDNA samples were subjected to PCR amplification using sequence-specific oligonucleotide primers (see Table 1). Reactions were performed in 0.2 ml thin-walled reaction tubes in a PerkinElmer (Wellesley, MA) GeneAmp thermocycler 9600. Reagents were assembled in a final volume of 50 μl with 2 μl of first-strand cDNA, 2 μm forward primer, 2 μm reverse primer, 1× PCR buffer [10× PCR buffer (in mm): 200 Tris-HCl, pH 8.4, and 500 KCl; Invitrogen], 2.5 mm MgCl2, and 0.1 mm each of dNTPs and RNase-free water to 49.5 μl. cDNA was denatured at 93°C for 5 min; the temperature was then lowered to 72°C. TaqDNA polymerase (0.5 μl, 2.5 U; Invitrogen) was added, and cDNA was amplified according to a standard protocol (94°C, 30 s; 58°C, 60 s; 72°C, 30 s). Twelve microliters of each PCR were run on agarose gels (Invitrogen) in 1× TAE buffer (0.04 m Tris-acetate, 0.001 m EDTA), and reaction products were visualized, after soaking gels in 0.5 μg/ml ethidium bromide solution in 1× TAE for 10 min, with transilluminator (Renner, Darmstadt, Germany). Pictures were taken by a computer-assisted gel documentation system (Intas, Göttingen, Germany). Possible genomic DNA contaminations were evaluated in parallel reactions without reverse transcriptase. All RT-PCR experiments were reproduced at least three times, of which one representative experiment is depicted here.
Table 1.
Oligonucleotide primers (primer sequences 5′–3′)
| Gene name | Cycles | Oligonucleotide | |
|---|---|---|---|
| β-Actin | 20 | Upstream | TCATGAAGTGTGACGTTGACATCCGT |
| Downstream | CCTAGAAGCATTTGCGGTGCACGAT | ||
| CNTF | 32 | Upstream | CAGGCTTACCGTACCTTCCA |
| Downstream | TGACACGGAGGTCATGGATA | ||
| CNTFRα | 32 | Upstream | CTGTTTCCACCGTGACTCCT |
| Downstream | CAAGGCGTTGCTGACACTTA | ||
| LIF | 32 | Upstream | CTCTTCCCATCACCCCTGTA |
| Downstream | AGTGGGGTTCAGGACCTTCT | ||
| LIFRβ | 32 | Upstream | TTTCAGAAACGGGGATGTTC |
| Downstream | CTGTTTCCACCGTGACTCCT | ||
| CT-1 | 32 | Upstream | GAGGGATACGTGCAGCAACA |
| Downstream | GGCCAGCACTGTCTCCAC |
CNTFRα, CNTF receptor α.
FluoroGold labeling of PSNs.
Forty-eight hours after intra-adrenal injection of 5 μl of FluoroGold (0.2%; Fluorochrome, Englewood, NJ), adult rats were deeply anesthetized with diethylether and transcardially perfused with freshly prepared 4% paraformaldehyde (PFA). Thereafter, the whole thoracic spinal cord was exposed, and spinal cord segments T7–T10 were removed. These spinal cord segments contain the majority of PSNs, which project to the adrenal medulla (Strack et al., 1988; Pyner and Coote, 1994). After 12 h of postfixation (4% PFA), serial cross sections of the spinal cord (25 μm) were performed using a vibrating blade microtome (VT 1000 E; Leica, Bensheim, Germany); sections were collected and mounted on chromalaun-coated slides. Sections were further processed for immunohistochemistry as described below and examined in a Zeiss (Oberkochen, Germany) Axiophot fluorescent microscope using a UV filter set (excitation filter, 390–420 nm; barrier filter, 425–450 nm; Zeiss).
Determination of numbers of neurons in the intermediolateral column of the spinal cord by acetylcholinesterase and Nissl staining.
Nissl-stained cryostat sections (16 μm) of the thoracic spinal cord [E18.5, wild-type; LIFRβ (−/−); CT-1 (−/−), segments T7–T11] were used for determining numbers of neurons in the intermediolateral column (100 adjacent cross sections per animal, 16 μm). To identify these neurons reliably, we counterstained Nissl-stained sections weakly by acetylcholinesterase (AChE) histochemistry (Andrä and Lojda, 1986; Roosen et al., 2001). Sections were stained for 45 min at 37°C in the following solution (60 ml): 30.0 mg of acetylthiocholine iodide (SERVA Electrophoresis, Heidelberg, Germany), 44.4 ml of 0.1 m Tris-maleate buffer, pH 5.0 (containing 0.1% Triton X-100), 6.0 ml of 0.4 m sodium citrate, 6.0 ml of 0.12 m copper citrate, 3.0 ml of 0.16 m potassium ferricyanide, and 0.6 ml of tetraisopropylpyrophosphoramide 10−3 m (Sigma, St. Louis, MO). Sections from knock-out and wild-type animals were always processed in parallel. A grid (120 × 240 μm2) was centered over the AChE-positive intermediolateral column, and all neurons within with a clearly visible nucleus were counted.
Immunohistochemistry and cell counts.
Mice and rats at different embryonic and postnatal ages were transcardially perfused, using 4% PFA, according to a standard protocol. Embryos at E18.5 were also perfused transcardially. Tissue samples (spinal cord, adrenal gland) were removed and postfixed overnight in 4% PFA. Samples were then placed in 15% sucrose for 6–15 h for cryoprotection and finally were frozen on dry ice. Cryosections (20 μm) were cut on a cryostat (Leica) and mounted on chromalaun-precoated slides. Sections were processed for immunohistochemistry using standard ABC staining protocols. As blocking agents, normal goat or horse serum with 0.1% Triton X-100 was used for 2 h at room temperature. After washing in phosphate buffer (PB), pH 7.4, sections were incubated for 48 h at 4°C with the following primary antibodies: LIFRβ, rabbit anti-mouse, 1:200 (sc-659; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-rat, 1:400; tyrosine hydroxylase (TH), sheep anti-mouse, 1:400 (Chemicon, Temecula, CA); phenylethanolamine N-methyltransferase (PNMT), rabbit anti-mouse, 1:2000 (DiaSorin, Stillwater, MN); CNTF, rabbit anti-rat CNTF, 1:500, K10 (M. Sendtner, University of Würzburg, Würzburg, Germany); S100B, monoclonal (mouse), 1:1000 (Dako, High Wycombe, UK); SMI312, monoclonal (mouse) anti-neurofilament (phosphorylated), 1:1000 (Sternberger Monoclonals, Lutherville, MD). After multiple washing steps (PB), a secondary antibody [biotinylated anti-rabbit IgG, 1:200 (applied for LIFRβ, PNMT, CNTF); biotinylated anti-sheep IgG, 1:100 (for TH); cyanine 3 (Cy3)-conjugated anti-mouse IgG, 1:200 (for SMI312); all from Vector Laboratories, Burlingame, CA] was applied at room temperature for 2 h. For visualization of biotinylated antibodies, Cy2- (1:500; PNMT, TH, S100B) or Cy3- (1:2000; LIFRβ, TH, CNTF) conjugated streptavidin (Jackson ImmunoResearch, West Grove, PA) were applied for 2 h at room temperature. Sections were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, Steinheim, Germany). After final washing steps, coverslips were placed on the slides using fluorescent mounting medium (Dako S3023, or Moviol). All dilutions were made in PB. Serial sections of whole adrenal glands of LIFRβ-deficient mice and wild-type littermates (E18.5) were alternately mounted on chromalaun-precoated slides, resulting in two groups of slides containing every second section. One set was stained for TH and the other for PNMT. Only cells positive for TH-immunoreactivity (IR) and PNMT-IR showing a clearly visible nucleus were counted.
