

Abstract
Enhanced oxidative stress has been implicated in the excitotoxicity of the CNS, and 8-oxo-7,8-dihydro-guanine (8-oxoG), a major type of oxidative damage in nucleic acids, was reported to be accumulated in the rat hippocampus after kainate administration. We herein showed that the 8-oxoG levels in mitochondrial DNA and cellular RNA increased significantly in the CA3 subregion of the mouse hippocampus 6–12 h after kainate administration but returned to basal levels within a few days. Laser-scanning confocal microscopy revealed the 8-oxoG accumulation in mitochondrial DNA to be remarkable in CA3 microglia, whereas that in nuclear DNA or cellular RNA was also detected in the CA3 pyramidal cells and astrocytes. 8-oxoG accumulation in cellular DNA or RNA should be suppressed by MutT homolog 1 (MTH1) with 8-oxo-dGTPase (8-oxo-7,8-dihydro-2′-deoxyguanosine triphosphatase) activity and 8-oxoG-DNA glycosylase 1 (OGG1) with 8-oxoG DNA glycosylase activity. We thus examined the expression level of MTH1 and OGG1 in the mouse hippocampus after kainate administration. The Mth1 mRNA level decreased soon after kainate administration and then quickly recovered beyond the basal level, and a continuously increased MTH1 protein level was observed, whereas the Ogg1 mRNA level remained constant. MTH1-null and wild-type mice exhibited a similar degree of CA3 neuron loss after kainate administration; however, the 8-oxoG levels that accumulated in mitochondrial DNA and cellular RNA in the CA3 microglia significantly increased in the MTH1-null mice in comparison with wild-type mice, thus demonstrating that MTH1 efficiently suppresses the accumulation of 8-oxoG in both cellular DNA and RNA in the hippocampus, especially in microglia, caused by excitotoxicity.
Keywords: hippocampus, repair, knock-out mice, oxidation, nucleotide, mitochondria, microglia, kainate
Introduction
Glutamate, an excitatory neurotransmitter, plays an important role in brain function; however, excessive amounts of glutamate can cause neuronal damage through the massive activation of its receptors (Olney, 1969; Nakanishi, 1992; Hollmann and Heinemann, 1994; Lerma, 2003). The molecular mechanism of the excitotoxicity has been investigated using a potent agonist, kainate (Shinozaki and Konishi, 1970; Olney et al., 1974). Kainate administration to rodents produces epileptiform seizures followed by a delayed loss of pyramidal cells in the CA3 subregion of hippocampus (Nadler, 1981; Ben-Ari, 1985).
The activation of kainate receptors induces various responses, such as an increase of intracellular Ca2+ and the production of reactive oxygen species (ROS) (Coyle and Puttfarcken, 1993; Wang et al., 2005). Lipid peroxidation has been implicated as a major type of neuronal damage by ROS under excitotoxicity (Bruce and Baudry, 1995; Waterfall et al., 1995); however, 8-oxo-7,8-dihydro-guanine (8-oxoG), a major type of oxidative damage in nucleic acids (Kasai and Nishimura, 1984), is also known to accumulate in the rat brain after kainate administration (Lan et al., 2000; Patel and Li, 2003). Oxidative damage in nucleic acids, therefore, is also implicated to play a role in excitotoxicity.
8-oxoG can pair with adenine as well as cytosine, and thus alter the genetic information in DNA or RNA (Tajiri et al., 1995; Taddei et al., 1997). 8-oxoG in DNA or RNA is derived not only from their direct oxidation but also from the incorporation of 8-oxo-7,8-dihydro-2′-deoxyguanosine triphosphate (8-oxo-dGTP) or 8-oxo-GTP generated in nucleotide pools. In Escherichia coli, MutM excises 8-oxoG opposite cytosine in DNA (Michaels et al., 1992), and MutT hydrolyzes 8-oxo-dGTP and 8-oxo-GTP to monophosphates (Maki and Sekiguchi, 1992; Taddei et al., 1997). In mammals, 8-oxoG-DNA glycosylase 1 (OGG1), as a functional homolog for MutM (Boiteux and Radicella, 1999; Nishioka et al., 1999), and MutT homolog 1 (MTH1), as a MutT homolog (Sakumi et al., 1993; Furuichi et al., 1994; Kakuma et al., 1995), were identified.
A high degree of 8-oxoG accumulation occurs in the damaged neurons of patients with various neurodegenerative diseases, such as Parkinson’s disease (PD) (Shimura-Miura et al., 1999; Zhang et al., 1999), Alzheimer’s disease (AD) (Nunomura et al., 2001), or amyotrophic lateral sclerosis (Kikuchi et al., 2002). Furthermore, the expression of MTH1 and OGG1 significantly increases in the substantia nigral neurons of PD patients (Shimura-Miura et al., 1999; Fukae et al., 2005) or in the entorhinal cortex of AD patients (Furuta et al., 2001).
We recently showed that MTH1-null mice exhibited a greater accumulation of 8-oxoG in the terminal fibers of dopamine neurons in the striatum accompanied with an accelerated dysfunction of the terminal fibers after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration, than wild-type mice (Yamaguchi et al., 2005).
In the present study, we examined both the temporal and spatial dynamics of 8-oxoG accumulation in the mouse hippocampus after the systemic administration of kainate, and thus found kainate to alter the MTH1 expression along with inducing a significant increase in the 8-oxoG accumulation in CA3 microglia. We further compared the extent of hippocampal damage between MTH1-null and wild-type mice after kainate administration.
Materials and Methods
Antibodies.
Mouse N45.1 mAb (1:100), which preferentially recognizes 8-oxoG in DNA, was obtained from JaICa (Fukuroi, Japan), and mouse 15A3 mAb (1:2400), which recognizes 8-oxoG in both DNA and RNA was obtained from QED Bioscience (San Diego, CA). Rabbit anti-glial fibrillary acidic protein (GFAP) polyclonal antibodies (1:15000), which were used to identify astrocytes, were purchased from DakoCytomation (Kyoto, Japan). Rabbit anti-voltage-dependent anion-selective channels (VDAC) polyclonal antibodies (1:500) have been prepared previously (Alam et al., 2003) and were used to identify mitochondria. Rat anti-F4/80 mAb (CI:A3-1; 1:500), which were used to identify microglia, was purchased from Serotec (Oxford, UK). Mouse anti-NeuN mAb (A60; 1:250) was purchased from Chemicon (Temecula, CA), and it was used to identify neurons. Alexa Fluor-labeled second antibodies were obtained from Invitrogen (Tokyo, Japan).
Animals.
We previously established Mth1 gene knock-out mice (Tsuzuki et al., 2001; Sakumi et al., 2003). Heterozygous mice (Mth+/−) have been backcrossed to C57BL/6J (Clea Japan, Tokyo, Japan) for >15 generations, thereby ensuring a standard C57BL/6J genetic background. For the experiment examining pyramidal cell loss in hippocampus, Mth1−/− (MTH1-null) and Mth1+/+ (wild-type) male mice were obtained by mating between the heterozygous mice. For the other experiments, the MTH1-null mice that were obtained by the mating of the heterozygous mice, were bred only one generation to yield MTH1-null offspring, and C57BL/6J mice were used as controls. All animals were maintained in an air-conditioned, light time-controlled, specific pathogen-free room. The handling and killing of all animals were done in accordance with national prescribed guidelines, and ethical approval for the studies was granted by the Animal Care and Use Committee of Kyushu University.
Experimental design and kainate treatment.
Eight- to 10-week-old male mice were used for this study. The mice were injected intraperitoneally with either saline (vehicle) or kainate (Wako, Osaka, Japan) dissolved in saline and then killed at various time points, ranging from 6, 12, 24, 72 h, 1 week, to 2 weeks after the injection. Kainate was administered at a dose of 25 mg/kg for the experiment examining pyramidal cell loss in the hippocampus, whereas it was administered at a dose of 30 mg/kg for the other experiments. To compare the degree of epileptiform seizures, we recorded the seizure score for 1 h after kainate administration as follows: 0, no reaction; 1, arrest of motion; 2, myoclonic jerks of the head and neck, with brief twitching movements; 3, unilateral clonic activity; 4, bilateral forelimb tonic and clonic activity; 5, generalized tonic–clonic activity with loss of postural tone including death from continuous convulsion, according to the previously described criteria (Yang et al., 1997).
Tissue processing.
The animals deeply anesthetized with pentobarbital (30 mg/kg, i.p.) were perfused intracardially with saline followed by cold 4% paraformaldehyde (PFA) in 0.1 m PBS. The brains were removed, immersed for 12 h in the same 4% PFA fixative at 4°C, and cryoprotected in 20, 30% sucrose in PBS for 48 h at 4°C. The brains were then frozen and stored at −80°C until use. Serial coronal sections (40 μm thickness) were cut on a cryostat, collected as free-floating sections in PBS, and then were processed immediately for immunohistochemistry (IHC). For the immunohistochemical detection of NeuN, brain samples were embedded in paraffin, and serial coronal sections (6 μm thickness) were cut.
