

Abstract
Reelin and Disabled 1 (Dab1) are essential for positioning migrating neurons in the developing neocortex. Cell-autonomous RNA interference-mediated suppression of Dab1 in migrating neurons destined for layer 2/3 shifted the median position of these cells to deeper positions within the cortex. At the time of migration arrest [embryonic day 20 (E20) to E21], Dab1-suppressed cells were underrepresented in the upper ∼40 μm of the cortex compared with controls, suggesting that Dab1 is essential for somal translocation through the cell-dense cortical plate. Closer examination of the morphology of Dab1-suppressed neurons at E20 revealed simplified leading processes that are less likely to contact the marginal zone (MZ), in which high levels of Reelin are expressed. Examination of Dab1-suppressed cells 3 d later (postnatal day 2) revealed simplified dendrites that are also less likely to contact the MZ. These data reveal a cell-autonomous role of Dab1 in dendritogenesis in the neocortex and suggest that remodeling of the leading process of a migrating neuron into a nascent dendrite by Reelin/Dab1 signaling plays an important role in cell positioning.
Keywords: Reelin, lamination, Dab1, migration, branching, dendrite
Introduction
The secreted protein Reelin initiates a signaling cascade that positions neurons in the developing mammalian brain (D’Arcangelo et al., 1995). In human, absence of Reelin causes lissencephaly (smooth brain) with cerebellar hypoplasia (Hong et al., 2000), a recessive syndrome characterized by ataxia, mental retardation, and mild epilepsy. The human syndrome parallels the anatomical disruptions observed previously in the reeler mouse (Falconer, 1951; Caviness and Sidman, 1973; Goffinet, 1980; Goffinet et al., 1984). The cellular architecture of the reeler mouse brain is dramatically disrupted: the cellular layers of the cerebral cortex are disorganized and approximately inverted (Caviness and Sidman, 1973).
Reelin is highly expressed by Cajal-Retzius cells located in the outermost layer of the developing cortex (D’Arcangelo et al., 1995) in which Reelin could regulate the position of newly forming cortical layers. Reelin binds the receptors VLDLR (very-low-density lipoprotein receptor) and ApoER2 (apolipoprotein E receptor 2) (D’Arcangelo et al., 1999; Hiesberger et al., 1999; Trommsdorff et al., 1999), causing the tyrosine phosphorylation of Disabled 1 (Dab1), a cytoplasmic adaptor protein (Howell et al., 1997a, 1999). Mice lacking Dab1 (Gonzalez et al., 1997) or Reelin (Caviness and Sidman, 1973) show essentially indistinguishable histological defects.
In one model of Reelin function, neocortical lamination is produced by alteration of neuronal glial adhesion, such that migrating neurons detach from the radial glial fiber substrate as they reach the end of their migration route underneath the marginal zone (MZ) (Pinto Lord et al., 1982). Subsequent in vitro analysis of glial-directed neuronal migration showed that recombinant Reelin inhibited migration and led to detachment of migrating neurons from the glial fiber (Dulabon et al., 2000), a finding supported by examination of migrating neurons in the scrambler mouse, which is also deficient in Reelin signaling (Sanada et al., 2004).
The model that Reelin-induced radial glial detachment leads to neuronal lamination has been challenged by studies that emphasize the apparent ability of Reelin to act as diffusible stimulant or cofactor for migration. For example, ablation of the Reelin-producing cells (Cajal-Retzius cells) by domoic acid causes premature migration arrest of cells fated for layers 2/3 (Super et al., 2000), and ectopic expression of Reelin in proliferative areas of the developing brain is sufficient to rescue cerebellar development and early, but not later, aspects of neocortical development in reeler mice (Magdaleno et al., 2002). These findings are further complicated by the observations that Reelin signaling can directly stimulate radial glial cell growth and maturation (Hartfuss et al., 2003; Luque et al., 2003).
To understand how Reelin signaling controls the positioning of neurons in vivo, we used RNA interference (RNAi) to suppress expression of Dab1 in a subset of migrating neurons destined for cortical layer 2/3. We find that Dab1-suppressed cells are malpositioned ∼40 μm below the MZ border, apparently unable to translocate normally through the cell-dense cortical plate (CP). The leading process and postnatal dendrite of RNAi-treated cells are also simplified and show less contact with their normal target area, the MZ. These results suggest that Dab1-dependent elaboration of the nascent dendrite is essential for both terminal translocation and cell positioning.
Materials and Methods
Mice.
Mice were cared for according to animal protocols approved by the Institutional Animal Care and Use Committees of the Beth Israel Deaconess Medical Center, Harvard Medical School, and Upstate Medical University. Mice were housed in a virus-free colony according to standard methods. Timed-pregnant Swiss Webster mice were obtained from Taconic Farms (Germantown, NY). scrambler mice were derived from continued inbreeding of a previous outcross of scrambler mice obtained from The Jackson Laboratory (Bar Harbor, ME) (Sweet et al., 1996) to wild-type C57BL/6J.
PCR typing of mice.
DNA was obtained from proteinase K digests of mouse tail tips. scrambler mice are spontaneous mutants that are Dab1 null. Mice were typed using microsatellite markers D4Mit31 and D4Mit331. As described previously (Ware et al., 1997), these markers flank the scrambler locus and can be used to determine the presence of the scrambler mutation in the mixed B6/C3H background.
Explant cultures.
Embryonic day 15 (E15) and E16 embryos were obtained from Dab1+/− matings. The day of plug discovery was designated E0. Pregnant females were killed with CO2 narcosis, followed by cervical dislocation. Embryonic brains were removed from the skull, and each cerebral hemisphere was separated from the diencephalon and placed medial side down on a floating 0.2 μm Nuclepore filter (Whatman, Clifton, NJ). The filters were floated on Neurobasal media containing 2% B27 neuronal supplement, 1× glutamax, 3 mg/ml additional glucose, and 1× penicillin/streptomycin. All cell culture reagents were from Invitrogen (Carlsbad, CA). Explant cultures were maintained at 37°C at 5% CO2, except for the brief viral labeling periods (<10 min), which were performed at room temperature.
Labeling.
Wiretrol 10 μl pipettes (Drummond Scientific, Broomall, PA) were pulled to a fine point with a vertical electrode puller (Narishige, Tokyo, Japan), and the tip was snapped off with jewelers forceps. One to 2 μl of 106 infectious particles/ml replication-incompetent Sindbis virus (pSinRep5; catalog #K750-01; Invitrogen) expressing enhanced green fluorescence protein (eGFP) from the subgenomic promoter (D’Apuzzo et al., 2001) was injected into multiple lateral and dorsal points in each explant. The explants were then cultured on floating filters for an additional period between 8 and 18 h, fixed in 4% paraformaldehyde/PBS, and vibratome sectioned. The maximum total duration of all experiments from dissection to fixation was 24 h.
Electroporations.