In situ hybridization.
Riboprobes for mouse CNTF (GenBank accession number NM_170786, base pair 126–942), LIF (NM_008501, 138–1108), CT-1 (NM_007795, 519–1240), and OsM (NM_001013365, 501–1527) were subcloned and processed using pGEM-T Easy Vector (A1360; Promega) and TOP10 competent cells (C4040-10; Invitrogen). Plasmids for mouse CT-2 (NM_198858, full length), CLC (NM_019952, 203 to poly A tail), and CLF (NM_018827, 377 to poly A tail) were kindly provided by Dr. Hermann Rohrer (Max Planck Institute for Brain Research, Frankfurt/Main, Germany). Riboprobes for mouse choline acetyltransferase (ChAT) were generated as described by Ishii et al. (1990). In situ hybridizations were performed as described by Ernsberger et al. (1997, 2000). Specificity of the reaction products was confirmed with corresponding sense riboprobes. In addition, all riboprobes were sequenced and were found to be consistent with the expected sequences (MWG Biotech, Ebersberg, Germany). Mice were transcardially perfused (E18.5) according to a standard protocol or were immersion fixed (E13.5) using 4% PFA. Tissue samples (E18.5) and complete embryos (E13.5) were postfixed overnight in 4% PFA. Samples were then placed in 15% sucrose overnight for cryoprotection and finally frozen on dry ice. Cryosections (16 μm) were cut on a cryostat (Leica), and mounted on precoated slides (SuperFrost Plus; Menzel-Gläser, Braunschweig, Germany). Because of the staining pattern within the cells, nuclei were not always clearly distinguishable. Therefore, numbers of all ChAT-positive PSNs (all positive cells dorsally of the somatic motoneurons including those heading toward the central canal) were determined in a series of 100 adjacent sections (16 μm) between spinal cord segments T7 and T11.
DiI tracing and counting of PSNs.
To determine numbers of PSNs that innervate adrenal chromaffin cells, a small piece of solid DiI (Invitrogen, Eugene, OR), a lipophilic fluorescent dye, which is passively retrogradely transported even in membranes of fixed tissue, was implanted into the left adrenal medulla of perfusion-fixed mice (E18.5). DiI-injected embryos were stored for 5 months in 4% PFA at room temperature. After this tracing period, the complete spinal cord was removed, cryoprotected (30% sucrose), and frozen on dry ice. Serial sections (20 μm) were collected on chromalaun-coated slides and finally counterstained with DAPI. Only brightly labeled PSNs containing a clearly visible DAPI-stained nucleus were counted. Counts were performed throughout the complete spinal cord, resulting in a complete registration of all traced cells.
Electron microscopy.
E18.5 mice were immersion fixed or transcardially perfused with a mixture consisting of 1.5% glutaraldehyde and 1.5% PFA and postfixed in the same fixative overnight at 4°C. Adrenal glands were embedded in Epon according to standard protocols. Ultrathin sections were collected on formvar-coated single-slot copper grids and stained with 5% uranylacetate, followed by lead citrate (Reynolds, 1963). Numbers of axon terminals (identified by containing small clear synaptic vesicles) on chromaffin cells were evaluated in electron micrographs (magnification, 6300×) taken from two sections through the complete adrenal medulla, with a distance of 200 μm. Eight LIFRβ (−/−) mice and six wild-type littermates were analyzed.
Determination of catecholamine content.
E18.5 LIFRβ-, CT-1-, and CNTF-deficient and wild-type mice were killed by CO2 asphyxation, and adrenal glands were quickly removed. Catecholamines were quantified by HPLC electrochemical detection, as described by Müller and Unsicker (1981).
Cell culture.
Adrenal medullary cells were dissociated from postnatal day 6 (P6) rat adrenal glands as described previously (Unsicker et al., 1987). After microdissection, pieces of chromaffin tissue were collected in Ca2+-free, Mg2+-free HBSS (CMF) and then incubated for 30 min in 0.1% collagenase (Worthington Biochemical, Lakewood, NJ) in CMF. Tissues were then treated with two to three subsequent cycles of trypsin (0.125% in CMF, 15 min, 37°C), followed by enzyme inactivation in ice-cold fetal calf serum (Invitrogen) and trituration using fire-polished Pasteur pipettes. Cells were seeded on polyornithine–laminin-coated coverslips at a density of 250 cells/mm2 in 500 μl of culture medium. Culture medium was DMEM supplemented with the N1 additives 0.25% BSA, penicillin, streptomycin, and neomycin. Cells were maintained in a 5% CO2/95% air atmosphere for 48 h. PC12 (pheochromocytoma 12) cells obtained from American Type Culture Collection (Manassas, VA) were maintained in Roswell Park Memorial Institute 1640 medium (Sigma) supplemented with 10% heat-inactivated horse serum, 5% fetal bovine serum, 100 U/ml penicillin, and 50 U/ml streptomycin (Sigma). For immunocytochemistry, cultured chromaffin cells were fixed with −20°C acetone for 10 min, blocked with 3% BSA in PBS, and subsequently incubated with the polyclonal CNTF antiserum (1:400) (Meyer and Unsicker, 1994) overnight at 4°C. CNTF-immunoreactive cells were identified as chromaffin cells by double-labeling with tyrosine hydroxylase antibodies (monoclonal; Roche Diagnostics). A tetramethylrhodamine-isothiocyanate-conjugated anti-rabbit IgG (Invitrogen) was used for CNTF detection, whereas TH was visualized with FITC-conjugated anti-mouse IgG (Invitrogen). The specificity of the CNTF antiserum was tested by omitting the primary antibody and by preabsorbing the CNTF antiserum with a 50-fold excess of CNTF protein.
Statistics.
All results are given as mean ± SEM, and differences were tested for statistical significance using Student’s t test and Origin 6.1 software (OriginLab, Northampton, MA).
Results
PSNs display immunoreactivity for LIFRβ
To identify PSNs in the intermediolateral column of the spinal cord as a putative target cell population for ligands of the LIFRβ, we first localized LIFRβ immunoreactivity in the adult mouse and rat thoracic spinal cord. Figure 1 A–C (mouse) and D–F (rat) shows LIFRβ-IR in somatic motoneurons of the ventral horn and in neurons within the intermediolateral column, as well as in other neurons including interneurons and white matter glial cells. Similar results were obtained for the E18.5 mouse spinal cord (data not shown). The identity of PSNs specifically projecting to one of their major target regions, the adrenal medulla, was verified by retrograde FluoroGold labeling. For technical reasons, this was performed in adult animals (rat). Figure 1, E and F, show colocalization of LIFRβ-IR and FluoroGold, corroborating the notion that PSNs projecting to the adrenal medulla express the LIFRβ.
Figure 1.

LIFRβ-IR in the adult rat and mouse spinal cord. A, Overview showing the distribution of LIFRβ in the ventral horn (circled), dorsal horn, and region of the mouse intermediolateral column (circled). Scale bar, 500 μm. B, C, Higher magnifications of the circled areas in A show LIFRβ-positive neurons in both locations. Scale bar, 50 μm. D, Overview showing the distribution of LIFRβ-IR in the ventral horn, dorsal horn, and region of the rat intermediolateral column (circled). Scale bar, 1000 μm. E, LIFRβ immunostaining (red) and DAPI staining of neurons within the IML. F, Retrograde tracing with FluoroGold (FG; green) from the adrenal medulla shows positive neurons combined with DAPI labeling. Comparison of E and F suggests that cells marked by white * and # are LIFRβ-positive PSNs projecting to adrenal chromaffin cells. Scale bars: E, F, 20 μm. VH, Ventral horn; DH, dorsal horn; cc, central canal; IML, intermediolateral column.