Immunohistochemistry.
Free-floating sections with an appropriate pretreatment were incubated in Block Ace (Dainippon Pharmaceutical, Osaka, Japan) for 30 min at room temperature (RT), and then were incubated with each appropriately diluted primary antibody in 10% Block Ace, at 4°C overnight. The rinsed sections were immersed in a solution of 3% H2O2 in methanol/PBS (1:1) for 5 min at RT, and then were processed using a Vectastain ABC kit (Vector Laboratories, Burlingame, CA) with the proper biotinylated secondary antibody, and then the peroxidase reaction product was detected using 3′3′-diaminobenzidine-tetrahydrochloride (DAB) (Vector Laboratories). Paraffin sections were deparaffinized and then were processed with the same treatment as free-floating sections. Digital images were acquired using Axioskop2 Plus (Carl Zeiss, Tokyo, Japan) equipped with an AxioCam.
Quantitative detection of 8-oxoG.
To detect 8-oxoG in nuclear or mitochondrial DNA and in cellular RNA, free-floating sections (40 μm thickness) were pretreated as described previously (Yamaguchi et al., 2005). Briefly, to detect 8-oxoG in RNA, free-floating sections were directly subjected to IHC with the 15A3, without any pretreatment. To detect 8-oxoG in the mitochondrial DNA rather than nuclear DNA, the 5 mg/ml RNase (Sigma-Aldrich, St. Louis, MO)-treated sections were directly subjected to IHC with the N45.1 mAb. To detect 8-oxoG in nuclear DNA, the RNase-treated sections were washed in PBD (0.3% Triton X in PBS) and immersed in H2O for 10 min. Furthermore, the sections were treated with 2N HCl at RT for 30 min, thus denaturing the nuclear DNA, and then the sections were treated with Tris-HCl, pH 7.5, for 10 min and subjected to IHC with the N45.1 mAb. IHC for 8-oxoG was performed with four coronal sections from each brain sample corresponding to every first section of 10 serial sections (bregma, −2.54 mm to approximately −1.70 mm) (Franklin and Paxinos, 2001). All acquired digital images were processed uniformly at a threshold in a grayscale mode to subtract any background corresponding to the area without tissue by Adobe Photoshop, version 7.0 (Adobe Systems, San Jose, CA). The intensity of 8-oxoG immunoreactivity (IR) in a given area of CA3 and CA1 subregions was measured using the Image Gauge, version 3.2 (Fujifilm, Tokyo, Japan). From each individual animal, four representative coronal sections with two hippocampus formations corresponding to every first section of 10 serial sections (bregma, −2.54 mm to approximately −1.70 mm) were measured, and the mean intensity of 8-oxoG IR per pixel was calculated as an 8-oxoG index for each animal.
Counting NeuN-positive cells.
The damage of pyramidal cells in hippocampus was quantified by counting NeuN-positive cells in paraffin sections (6 μm thickness). NeuN immunostaining was performed with four coronal sections from each brain sample, corresponding to every first section of 10 serial sections (bregma, −2.54 mm to approximately −1.70 mm). All acquired digital images were processed uniformly at a threshold in a grayscale mode to subtract any background corresponding to the area without neurons by Adobe Photoshop, version 7.0, and then immunoreactive nuclei in CA3 subregion were counted as NeuN-positive pyramidal cells. A quantitative analysis was performed by an individual unaware of the experimental treatments.
Laser-scanning confocal microscopy.
Free-floating sections incubated with a proper primary antibody were then further incubated with a proper Alexa Fluor-labeled second antibody for 45 min at RT. The sections were incubated in a solution containing 0.05 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich) for 10 min at RT, and mounted on slides with Vectashield (Vector Laboratories). Confocal images were acquired under Eclipse TE300 (Nikon, Kanagawa, Japan) equipped with the Radiance 2100 laser-scanning confocal microscope system (Bio-Rad Laboratories, Hercules, CA). All sections from each experimental animal and group to be compared were processed in parallel.
Reverse transcription-PCR.
Total RNA from the hippocampus of C57BL/6J mice was prepared using ISOGEN (Nippongene, Tokyo, Japan). Reverse transcription-PCR (RT-PCR) for Mth1, Ogg1, and Gapdh mRNAs was performed as follows. First-strand cDNA, which was synthesized using the First-Strand cDNA Synthesis kits (Amersham Biosystems, Tokyo, Japan) according to the manufacturer’s instructions, was subjected to PCR. PCR was performed according to a previously described method (Ichinoe et al., 2004). Each PCR was performed under the conditions of linear amplification. The primer sequences used for PCR were as follows: Mth1, mtYS-1, CTCCGCCCCGGGAAACT-TTG, mt5-3, AACCACTGAGGGCGCATTTC; Ogg1, mO5-2, CCAGCTCTATTGCACTGTGTA, mO3-3, GCCATACATGGACATCCAC; Gapdh, mGA5-1, CTGCCATTTGCAGTGGCAAAG, mGA3-1, TGGTATTCAAGAGAGTAGGGA. The primers were obtained from either Greiner Japan (Tokyo, Japan) or Genenet (Fukuoka, Japan). PCR products were subjected to agarose gel electrophoresis, and the band intensity on the gel stained with ethidium bromide was measured using Image Gauge, version 3.2.
Western blotting.
Mouse hippocampi were homogenized on ice in 200–400 μl of lysis buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and protease inhibitor mixture (Nacalai tesque, Kyoto, Japan) using Potter-type homogenizer. The lysates were centrifuged at 100,000 rpm for 30 min, and then the supernatant was recovered as hippocampal lysates. The lysates were subjected to SDS-PAGE (15%) followed by Western blotting using anti-MTH1 (3.0 μg/ml) (Kakuma et al., 1995), according to the previously described method (Tsuchimoto et al., 2001). After blotting, the membranes were incubated with 0.5% Ponceau S (MP Biomedicals, Ecshwege, Germany) to quantitate the amount of protein on the membranes. Recombinant mouse MTH1 (mMTH1) expressed in E. coli BL21T cells carrying pET3a:mMTH1 was used as a standard. The amount of protein was measured using Image Gauge, version 3.2.
HPLC with tandem mass spectrometry analysis.
The nuclear DNA prepared from the tissue specimens was subjected to an HPLC with tandem mass spectrometry (HPLC-MS/MS) analysis of 8-oxoG, according to a previously described method (Tsuruya et al., 2003). The brain, except for the cerebellum and olfactory bulb, was separated into two regions, namely, the cerebral cortex containing hippocampus and another region containing the thalamus, midbrain, and brainstem, before DNA extraction.
Statistical analysis.
The data are expressed as means ± SEM. All data were compared using the Mann–Whitney U test. Statistical significance was accepted at a level of p < 0.05.
Results
Systemic administration of kainate induces 8-oxoG accumulation in cellular DNA and RNA in CA3 subregion of mouse hippocampus
Accumulation of 8-oxoG detected by HPLC-electrochemical detector, was reported to be observed in DNA extracted from the rat brains after kainate administration, thus indicating that the oxidation of nucleic acid might play a role in the excitotoxicity (Lan et al., 2000; Patel and Li, 2003). To determine the temporal and spatial accumulation of 8-oxoG in the mouse brain after systemic kainate administration, which produced epileptiform seizures followed by a delayed loss of pyramidal cells in the CA3 subregion of hippocampus as reported previously (Nadler, 1981; Ben-Ari, 1985) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), we performed quantitative IHC for 8-oxoG.
C57BL/6J male mice were injected i.p. with kainate (30 mg/kg) to induce epileptiform seizures, and then were killed at various time points, and the brain sections were subjected to IHC to detect 8-oxoG in cellular DNA and RNA (Fig. 1). To distinguish 8-oxoG accumulated in nuclear DNA, in mitochondrial DNA, and in cellular RNA, we applied three different pretreatments of brain sections as we previously established (Yamaguchi et al., 2005) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In samples pretreated with RNase and HCl, the N45.1 mAb exhibited 8-oxoG IR in DNA, preferentially in nuclear DNA (Fig. 1A). Weak but distinct IR for 8-oxoG in nuclei was observed in the control hippocampus, and its intensity was increased across the whole hippocampus 6 h after kainate administration. Twelve hours after kainate administration, however, the intensity of 8-oxoG IR apparently increased in regions other than the nuclei of the pyramidal cell layer in the CA3 subregion, thus indicating that 8-oxoG accumulated highly in cytoplasmic DNA, such as the mitochondrial DNA, in addition to nuclear DNA. Such 8-oxoG IR began to decrease after 24 h, and it returned to control levels after 72 h. In samples pretreated with RNase but not HCl, the N45.1 mAb preferentially detected 8-oxoG in mitochondrial DNA (Fig. 1B). 8-oxoG accumulation in mitochondrial DNA was the most evident in the CA3 subregion 6–12 h after kainate administration. Under such conditions, the cytoplasmic IR for 8-oxoG was likely localized mostly in regions adjacent to the pyramidal cell layer in the CA3 subregion, and there was no apparent increase of 8-oxoG IR in the nuclei of CA3 pyramidal cells. A strong 8-oxoG IR was detected in the cells with an activated microglia-like shape, which were distributed in the vicinity of the CA3 subregion 12–24 h after kainate administration (Fig. 1B, bottom panel). Such a strong 8-oxoG IR with microglia-like shape was also observed in Fig. 1A (12, 24 h, bottom panel). Finally, 8-oxoG accumulated in RNA detected by 15A3 mAb was the most remarkable in the CA3 pyramidal cells and in the microglia-like cells in the vicinity of CA3 subregion at 6 h after kainate administration (Fig. 1C).