In utero electroporations were performed as described previously (Tabata and Nakajima, 2001) with minor modification. Swiss Webster pregnant dams were deeply anesthetized by intraperitoneal Avertin injection at 0.02 ml/g body weight. A laparotomy was performed to expose the uterine horns. The cerebral ventricles of individual embryos were injected through the uterine wall using a Hamilton syringe fitted with a 30 gauge needle (Hamilton, Reno, NV). The injection solution contained control or RNAi plasmids mixed with 0.2 mg/ml pCAG-GFP (Matsuda and Cepko, 2004) to visualize transfected neurons and ∼0.01% fast green (Sigma, St. Louis, MO) to aid targeting. Tweezertrodes were placed on either side of the uterus at the level of the injected embryo’s head, and five 50 ms square pulses at 50 V were administered at 950 ms intervals with BTX830 square-wave pulse generator (Harvard Apparatus, Holliston, MA) to electroporate plasmid DNA. Three to four embryos were typically electroporated per dam. Embryos were allowed to develop after surgery for defined periods and then were killed and screened for GFP expression.
RNA interference constructs.
RNAi constructs were constructed by annealing 68-mer oligonucleotides containing 21–23 nt complementary regions to Dab1, cloning these annealed oligomers first into the pSilencer vector (Ambion, Austin, TX), and finally into pSuper.retro.neo (OligoEngine, Seattle, WA). Oligomers were based on Dab1 sequence (GenBank accession number Y08381). Oligomers A and C target isoforms Dab1 555, Dab1 271, and Dab1 217, whereas oligomer D targets sequence specific to Dab1 217. Oligomer A targets nucleotides 257–277, oligomer C targets nucleotides 666–686, and oligomer D targets nucleotides 863–885 (A1, 5′-GATCCCAAGTAGGATGTCAACTGAGACTTCAAGAGA-GTCTCAGTTGACATCCTACTTTTTTTTGGAAA; A2, AGCTTTTC-CAAAAAAAAGTAGGATGTCAACTGAGACTCTCTTGAAGTCTCA-GTTGACATCCTACTTGG-3′; C1, 5′-GATCCCAATCACAGATTTGTGGCCATCTTCAAGAGAGATGGCCACAAATCTGTGATT TTTTTTGGAAA-3′; C2, 5′-AGCTTTTCCAAAAAAAATCACAGATTTGTGGCCATCTCTCTTGAAGATGGCCACAAATCTGTGATTGG-3′; D1, 5′-GATCCCAATTTCTGAACCACGTCAGGGTTCAAGAGA-CCCTGACGTGGTTCAGAAATT TTTTTTGGAAA-3′; and D2, 5′-AGCTTTTCCAAAAAAAATTTCTGAACCACGTCAGGGTCTCT-TGAACCCTGACGTGGTTCAGAAATTGG-3′).
Full-length Dab1 was PCR amplified and cloned in-frame into PCR4 TOPO CT-GFP (Invitrogen) to create a Dab1::GFP fusion, which was then used as a target for the initial screen pSilencer-based (anti-Dab1) RNAi plasmids. RNA interference plasmids were mixed at 2 mg/ml each, combined with 0.2 mg/ml Dab1::GFP and 0.2 mg/ml pDsRed (Clontech, Mountain View, CA), and lipofected using Lipofectamine2000 (Invitrogen) into human embryonic kidney HEK293 cells. Efficacy of the distinct RNAi constructs (A–D) was determined by the amount of Dab1::GFP suppression (green fluorescence) compared with control pDsRed (red fluorescence). Difficulties in generating sufficient amounts of plasmid DNA from maxipreps of pSilencer vectors led us to clone oligos C and D into the vector (RV) (pSuper.retro.neo plasmid). Oligos C1 and C2 and D1 and D2 were annealed and cloned into HindIII- and BglII-digested RV. RV+C (RVC) and RV+D (RVD) were combined at 0.3–0.5 mg/ml each, with 0.2 mg/ml pCAG-GFP and 0.01% fast green for electroporation. Constructs C and D were screened for activity, and construct C showed consistent suppression of Dab1::GFP by fluorescence (n = 3 trials). Initial immunocytochemistry and in vivo electroporations suggested that a combination of C plus D was more effective than C alone, and therefore a mixture of these constructs was used in all additional experiments. Control electroporations were performed with 0.6–1.0 mg/ml empty pSuper.retro.neo vector (RVN) with 0.2 mg/ml pCAG-GFP and 0.01% fast green.
Histology/histochemistry.
Embryonic brains that were E20 or older were perfused with PBS and then with fresh 4% paraformaldehyde/PBS. The brains were then removed and further fixed overnight in 4% paraformaldehyde/PBS. Explants and brains to age E19 were drop fixed overnight in 4% paraformaldehyde/PBS. The fixed tissue was then embedded in 10% calf gelatin (Aldrich Chemical Company, Milwaukee, WI), postfixed for a minimum of 24 h, and then vibratome sectioned at 100 μm. Floating sections were immunostained overnight in primary antibody diluted into PBST (PBS plus 0.1% Triton X-100) with 2% BSA. The primary antibodies used were as follows: 1:40 dilution of monoclonal antibody (mAb) CR50 anti-Reelin hybridoma supernatant (kindly provided by M. Ogawa, RIKEN Brain Science Institute, Wako, Japan), 1:50 dilution of mAb RC2 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), 1:500 DCX 88 anti-doublecortin polyclonal sera (Gleeson et al., 1999), 1:250 dilution of Cux1 polyclonal sera (kindly provided by J. Cunningham, Brigham and Women’s Hospital, Boston, MA), 1:500 dilution of anti-Brn1 polyclonal sera (Santa Cruz Biotechnology, Santa Cruz, CA), and 1:250 dilution of B3 anti-Dab1 polyclonal sera (kindly provided by B. Howell, National Institute of Neurological Disorders and Stroke, Bethesda, MD). Nuclear staining used propidium iodide (2.5 mg/ml) or Hoechst 33258 (2 μg/ml), and sections were mounted in Citifluor antifade (Ted Pella, Redding, CA). Explant sections were examined with an Olympus Optical (Tokyo, Japan) AX70 microscope fitted with a SPOT-RT CCD camera. Confocal images were recorded on either a Zeiss (Oberkochen, Germany) LSM 510 or Bio-Rad (Hercules, CA) 1024 laser scanning confocal microscopes (State University of New York Upstate Medical University Center for Bioresearch Imaging).
Neuronal morphology.
To determine the structure of GFP-positive (GFP+) cells, z-series of RVN- and RVC+RVD-treated brains were acquired at 1 μm intervals through 21 μm of tissue using a Zeiss LSM510 confocal and a 63×, 1.4 numerical aperture objective. These z-series were then reconstructed using LSM 510 software, and cell morphology was examined. To quantify the relative amounts of GFP+ neurites in the MZ, single optical sections (optical depth of 2.5 μm, full-width at half-maximum) were acquired with constant laser intensity and photomultiplier settings. The images were then imported into NIH ImageJ (W. Rasband, National Institutes of Health, Bethesda, MD) and thresholded to a value of 80 (∼30% over baseline; full scale of 255). Regions of interest corresponding to the MZ (containing the neurites) and subadjacent 70 μm of the CP (containing the cell bodies) were defined, and the above-threshold pixels were measured. The ratio of the number of MZ GFP+ pixels to the number of CP GFP+ pixels was taken for each image and used for comparison between experimental conditions.