The LIFRβ ligands CNTF, LIF, and CT-1 are expressed in the embryonic and adult mouse spinal cord and adrenal gland
We next investigated whether the LIFRβ ligands CNTF, LIF, and CT-1 are expressed in mouse spinal cord and adrenal gland. Figure 2 A shows that mRNAs of these three ligands can be detected in both thoracic spinal cord and adrenal gland from E14 through to adulthood. Attempts to reveal the cellular localization of CNTF, LIF, and CT-1 provided evidence for CNTF-IR in S100-positive Schwann cells of the postnatal mouse adrenal medulla (Fig. 2B–D). Other adrenal medullary cell types, including chromaffin cells, were negative. However, CNTF-IR became detectable when dissociated chromaffin cells, identified by TH-IR, were grown in culture (Fig. 2 E,F). In situ hybridizations for CNTF, LIF, CT-1, CT-2, CLC/CLF, and OsM mRNAs failed to reveal specific signals in embryonic adrenal gland and spinal cord (data not shown), suggesting that levels of the respective mRNAs were below detectability by in situ hybridization. However, CNTF and CT-1 mRNAs could be detected in tissues taken as positive controls (CNTF, Schwann cells of adult peripheral nerves; CT-1, striated muscle). Together, these data suggest that CNTF, LIF, and CT-1 mRNAs are expressed at low levels in embryonic as well as adult spinal cord and adrenal gland. Their cellular localization remains to be revealed.
Figure 2.
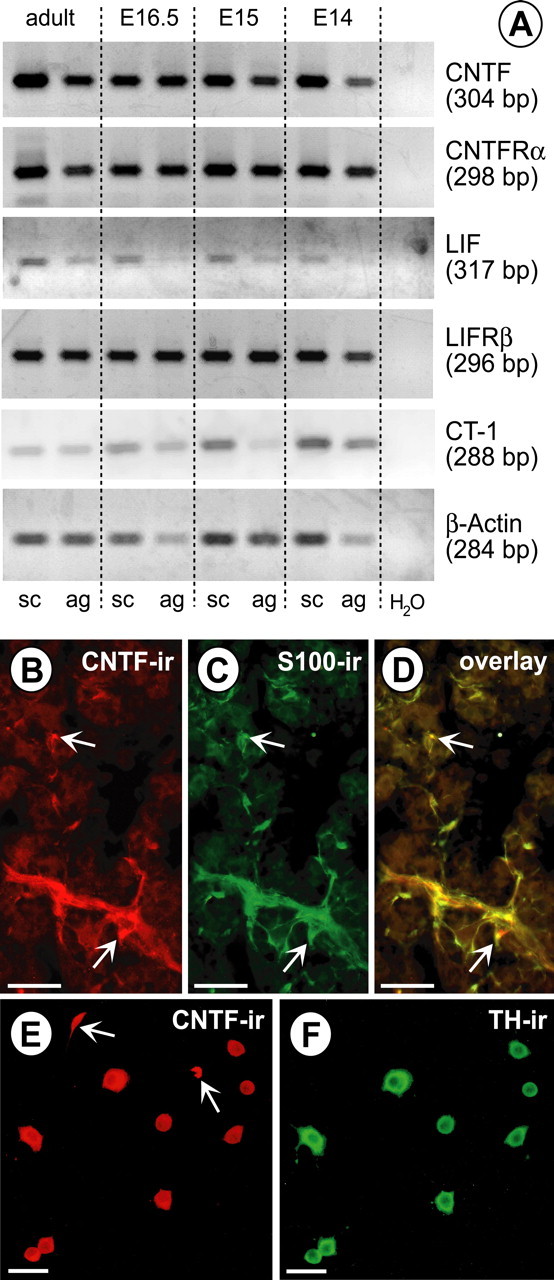
Expression of LIFRβ ligands in the mouse adrenal gland and spinal cord. A, Expression of CNTF, LIF, CT-1, CNTF receptor α (CNTFRα), and LIFRβ mRNAs, revealed by RT-PCR, in the adrenal gland (ag) and spinal cord (sc) at E14, E15, E16.5, and adult. B–D, CNTF expression in Schwann cells of the adult mouse adrenal medulla. The arrows identify two examples of clearly double-labeled cells. B, CNTF-IR. C, S100-IR. D, Overlay. Scale bars, 100 μm. E, CNTF-IR in adrenal medullary cells isolated from P6 rat adrenal glands. F, TH-IR. Most cells that are immunoreactive for CNTF express TH-IR, indicating that they are chromaffin cells. Two cells with CNTF-IR (arrows) are TH negative and possibly represent Schwann cells. Scale bars, 20 μm.
Numbers of PSNs are decreased in LIFRβ- and CT-1-deficient mice
To investigate a putative physiological role of LIFRβ signaling for the development and maintenance of embryonic PSNs, neurons at thoracic levels T7–T11 stained by a combination of Nissl and AChE histochemistry were counted at E18.5 in LIFRβ- and CT-1-deficient mice, respectively, and wild-type littermates. Spinal cord levels T7–T11 were analyzed, because they contain the highest density of PSNs projecting to the adrenal medulla, a well established and thoroughly investigated target organ of the PSN (Schober and Unsicker, 2001). Figure 3 documents a 22.5% loss of PSNs in LIFRβ (−/−) mice compared with wild-type littermates. A similar loss (−18.5%) was found in CT-1-deficient mice at the same age (Fig. 3), suggesting CT-1 as a candidate factor for LIFRβ-expressing PSNs. To determine the time interval at which these neuronal losses occur, PSNs were identified in LIFRβ (−/−) mice and wild-type littermates at E13.5 using ChAT in situ hybridization. As shown in Figure 4, at this age, the population of PSNs has already completely separated from somatic motoneurons. LIFRβ-deficient E13.5 embryos revealed a small, but significant 9% loss in this neuronal population, which may be attributable to a reduction in cell number or reduction in ChAT mRNA levels.
Figure 3.
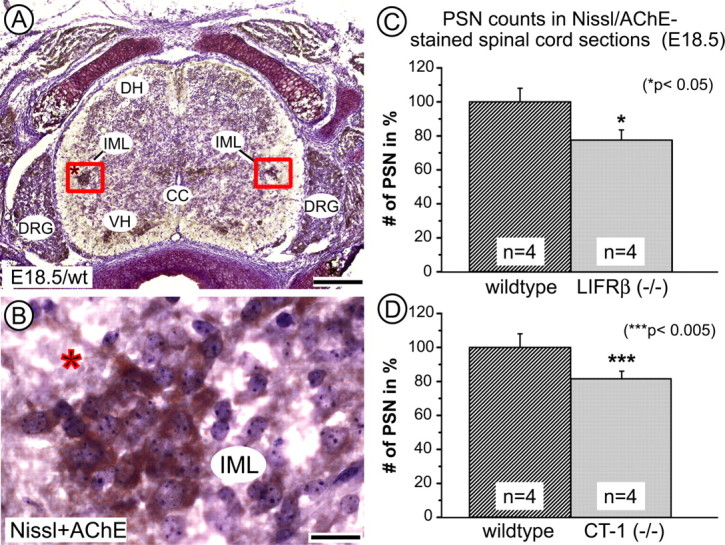
Quantification of AChE-positive, Nissl-stained PSNs within an area of 120 × 240 μm of the intermediate gray in the spinal cord at E18.5. A, Overview showing AChE-positive neurons (brown) in the intermediolateral column (IML), ventral horn (VH), and dorsal root ganglia (DRG). DH, Dorsal horn; CC, central canal. Scale bar, 200 μm. B, Higher magnification of the IML indicated by the asterisk in A. Scale bar, 25 μm. C, D, Quantification of PSNs in LIFRβ-deficient (C) and CT-1-deficient mice (D) reveals reductions of −22.5 and −18.5%, respectively. Data are given as mean ± SEM.