Figure 1.
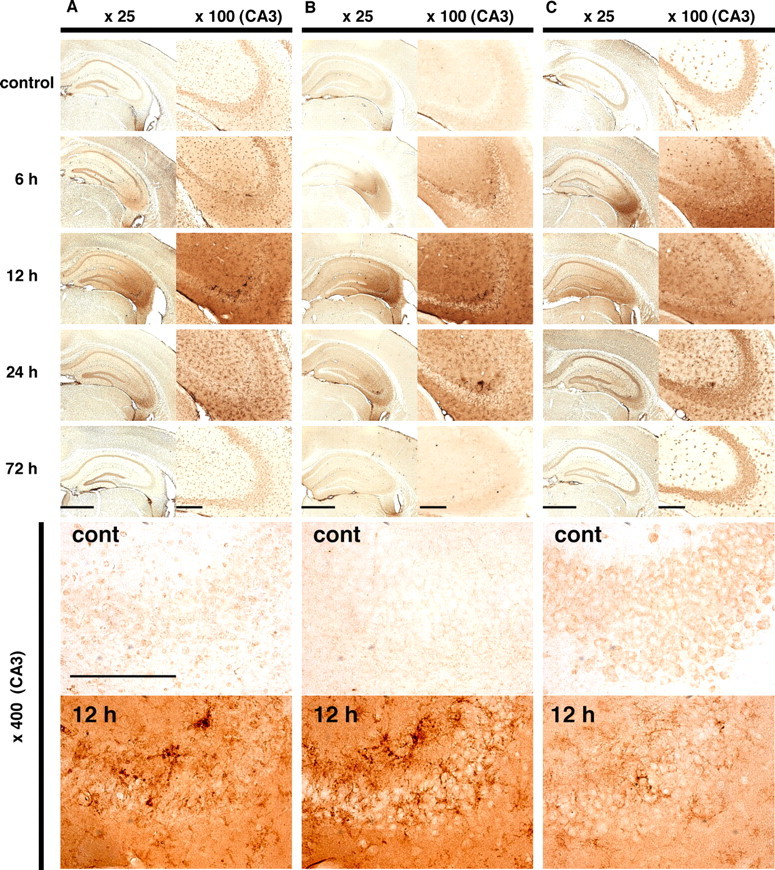
Immunohistochemical detection of 8-oxoG in the hippocampus under excitotoxicity. Male C57BL/6J mice were injected with 30 mg/kg kainate and then were killed at 6, 12, 24, and 72 h after the injection, and the brains were prepared for IHC to detect 8-oxoG. A, The detection of 8-oxoG in nuclear DNA. Free-floating sections were treated with RNase and HCl before incubation with N45.1 mAb to detect 8-oxoG in nuclear DNA. B, The detection of 8-oxoG in mitochondrial DNA. Free-floating sections were treated only with RNase before incubation with N45.1 mAb to detect 8-oxoG in mitochondrial DNA. C, The detection of 8-oxoG in cellular RNA. Free-floating sections were incubated with 15A3 mAb without any pretreatment to detect 8-oxoG in RNA. Control mice were injected with saline. The panels on the left show a lower magnification (25×), whereas a magnified view of each CA3 subregion (100×) is shown in the panels on the right. Further magnified views (400×) of the control (cont) and 12 h samples are shown in the bottom panels. Scale bars: left panels, 1 mm; right panels, 200 μm; bottom panels, 200 μm.
The intensities of 8-oxoG IR in the CA1 and CA3 subregions shown in Figure 1, B and C, were separately digitized and the 8-oxoG index in each region is shown as a bar graph (Fig. 2). As seen in Figure 1A, the IR for 8-oxoG in nuclear DNA could hardly be digitized because more prominent IR was found in regions other than the nuclei of CA3 pyramidal cells. The accumulation of 8-oxoG in the mitochondrial DNA was more evident in CA3 than in the CA1 subregion, and the 8-oxoG index in the former, which peaked 12 h after kainate administration, was >150% of the level seen in the control (Fig. 2A). The accumulation of 8-oxoG in RNA peaked 6–12 h after kainate administration, and the 8-oxoG index increased to >120% of the control level (Fig. 2C). Again, the accumulation of 8-oxoG in RNA was also more evident in the CA3 than in the CA1 subregion.
Figure 2.

Kinetics of 8-oxoG accumulation in hippocampus after kainate administration. IHC for 8-oxoG was performed with four coronal sections from each brain sample corresponding to every first section of 10 serial sections (bregma, −2.54 mm to approximately −1.70 mm) as shown in Figure 1, and 8-oxoG indexes in the CA3 and CA1 subregions of bilateral hippocampi were determined. A, The accumulation of 8-oxoG in mitochondrial (Mt) DNA in the CA3 subregion. B, The accumulation of 8-oxoG in mitochondrial DNA in the CA1 subregion. C, The accumulation of 8-oxoG in the cellular RNA in CA3 subregion. D, The accumulation of 8-oxoG in cellular RNA in CA1 subregion. The relative value of each 8-oxoG index to that of the control is shown as a bar graph with the means ± SEM (n = 4 for each time point, except n = 5 for 24 h). *p < 0.05, Mann–Whitney U test.
We next performed multi-immunofluorescence microscopy with a laser-scanning confocal microscope using brain samples from control mice and those 12 h after kainate administration, to specify the types of cells that accumulate 8-oxoG in DNA or RNA under the excitotoxicity. As shown in Fig. 3A, in CA3 subregion of the control brain, a higher level of 8-oxoG IR was observed in the nuclei of GFAP-positive astrocytes than in pyramidal cells. After kainate administration, the level of 8-oxoG IR apparently increased in the nuclei of both the astrocytes and pyramidal cells. 8-oxoG IR in the mitochondrial DNA was barely detectable in the control mice, and the kainate administration significantly increased the level of 8-oxoG IR in mitochondrial DNA (Fig. 3B). As seen in Figure 3Bf, the distribution of 8-oxoG IR appears to be in cells with processes such as activated microglia. Strong VDAC IR was detected in the soma of the CA3 pyramidal cells, thus indicating an abundance of mitochondria in the cells; however, the strong 8-oxoG IR tends to colocalize with VDAC IR in regions other than the soma of CA3 pyramidal cells (Fig. 3Bh,Bj). F4/80-positive microglia were rarely observed in the control brain; however, F4/80 IR apparently increased within the CA3 subregion after kainate administration, thus indicating the activation of microglia. As shown in Figure 3, Ch and Cj, 8-oxoG IR in mitochondrial DNA was largely colocalized with the F4/80 IR, thus indicating that 8-oxoG accumulates mostly in the mitochondrial DNA in the microglia under excitotoxicity. A weak 8-oxoG IR in cellular RNA was detected in the soma of CA3 pyramidal cells in the control brain, and the level also increased after kainate administration (Fig. 3D). Furthermore, the increased 8-oxoG IR in the cellular RNA was partially colocalized with VDAC IR, thus suggesting that 8-oxoG also accumulates in mitochondrial RNA. The GFAP-positive structures, namely, processes of astrocytes, were rarely merged with 8-oxoG IR in the mitochondrial DNA or cellular RNA (data not shown).
Figure 3.