Results
To label migrating neurons in vivo, neuronal precursors in the mouse cortical ventricular zone were transfected with a GFP expression vector (pCAG-GFP) on E16. The expression vector was introduced by in utero electroporation immediately after DNA injection into the embryonic cerebral ventricle (Tabata and Nakajima, 2001). Neurons born on E16 migrate into the CP and populate layers 2/3 and a small population in layer 4 in the cortical plate (Takahashi et al., 1999). To confirm that electroporated cells adopted layer-appropriate identity, embryos were electroporated at E16 and examined at P0 after fixation and vibratome sectioning. Consistent with previous birthdating studies, the majority of GFP+ cells were found in the upper layers of the developing cortex (Fig. 1). Immunostaining with antiserum directed against Brn1, a POU domain transcription factor expressed in layers 2–5 (McEvilly et al., 2002) indicates that GFP+ cells have adopted appropriate layer 2/3 neuronal identity (Fig. 1A,B). Similar results were obtained with Cux1, a Cut-related transcription factor (Neufeld et al., 1992) expressed in layers 2–4 (Nieto et al., 2004) (Fig. 1C,D). In addition, GFP+ cells in the CP were positive for the early neuronal markers DCX+ (Gleeson et al., 1999) (Fig. 1E,F) and Tuj1+ (data not shown), but GFP+ cells in the cortical plate were negative for the neuronal precursor marker RC2 (Misson et al., 1988) and Nestin (Hockfield and McKay, 1985) (data not shown). These electroporation conditions appear to permit normal differentiation and positioning of layer 2/3 fated neurons.
Figure 1.
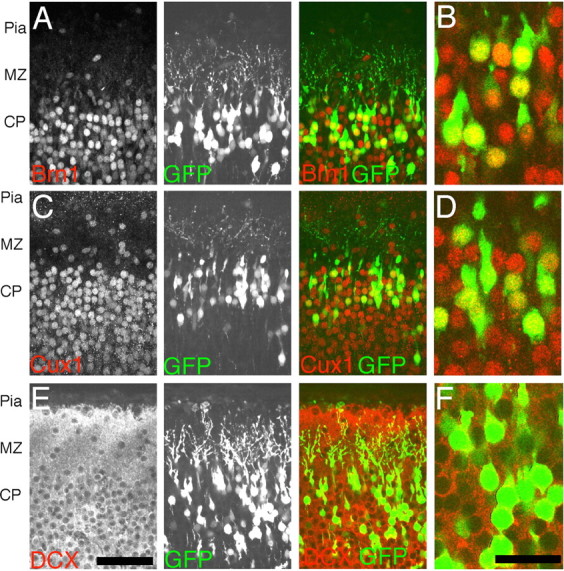
Expression of layer-appropriate markers in electroporated cells. P0 coronal sections of cerebral cortex 5 d after E16 pCAG-GFP electroporations show appropriate expression of layer-specific marker. GFP+ cells in the P0 cortex immunolocalize with the POU domain transcription factor Brn1, a marker of layer 2–5 cells (A, B), and the Cut-related transcription factor Cux1, a marker of layer 2–4 cells (C, D) in the mouse cortex. In addition, GFP+ cells are positive for DCX, a marker of immature postmitotic neurons (E, F). B, D, and F are single confocal sections that have been digitally zoomed three times, whereas A, C, and E are flattened z-series through 10 μm of tissue. Scale bars: E, 50 μm; F, 20 μm.
Morphological changes in processes at the CP–MZ junction
During normal cortical development, migrating neurons arrest at the top of the CP just below the MZ (His, 1904; Ramón y Cajal, 1995; Marin-Padilla, 1978). To examine the morphological transformation that neurons undergo as they contact the MZ in vivo, electroporations with pCAG-GFP were performed at E16 and analyzed at E17, E19, E20, and P0 in fixed and sectioned tissue (Fig. 2A–D). This analysis allows us to examine the leading population of GFP+ migrating neurons contacting the MZ. Optical z-series through 40 μm were taken and then projected into a single plane (flattened) using the maximum pixel intensity algorithm (Bio-Rad Lasersharp 2000 software) to capture GFP+ cell morphology (Fig. 2E–G). Our analysis focused on two populations: migrating cells with their cell body and leading process in the CP, and migrating cells with their cell body in the CP but their leading process in the MZ.
Figure 2.

In vivo morphological changes in migrating neurons in contact with the marginal zone. Time course of cell migration after E16 electroporation. A, At E17, 1 d after electroporation, GFP+ cells in sections of the neocortex are found primarily in the ventricular zone (VZ). By E19 (B) and E20 (C), GFP+ cells have entered the cortex and are approaching the MZ. D, By P0, typically 5 d after electroporation, GFP+ cells are found in a tight layer underneath the MZ. E, F, Examples of the first group of GFP+ cells contacting the MZ from an E19 section of E16 pCAG-GFP-electroporated (green) and E16 bromodeoxyuridine (BrdU)-labeled (red) cortex. E, The white box identifies the migrating cells at the leading edge of the GFP+ cells that are examined at higher magnification in F. The cell on the left has a clear branch (yellow arrow) approximately two cell diameters above the CP, but the cell on the right is not branched. G, Arrows identify the BrdU+ nuclei from the cells shown in F. H, A higher percentage of cells show leading process branching if the process was in the MZ as opposed to the cortex (CP) (***p < 0.001, χ2 test; n = 30 leading processes in MZ; n = 25 leading processes in CP). I, Histogram of the location of the principal branch point with respect to the CP–MZ boundary (red line, x = 0). The x-axis is in nuclear diameters (7 μm). A majority of branch points in MZ-contacting cells were found in the first two cell diameters above the CP–MZ boundary. The leading cell population (n = 55 cells) was derived from z-series from four E16 electroporated brains fixed at E19 (n = 1) or E20 (n = 3). Scale bars: D, 500 μm; G, 25 μm.
In the leading cell population, a difference in branching is observed between cells with their leading process in the MZ compared with cells with their leading process in the CP. A greater percentage of the leading processes in the MZ are branched compared with leading processes still in the CP (Fig. 2H) (80 vs 37%; p < 0.001, χ2 test). In addition, the majority of primary branch points in these processes is found within the first 14 μm (two cell diameters) of the MZ–CP boundary (Fig. 2I) regardless of the position of the cell body, suggesting that the branch point is relatively stable despite somal translocation. Similar branched structures were observed in explant studies of migrating neurons coincident with cell-body terminal translocation along the leading process (Nadarajah et al., 2001), confirming that the leading process contacts the MZ and can be transformed into a branched leading process before final somal positioning. These findings suggest that the leading process of migrating neurons is rapidly transformed into a branched process on contact with the MZ, in some cases before the soma has stopped translocation.