Figure 4.
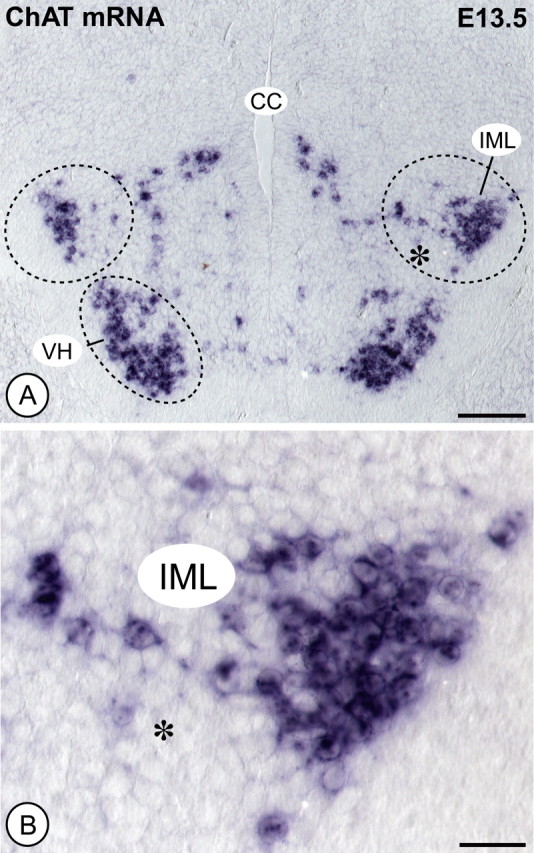
Counts of ChAT mRNA-positive PSNs in E13.5 spinal cord. A, ChAT in situ shows positive motoneurons in the ventral horn (VH) and, more dorsally, PSNs predominantly located in the intermediolateral column (IML). CC, Central canal. Scale bar, 100 μm. B, Higher magnification of the IML indicated by the asterisk in A. Scale bar, 25 μm. Quantification of PSNs in LIFRβ-deficient mice reveals a 9% loss compared with wild-type littermates.
PSNs projecting to the adrenal gland are more severely affected than the average PSN population in E18.5 LIFRβ-deficient embryos
To determine whether or not losses of PSNs projecting to the adrenal gland matched losses in the overall PSN population, we specifically labeled PSNs that are preganglionic to the adrenal medulla by retrograde tracing with the lipophilic dye DiI. A DiI crystal was implanted into the left adrenal gland of perfusion-fixed E18.5 embryos (Fig. 5A), and all labeled neurons throughout the spinal cord were counted after 5 months. Figure 5C–F documents that perikarya of PSNs were labeled together with their processes extending into the regions of the autonomic nucleus centralis and nucleus intercalatus. Cell counts (Fig. 5B) revealed a 55% loss of PSNs that specifically projected to the adrenal medulla in LIFRβ (−/−) mice. Together, these data indicate that LIFRβ deficiency causes a significant loss in the number of PSNs during their embryonic development. However, the PSN population innervating the adrenal medulla seems to be more severely affected, suggesting that the requirement for LIFRβ ligands may vary among specific pools of PSNs.
Figure 5.
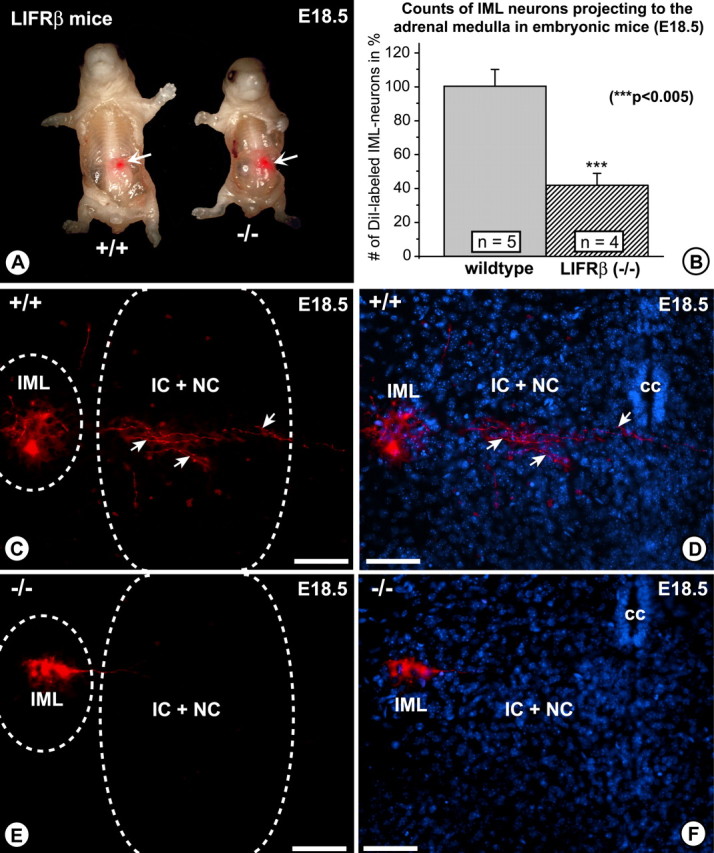
Retrograde tracing with the lipophilic dye DiI from the adrenal gland to the spinal cord in E18.5 LIFRβ (+/+) and LIFRβ (−/−) mice. A, The arrows point to the site where the DiI crystal was inserted. B, Quantitative determination of DiI-traced PSN numbers of LIFRβ (+/+) and LIFRβ (−/−) mice. The numbers of positive PSNs are reduced by 55% in LIFRβ mutants. C, E, DiI-traced PSNs. Positive neurons are located within the intermediolateral column. They extend processes into the area of the intercalated nucleus and central nucleus (arrows). D, F, DAPI staining of the area shown in C and E. Scale bars: C–F, 100 μm. Data are given as mean ± SEM. IML, Intermediolateral column; IC, intercalated nucleus; NC, central nucleus; cc, central canal.
Losses of PSNs projecting to the adrenal medulla are not caused by a numerical deficit in postganglionic adrenal chromaffin cells
A numerical decrease in PSNs might result from a specific loss of their postganglionic targets, e.g., adrenal medullary chromaffin cells, which have already been contacted by PSN axons at this age. Losses of PSNs have been documented in mice with a targeted deletion of the NGF receptor tyrosine kinase A (trkA) (Schober et al., 1997), probably resulting from the death of NGF-deprived postganglionic paravertebral sympathetic neurons (Smeyne et al., 1994). Regardless of the PSN phenotype, however, the possibility of an adrenal medullary phenotype also had to be considered in light of the expression of LIFRβ, CNTF, LIF, and CT-1 mRNAs in the embryonic adrenal gland. Figure 6, A and B, show, however, that there were no overt differences in numbers of TH-immunoreactive cells in E18.5 adrenal glands of LIFRβ-deficient mice compared with wild-type littermates. Cell counts (Fig. 6E) verified equal numbers of TH-positive chromaffin cells in knock-out and wild-type animals. These results suggest that losses of PSNs encountered in LIFRβ-deficient mice do not result from a numerical deficit of their postganglionic target cells.
Figure 6.