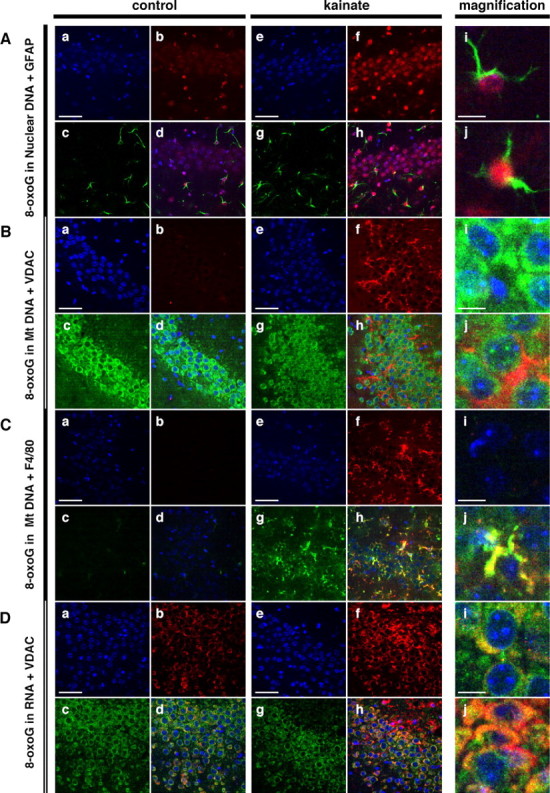
Localization of 8-oxoG accumulated in the CA3 subregion under the excitotoxicity. Male C57BL/6J mice were injected with saline (control; a–d, i) or 30 mg/kg kainate (kainate; e–h, j) and then were killed at 12 h after the injection, and brain sections were subjected to multi-immunofluorescence microscopy with a laser-scanning confocal microscope. A, The localization of nuclear 8-oxoG in astrocytes and CA3 pyramidal cells. Free-floating sections treated with RNase and HCl were incubated with DAPI (a, e; blue), N45.1 mAb (b, f; red), and anti-GFAP antibody (c, g; green), and their merged images are shown (d, h, i, j). B, The localization of mitochondrial (Mt) 8-oxoG in the CA3 subregion. Free-floating sections treated with RNase were incubated with DAPI (a, e; blue), N45.1mAb (b, f; red), and anti-VDAC antibody (c, g; green), and their merged images are shown (d, h, i, j). C, The localization of mitochondrial 8-oxoG in microglia. Free-floating sections treated with RNase were incubated with DAPI (a, e; blue), N45.1mAb (b, f; red), and anti-F4/80 antibody (c, g; green), and their merged images are shown (d, h, i, j). D, The localization of 8-oxoG in cellular RNA. Free-floating sections without any treatment were incubated with DAPI (a, e; blue), 15A3 mAb (b, f; red), and anti-VDAC antibody (c, g; green), and their merged images are shown (d, h, i, j). Scale bars: a–h, 50 μm; i, j (magnification), 10 μm.
From these results, we concluded that the excitotoxicity, caused by the systemic administration of kainate, increases the oxidative stress restricted to the CA3 subregion in the hippocampus. Consequently, 8-oxoG accumulates mostly in mitochondrial DNA and cellular RNA and to a lesser extent in nuclear DNA and in microglia, astrocytes, as well as pyramidal cells in the CA3 subregion.
Alteration of Mth1 gene expression in the mouse hippocampus after kainate administration
The level of 8-oxoG accumulating in the CA3 subregion, especially in cellular DNA after kainate administration apparently decreased after 24 h, and then it returned to the control level after 72 h, thus suggesting that the 8-oxoG accumulating in cellular DNA was thus efficiently removed by such repair enzymes as OGG1 with 8-oxoG DNA glycosylase. In situ hybridization revealed that Mth1 mRNA, encoding an enzyme that hydrolyzes 8-oxo-dGTP or 8-oxoGTP to monophosphate form, is expressed in the mouse hippocampus (Yamaguchi et al., 2005). To examine the expression levels of Mth1 and Ogg1 mRNA in the hippocampus with or without excitotoxicity, we performed semiquantitative RT-PCR analysis using RNA isolated from a dissected hippocampus (Fig. 4A). The level of Mth1 mRNA rapidly decreased to the 50% level seen in the control from 4 to 12 h after kainate administration; thereafter, it gradually recovered until it exceeded the control level after 72 h. In contrast, the expression level of Ogg1 mRNA was barely altered after kainate administration (Fig. 4B,C). These results suggest that the expression of the Ogg1 and Mth1 genes in the hippocampus were differentially regulated in response to the excitotoxicity caused by kainate.
Figure 4.
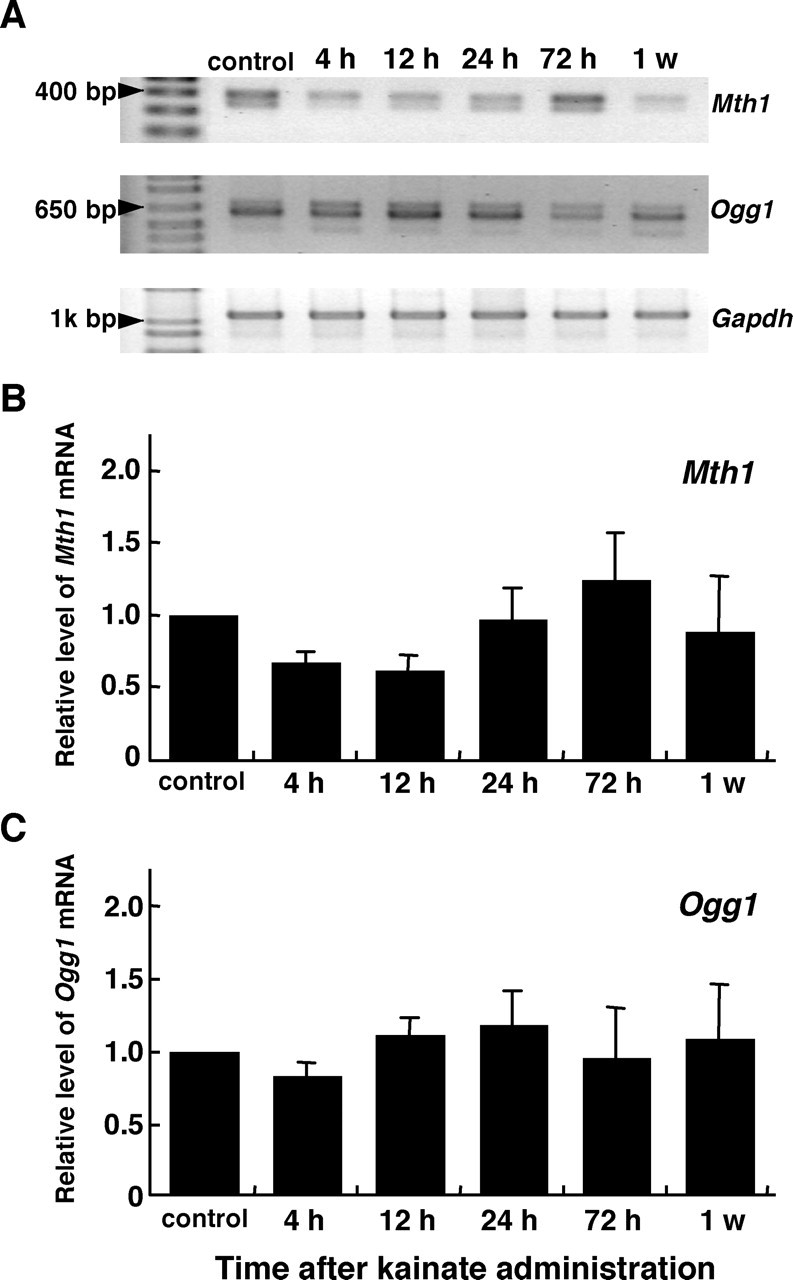
Altered expression of Mth1 and Ogg1 mRNA in the mouse hippocampus under excitotoxicity. A, RT-PCR analyses of Mth1 and Ogg1 mRNA. RNA was isolated from a dissected hippocampus from the mice at the time noted after the injection of kainate (30 mg/kg) or saline (control). RT-PCR for Mth1, Ogg1, and Gapdh mRNA was performed as described in Materials and Methods. B, The alteration of Mth1 mRNA level after kainate administration. The amount of Mth1 mRNA determined by RT-PCR was normalized by that of Gapdh mRNA at each time point, and the relative amount of Mth1 mRNA at each time point to that of the control is shown in the bar graph with mean ± SEM (n = 3 mice per group). C, The alteration of Ogg1 mRNA level after kainate administration. The relative amount of Ogg1 mRNA determined as described in B is shown in the bar graph.
Next, the expression levels of MTH1 protein were examined by Western blotting of the hippocampal extracts prepared from mice with or without excitotoxicity (Fig. 5). An 18 kDa MTH1 protein, which comigrated with the recombinant mouse MTH1 protein, was detected in wild-type but not in the MTH1-null mice samples. In the control samples, MTH1 protein was barely detectable; however, its expression level gradually increased to more than fivefold within 72 h after kainate administration and then remained high for a week. We thus concluded that the excitotoxicity initiated by kainate administration results in an increase of MTH1 protein expression in the mouse hippocampus.
Figure 5.