Normal neurite targeting requires Reelin signaling
Previous work demonstrated that the leading process of migrating neurons is blunted as a result of cellular obstruction in the developing reeler cortex (Pinto Lord et al., 1982). We undertook an analysis of the morphology of neurons in explants of the developing scrambler mouse cortex. scrambler mice (Sweet et al., 1996) lack the cytoplasmic adaptor protein Dab1 that is essential for Reelin signaling (Howell et al., 1997b; Sheldon et al., 1997; Ware et al., 1997) yet still express Reelin. Thus, the scrambler cortex presents the opportunity to determine the morphology of Reelin-insensitive cells in the presence of Reelin. Examination of apical (pial-directed) processes of viral GFP-labeled cells in explants of scrambler (−/−) or control (+/+ or +/−) embryonic cortex confirmed previous findings from the reeler mouse (Pinto Lord et al., 1982): namely, that apical processes in the −/− explants appear disorganized and imprecisely targeted. Although the majority (87%) of cells in control explants contact the Reelin expression domain (MZ), only 43% of cells in −/− explants contact the Reelin expression domain (p = 0.03, Student’s t test, single tailed; df = 4) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These results establish that Dab1 deficiency results in altered neurite morphology and targeting in the presence of Reelin but do not distinguish between the cell-autonomous and non-cell-autonomous effects of Dab1 deficiency.
Cell-autonomous function of Dab1 in neuronal migration and neurite formation
To determine the cell-autonomous function of Dab1, an RNAi protocol was used. Four RNAi constructs were initially screened, and a combination two, RVC and RVD, was identified that suppressed a Dab1::GFP fusion in HEK293 cells (Fig. 3A). Western blot analysis (Fig. 3B) showed that the Dab1::GFP fusion protein was undetectable by anti-GFP immunoprobe and faintly detectable by probing with an affinity-purified anti-Dab1 antiserum B3 (Howell et al., 1997a). The RVC+RVD mixture was combined with pCAG-GFP and then tested for suppression of endogenous Dab1 in cortical tissue, as assayed by immunocytochemistry with the B3 anti-Dab1 antiserum (Fig. 3C). An examination of randomly selected GFP+ population of cells in the RVC+RVD cultures at E20, 4 d after electroporation, showed significantly lower levels of Dab1 immunoreactivity compared with the RVN control cultures, indicating the effectiveness of RVC+RVD constructs in downregulation of endogenous Dab1 protein levels over a 4 d period (Fig. 3C).
Figure 3.

Suppression of Dab1 protein expression by RNAi directed against Dab1 mRNA. A, HEK293 cells cotransfected with pSuper.retro.neo plasmids directed against Dab1 (RVC+RVD) and with a plasmid encoding Dab1::GFP show reduced the amount of Dab1::GFP expression compared with control transfection (RVN with Dab1::GFP). A plasmid encoding the red fluorescent protein DsRed (pDsRed) was included in all transfections as a transfection efficiency control. Three independent transfections of HEK293 cell cultures were performed to test Dab1::GFP suppression by RVN, RVC, and RVD. Green to red fluorescence ratios within regions of interest was 1.05 ± 0.25 for RVN-treated cultures (n = 3) compared with 0.20 ± 0.23 for RVC+RVD-treated cultures (n = 3) (means ± SD; unpaired t test, p = 0.012; df = 4). B, Western blot analysis of whole-cell lysates from HEK293 cell transfections confirms Dab1::GFP suppression after probing with anti-Dab1 antisera (B3) or with an anti-GFP antibody. Reprobing the immunoblot with an anti-α-tubulin antibody confirms approximately equal loading of whole-cell lysates into each lane. C, Suppression of endogenous Dab1 in primary cortical cultures after electroporation of RNAi constructs with pCAG-GFP. Quantification of Dab1 immunostaining in the GFP+ populations in control (RVN) and (RVC+RVD) electroporated cell cultures. Mean pixel intensities of the cell soma are expressed in arbitrary units (au) after background correction (subtraction of the mean pixel intensity from a cell-free region of the same culture). A difference in mean pixel intensity is observed between RVN- and RVC+RVD-treated neuronal cultures (p = 0.001, unpaired t test; df = 58; n = 30 GFP+ cells in each condition). Neurons derived from three electroporated and pooled brains were quantified in each condition in C. Scale bar, 230 μm.
Cell-autonomous function of Dab1 in cell positioning
Reelin signaling is essential for appropriate positioning of migrating neurons in the developing cortex. Embryos electroporated in utero at E16 with CAG-GFP in combination with RVC+RVD or control RVN vectors were examined at E20. The position of GFP+ cells in RVC+RVD-treated brains was compared with RVN controls in two areas, cingulate cortex (Fig. 4A) and lateral neocortex (Fig. 4B). Cell position was measured from the CP–MZ boundary to the cell somata of the GFP+ labeled cells. Cells within 320 μm of the CP–MZ boundary were included in this analysis. We examined cells in the lateral neocortex, measuring the soma position of ∼60 GFP+ cells per brain. At E20, the median position of cell somata in control RVN-treated brains was 32 μm below the CP–MZ boundary (average ± SD of median positions, 32 ± 12.5 μm; n = 8 brains). In contrast, the median position of cell somata in RVC+RVD-treated embryos was significantly different (mean ± SD, 113 ± 38 μm; n = 9 brains; p < 0.0001, unpaired t test; df = 15). Examination of a smaller number of brains, in which the electroporation was targeted toward the cingulate cortex, confirmed that RVC+RVD treatment altered the median position of migrating cells in other areas of developing cortex. Whereas control RVN-treated cells in the cingulate showed a median position of 22 ± 3 μm (n = 3 brains) below the CP–MZ, RVC+RVD-treated brains showed a median position of 64 ± 5 μm (n = 3 brains; p < 0.0001, unpaired t test; df = 4).
Figure 4.