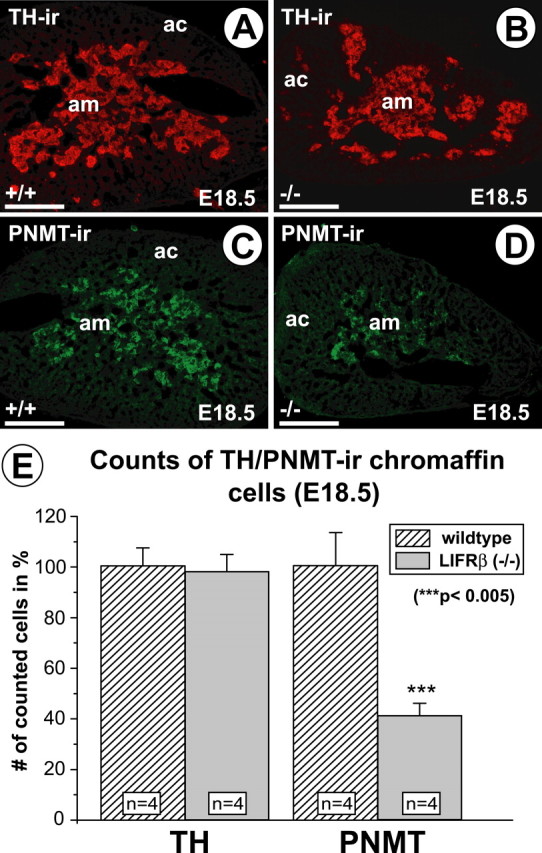
TH- and PNMT-immunoreactive cells in the adrenal gland of E18.5 LIFRβ (+/+) and LIFRβ (−/−) mice. Although the numbers of TH-positive cells (A, B) do not seem to be overtly altered, the numbers of PNMT-positive cells (C, D) are clearly reduced. am, Adrenal medulla; ac, adrenal cortex. Scale bars, 100 μm. E, Quantification of TH- and PNMT-immunoreactive cells in LIFRβ (+/+)- and LIFRβ-deficient mice. Although the numbers of TH-positive cells are not altered, the numbers of PNMT-positive cells are reduced by >50%. Data are given as mean ± SEM.
LIFRβ (−/−), in contrast to CT-1 (−/−) mice, reveal a reduction of PNMT-positive chromaffin cells and adrenaline levels
Despite normal numbers of adrenal chromaffin cells in LIFRβ (−/−) mice, we were interested to identify a putative deficit in chromaffin cell differentiation possibly caused by the substantial loss of PSNs in LIFRβ-deficient mice and/or deficient LIFRβ signaling within the adrenal gland. Figure 6, C and D, reveal a substantial reduction in PNMT-IR in the adrenal medulla of LIFRβ (−/−) mice. Cell counts (Fig. 6E) confirmed a reduction in numbers of PNMT-positive cells by >50%. Furthermore, quantitative determinations of adrenaline (Fig. 7A) showed a 65% loss in adrenaline levels of LIFRβ (−/−) adrenal glands. Levels of noradrenaline were unchanged. Together, these results suggest a substantial deficit in adrenaline synthesis and differentiation of chromaffin cells toward the PNMT-positive phenotype in mice lacking LIFRβ. In contrast, animals lacking only CT-1 did not show a detectable reduction of adrenaline levels (Fig. 7B).
Figure 7.

Quantitative determinations of catecholamines by HPLC and electrochemical detection in adrenal glands of E18.5 LIFRβ, CT-1, and CNTF knock-out mice and respective wild-type littermates. A, Quantification of adrenal catecholamines LIFRβ (+/+) and LIFRβ (−/−) mice. Although the levels of noradrenaline are not altered, the levels of adrenaline are decreased by ∼65%. B, Quantifications of noradrenaline and adrenaline in CT-1- and CNTF-deficient mice fail to reveal significant changes compared with wild-type littermates. Data are given as mean ± SEM.
LIFRβ-deficient mice fail to reveal defects in adrenal innervation patterns and numbers of nerve terminals
A numerical reduction in numbers of PSNs visualized by retrograde DiI tracing might reflect degeneration of PSN axons within the target and concomitant failure of DiI uptake rather than a real loss of PSN cell bodies. To investigate this possibility and visualize nerve fibers within the adrenal gland, we used SMI312, a monoclonal antibody against phosphorylated neurofilaments. Adrenal glands of E18.5 LIFRβ (−/−) mice and wild-type littermates failed to reveal overt differences in staining patterns (data not shown). Because this method did not permit an exact quantification of axon numbers, electron microscopy (EM) was used to determine numbers of axon terminals abutting chromaffin cells (Fig. 8). Numbers of nerve terminals on chromaffin cells were not significantly reduced. This suggests that the numerical reduction of PSN cell bodies in the spinal cord visualized by DiI tracing is unlikely attributable to a retraction of terminal axons from their target and must rather reflect a real loss of PSNs. Furthermore, normal numbers of axon terminals on adrenal chromaffin cells suggest that reduced numbers of PSNs in LIFRβ-deficient mice are capable of maintaining a normal terminal arborization, possibly resulting from axonal sprouting. The morphology of nerve terminals was not different between wild-type and LIFRβ (−/−) mice, indicating that LIFRβ signaling does not play a role in formation and morphological maturation of synapses (Fig. 8).
Figure 8.
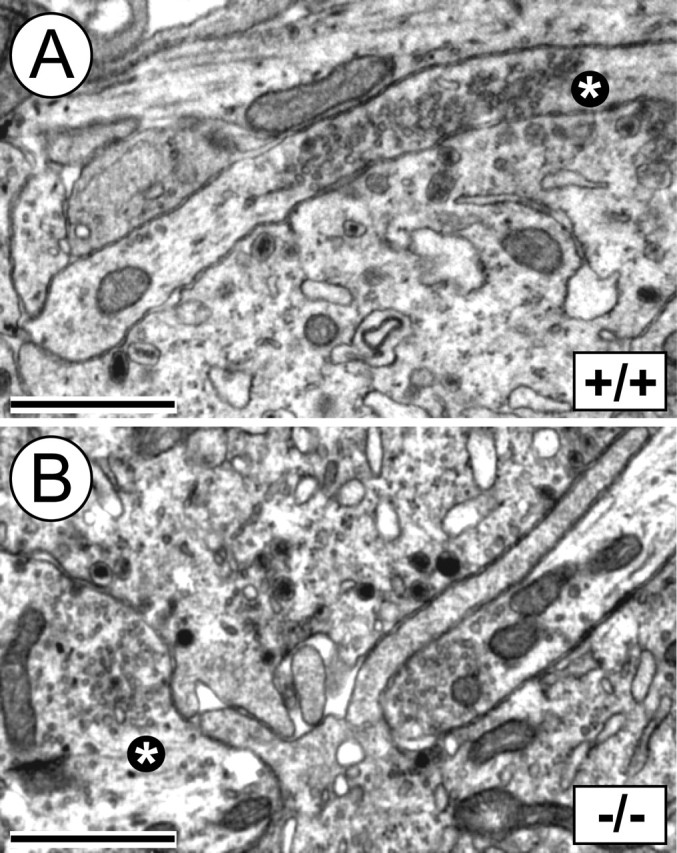
Electron microscopy of nerve terminals on adrenal chromaffin cells in E18.5 LIFRβ (+/+) and LIFRβ (−/−) mice. A, B, Axon terminals abutting chromaffin cells show clear synaptic vesicles and no overt morphological differences when comparing LIFRβ (+/+) (A) and LIFRβ (−/−) (B) adrenal medullae. The asterisks mark evaluated axonal terminals. Scale bars, 1 μm. Quantification of nerve terminals on chromaffin cells failed to reveal any significant difference between LIFRβ (−/−) mice and wild-type littermates. Six wild-type and eight LIFRβ knock-out mice were analyzed.
Discussion
The present study provides evidence that establishment and maintenance of a correct number of PSNs during embryonic development requires signaling via LIFRβ and expression of CT-1. The LIFRβ (−/−) and CT-1 (−/−) phenotypes each reveal a numerically similar deficit of ∼20% of PSNs at E18.5. The numerical decrease of PSNs monitored in LIFRβ (−/−) mice has apparently commenced at E13.5 and is progressive until E18.5, i.e., shortly before LIFRβ-deficient animals die.