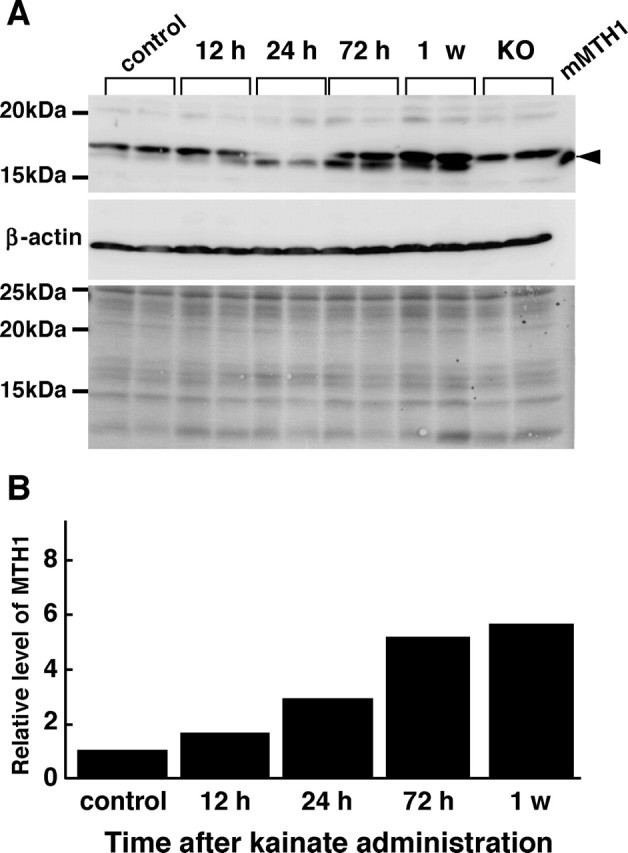
Increased expression of MTH1 protein in the hippocampus under the excitotoxicity. A, Western blotting analysis of MTH1 protein. Whole-cell extracts prepared from the mouse hippocampus prepared at 12, 24, 72 h, and 1 week after kainate administration were subjected to a Western blotting analysis with anti-MTH1 and anti-β-actin. The Western blotting results are shown in the top (MTH1) and middle (β-actin) panels. The bottom panel is a Ponceau S-stained filter before incubation with the antibodies to confirm sample loading. Extracts from MTH1-null mouse (KO) and recombinant mouse MTH1 (mMTH1) were also subjected to Western blotting. The arrowhead indicates mMTH1. B, The increased expression of MTH1 protein in the hippocampus after kainate administration. The amount of MTH1 protein determined by Western blotting was normalized by that of β-actin at each time point, and the mean value of the relative amount of MTH1 protein at each time point to that of the control is shown in the bar graph. n = 2 mice per group.
MTH1 deficiency does not aggravate the degeneration of CA3 pyramidal cells in the hippocampus induced by kainate administration
To clarify the significance of an increased expression of MTH1 in hippocampus during excitotoxicity, we compared the extent of the degeneration or loss of the CA3 pyramidal cells in hippocampus between wild-type and MTH1-null mice after kainate administration. Wild-type and MTH1-null mice were administered with 25 mg/kg kainate, and the extent of their epileptiform seizure was scored for 1 h. As shown in Fig. 6A, wild-type and MTH1-null mice exhibited almost an identical time course and extent of seizure after kainate administration. Furthermore, the two groups of mice exhibited the same survival rate (89%) under excitotoxicity within a week. We next examined the number of NeuN-positive cells in the CA3 subregion, which represent undamaged pyramidal cells, 1 week after kainate administration. The number of NeuN-positive cells decreased to the 60% level of the control both in wild-type and MTH1-null mice (Fig. 6B). Cresyl violet staining also revealed a similar extent of degeneration of the CA3 pyramidal cells between the two groups (data not shown), and we thus concluded that MTH1 deficiency does not aggravate the degeneration of CA3 pyramidal cells in the hippocampus under excitotoxicity.
Figure 6.
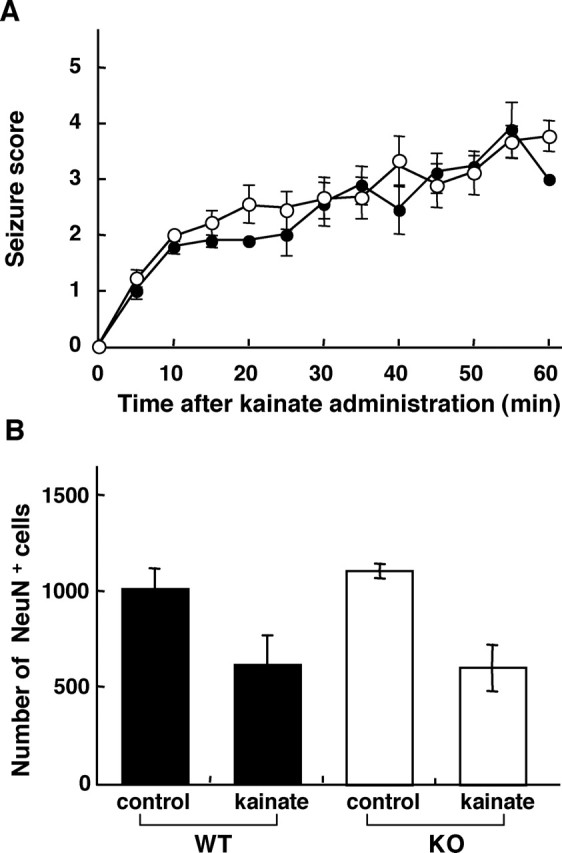
MTH1 deficiency does not aggravate the degeneration of CA3 pyramidal cells in hippocampus induced under the excitotoxicity. A, Seizure responses of MTH1-null and wild-type mice after kainate administration. Every 5 min for 1 h after kainate administration (25 mg/kg, i.p.), observed seizures were scored as described in Materials and Methods, and their mean values with SEM at each time point are plotted. Nine mice in each group were observed and scored. Closed circle, Wild-type mice; open circle, MTH1-null mice. One of nine mice in each group died within 2 h. B, MTH1-null mice did not exhibit an accelerated degeneration of CA3 pyramidal cells under excitotoxicity. The mice were killed 7 d after kainate administration (25 mg/kg, i.p.), and then brain sections were prepared for NeuN immunohistochemistry. The mean number of NeuN-positive pyramidal cells in CA3 subregion of four coronal sections from each brain sample, corresponding to every first section of 10 serial sections (bregma, −2.54 mm to approximately −1.70 mm) are shown in the bar graph with SEM. Filled bar, Wild-type mice; open bar, MTH1-null mice. Control mice injected with saline, n = 4; kainate-injected mice, n = 7 for each group.
MTH1 deficiency augments the kainate-induced accumulation of 8-oxoG in the hippocampus
To evaluate the role of MTH1 against the accumulation of 8-oxoG in mouse hippocampus under the excitotoxicity, we compared the levels of 8-oxoG in brains from wild-type and MTH1-null mice after kainate administration (30 mg/kg). First, nuclear DNA was prepared from two regions of the brains, the cerebral cortex containing hippocampus and another region that contains the thalamus, midbrain, and brainstem, and then it was subjected to an HPLC-MS/MS analysis to quantify the amount of 8-oxoG in nuclear DNA (Fig. 7). In cerebral cortex with hippocampus, the wild-type mice exhibited a 1.5-fold increase in the contents of 8-oxoG 24 h after kainate administration, whereas the MTH1-null mice exhibited a twofold increase in the contents of 8-oxoG at the same time. The increased contents of 8-oxoG in both groups decreased to the control levels within 2 weeks. Similar results were obtained from the other brain region containing thalamus, midbrain, and brainstem; however, no such increase in the 8-oxoG contents was observed in the nuclear DNA prepared from the cerebellum and liver after kainate administration (data not shown).
Figure 7.
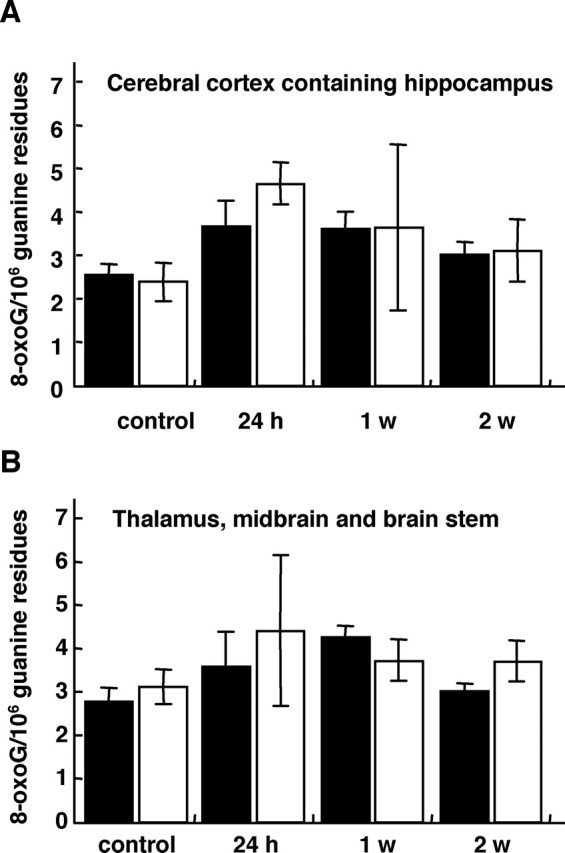
HPLC-MS/MS analyses of 8-oxoG accumulated in nuclear DNA prepared from brain tissues after kainate administration. MTH1-null and wild-type mice were killed at 24 h, 1 week, and 2 weeks after kainate administration (30 mg/kg, i.p.), and nuclear DNA was prepared from the cerebral cortex containing the hippocampus (A) and another brain region containing the thalamus, midbrain, and brainstem (B). The 8-oxoG content in each DNA sample was determined by an HPLC-MS/MS analysis, and the mean number of 8-oxoG residues per 106 guanine residues is shown in the bar graph with SEM. There was no statistically significant difference between the data from the wild-type and MTH1-null mice. Filled bar, Wild-type mice; open bar, MTH1-null mice. Control mice injected with saline, n = 11; kainate-injected mice, n = 3, for each time point.