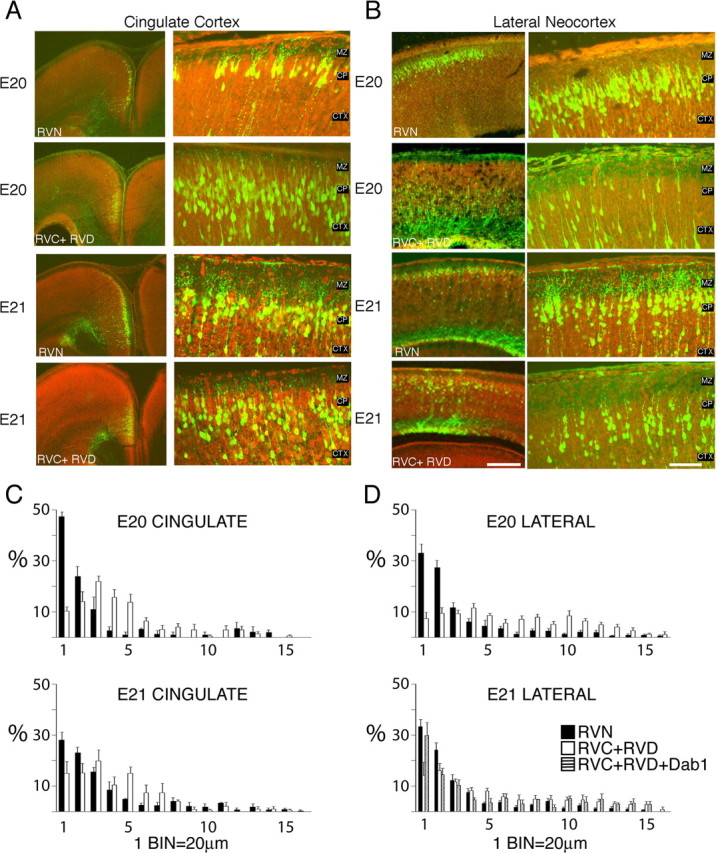
Dispersed cellular layering after treatment with RNAi directed against Dab1. Sections stained with the nucleic acid dye propidium iodide (red) from embryos electroporated on E16 and examined on E20 or E21 in the cingulate (A) or lateral neocortex (B). RVN control electroporated brains show precise layering by E20 and E21. B, Embryos electroporated with RNAi plasmids (RVC+RVD) directed against Dab1 show more cellular dispersion and fewer cells at the CP–MZ boundary. Scale bars: low-magnification images, 250 μm; high-magnification images, 50 μm. C, D, Binned histogram of the distribution of GFP+ cell bodies in the upper 320 μm of the cingulate cortex (C) and lateral neocortex (D). Control RVN electroporations (black bars) show more cells in the upper bins (bins 1–2) than RVC+RVD (white bars). Coelectroporation of a cDNA encoding full-length Dab1 (cross-hatched bars) partially rescues RVC+RVD malpositioning. A total of 48 electroporated brains were examined, six to nine brains for each condition at each time point in the lateral neocortex, two to four brains for each condition at each time point in the cingulate cortex, and four brains for the Dab1 rescue in the lateral neocortex at E21. The soma positions for a total of 2792 cells or an average of 58 cells per brain were measured. CTX, Cortex.
The median cell somata position differences observed between RVN- and RVC+RVD-treated cells might be attributable to a general developmental delay rather than a specific migration defect through the cortex. To distinguish between these possibilities, the experiment was repeated, but, in this case, the embryos were allowed to develop 1 additional day to E21. Estimates of the rate of migration in the developing cerebral cortex vary between 10 and 35 μm/h (O’Rourke et al., 1992; Nadarajah et al., 2001; Sanada et al., 2004). We reasoned that an additional 24 h of development should allow ample time for the RVC+RVD-treated cells to “catch up” the 81 μm difference in their median position compared with control. Examination of lateral neocortex at E21 suggests that the differences in RVN- vs RVC+RVD-treated cells are persistent (Fig. 4A,B). At E21, RVN-treated cells had a median position of 34 ± 14 μm (n = 9 brains) compared with a median position of 93 ± 59 μm (n = 6 brains; p = 0.012, unpaired t test; df = 13). This is a shift of 20 μm during 1 d in the RVC+RVD population but is not significantly different from the median position of the RVC+RVD on the preceding day (113 μm; p = 0.42, unpaired t test; df = 13). This suggests that the RNAi effect is persistent and cellular malpositioning does not recover with an additional day of development.
To control for potential “off-target” effects of the RNAi, a pCAG-Dab1 expression vector was included in the RVC+RVD electroporation (Fig. 4D). The median position of CAG-Dab1-rescued cells in the lateral neocortex on E21 was 54.2 ± 28.9 μm, an intermediate value between RVN-treated (34 μm) and RVC+RVD-treated (93 μm) cells and not significantly different from RVN-treated cells (p = 0.112, unpaired t test; df = 11). The ability of exogenous Dab1 to partially rescue the RNAi effect suggests that the RVC+RVD effect on cell positioning occurs primarily through downregulation of endogenous Dab1 mRNA and not by interference with other mRNAs within the migrating neuron.
RVC+RVD-treated cells appeared to arrest underneath the cell-dense CP, ∼30–50 μm below the CP–MZ boundary in both lateral neocortex and cingulate cortex (Fig. 4A,B). This qualitative assessment was further explored by binning cell positions in 20-μm-wide bins relative to the CP–MZ boundary (Fig. 4C,D). We then compared the percentage of cells in the uppermost two bins (40 μm) of cortex in both conditions. At E20, 60.3% of control cells but only 16.9% of RVC+RVD-treated cells are found in the upper 40 μm of lateral neocortex (p < 0.0001, unpaired t test; df = 15). One day later, 57.7% of control cells and 30.3% of RVC+RVD-treated cells are found in the upper 40 μm (p = 0.003, unpaired t test; df = 13). Similarly, in the cingulate cortex, the percentage of cells in the upper 40 μm is significantly different between control and RVC+RVD-treated cells at E20 (71.1 vs 24.2%; p = 0.001, unpaired t test; df = 4) and at E21 (51.2 vs 30.1%; p = 0.002, unpaired t test; df = 4). This result suggests that migration through the cell-dense CP may require Dab1 and Reelin signaling.
RVC+RVD treatment also lowers the amount of GFP+ processes in the MZ (Fig. 4A,B). To quantify the amount of neurite growth in the MZ, we measured the number of above-threshold GFP+ pixels in the MZ and normalized that value to the number of GFP+ pixels in the underlying CP in which the cell bodies are located (see Materials and Methods). At E20 and P2, RVN control sections showed an approximately twofold higher ratio of MZ GFP+ pixel area/CP GFP+ pixel area than RVC+RVD sections at both E20 (p = 0.014, unpaired t test; df = 6) and P2 (p = 0.025, unpaired t test; df = 7) (Fig. 5I). These finding suggest that a Dab1 deficiency leads to neurite deficiency in the MZ and raises the possibility that Reelin signaling is required for the initiation and/or promotion of neurite growth in the MZ.
Figure 5.