Numbers of PSNs are known to be regulated by their postganglionic targets. For example, PSN losses of ∼40% (Schober et al., 1997) have been shown to occur during destruction of sympathetic ganglia in mice lacking the NGF receptor trkA (Smeyne et al., 1994). Along the same line, administration of exogenous NGF to chick embryos was shown to increase numbers of both postganglionic and preganglionic sympathetic neurons (Oppenheim et al., 1982). Whether losses of PSNs in LIFRβ-deficient mice might be attributable to alterations in their postganglionic targets was tested in our study by investigating one major target, the chromaffin cells of the adrenal medulla. Our data suggest that PSNs are not lost in LIFRβ deficiency because of a reduction in numbers of chromaffin cells. Furthermore, it should be noted that PSNs in LIFRβ (−/−) mice disappear before the period of ontogenetic cell death in the postganglionic peripheral sympathetic system, which occurs after birth (Parker et al., 1988; Lawson et al., 1997).
The decrease in numbers of PSNs (−20%) occurs in analogy to, but seems to be less dramatic than the loss of somatic motoneurons found in LIFRβ-deficient mice (>35%, facial motoneurons; >40%, lumbar motoneurons; >50%, nucleus ambiguus) (Li et al., 1995). This difference might be accounted for by a putative small delay in maturation of PSNs compared with somatic motoneurons. Moreover, given that somatic motoneurons, in contrast to PSNs (cf. Wetts and Vaughn, 1998), undergo programmed cell death during their embryonic development, distinct trophic factor requirements of PSNs and somatic motoneurons might be also conceivable. Somatic motoneurons and PSNs originate from a common pool of progenitor cells, which, in the mouse, proliferate from E9.5 to E11 (for review, see Schober and Unsicker, 2001). The cells subsequently migrate into the developing ventral horn and intermediolateral column of the spinal cord. Somatic motoneurons and PSNs separate until E14 (E16 in rat) (Markham and Vaughn, 1991) (for review, see Schober and Unsicker, 2001). This is consistent with our observation that somatic motoneurons and PSNs are well separated at E13.5 in the mouse (compare Fig. 4). The delay of PSNs, compared with the somatic motoneurons, in reaching their final position within the spinal cord, might be linked to a delay in maturation and account for the smaller loss of PSNs (−20%) than somatic motoneurons (cf. Li et al., 1995). Because LIFRβ (−/−) mice die at birth, it is not possible to investigate the hypothesis that PSN death might further progress and finally match, in magnitude, death of somatic motoneurons. Nevertheless, losses of somatic motoneurons and PSNs, which both carry receptors for LIFRβ, in LIFRβ-deficient mice, likely suggest that similar LIFRβ-dependent mechanisms may operate during development of both neuron populations. Both local and retrograde mechanisms may underlie regulation of survival. Retrograde mechanisms are likely to govern somatic motoneuron survival during their embryonic development. For PSNs, both local and retrograde mechanisms are conceivable, given the dual locations of LIFRβ ligands in the spinal cord and adrenal gland. A deficit in proliferation of motoneuron progenitors attributable to LIFRβ deficiency has not been reported, but cannot be completely ruled out in light of the small (−9%) reduction of PSN numbers at E13.5 found in our study.
The coincidence concerning an identical magnitude of PSN losses in LIFRβ- and CT-1-deficient mice, together with the expression of CT-1 in both spinal cord and adrenal gland, may suggest that CT-1 could be one physiologically relevant ligand for the maintenance of embryonic LIFRβ expressing PSNs. CT-1 is a well documented muscle-derived trophic factor (Oppenheim et al., 2001). Its deficiency causes increased motoneuron cell loss in spinal cord and brainstem between E13.5 and the first postnatal week, further underscoring the physiological relevance of CT-1 as a trophic factor for embryonic motoneurons. Retrograde axonal transport of CT-1 has been documented (Rind et al., 2005). Whether CT-1 acts on PSNs as a target-derived or local factor remains to be investigated. By E13, axon terminals from PSNs expressing LIFRβ have invaded the adrenal gland (rat) (Daikoku et al., 1977), providing a substrate for putative transport of LIFRβ ligands to the spinal cord.
Using a model of target deprivation, i.e., adrenomedullectomy, we showed previously that many neurotrophic factors, when substituted to the medullectomized adrenal gland, can rescue PSNs from death, including CNTF, FGF-2, TGF-β, GDNF, and NT-4 (Blottner et al., 1989, 1996; Blottner and Unsicker, 1990; Schober et al., 1998, 1999). However, only NT-4 has thus far proven its physiological relevance, because NT-4 mutant mice show significant losses of PSNs projecting to the adrenal medulla and the paravertebral and prevertebral sympathetic ganglia (Schober et al., 1998; Roosen et al., 2001). Interestingly, losses of PSNs in NT-4 (−/−) mice start after postnatal day 6, i.e., they do not occur during embryonic development. The full manifestation of PSN losses is seen in adult NT-4 (−/−) mice, suggesting that NT-4 serves as a neuronal maintenance factor after birth. In contrast to NT-4, LIFRβ ligands must be important for an earlier phase in the development of PSNs.
The 55% loss in the total number of PSNs specifically projecting to the adrenal medulla, compared with a 22.5% loss in the total number of PSNs may be interpreted in several directions. Our first concern had been that growth of preganglionic fibers to the adrenal medulla was impaired in the absence of LIFRβ signaling, causing a deficit in retrograde DiI labeling. However, this possibility is unlikely, because numbers of axon terminals on chromaffin cells identified by EM were normal in LIFRβ (−/−) mice. Another possibility may be a specific loss of those PSNs that project to the adrenal medulla, compared with PSNs projecting to other targets, i.e., prevertebral and paravertebral sympathetic ganglia. Differences concerning trophic requirements of distinct pools of somatic motoneurons have been reported (Gould and Oppenheim, 2004). However, the 55% loss seen in DiI traced embryos versus the 22.5% loss evaluated by Nissl/AChE staining might even be numerically fully consistent. It has been shown previously that 25% of the total number of PSNs in segments Th7–Th10 project to the adrenal medulla, whereas the remaining 75% project to other targets (Blottner and Baumgarten, 1992). It is conceivable, therefore, that a 20% loss seen in Nissl/AChE stainings may almost match the 55% loss seen after DiI tracing.
In addition to the PSN phenotype, LIFRβ knock-out mice show deficits in differentiation of at least one subset of adrenal chromaffin cells, which synthesizes adrenaline and can be identified by PNMT, the adrenaline synthesizing enzyme, as a marker. PNMT is widely thought to be exclusively regulated by corticosteroid hormones (Wurtman and Axelrod, 1965; Wurtman et al., 1968). However, more recent evidence points to the possibility that PNMT activity is also neurally controlled (Wong et al., 1993; Morita and Wong, 1996). In rat, preganglionic nerve endings invade the adrenal gland at E13.5 (Daikoku et al., 1977). Although innervation of chromaffin cells in terms of eliciting release of catecholamines has been reported to occur not before the second postnatal week in the rat (Seidler and Slotkin, 1985), it cannot be excluded that preganglionic axons might influence chromaffin cell differentiation by mechanisms other than nerve impulse activity. In an alternative scenario, one might conceive, in light of LIFRβ expression in the developing adrenal gland, that chromaffin cells may respond to local sources of the respective ligands. Whatever explanation may be correct, the LIFRβ mutant phenotype suggests that the regulation of PNMT and adrenaline synthesis may be more complex than assumed previously. Interestingly, losses of PNMT affected only approximately one-half of the PNMT-positive chromaffin cell population, adding another aspect to the heterogeneity of adrenal chromaffin cells (cf. Huber et al., 2002). A partial loss in the capacity of the adrenal medulla to synthesize adrenaline is also a feature of the NT-4 (−/−) phenotype (Roosen et al., 2001). Interestingly, deficiency in CT-1, in contrast to LIFRβ, did not affect levels of adrenaline in the adrenal gland, raising the possibility that additional ligands to the LIFRβ are implied in the regulation of adrenal hormones.