Based on the results of the HPLC-MS/MS analysis, we next immunohistochemically compared the extent of 8-oxoG accumulation in the mitochondrial DNA and cellular RNA in the hippocampus between wild-type and MTH1-null mice (Figs. 8, 9). As shown in Figures 8A and 9A, 24 h after kainate administration, the 8-oxoG index in mitochondrial DNA in wild-type CA3 subregion increased to 140% of the level seen in the control and returned to near control levels within 72 h, whereas that in MTH1-null mice reached 200% of the level seen in the control, and this level was significantly higher than the level in wild type (p < 0.05). Seventy-two hours after kainate administration, the 8-oxoG index in MTH1-null mice still remained significantly higher than those in the wild-type or the control mice. Essentially similar results were observed in the CA1 subregion. As shown in Figures 8B and 9B, the 8-oxoG index in cellular RNA in wild-type CA3 subregion slightly increased to 120% of the level seen in the control 24 h after kainate administration and returned to the control level within 72 h, whereas the MTH1-null mice reached 150% of the level seen in the control, and this level was significantly higher than the level in wild type (p < 0.05). Again, 72 h after kainate administration, the 8-oxoG index in MTH1-null mice remained higher than those in the wild-type or the control mice. Similar results were observed in the CA1 subregion. It is noteworthy that the 8-oxoG IRs in both mitochondrial DNA and cellular RNA observed in MTH1-null mice apparently exhibited an activated microglia-like shape with multiple processes (Fig. 8C). In the CA3 subregion, there was no significant difference in the density of the F4/80-positive microglia between the MTH1-null and wild-type mice, at 72 h after kainate administration (data not shown). As a result, we concluded that MTH1 efficiently suppresses the accumulation of 8-oxoG in cellular DNA and RNA in the hippocampus under excitotoxicity, and such suppression occurred most efficiently in the mitochondrial DNA and cellular RNA of the CA3 microglia.
Figure 8.
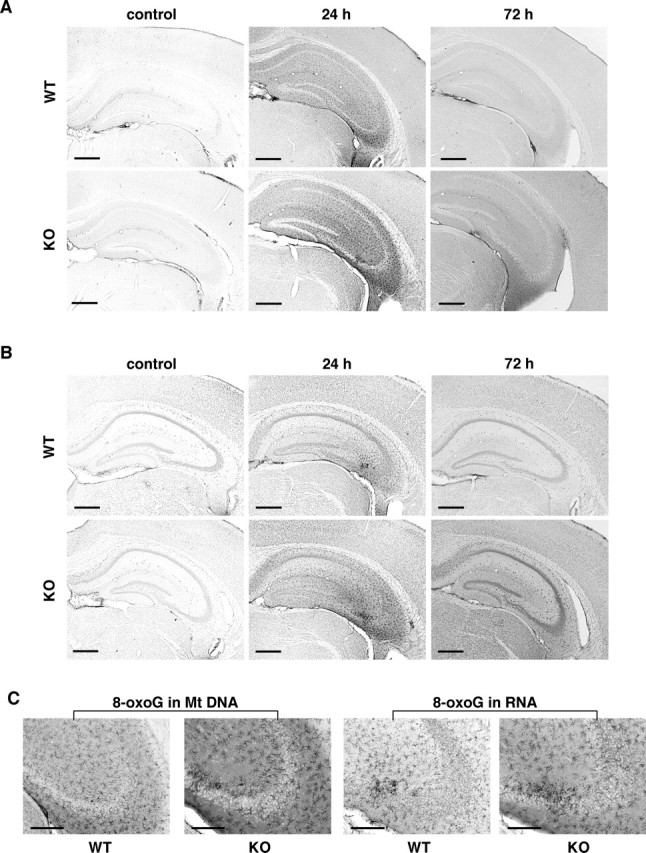
MTH1 deficiency augments the kainate-induced accumulation of 8-oxoG in the hippocampus. Wild-type (WT) and MTH1-null mice (KO) were injected with kainate (30 mg/kg, i.p.) or saline (control), and at 24 h and 72 h after kainate administration, brains were removed and coronal sections were subjected to IHC. A, The accumulation of 8-oxoG in mitochondrial DNA in the hippocampus. Free-floating sections treated with only RNase were subjected to IHC with N45.1 mAb. B, The accumulation of 8-oxoG in cellular RNA in the hippocampus. Free-floating sections without pretreatment were subjected to IHC with 15A3 mAb. C, The accumulation of 8-oxoG in microglia-like cells in CA3 under excitotoxicity. Magnified views (100×) of the CA3 subregions from A (24 h) [8-oxoG in mitochondrial (Mt) DNA] and B (24 h) (8-oxoG in RNA) are shown. Scale bars: A, B, 500 μm; C, 200 μm.
Figure 9.

Kinetics of 8-oxoG accumulation in hippocampus of MTH1-null (KO) mice and wild-type (WT) mice after kainate administration. A, The 8-oxoG indexes in CA3 and CA1 subregions of hippocampi were determined from data shown in Fig. 8A, and the relative value of each 8-oxoG index to that of the control is shown as a bar graph with the means ± SEM [control mice injected with saline (n = 4); kainate-injected mice (n = 5–6 mice)]. B, The 8-oxoG indexes in CA3 and CA1 subregions of bilateral hippocampi were determined from data shown in Fig. 8B, and the relative value of each 8-oxoG index to that of the control is shown as a bar graph with the means ± SEM [control mice injected with saline (n = 4); kainate-injected mice (n = 5–6 mice)]. *p < 0.05, Mann–Whitney U test. Mt, Mitochondrial.
Discussion
In the present study, we demonstrated that the accumulation of 8-oxoG in the nuclear and mitochondrial DNA or cellular RNA significantly and transiently increased in the hippocampus after the systemic administration of kainate. Accumulation of 8-oxoG was observed in the microglia and astrocytes as well as in CA3 pyramidal cells in the hippocampus, and the most significant accumulation was seen in the mitochondrial DNA of the CA3 microglia. In the hippocampus, the expression of MTH1 first transiently decreases, and then it increases after the accumulation of 8-oxoG after kainate administration. MTH1 deficiency does not aggravate the kainate-induced degeneration of the CA3 pyramidal cells; however, it significantly accelerates the accumulation of 8-oxoG in the hippocampus, especially in the CA3 microglia under excitotoxicity. The present study thus demonstrated that MTH1 suppresses the accumulation of 8-oxoG in the hippocampus under excitotoxicity.
DNA fragmentation has been observed in the hippocampal neurons after kainate administration, and thus ROS generated by excitotoxicity has been implicated in DNA damage (Kasof et al., 1995; Weiss et al., 1996). Until today, however, there have been only two previous studies reporting the 8-oxoG contents in total cellular DNA extracted from rat hippocampus, amygdala/pyriform cortex, and thalamus to increase by severalfold in comparison with the control (5–10 residues of 8-oxoG per 105 guanine residues) from several hours to 72 h after kainate administration (Lan et al., 2000; Patel and Li, 2003). In the present study, we determined the contents of 8-oxoG in nuclear DNA prepared from the cerebral cortex containing the hippocampus by HPLC-MS/MS analysis. In the nuclear DNA, a 1.5- to 2-fold increase of 8-oxoG contents (four to five residues of 8-oxoG per 106 guanine residues) were detected 24 h after kainate administration; however, the level of 8-oxoG was much lower than that seen in previous reports. Because we detected more 8-oxoG IR in the mitochondrial DNA than in nuclear DNA in the hippocampus after kainate administration, the higher 8-oxoG contents in the previous reports most likely reflect the accumulation of 8-oxoG in the mitochondrial DNA.
With refined immunohistochemical approaches, we demonstrated that 8-oxoG transiently accumulated in the microglia and astrocytes as well as in CA3 pyramidal cells in the hippocampus under excitotoxicity, and among them the 8-oxoG accumulation was the most significant in the mitochondrial DNA and cellular RNA in activated microglia. 8-oxoG accumulation in mitochondrial DNA and cellular RNA under excitotoxicity is significantly higher in MTH1-null than in wild-type hippocampus, and it is the most remarkable in microglia (Fig. 8), whereas the nuclear 8-oxoG accumulation in the MTH1-null cerebral cortex was slightly higher than that in wild type (Fig. 7). Because MTH1 efficiently hydrolyzes 8-oxo-GTP, as well as 8-oxo-dGTP, to eliminate them from the nucleotide pools (Hayakawa et al., 1999), the increased accumulation of 8-oxoG in MTH1-null mice must result from the incorporation of 8-oxo-dGTP into DNA or 8-oxo-GTP into RNA, respectively. As a result, the slightly increased nuclear 8-oxoG in the MTH1-null cerebral cortex is thought to be derived from such mitotic cells as astrocytes and microglia but not postmitotic neurons. In contrast, mitochondrial DNA is replicated even in the postmitotic neurons as well as in the astrocytes and microglia, and RNA is always synthesized in living cells, therefore, the predominant accumulation of 8-oxoG in mitochondrial DNA and cellular RNA especially in microglia of MTH1-null mice indicates that kainate may increase the ROS production in microglia as well as in neurons.