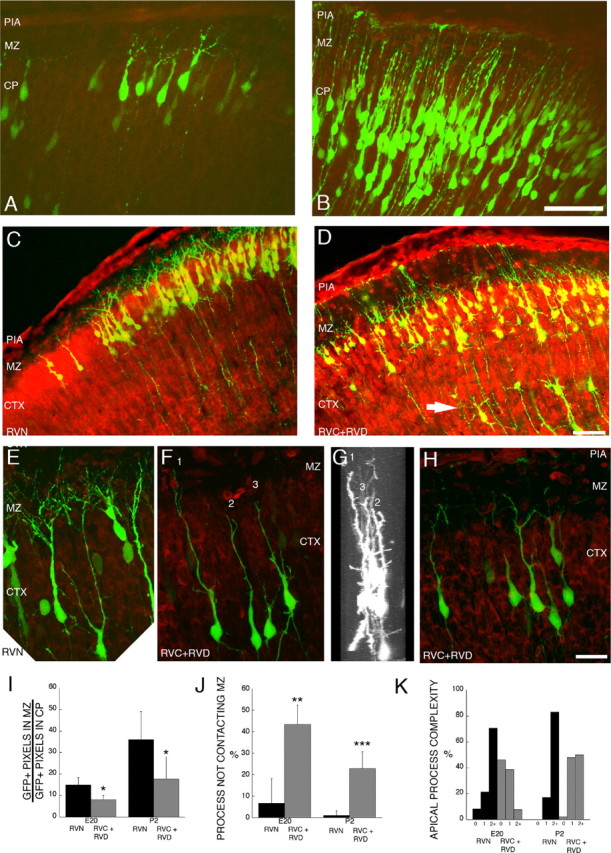
Alterations of leading process and apical dendrite morphology after treatment with RNAi directed against Dab1. Sections stained with the nucleic acid dye propidium iodide (red) from embryos electroporated at E16 and examined at E20 and P2. A, Control (RVN) neurons electroporated at E16 show branched neurites at E20. B, RVC+RVD-treated neurons show less branching in the apical process contacting the MZ. C, D, Low-magnification images of layer 2/3 cortical neurons on P2, 7 d after in utero electroporation on E16. Control RVN electroporated neurons show precise lamination and exuberant dendritic growth in the MZ on P2 (C), whereas Dab1-suppressed cells (RVC+RVD) show disrupted lamination with occasional ectopic deep cells (arrow) and sparse dendrites in the MZ (D). C–F, Flattened z-series of P2 layer 2/3 neurons revealing extensive dendrites in the RVN-treated cells (E) and stunted dendrites that either do not penetrate the MZ (cells 2 and 3) (F–H) or stunted dendrites that do not show extensive secondary and tertiary branching in the MZ (H). G, A 90° rotation of D reveals that cells 1–3 are entirely contained in the optical section and that the stunted dendrites are not attributable to sectioning artifact. Scale bars: B, D, 50 μm; H, 20 μm. I, Quantification of GFP+ pixels in the MZ at E20 and P2, after normalization (see Materials and Methods). Control (RVN) embryos showed approximately twofold more neurites in the MZ compared with RVC+RVD-treated embryos at both E20 (p = 0.014, unpaired t test; df = 6) and P2 (p = 0.025, unpaired t test; df = 7). Single sections from nine RVC+RVD embryos (n = 4 at E20; n = 5 at P2) and eight RVN embryos (n = 4 at E20; n = 4 at P2) were analyzed. J, Quantification of noncontacting apical processes after RVC+RVD treatment shows RNAi-mediated difference at both E20 (p = 0.012, unpaired t test; df = 4) and P2 (p = 0.001, unpaired t test; df = 6). K, Quantification of apical process branching complexity from unbranched (0th order) to second-order and above (2+) branching in RVN- and RVC+RVD-treated cells at E20 and P2. RVC+RVD-treated cells showed less branching than RVN-treated cells at both time points. At E20, morphological analysis was performed on 24 cells in three RVN-treated brains and 26 cells in three RVC+RVD-treated brains. At P2, morphological analysis was performed on 47 cells in five RVN-treated brains and 44 cells in three RVC+RVD-treated brains. Values in I and J are means ± SD. CTX, Cortex.
Closer examination of the morphology of RVC+RVD-treated cells showed two types of apical (pial-directed) process abnormalities that contribute to the lower amount of GFP+ processes observed in the MZ (Fig. 5A–H). A lower percentage of RVC+RVD-treated cells contacted the MZ with their apical process, and the processes were themselves less branched (Fig. 5J, K). At E20, 6.7% of control RVN cells did not contact the MZ (n = 24 cells in 3 brains) compared 43.3% of the RVC+RVD-treated cells (n = 26 cells in 3 brains; p = 0.012, unpaired t test; df = 4). Similarly, at P2, an average of 1.0% of the control RVN cells (n = 47 cells in 5 brains) did not contact the MZ compared with 22.9% of the RVC+RVD-treated cells (n = 44 cells in 3 brains; p = 0.001, unpaired t test; df = 6). Differences between control and experimental morphologies are probably not attributable to a general developmental delay caused by RNAi treatment because the RNAi-treated cells are not morphologically similar to less mature control cells, which overwhelmingly (>98%) contact the MZ with their apical processes at P2. The 43.3% (E20) and 22.9% (P2) values for the percentage of noncontacting RVC+RVD-treated cells are different from the 57.0% of noncontacting cells in the cortical explants from the scrambler (Dab1-deficient) embryos (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This difference may reflect (1) the incomplete suppression of Dab1 by RNAi and (2) possible non-cell-autonomous effects attributable to the significant cortical disorganization of the scrambler mouse explants compared with the minor disorganization of the RVC+RVD-treated cortices.
As mentioned, the branching complexity of RNAi-treated cells appeared reduced. Although it was usually not possible to unambiguously identify all of the dendritic arbors of individual cells because of the density of labeled cells, it was possible to trace the apical arbor to the first branch point and often to the second. Therefore, we quantified the neurite branching in the same sets of z-series by determining the percentage of apical processes without detectable branching (0th order), those with detectable single branch point (first order), and those with detectable higher-order branching (second order and above). At E20, RVC+RVD-treated cells showed an approximate fivefold higher percentage of 0th-order apical processes (no branch detected) (Fig. 5K) compared with control cells (46 vs 8.3%, respectively). By P2, all cells showed at least one apical branch, but only 50% of the RVC+RVD-treated cells showed higher-order branching (second order and above) compared with the majority (83%) of RVN-treated cells. The observation that many of the RNAi-treated cells had simplified dendrites that did not enter the MZ suggests that Reelin signaling may be specifically required for normal dendritic elaboration in the MZ.
Discussion
Our finding of the arrest of Dab1-deficient cells ∼40 μm beneath normal cells is consistent with the previous observation that mutant cortical cells arrest beneath wild-type cortical cells in chimeric animals composed of Dab1 (−/−) and wild-type (+/+) cells (Hammond et al., 2001). If the cell-dense CP represents a physical barrier to terminal translocation, this might contribute to the approximate inversion of cortical layers observed in the scrambler and reeler mice. In the absence of Reelin signaling, successively generated neurons may accrete beneath cell-dense layers, resulting in a cellular “traffic jam” similar to that proposed for reeler mice (Pinto Lord et al., 1982). In both models, cells that are superficial to migrating neurons obstruct further migration, but the obstructing cells need not be mutant because the wild-type CP appears to also provide an obstructive layer that can be surmounted only with appropriate levels of Reelin signaling.
The inversion and malpositioning of cells in the reeler mouse cortex is likely exacerbated by fascicles of thalamocortical afferents, termed the internal plexiform zone (IPZ) (Pinto Lord and Caviness, 1979). In reeler mice, the IPZ fibers inappropriately pass through the cortex, targeting the superficially displaced subplate. These IPZ fibers may obstruct radially migrating neurons and thus effectively “trap” a subset of migrating neurons at abnormally deep positions within the cortex (Tabata and Nakajima, 2002). Because IPZ fibers are not observed in our electroporation experiments that targeted layer 2/3-fated cells, they are unlikely to contribute to the cellular malpositioning reported here.