In conclusion, the present study adds ligands to the LIFRβ, most likely CT-1, next to NT-4, to the list of molecules that have a physiological relevance for the development and maintenance of the structural and functional integrity of the sympathetic preganglionic and postganglionic circuitry. LIFRβ ligands and NT-4 appear to complement each other insofar as the former affect development and maintenance during the embryonic period, whereas NT-4 serves as a maintenance factor for adult PSN neurons.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grants SFB 581 (M.S.) and SFB 488/A6 (K.U.). We thank Richard Hertel, Ingrid Stenull, Gerald Bendner, Nicole Karch, and Barbara Brühl for valuable technical assistance. We also thank Dr. Jens Strelau for helpful suggestions and comments on the RT-PCR.
References
- Andrä J, Lojda Z (1986). A histochemical method for the demonstration of acetylcholinesterase activity using semipermeable membranes. Histochemistry 84:575–579. [DOI] [PubMed] [Google Scholar]
- Arakawa Y, Sendtner M, Thoenen H (1990). Survival effect of ciliary neurotrophic factor (CNTF) on chick embryonic motoneurons in culture: comparison with other neurotrophic factors and cytokines. J Neurosci 10:3507–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazan JF (1991). Neuropoietic cytokines in the hematopoietic fold. Neuron 7:197–208. [DOI] [PubMed] [Google Scholar]
- Blottner D, Baumgarten H-G (1992). Nitric oxides synthas (NOS)-containing sympathoadrenal cholinergic neurons of the rat IML-cell column: evidence from histochemistry, and retrograde labeling. J Comp Neurol 316:45–55. [DOI] [PubMed] [Google Scholar]
- Blottner D, Unsicker K (1990). Maintenance of intermediolateral spinal cord neurons by fibroblast growth factor administered to the medullectomized rat adrenal gland: dependence on intact organ innervation and cellular organization of implants. Eur J Neurosci 2:378–382. [DOI] [PubMed] [Google Scholar]
- Blottner D, Brüggemann W, Unsicker K (1989). Ciliary neurotrophic factor supports target-deprived preganglionic sympathetic spinal cord neurons. Neurosci Lett 105:316–320. [DOI] [PubMed] [Google Scholar]
- Blottner D, Wolf N, Lachmund A, Flanders KC, Unsicker K (1996). TGF-β rescues target deprived preganglionic sympathetic neurons in the spinal cord. Eur J Neurosci 8:202–210. [DOI] [PubMed] [Google Scholar]
- Cheema SS, Richards LJ, Murphy M, Bartlett PF (1994). Leukaemia inhibitory factor rescues motoneurones from axotomy-induced cell death. NeuroReport 5:989–992. [DOI] [PubMed] [Google Scholar]
- Daikoku S, Kinutani M, Sako M (1977). Development of the adrenal medullary cells in rats with reference to synaptogenesis. Cell Tissue Res 179:77–86. [DOI] [PubMed] [Google Scholar]
- Derouet D, Rousseau F, Alfonsi F, Froger J, Hermann J, Barbier F, Perret D, Diveu C, Guillet C, Preisser L, Dumont A, Barbado M, Morel A, deLapeyriere O, Gascan H, Chevalier S (2004). Neuropoietin, a new IL-6-related cytokine signaling through the ciliary neurotrophic factor receptor. Proc Natl Acad Sci USA 101:4827–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernsberger U, Patzke H, Rohrer H (1997). The developmental expression of choline acetyltransferase (ChAT) and the neuropeptide VIP in chick sympathetic neurons: evidence for different regulatory events in cholinergic differentiation. Mech Dev 68:115–126. [DOI] [PubMed] [Google Scholar]
- Ernsberger U, Reissmann E, Mason I, Rohrer H (2000). The expression of dopamine beta-hydroxylase, tyrosine hydroxylase, and Phox2 transcription factors in sympathetic neurons: evidence for common regulation during noradrenergic induction and diverging regulation later in development. Mech Dev 92:169–177. [DOI] [PubMed] [Google Scholar]
- Forger NG, Prevette D, deLapeyriere O, de Bovis B, Wang S, Bartlett P, Oppenheim RW (2003). Cardiotrophin-like cytokine/cytokine-like factor 1 is an essential trophic factor for lumbar and facial motoneurons in vivo. J Neurosci 23:8854–8858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearing DP, Gough NM, King JA, Hilton DJ, Nicola NA, Simpson RJ, Nice EC, Kelso A, Metcalf D (1987). Molecular cloning and expression of cDNA encoding a murine myeloid leukaemia inhibitory factor (LIF). EMBO J 6:3995–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearing DP, Thut CJ, VandeBos T, Gimpel SD, Delaney PB, King J, Price V, Cosman D, Beckmann MP (1991). Leukemia inhibitory factor receptor is structurally related to the IL-6 signal transducer, gp130. EMBO J 10:2839–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearing DP, Comeau MR, Friend DJ, Gimpel SD, Thut CJ, McGourty J, Brasher KK, King JA, Gillis S, Mosley B, Zeigler SF, Cosman D (1992). The IL-6 signal transducer, gp130: an oncostatin M receptor and affinity converter for the LIF receptor. Science 255:1434–1437. [DOI] [PubMed] [Google Scholar]
- Gould TW, Oppenheim RW (2004). The function of neurotrophic factor receptors expressed by the developing adductor motor pool in vivo. J Neurosci 12:4668–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F (2003). Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J 374:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmann B, Wiese S, Samsam M, Grohmann K, Pennica D, Martini R, Sendtner M (2005). Triple knock-out of CNTF, LIF, and CT-1 defines cooperative and distinct roles of these neurotrophic factors for motoneuron maintenance and function. J Neurosci 25:1778–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber K, Brühl B, Guillemot F, Olson EN, Ernsberger U, Unsicker K (2002). Development of chromaffin cells depends on MASH1 function. Development 129:4729–4738. [DOI] [PubMed] [Google Scholar]
- Ishii K, Oda Y, Ichikawa T, Deguchi T (1990). Complementary DNAs for choline acetyltransferase from spinal cords of rat and mouse: nucleotide sequences, expression in mammalian cells, and in situ hybridization. Brain Res Mol Brain Res 7:151–159. [DOI] [PubMed] [Google Scholar]
- Lawson SJ, Davies HJ, Bennett JP, Lowrie MB (1997). Evidence that spinal interneurons undergo programmed cell death postnatally in the rat. Eur J Neurosci 9:794–799. [DOI] [PubMed] [Google Scholar]
- Leung DW, Parent AS, Cachianes G, Esch F, Coulombe JN, Nikolics K, Eckenstein FP, Nishi R (1992). Cloning, expression during development, and evidence for release of a trophic factor for ciliary ganglion neurons. Neuron 8:1045–1053. [DOI] [PubMed] [Google Scholar]
- Li M, Sendtner M, Smith A (1995). Essential function of LIF receptor in motor neurons. Nature 378:724–727. [DOI] [PubMed] [Google Scholar]
- Lin LF, Mismer D, Lile JD, Armes LG, Butler ET, Vannice JL, Collins F (1989). Purification, cloning, and expression of ciliary neurotrophic factor (CNTF). Science 246:1023–1025. [DOI] [PubMed] [Google Scholar]
- Markham JA, Vaughn JE (1991). Migration patterns of sympathetic preganglionic neurons in embryonic rat spinal cord. J Neurobiol 22:811–822. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Martinou I, Kato AC (1992). Cholinergic differentiation factor (CDF/LIF) promotes survival of isolated rat embryonic motoneurons in vitro. Neuron 8:737–744. [DOI] [PubMed] [Google Scholar]
- Meyer V, Unsicker K (1994). Cell density and exogenous CNTF affect CNTF mRNA levels in glial cell cultures. NeuroReport 5:1946–1948. [DOI] [PubMed] [Google Scholar]
- Morikawa Y (2005). Oncostatin M in the development of the nervous system. Anat Sci Int 80:53–59. [DOI] [PubMed] [Google Scholar]
- Morita K, Wong DL (1996). Role of Egr-1 in cholinergic stimulation of phenylethanolamine N-methyltransferase promoter. J Neurochem 67:1344–1351. [DOI] [PubMed] [Google Scholar]
- Müller TH, Unsicker K (1981). High-performance liquid chromatography with electrochemical detection as highly efficient tool for studying catecholaminergic systems. I. Quantification of noradrenaline, adrenaline and dopamine in cultured adrenal medullary cells. J Neurosci Methods 4:39–52. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW (1991). Cell death during development of the nervous system. Annu Rev Neurosci 14:453–501. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Maderdrut JL, Wells DJ (1982). Cell death of motoneurons in the chick embryo spinal cord. VI. Reduction of naturally occurring cell death in the thoracolumbar column of Terni by nerve growth factor. J Comp Neurol 210:174–189. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Wiese S, Prevette D, Armanini M, Wang S, Houenou LJ, Holtmann B, Gotz R, Pennica D, Sendtner M (2001). Cardiotrophin-1, a muscle-derived cytokine, is required for the survival of subpopulations of developing motoneurons. J Neurosci 21:1283–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker TL, Kesse WK, Tomlinson A, Coupland RE (1988). Ontogenesis of preganglionic sympathetic innervation of rat adrenal chromaffin cells. In: Progress in catecholamine research, part A, basic aspects and peripheral mechanisms (Dahlström A, Belmaker RH, Sandler M, eds) , pp. 227–232. New York: Alan R. Liss.