Three pathways have been proposed for the production of ROS in the CA3 subregion under the kainate-induced excitotoxicity; activated kainate receptor induces the influx of cations, and especially Ca2+ activates nitric oxide synthase, whose product, NO, reacts with O2− to produce ONOO−, whereas kainate itself reduces the intracellular level of glutathione, an antioxidant. Finally, kainate induces the release of glutamate, which also enhances the cellular responses induced by kainate (Coyle and Puttfarcken, 1993; Floreani et al., 1997; Wang et al., 2005). Such production of ROS in the CA3 subregions of hippocampus was confirmed from 6 to 12 h after kainate administration by the use of dihydroethidium (K. Kajitani and Y. Nakabeppu, unpublished observations).
It has been hypothesized that kainate exerts its excitotoxicity through the AMPA/kainate receptors in neurons of the hippocampus. It has recently been reported, however, that microglia also equip AMPA/kainate receptors (GluR2–5, -7), which induce an inward current and production of cytokine such as TNF-α by binding with kainate (Streit et al., 1999; Noda et al., 2000). Because CA3 pyramidal cells are not damaged at 6 h after kainate administration (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), when the microglia started accumulating 8-oxoG, kainate may directly activate the microglia through their AMPA/kainate receptors to produce ROS, and then microglia may later proliferate as an inflammatory response to the damaged CA3 neurons. The remarkable accumulation of 8-oxoG in the microglia is therefore likely caused by ROS produced downstream of the AMPA/kainate receptors accompanied by the proliferation of microglia. The responses of non-neuronal cells such as microglia, astrocytes, and endothelial cells in the early stages of neurodegeneration may be neuroprotective; however, in the late phases, they may lead to the entry of peripheral inflammatory cells into the brain and promote neurodegeneration (Ke and Gibson, 2004). In the present study, we could find no accelerated dysfunction or cell death in the hippocampus attributable to the increased accumulation of 8-oxoG in MTH1-null microglia as well as in the CA3 pyramidal cells a week after kainate administration. Because it has been shown that kainate-induced excitotoxicity often results in hippocampal sclerosis with recurrent spontaneous seizures within a couple months, in which gliosis may play an important role (Nadler, 1981; Ben-Ari, 1985; Fisher et al., 1998), it is thus necessary to conduct a long-term observation after kainate administration, to evaluate the effects of an increased accumulation of 8-oxoG in the MTH1-null hippocampus, especially in microglia.
The level of Mth1 mRNA transiently decreased soon after kainate administration; however, its level increased when the contents of 8-oxoG in the DNA and RNA decreased. The level of mRNA for OGG1, which excises 8-oxoG in DNA, remained constant throughout the experiment. Kainate has been reported to transiently decrease the levels of several types of mRNAs (Garcia et al., 1997; Kofler et al., 1997; Tsunashima et al., 1997; Gorecki et al., 1998). The biological significance of the transient decrease of such mRNAs is not certain; however, it is plausible that oxidative stress under the excitotoxicity accelerates the degradation of certain types of mRNAs. In contrast, the level of MTH1 protein slowly and continuously increased in the hippocampus, and the increased level of MTH1 protein persisted for a week. The increase of the MTH1 protein level may be attributed to the suppression of its degradation or an increased translation of Mth1 mRNA. Our results indicate that the excitotoxicity preferentially causes the oxidation of dGTP and GTP in the nucleotide pools in comparison with the direct oxidation of guanine in DNA or RNA. Because GTP functions as an essential regulatory molecule in various biochemical reactions such as translation or signal transduction (Sprang, 1997), the oxidation of GTP in the nucleotide pool may alter the stability of a subset of mRNAs or the efficiency of their transcription or translation.
We reported that human MTH1 suppresses mitochondrial dysfunction and cell death caused by H2O2 (Yoshimura et al., 2003); furthermore, we demonstrated that MTH1 suppresses the transient increase of 8-oxoG in mitochondrial DNA in the dopaminergic nerve terminals in mouse striatum after MPTP administration, and it protects the nerve terminals (Yamaguchi et al., 2005). In the present study, however, we barely observed the protective role of MTH1 against the degeneration of CA3 pyramidal cells under excitotoxicity. The neurons are postmitotic; thus, there is little chance to incorporate 8-oxo-dGTP into their nuclear DNA. Even if there is a direct oxidation of guanine in DNA, OGG1 can efficiently repair the oxidized base (Boiteux and Radicella, 1999; Nishioka et al., 1999). Furthermore, 8-oxoG, which accumulated in the mitochondrial DNA of microglia in the MTH1-null hippocampus, significantly decreased within 72 h after kainate administration, thus suggesting that 8-oxoG in the mitochondrial DNA incorporated from the nucleotide pool can be efficiently repaired by the OGG1-initiated base excision repair.
We previously reported that lung tumors spontaneously developed in OGG1-null mice, in which 8-oxoG was found to accumulate in their genomes. We later found that no tumor was formed in the lungs of mice lacking both OGG1 and MTH1, despite the increased accumulation of 8-oxoG in the mice. These results suggest that an accumulation of large amount of 8-oxo-dGTP and 8-oxo-GTP in nucleotide pools or 8-oxoG in cellular DNA and RNA attributable to a lack of both MTH1 and OGG1 may accelerate cell death, and thus cancer stem cells lacking both OGG1 and MTH1 may not survive to produce large tumors (Sakumi et al., 2003). It is expected that neurons or microglia lacking both OGG1 and MTH1 are therefore more vulnerable to excitotoxicity, and such experiments are now underway in our laboratory.
Footnotes
This work was supported by grants from Core Research for Evolutional Science and Technology, Japan Science and Technology Agency, the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Grant 16012248), and the Japan Society for the Promotion of Science (Grants 15590347 and 16390119). We thank Drs. Yoshimichi Nakatsu, Kunihiko Sakumi, Daisuke Tsuchimoto, and Mizuki Ohno for their helpful discussions; Setsuko Kitamura, Naomi Adachi, Akemi Matsuyama, Keiko Aiura, and Yukiko Kajitani for their technical assistance; Drs. Shigenobu Kanba and Nobutada Tashiro for providing us with the opportunity to conduct this study; and Dr. Brian Quinn for comments on this manuscript.
References
- Alam TI, Kanki T, Muta T, Ukaji K, Abe Y, Nakayama H, Takio K, Hamasaki N, Kang D (2003). Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res 31:1640–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y (1985). Limbic seizure and brain damage produced by kainic acid: mechanisms and relevance to human temporal lobe epilepsy. Neuroscience 14:375–403. [DOI] [PubMed] [Google Scholar]
- Boiteux S, Radicella JP (1999). Base excision repair of 8-hydroxyguanine protects DNA from endogenous oxidative stress. Biochimie 81:59–67. [DOI] [PubMed] [Google Scholar]
- Bruce AJ, Baudry M (1995). Oxygen free radicals in rat limbic structures after kainate-induced seizures. Free Radic Biol Med 18:993–1002. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P (1993). Oxidative stress, glutamate, and neurodegenerative disorders. Science 262:689–695. [DOI] [PubMed] [Google Scholar]
- Fisher PD, Sperber EF, Moshe SL (1998). Hippocampal sclerosis revisited. Brain Dev 20:563–573. [DOI] [PubMed] [Google Scholar]
- Floreani M, Skaper SD, Facci L, Lipartiti M, Giusti P (1997). Melatonin maintains glutathione homeostasis in kainic acid-exposed rat brain tissues. FASEB J 11:1309–1315. [DOI] [PubMed] [Google Scholar]
- Franklin KB, Paxinos G (2001). In: The mouse brain in stereotaxic coordinates New York: Academic.