Previous analysis of the modes of neuronal migration in slices of embryonic cortex suggested that the terminal 30–50 μm of migration displays kinetic properties that are distinct from the intermittent (saltatory) soma movement observed in the deeper cortex (Nadarajah et al., 2001). On leading process (mean length of 30–50 μm) contact with the MZ, soma movement switches from saltatory to translocative (continuous), and the mean soma migration rate increases from 35 to 60 μm/h (Nadarajah et al., 2001). Our study identifies the final stage of migration through the upper ∼40 μm of cortex, or the approximate length of the leading process, as a critical Dab1-dependent event. This observation is consistent with a previous study of the scrambler mutant, in which inappropriate glial attachment of the migrating neuron was observed in the upper CP (Sanada et al., 2004). It is therefore possible that Reelin signaling stimulates the transition from saltatory migration to terminal translocation through the CP and that this transition in the mode of migration is required for cells to penetrate the cell-dense CP.
In addition to forming layers underneath the Reelin-expressing cells, the dendritic arbors of Dab1-expressing cells are often localized to the layer of cells containing Reelin-expressing cells. In reeler mice, these dendritic arbors are substantially stunted and disorganized (Pinto Lord et al., 1982; Tabata and Nakajima, 2002; Niu et al., 2004). The functional correlation between Reelin signaling and dendritic arborization was established in studies of cultured hippocampal neurons, in which Reelin application caused an approximate fourfold enhancement of dendritic length and an approximate threefold enhancement in dendritic branching (Niu et al., 2004). Reelin signaling also is known to affect neuronal precursor cells (Hartfuss et al., 2003; Luque et al., 2003) and radial glial cells, which in reeler mice possess short processes (Hartfuss et al., 2003) and display less branching at the pial surface (Pinto Lord et al., 1982; Hunter-Schaedle, 1997). These observations point to a common function of Reelin signaling in the establishment and/or elaboration of processes in both the neuronal precursor and the postmigratory neuron.
In our study, we present evidence that Reelin signaling has an important function in the establishment or elaboration of neurites in the intervening, developmental stage between neuronal precursor and postmigratory neurons. The simplest interpretation is that the abnormalities associated with the leading process and dendrites are secondary to a migration defect and the resultant malpositioning of the neuronal soma. Within the context of the previously mentioned cell culture studies that reported a direct effect of Reelin on radial glial processes and cultured hippocampal dendrites, a direct Reelin-dependent effect on the leading process and dendrite formation is also possible. Dendrites form preferentially in axon-rich strata such as the MZ (Pinto Lord and Caviness, 1979), and Cajal-Retzius cells display extensive axonal ramifications primarily in the MZ (Marin-Padilla, 1990). The observation that Reelin is axonally transported and secreted by the Cajal-Retzius cells (Derer et al., 2001) makes Reelin a logical candidate for an axon-derived factor that stimulates early dendritogenesis of neocortical pyramidal neurons.
The idea that the leading process is an immature dendrite finds support in previous studies of the developing cortex and cerebellum. Golgi analysis of developing neocortex suggested to Pinto Lord and Caviness (1979) that the apical dendrite of a cortical pyramidal cell may represent “a transformation of the leading migratory process of the cell.” Evidence for leading process transformation is found also in time-lapse analysis of neocortical explants; somal translocation (migration) is observed in some cells that possess a branched apical process projecting into the MZ (Nadarajah et al., 2001). Similarly, cerebellar granule cells are also thought to translocate from the external granule layer to the internal granule layer using a leading, unbranched dendrite (Ramón y Cajal, 1995; Rivas and Hatten, 1995). If the leading process is dendritic in character and responsive to dendritic growth and remodeling cues, this observation provides a potential mechanistic link between the processes of migration, lamination, and dendritogenesis. The specific actions of Reelin during migration and dendritogenesis, however, remain somewhat unclear. The leading process may respond to Reelin by detaching from its radial glial substrate, as has been previously suggested (Pinto Lord et al., 1982; Dulabon et al., 2000; Sanada et al., 2004), a phenomenon that would permit subsequent signaling events to stimulate dendritogenesis. Alternatively, Reelin signaling might stimulate dendritogenesis directly (Niu et al., 2004) or coordinate both glial de-adhesion and dendritogenesis simultaneously.
One of the most striking observations in these studies is the precision of branching in the MZ. In our analysis, the majority of principal branch points of the leading process occurred within the first 14 μm, or approximately two somata diameters, of the MZ. Cells with branched trailing processes were rarely observed, suggesting that cell bodies do not translocate or migrate through branch points and that the cell body would stop at a stabilized leading branch point. The location of the branch point one to two cell diameters above the CP may position the newly arrived cell body in a manner that is consistent with an inside-out pattern of neocortical development. These results outline a model in which the nascent dendrite has a critical role both in terminal translocation and in migration arrest (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Footnotes
E. C. Olson’s present address: Department of Neuroscience and Physiology, State University of New York, Upstate Medical University, Syracuse, NY 13210.
E.C.O. was supported by a Goldenson Berenberg Fellowship. S.K. was supported by a Helen Hay Whitney Postdoctoral Fellowship. C.A.W. was supported by National Institute of Neurological Disorders and Stroke Grant P01 NS40043. C.A.W. is an Investigator of the Howard Hughes Medical Institute. We thank members of the Walsh laboratory and Mary Beth Hatten for helpful discussions, Anjen Chenn and David Cameron for comments on this manuscript, Brian Howell for providing the affinity-purified B3 antibody, Dave Feldheim for the GFP-expressing Sindbis virus, Takahiko Matsuda and Constance Cepko for the CAG-GFP expression vector, and Masaharu Ogawa for CR50 antibody. We thank Paul Sheehe for assistance with statistical analysis of cell position and Julie Ritchie for technical support.
References
- Caviness VS Jr, Sidman RL (1973). Time of origin or corresponding cell classes in the cerebral cortex of normal and reeler mutant mice: an autoradiographic analysis. J Comp Neurol 148:141–151. [DOI] [PubMed] [Google Scholar]
- D’Apuzzo M, Mandolesi G, Reis G, Schuman EM (2001). Abundant GFP expression and LTP in hippocampal acute slices by in vivo injection of sindbis virus. J Neurophysiol 86:1037–1042. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T (1995). A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374:719–723. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T (1999). Reelin is a ligand for lipoprotein receptors. Neuron 24:471–479. [DOI] [PubMed] [Google Scholar]
- Derer P, Derer M, Goffinet A (2001). Axonal secretion of Reelin by Cajal-Retzius cells: evidence from comparison of normal and Reln(Orl) mutant mice. J Comp Neurol 440:136–143. [DOI] [PubMed] [Google Scholar]
- Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, Kreidberg JA, Anton ES (2000). Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron 27:33–44. [DOI] [PubMed] [Google Scholar]
- Falconer D (1951). Two new mutants, “trembler” and “reeler,” with neurological actions in the house mouse. J Genet 192–201. [DOI] [PubMed]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA (1999). Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 23:257–271. [DOI] [PubMed] [Google Scholar]
- Goffinet AM (1980). The cerebral cortex of the reeler mouse embryo. An electron microscopic analysis. Anat Embryol (Berl) 159:199–210. [DOI] [PubMed] [Google Scholar]
- Goffinet AM, So KF, Yamamoto M, Edwards M, Caviness VS Jr (1984). Architectonic and hodological organization of the cerebellum in reeler mutant mice. Brain Res 318:263–276. [DOI] [PubMed] [Google Scholar]
- Gonzalez JL, Russo CJ, Goldowitz D, Sweet HO, Davisson MT, Walsh CA (1997). Birthdate and cell marker analysis of scrambler: a novel mutation affecting cortical development with a reeler-like phenotype. J Neurosci 17:9204–9211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond V, Howell B, Godinho L, Tan SS (2001). disabled-1 functions cell autonomously during radial migration and cortical layering of pyramidal neurons. J Neurosci 21:8798–8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartfuss E, Forster E, Bock HH, Hack MA, Leprince P, Luque JM, Herz J, Frotscher M, Gotz M (2003). Reelin signaling directly affects radial glia morphology and biochemical maturation. Development 130:4597–4609. [DOI] [PubMed] [Google Scholar]
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J (1999). Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 24:481–489. [DOI] [PubMed] [Google Scholar]
- His W (1904). In: Die Entwicklung des Menschlichen Gehirns wahrend der estern Monate Liepzig, Germany: Hirzel.