- Pennica D, Shaw KJ, Swanson TA, Moore MW, Shelton DL, Zioncheck KA, Rosenthal A, Taga T, Paoni NF, Wood WI (1995). Cardiotrophin-1. Biological activities and binding to the leukemia inhibitory factor receptor/gp130 signaling complex. J Biol Chem 270:10915–10922. [DOI] [PubMed] [Google Scholar]
- Pennica D, Arce V, Swanson TA, Vejsada R, Pollock RA, Armanini M, Dudley K, Phillips HS, Rosenthal A, Kato AC, Henderson CE (1996). Cardiotrophin-1, a cytokine present in embryonic muscle, supports long-term survival of spinal motoneurons. Neuron 17:63–74. [DOI] [PubMed] [Google Scholar]
- Pyner S, Coote JH (1994). Evidence that sympathetic preganglionic neurones are arranged in target-specific columns in the thoracic spinal cord of the rat. J Comp Neurol 342:15–22. [DOI] [PubMed] [Google Scholar]
- Reynolds ES (1963). The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol 17:208–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rind HB, Butowt R, von Bartheld CS (2005). Synaptic targeting of retrogradely transported trophic factors in motoneurons: comparison of glial cell line-derived neurotrophic factor, brain-derived neurotrophic factor, and cardiotrophin-1 with tetanus toxin. J Neurosci 25:539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosen A, Schober A, Strelau J, Böttner M, Faulhaber J, Bendner G, Seller H, Ehmke H, McIllwarth SL, Lewin GR, Unsicker K (2001). Lack of NT-4 causes selective structural and chemical deficits in sympathetic ganglia and their preganglionic innervation. J Neurosci 21:3073–3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober A, Unsicker K (2001). Growth and neurotrophic factors regulating development and maintenance of sympathetic preganglionic neurons. Int Rev Cytol 205:37–76. [DOI] [PubMed] [Google Scholar]
- Schober A, Minichiello L, Keller M, Huber K, Layer PG, Roig-López JL, García-Arrarás JE, Klein R, Unsicker K (1997). Reduced acetylcholinesterase (AChE) activity in adrenal medulla and loss of sympathetic preganglionic neurons in TrkA-deficient, but not TrkB-deficient, mice. J Neurosci 17:891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober A, Wolf N, Huber K, Hertel R, Krieglstein K, Minichiello L, Kahane N, Widenfalk J, Kalcheim C, Olson L, Klein R, Lewin GR, Unsicker K (1998). TrkB and neurotrophin-4 are important for development and maintenance of sympathetic preganglionic neurons innervating the adrenal medulla. J Neurosci 18:7272–7284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober A, Hertel R, Arumae U, Farkas L, Jaszai J, Krieglstein K, Saarma M, Unsicker K (1999). Glial cell line-derived neurotrophic factor rescues target-deprived sympathetic spinal cord neurons but requires transforming growth factor-β as cofactor in vivo. J Neurosci 19:2008–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidler FJ, Slotkin TA (1985). Adrenomedullary function in the neonatal rat: response to acute hypoxia. J Physiol (Lond) 358:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendtner M, Kreutzberg GW, Thoenen H (1990). Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature 345:440–441. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira SA, Barbacid M (1994). Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature 368:193–194. [DOI] [PubMed] [Google Scholar]
- Stöckli KA, Lottspeich F, Sendtner M, Masiakowski P, Carroll P, Gotz R, Lindholm D, Thoenen H (1989). Molecular cloning, expression and regional distribution of rat ciliary neurotrophic factor. Nature 342:920–923. [DOI] [PubMed] [Google Scholar]
- Strack AM, Sawyer WB, Marubio LM, Loewy AD (1988). Spinal origin of sympathetic preganglionic neurons in the rat. Brain Res 455:187–191. [DOI] [PubMed] [Google Scholar]
- Unsicker K, Reichert-Preibsch H, Schmidt R, Pettmann B, Labourdette G, Sensenbrenner M (1987). Astroglial and fibroblast growth factors have neurotrophic functions for cultured peripheral and central nervous system neurons. Proc Natl Acad Sci USA 84:5459–5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware CB, Horowitz MC, Renshaw BR, Hunt JS, Liggitt D, Koblar SA, Gliniak BC, McKenna HJ, Papayannopoulou T, Thoma B, Cheng L, Danovan PJ, Peschon JJ, Bartlett PF, Willis CR, Wright BD, Carpenter MK, Davison BL, Gearing DP (1995). Targeted disruption of the low-affinity leukemia inhibitory factor receptor gene causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development 121:1283–1299. [DOI] [PubMed] [Google Scholar]
- Wetts R, Vaughn JE (1998). Differences in developmental cell death between somatic and autonomic motor neurons of rat spinal cord. J Comp Neurol 396:483–492. [DOI] [PubMed] [Google Scholar]
- Wong V, Arriaga R, Ip NY, Lindsay RM (1993). The neurotrophins BDNF, NT-3 and NT-4/5, but not NGF, up-regulate the cholinergic phenotype of developing motor neurons. Eur J Neurosci 5:466–474. [DOI] [PubMed] [Google Scholar]
- Wurtman RJ, Axelrod J (1965). Adrenaline synthesis: control by the pituitary gland and adrenal glucocorticoids. Science 150:1464–1465. [DOI] [PubMed] [Google Scholar]
- Wurtman RJ, Axelrod J, Vesell ES, Ross GT (1968). Species differences in inducibility of phenylethanolamine-N-methyl transferase. Endocrinology 82:584–590. [DOI] [PubMed] [Google Scholar]