- Fukae J, Takanashi M, Kubo S-i, Nishioka K-i, Nakabeppu Y, Mori H, Mizuno Y, Hattori N (2005). Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson’s disease and related neurodegenerative disorders. Acta Neuropathol (Berl) 109:256–262. [DOI] [PubMed] [Google Scholar]
- Furuichi M, Yoshida MC, Oda H, Tajiri T, Nakabeppu Y, Tsuzuki T, Sekiguchi M (1994). Genomic structure and chromosome location of the human mutT homologue gene MTH1 encoding 8-oxo-dGTPase for prevention of A:T to C:G transversion. Genomics 24:485–490. [DOI] [PubMed] [Google Scholar]
- Furuta A, Iida T, Nakabeppu Y, Iwaki T (2001). Expression of hMTH1 in the hippocampi of control and Alzheimer’s disease. NeuroReport 12:2895–2899. [DOI] [PubMed] [Google Scholar]
- Garcia ML, Murray KD, Garcia VB, Strehler EE, Isackson PJ (1997). Seizure-induced alterations of plasma membrane calcium ATPase isoforms 1, 2 and 3 mRNA and protein in rat hippocampus. Brain Res Mol Brain Res 45:230–238. [DOI] [PubMed] [Google Scholar]
- Gorecki DC, Lukasiuk K, Szklarczyk A, Kaczmarek L (1998). Kainate-evoked changes in dystrophin messenger RNA levels in the rat hippocampus. Neuroscience 84:467–477. [DOI] [PubMed] [Google Scholar]
- Hayakawa H, Hofer A, Thelander L, Kitajima S, Cai Y, Oshiro S, Yakushiji H, Nakabeppu Y, Kuwano M, Sekiguchi M (1999). Metabolic fate of oxidized guanine ribonucleotides in mammalian cells. Biochemistry 38:3610–3614. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S (1994). Cloned glutamate receptors. Annu Rev Neurosci 17:31–108. [DOI] [PubMed] [Google Scholar]
- Ichinoe A, Behmanesh M, Tominaga Y, Ushijima Y, Hirano S, Sakai Y, Tsuchimoto D, Sakumi K, Wake N, Nakabeppu Y (2004). Identification and characterization of two forms of mouse MUTYH proteins encoded by alternatively spliced transcripts. Nucleic Acids Res 32:477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuma T, Nishida J, Tsuzuki T, Sekiguchi M (1995). Mouse MTH1 protein with 8-oxo-7,8-dihydro-2′-deoxyguanosine 5′-triphosphatase activity that prevents transversion mutation. cDNA cloning and tissue distribution. J Biol Chem 270:25942–25948. [DOI] [PubMed] [Google Scholar]
- Kasai H, Nishimura S (1984). Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res 12:2137–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasof GM, Mandelzys A, Maika SD, Hammer RE, Curran T, Morgan JI (1995). Kainic acid-induced neuronal death is associated with DNA damage and a unique immediate-early gene response in c-fos-lacZ transgenic rats. J Neurosci 15:4238–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke ZJ, Gibson GE (2004). Selective response of various brain cell types during neurodegeneration induced by mild impairment of oxidative metabolism. Neurochem Int 45:361–369. [DOI] [PubMed] [Google Scholar]
- Kikuchi H, Furuta A, Nishioka K, Suzuki SO, Nakabeppu Y, Iwaki T (2002). Impairment of mitochondrial DNA repair enzymes against accumulation of 8-oxo-guanine in the spinal motor neurons of amyotrophic lateral sclerosis. Acta Neuropathol (Berl) 103:408–414. [DOI] [PubMed] [Google Scholar]
- Kofler N, Kirchmair E, Schwarzer C, Sperk G (1997). Altered expression of NPY-Y1 receptors in kainic acid induced epilepsy in rats. Neurosci Lett 230:129–132. [DOI] [PubMed] [Google Scholar]
- Lan J, Henshall DC, Simon RP, Chen J (2000). Formation of the base modification 8-hydroxyl-2′-deoxyguanosine and DNA fragmentation following seizures induced by systemic kainic acid in the rat. J Neurochem 74:302–309. [DOI] [PubMed] [Google Scholar]
- Lerma J (2003). Roles and rules of kainate receptors in synaptic transmission. Nat Rev Neurosci 4:481–495. [DOI] [PubMed] [Google Scholar]
- Maki H, Sekiguchi M (1992). MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 355:273–275. [DOI] [PubMed] [Google Scholar]
- Michaels ML, Cruz C, Grollman AP, Miller JH (1992). Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc Natl Acad Sci USA 89:7022–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler JV (1981). Minireview. Kainic acid as a tool for the study of temporal lobe epilepsy. Life Sci 29:2031–2042. [DOI] [PubMed] [Google Scholar]
- Nakanishi S (1992). Molecular diversity of glutamate receptors and implications for brain function. Science 258:597–603. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Ohtsubo T, Oda H, Fujiwara T, Kang D, Sugimachi K, Nakabeppu Y (1999). Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol Biol Cell 10:1637–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Nakanishi H, Nabekura J, Akaike N (2000). AMPA-kainate subtypes of glutamate receptor in rat cerebral microglia. J Neurosci 20:251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA (2001). Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60:759–767. [DOI] [PubMed] [Google Scholar]
- Olney JW (1969). Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 164:719–721. [DOI] [PubMed] [Google Scholar]
- Olney JW, Rhee V, Ho OL (1974). Kainic acid: a powerful neurotoxic analogue of glutamate. Brain Res 77:507–512. [DOI] [PubMed] [Google Scholar]
- Patel M, Li QY (2003). Age dependence of seizure-induced oxidative stress. Neuroscience 118:431–437. [DOI] [PubMed] [Google Scholar]
- Sakumi K, Furuichi M, Tsuzuki T, Kakuma T, Kawabata S, Maki H, Sekiguchi M (1993). Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J Biol Chem 268:23524–23530. [PubMed] [Google Scholar]
- Sakumi K, Tominaga Y, Furuichi M, Xu P, Tsuzuki T, Sekiguchi M, Nakabeppu Y (2003). Ogg1 knockout-associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Res 63:902–905. [PubMed] [Google Scholar]
- Shimura-Miura H, Hattori N, Kang D, Miyako K, Nakabeppu Y, Mizuno Y (1999). Increased 8-oxo-dGTPase in the mitochondria of substantia nigral neurons in Parkinson’s disease. Ann Neurol 46:920–924. [PubMed] [Google Scholar]
- Shinozaki H, Konishi S (1970). Actions of several anthelmintics and insecticides on rat cortical neurons. Brain Res 24:368–371. [DOI] [PubMed] [Google Scholar]
- Sprang SR (1997). G protein mechanisms: insights from structural analysis. Annu Rev Biochem 66:639–678. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA (1999). Reactive microgliosis. Prog Neurobiol 57:563–581. [DOI] [PubMed] [Google Scholar]
- Taddei F, Hayakawa H, Bouton M, Cirinesi A, Matic I, Sekiguchi M, Radman M (1997). Counteraction by MutT protein of transcriptional errors caused by oxidative damage. Science 278:128–130. [DOI] [PubMed] [Google Scholar]
- Tajiri T, Maki H, Sekiguchi M (1995). Functional cooperation of MutT, MutM and MutY proteins in preventing mutations caused by spontaneous oxidation of guanine nucleotide in Escherichia coli. Mutat Res 336:257–267. [DOI] [PubMed] [Google Scholar]
- Tsuchimoto D, Sakai Y, Sakumi K, Nishioka K, Sasaki M, Fujiwara T, Nakabeppu Y (2001). Human APE2 protein is mostly localized in the nuclei and to some extent in the mitochondria, while nuclear APE2 is partly associated with proliferating cell nuclear antigen. Nucleic Acids Res 29:2349–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunashima K, Schwarzer C, Kirchmair E, Sieghart W, Sperk G (1997). GABA(A) receptor subunits in the rat hippocampus III: altered messenger RNA expression in kainic acid-induced epilepsy. Neuroscience 80:1019–1032. [DOI] [PubMed] [Google Scholar]
- Tsuruya K, Furuichi M, Tominaga Y, Shinozaki M, Tokumoto M, Yoshimitsu T, Fukuda K, Kanai H, Hirakata H, Iida M, Nakabeppu Y (2003). Accumulation of 8-oxoguanine in the cellular DNA and the alteration of the OGG1 expression during ischemia-reperfusion injury in the rat kidney. DNA Repair 2:211–229. [DOI] [PubMed] [Google Scholar]
- Tsuzuki T, Egashira A, Igarashi H, Iwakuma T, Nakatsuru Y, Tominaga Y, Kawate H, Nakao K, Nakamura K, Ide F, Kura S, Nakabeppu Y, Katsuki M, Ishikawa T, Sekiguchi M (2001). Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proc Natl Acad Sci USA 98:11456–11461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Yu S, Simonyi A, Sun GY, Sun AY (2005). Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol 31:3–16. [DOI] [PubMed] [Google Scholar]
- Waterfall AH, Singh G, Fry JR, Marsden CA (1995). Detection of the lipid peroxidation product malonaldehyde in rat brain in vivo. Neurosci Lett 200:69–72. [DOI] [PubMed] [Google Scholar]
- Weiss S, Cataltepe O, Cole AJ (1996). Anatomical studies of DNA fragmentation in rat brain after systemic kainate administration. Neuroscience 74:541–551. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Kajitani K, Dan Y, Furuichi M, Ohno M, Sakumi K, Kang D, Nakabeppu Y (2005). MTH1, an oxidized purine nucleoside triphosphatase, protects the dopamine neurons from oxidative damage in nucleic acids caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Cell Death Differ in press. [DOI] [PubMed]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA (1997). Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 389:865–870. [DOI] [PubMed] [Google Scholar]
- Yoshimura D, Sakumi K, Ohno M, Sakai Y, Furuichi M, Iwai S, Nakabeppu Y (2003). An oxidized purine nucleoside triphosphatase, MTH1, suppresses cell death caused by oxidative stress. J Biol Chem 278:37965–37973. [DOI] [PubMed] [Google Scholar]
- Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG, Montine TJ (1999). Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol 154:1423–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]