- Hockfield S, McKay RD (1985). Identification of major cell classes in the developing mammalian nervous system. J Neurosci 5:3310–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, Martin ND, Walsh CA (2000). Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet 26:93–96. [DOI] [PubMed] [Google Scholar]
- Howell BW, Gertler FB, Cooper JA (1997a). Mouse disabled (mDab1): a Src binding protein implicated in neuronal development. EMBO J 16:121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P, Cooper JA (1997b). Neuronal position in the developing brain is regulated by mouse disabled-1. Nature 389:733–737. [DOI] [PubMed] [Google Scholar]
- Howell BW, Herrick TM, Cooper JA (1999). Reelin-induced tryosine phosphorylation of disabled 1 during neuronal positioning. Genes Dev 13:643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter-Schaedle KE (1997). Radial glial cell development and transformation are disturbed in reeler forebrain. J Neurobiol 33:459–472. [DOI] [PubMed] [Google Scholar]
- Luque JM, Morante-Oria J, Fairen A (2003). Localization of ApoER2, VLDLR and Dab1 in radial glia: groundwork for a new model of reelin action during cortical development. Brain Res Dev Brain Res 140:195–203. [DOI] [PubMed] [Google Scholar]
- Magdaleno S, Keshvara L, Curran T (2002). Rescue of ataxia and preplate splitting by ectopic expression of Reelin in reeler mice. Neuron 33:573–586. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M (1978). Dual origin of the mammalian neocortex and evolution of the cortical plate. Anat Embryol (Berl) 152:109–126. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M (1990). Three-dimensional structural organization of layer I of the human cerebral cortex: a Golgi study. J Comp Neurol 299:89–105. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL (2004). Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci USA 101:16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvilly RJ, de Diaz MO, Schonemann MD, Hooshmand F, Rosenfeld MG (2002). Transcriptional regulation of cortical neuron migration by POU domain factors. Science 295:1528–1532. [DOI] [PubMed] [Google Scholar]
- Misson JP, Edwards MA, Yamamoto M, Caviness VS Jr (1988). Identification of radial glial cells within the developing murine central nervous system: studies based upon a new immunohistochemical marker. Brain Res Dev Brain Res 44:95–108. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL (2001). Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci 4:143–150. [DOI] [PubMed] [Google Scholar]
- Neufeld EJ, Skalnik DG, Lievens PM, Orkin SH (1992). Human CCAAT displacement protein is homologous to the Drosophila homeoprotein, cut. Nat Genet 1:50–55. [DOI] [PubMed] [Google Scholar]
- Nieto M, Monuki ES, Tang H, Imitola J, Haubst N, Khoury SJ, Cunningham J, Gotz M, Walsh CA (2004). Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II-IV of the cerebral cortex. J Comp Neurol 479:168–180. [DOI] [PubMed] [Google Scholar]
- Niu S, Renfro A, Quattrocchi CC, Sheldon M, D’Arcangelo G (2004). Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron 41:71–84. [DOI] [PubMed] [Google Scholar]
- O’Rourke NA, Dailey ME, Smith SJ, McConnell SK (1992). Diverse migratory pathways in the developing cerebral cortex. Science 258:299–302. [DOI] [PubMed] [Google Scholar]
- Pinto Lord MC, Caviness VS Jr (1979). Determinants of cell shape and orientation: a comparative Golgi analysis of cell-axon interrelationships in the developing neocortex of normal and reeler mice. J Comp Neurol 187:49–69. [DOI] [PubMed] [Google Scholar]
- Pinto Lord MC, Evrard P, Caviness VS Jr (1982). Obstructed neuronal migration along radial glial fibers in the neocortex of the reeler mouse: a Golgi-EM analysis. Brain Res 256:379–393. [DOI] [PubMed] [Google Scholar]
- Ramón y Cajal S (1995). Histology of the nervous system of man and vertebrates. Vol II: Translated by N. Swanson and L. W. Swanson, Oxford: Oxford UP. [Google Scholar]
- Rivas RJ, Hatten ME (1995). Motility and cytoskeletal organization of migrating cerebellar granule neurons. J Neurosci 15:981–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanada K, Gupta A, Tsai LH (2004). Disabled-1-regulated adhesion of migrating neurons to radial glial fiber contributes to neuronal positioning during early corticogenesis. Neuron 42:197–211. [DOI] [PubMed] [Google Scholar]
- Sheldon M, Rice DS, D’Arcangelo G, Yoneshima H, Nakajima K, Mikoshiba K, Howell BW, Cooper JA, Goldowitz D, Curran T (1997). Scrambler and yotari disrupt the disabled gene and produce a reeler- like phenotype in mice. Nature 389:730–733. [DOI] [PubMed] [Google Scholar]
- Super H, Del Rio JA, Martinez A, Perez-Sust P, Soriano E (2000). Disruption of neuronal migration and radial glia in the developing cerebral cortex following ablation of Cajal-Retzius cells. Cereb Cortex 10:602–613. [DOI] [PubMed] [Google Scholar]
- Sweet HO, Bronson RT, Johnson KR, Cook SA, Davisson MT (1996). Scrambler, a new neurological mutation of the mouse with abnormalities of neuronal migration. Mamm Genome 7:798–802. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K (2001). Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience 103:865–872. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K (2002). Neurons tend to stop migration and differentiate along the cortical internal plexiform zones in the Reelin signal-deficient mice. J Neurosci Res 69:723–730. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Goto T, Miyama S, Nowakowski RS, Caviness VS Jr (1999). Sequence of neuron origin and neocortical laminar fate: relation to cell cycle of origin in the developing murine cerebral wall. J Neurosci 19:10357–10371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J (1999). Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97:689–701. [DOI] [PubMed] [Google Scholar]
- Ware ML, Fox JW, Gonzalez JL, Davis NM, Lambert de Rouvroit C, Russo CJ, Chua SC Jr, Goffinet AM, Walsh CA (1997). Aberrant splicing of a mouse disabled homolog, mdab1, in the scrambler mouse. Neuron 19:239–249. [DOI] [PubMed] [Google Scholar]